
Electrochemical Bottom-Up Synthesis of Chiral Carbon Dots from L-Proline and Their Application as Nano-Organocatalysts in a Stereoselective Aldol Reaction

 ,
, 


 and
and
Abstract
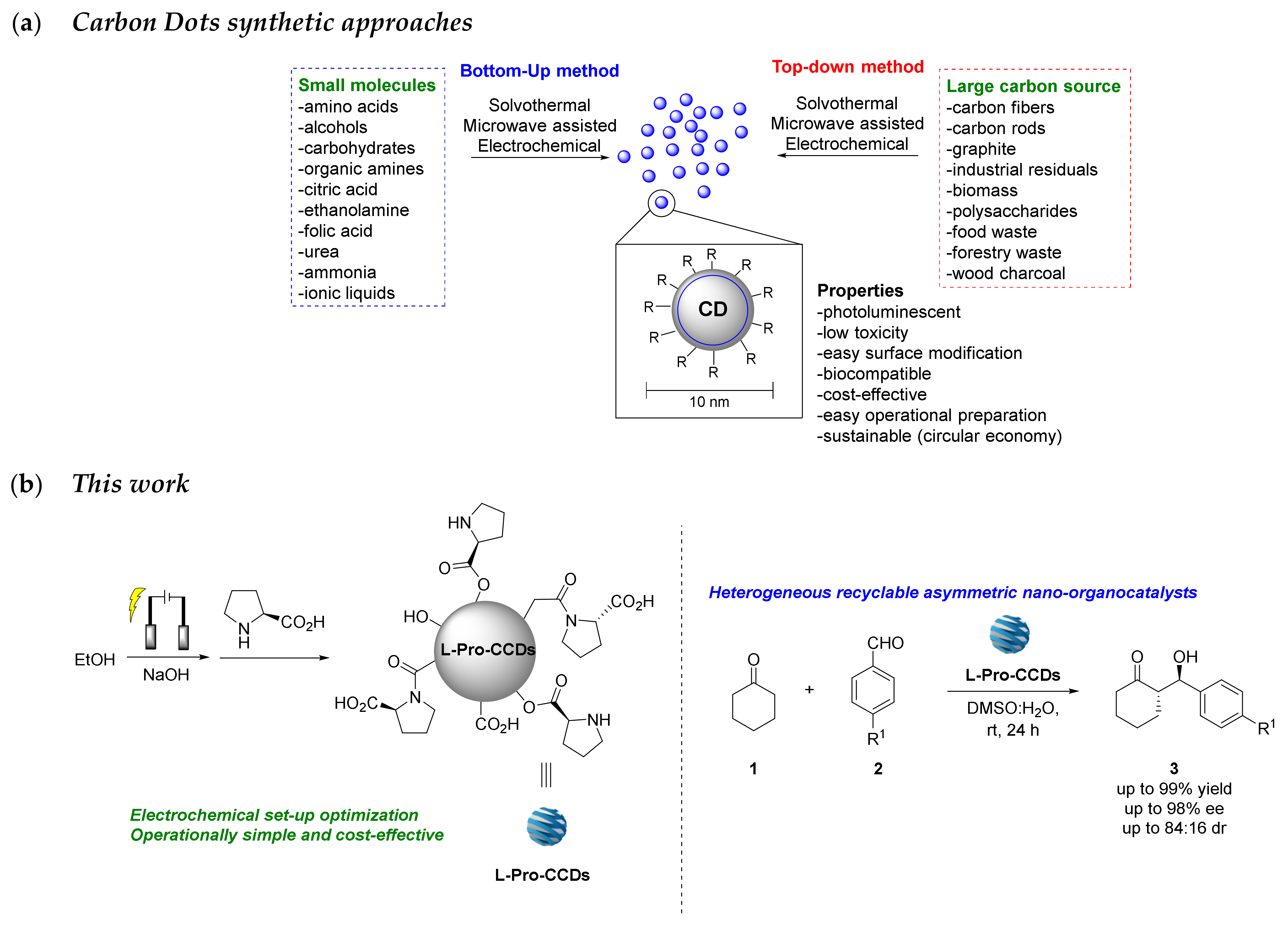
:1. Introduction
2. Results and Discussion
2.1. Optimisation of the Electrochemical CCDs Synthesis and Purification
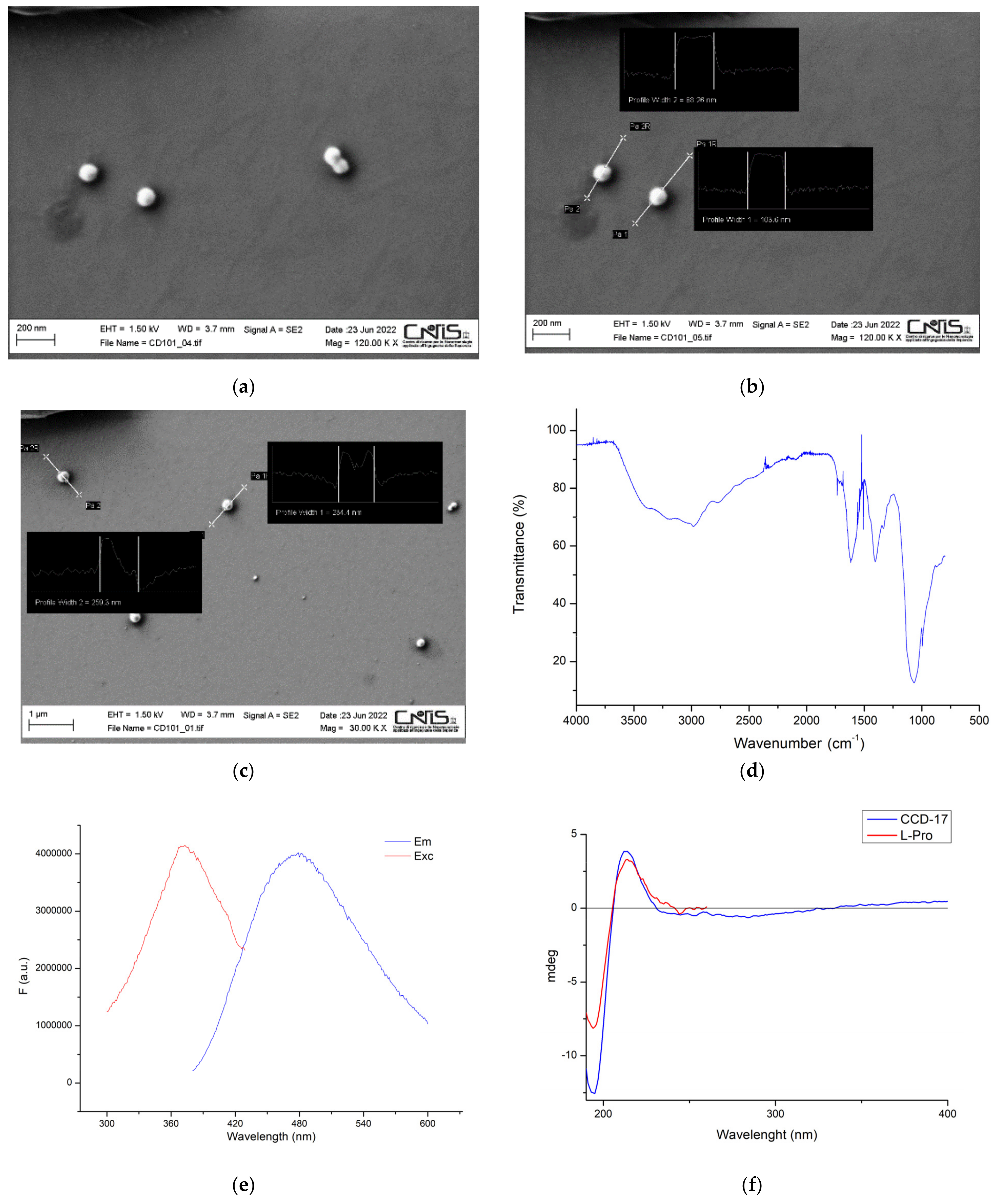
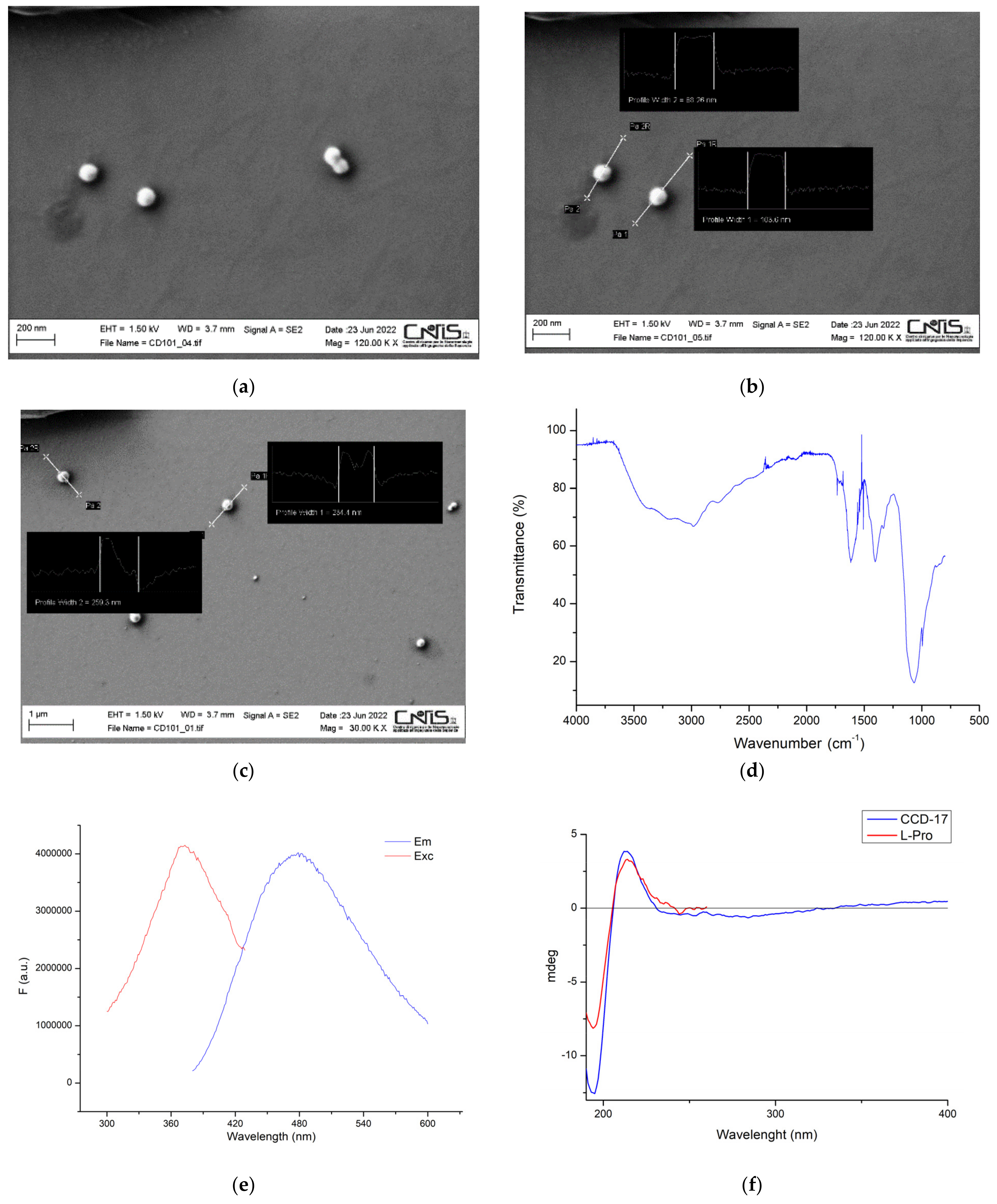
2.2. Chiral CDs Characterization
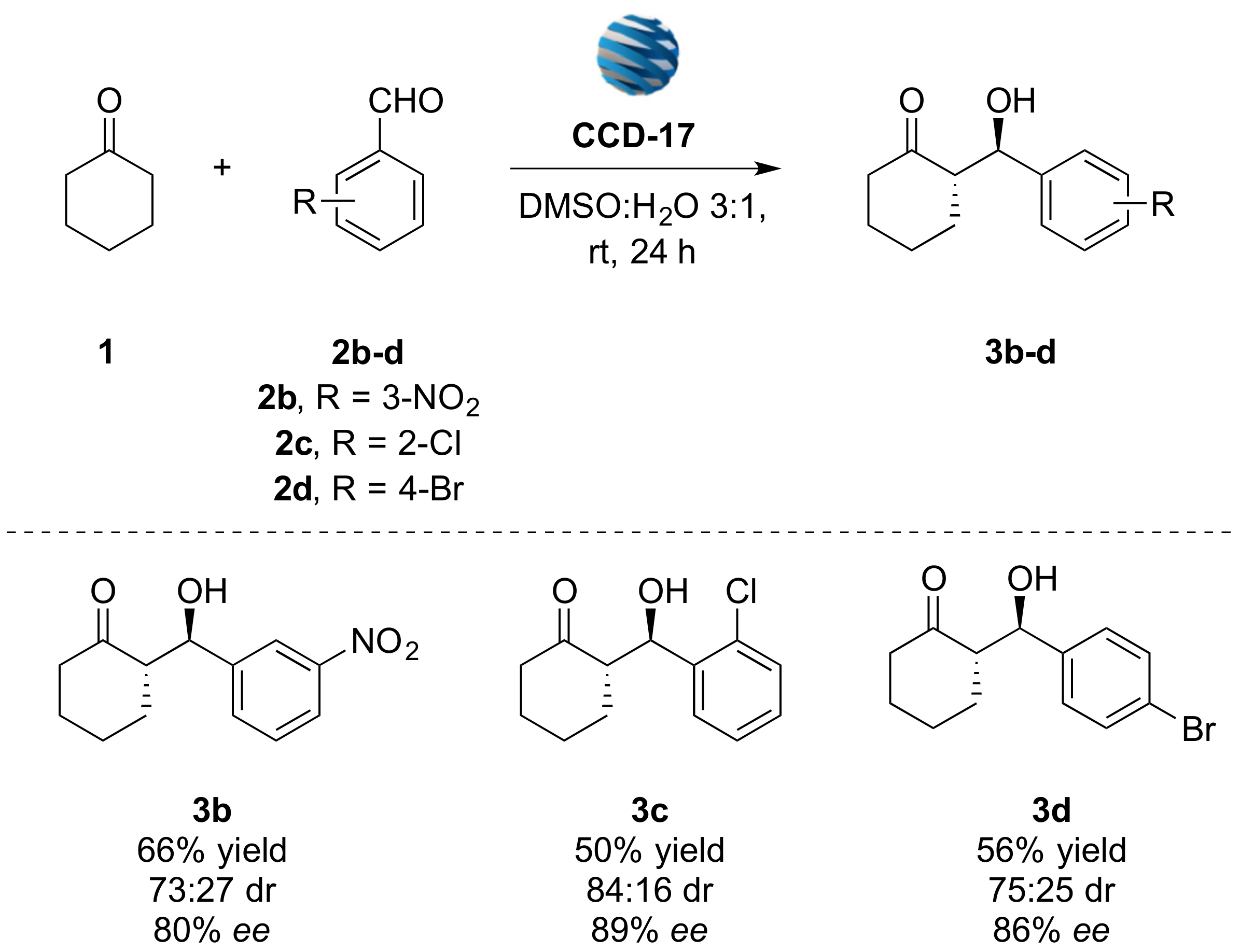
2.3. Application of CCDs as Nano-Organocatalysts
3. Materials and Methods
3.1. Experimental Procedures for the Synthesis of CCDs
3.1.1. Top-Down Electrochemical Synthesis
3.1.2. Bottom-Up Electrochemical Synthesis under Potentiostatic Conditions
3.1.3. Bottom-Up Electrochemical Synthesis under Constant Potential Difference Conditions
3.1.4. Bottom-Up Electrochemical Synthesis under Galvanostatic Conditions
3.1.5. Bottom-Up Electrochemical Synthesis under Potentiostatic Conditions from Ethanol
3.1.6. Functionalization of EtOH-Based CDs with L-Proline
3.1.7. CDs Purification
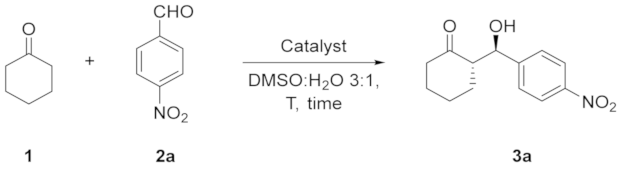
3.2. General Procedure for the Asymmetric Aldol Reaction–Synthesis of (S)-2-((R)-Hydroxy(4-nitrophenyl)methyl)cyclohexan-1-one (3a)
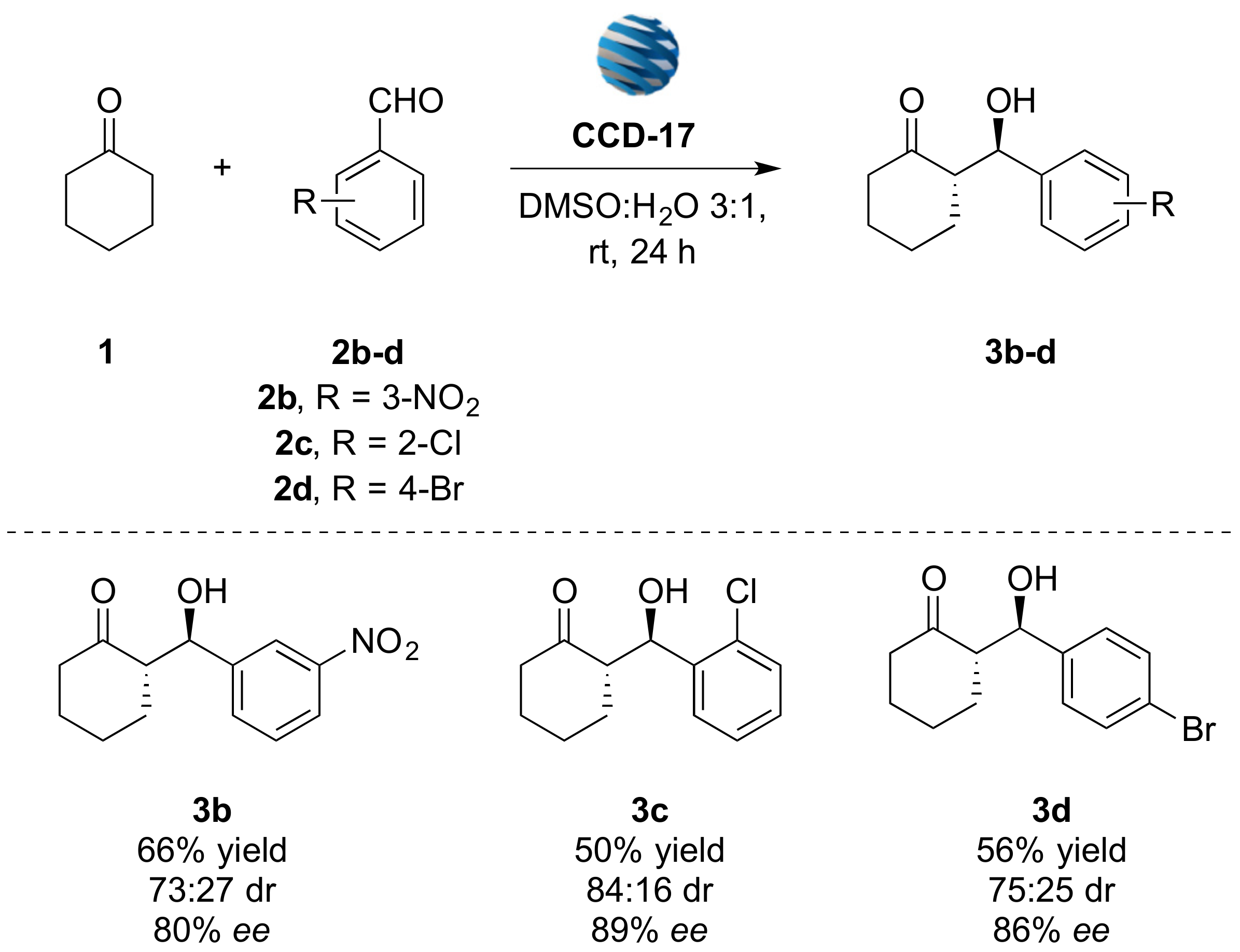
3.2.1. (S)-2-((R)-Hydroxy(3-nitrophenyl)methyl)cyclohexan-1-one (3b)
3.2.2. (S)-2-((R)-(2-chlorophenyl)(hydroxy)methyl)cyclohexan-1-one (3c)
3.2.3. (S)-2-((R)-(4-bromophenyl)(hydroxy)methyl)cyclohexan-1-one (3d)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, M.; Chen, T.; Gooding, J.J.; Liu, J. Review of Carbon and Graphene Quantum Dots for Sensing. ACS Sens. 2019, 4, 1732–1748. [Google Scholar] [CrossRef]
- Zhang, X.; Yin, J.; Yoon, J. Recent advances in development of chiral fluorescent and colorimetric sensors. Chem. Rev. 2014, 114, 4918–4959. [Google Scholar] [CrossRef]
- Zulfajri, M.; Sudewi, S.; Ismulyati, S.; Rasool, A.; Adlim, M.; Huang, G.G. Carbon Dot/Polymer Composites with Various Precursors and Their Sensing Applications: A Review. Coatings 2021, 11, 1100. [Google Scholar] [CrossRef]
- Barman, M.K.; Patra, A. Current status and prospects on chemical structure driven photoluminescence behaviour of carbon dots. J. Photochem. Photobiol. C Photochem. Rev. 2018, 37, 1–22. [Google Scholar] [CrossRef]
- Park, Y.; Kim, Y.; Chang, H.; Won, S.; Kim, H.; Kwon, W. Biocompatible nitrogen-doped carbon dots: Synthesis, characterization, and application. J. Mater. Chem. B 2020, 8, 8935–8951. [Google Scholar] [CrossRef]
- Wang, Y.; Sun, J.; He, B.; Feng, M. Synthesis and modification of biomass derived carbon dots in ionic liquids and their application: A mini review. Green Chem. Eng. 2020, 1, 94–108. [Google Scholar] [CrossRef]
- Kang, C.; Huang, Y.; Yang, H.; Yan, X.F.; Chen, Z.P. A Review of Carbon Dots Produced from Biomass Wastes. Nanomaterials 2020, 10, 2316. [Google Scholar] [CrossRef]
- Cui, L.; Ren, X.; Sun, M.; Liu, H.; Xia, L. Carbon Dots: Synthesis, Properties and Applications. Nanomaterials 2021, 11, 3419. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Zheng, D.; Wu, B.; Zhu, Y.; Zhu, L. Gel Systems Doped with Chiral Carbon Dots for Optical Combination. ACS Appl. Nano Mater. 2020, 3, 946–952. [Google Scholar] [CrossRef]
- Das, A.; Arefina, I.A.; Danilov, D.V.; Koroleva, A.V.; Zhizhin, E.V.; Parfenov, P.S.; Kuznetsova, V.A.; Ismagilov, A.O.; Litvin, A.P.; Fedorov, A.V.; et al. Chiral carbon dots based on L/D-cysteine produced via room temperature surface modification and one-pot carbonization. Nanoscale 2021, 13, 8058–8066. [Google Scholar] [CrossRef]
- Ðorđević, L.; Arcudi, F.; D’Urso, A.; Cacioppo, M.; Micali, N.; Bürgi, T.; Purrello, R.; Prato, M. Design principles of chiral carbon nanodots help convey chirality from molecular to nanoscale level. Nat. Commun. 2018, 9, 3442. [Google Scholar] [CrossRef]
- Victoria, F.; Manioudakis, J.; Zaroubi, L.; Findlay, B.; Naccache, R. Tuning residual chirality in carbon dots with anti-microbial properties. RSC Adv. 2020, 10, 32202–32210. [Google Scholar] [CrossRef] [PubMed]
- Rosso, C.; Filippini, G.; Prato, M. Carbon Dots as Nano-Organocatalysts for Synthetic Applications. ACS Catal. 2020, 10, 8090–8105. [Google Scholar] [CrossRef]
- Han, Y.; Huang, H.; Zhang, H.; Liu, Y.; Han, X.; Liu, R.; Li, H.; Kang, Z. Carbon Quantum Dots with Photoenhanced Hydrogen-Bond Catalytic Activity in Aldol Condensations. ACS Catal. 2014, 4, 781–787. [Google Scholar] [CrossRef]
- Li, H.; Sun, C.; Ali, M.; Zhou, F.; Zhang, X.; MacFarlane, D.R. Sulfated Carbon Quantum Dots as Efficient Visible-Light Switchable Acid Catalysts for Room-Temperature Ring-Opening Reactions. Angew. Chem. Int. Ed. 2015, 54, 8420–8424. [Google Scholar] [CrossRef]
- Gasperi, T.; Orsini, M.; Vetica, F.; de Figueiredo, R.M. Organocatalytic Asymmetric Multicomponent Reactions. In Multicomponent Reactions. Concepts and Applications for Design and Synthesis; Herrera, R.P., Marqués-López, E., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2015; pp. 16–71. [Google Scholar]
- Vetica, F.; de Figueiredo, R.M.; Orsini, M.; Tofani, D.; Gasperi, T. Recent Advances in Organocatalytic Cascade Reactions toward the Formation of Quaternary Stereocenters. Synthesis 2015, 47, 2139–2184. [Google Scholar] [CrossRef]
- Vetica, F.; Chauhan, P.; Dochain, S.; Enders, D. Asymmetric organocatalytic methods for the synthesis of tetrahydropyrans and their application in total synthesis. Chem. Soc. Rev. 2017, 46, 1661–1674. [Google Scholar] [CrossRef]
- Chen, X.Y.; Li, S.; Vetica, F.; Kumar, M.; Enders, D. N-Heterocyclic-Carbene-Catalyzed Domino Reactions via Two or More Activation Modes. iScience 2018, 2, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Bortolami, M.; Leonelli, F.; Feroci, M.; Vetica, F. Step economy in the Stereoselective Synthesis of Functionalized Oxindoles via Organocatalytic Domino/One-pot Reactions. Curr. Org. Chem. 2021, 25, 1321–1344. [Google Scholar] [CrossRef]
- Vetica, F.; Pandolfi, F.; Pettazzoni, L.; Leonelli, F.; Bortolami, M. Organocatalyst Design for the Stereoselective Annulation towards Bicyclic Diketones and Analogues. Symmetry 2022, 14, 355. [Google Scholar] [CrossRef]
- Enders, D.; Grondal, C.; Huettl, M.R.M. Asymmetric organocatalytic domino reactions. Angew. Chem. Int. Ed. 2007, 46, 1570–1581. [Google Scholar] [CrossRef]
- Grondal, C.; Jeanty, M.; Enders, D. Organocatalytic cascade reactions as a new tool in total synthesis. Nat. Chem. 2010, 2, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Volla, C.M.R.; Atodiresei, I.; Rueping, M. Catalytic C-C bond-forming multi-component cascade or domino reactions: Pushing the boundaries of complexity in asymmetric organocatalysis. Chem. Rev. 2014, 114, 2390–2431. [Google Scholar] [CrossRef]
- Filippini, G.; Amato, F.; Rosso, C.; Ragazzon, G.; Vega-Peñaloza, A.; Companyó, X.; Dell’Amico, L.; Bonchio, M.; Prato, M. Mapping the Surface Groups of Amine-Rich Carbon Dots Enables Covalent Catalysis in Aqueous Media. Chem 2020, 6, 3022–3037. [Google Scholar] [CrossRef]
- Zammataro, A.; Gangemi, C.M.A.; Pappalardo, A.; Toscano, R.M.; Puglisi, R.; Nicotra, G.; Fragala, M.E.; Tuccitto, N.; Sfrazzetto, G.T. Covalently functionalized carbon nanoparticles with a chiral Mn-Salen: A new nanocatalyst for enantioselective epoxidation of alkenes. Chem. Commun. 2019, 55, 5255–5258. [Google Scholar] [CrossRef]
- Liu, S.; He, Y.; Liu, Y.; Wang, S.; Jian, Y.; Li, B.; Xu, C. One-step hydrothermal synthesis of chiral carbon dots with high asymmetric catalytic activity for an enantioselective direct aldol reaction. Chem. Commun. 2021, 57, 3680–3683. [Google Scholar] [CrossRef] [PubMed]
- Pollok, D.; Waldvogel, S.R. Electro-organic synthesis—A 21st century technique. Chem. Sci. 2020, 11, 12386–12400. [Google Scholar] [CrossRef]
- Chiarotto, I.; Mattiello, L.; Feroci, M. The Electrogenerated Cyanomethyl Anion: An Old Base Still Smart. Acc. Chem. Res. 2019, 52, 3297–3308. [Google Scholar] [CrossRef]
- Cembellín, S.; Batanero, B. Organic Electrosynthesis towards Sustainability: Fundamentals and Greener Methodologies. Chem. Rec. 2021, 21, 2453–2471. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Niu, Y.; Zhang, G.; Xu, Y.; Li, J. Electrochemistry in Carbon-based Quantum Dots. Chem. Asian J. 2020, 15, 1214–1224. [Google Scholar] [CrossRef]
- Hu, L.; Li, H.; Liu, C.; Song, Y.; Zhang, M.; Huang, H.; Liu, Y.; Kang, Z. Chiral evolution of carbon dots and the tuning of laccase activity. Nanoscale 2018, 10, 2333–2340. [Google Scholar] [CrossRef]
- Niu, F.; Xu, Y.; Liu, J.; Song, Z.; Liu, M.; Liu, J. Controllable electrochemical/electroanalytical approach to generate nitrogen-doped carbon quantum dots from varied amino acids: Pinpointing the utmost quantum yield and the versatile photoluminescent and electrochemiluminescent applications. Electrochim. Acta 2017, 236, 239–251. [Google Scholar] [CrossRef]
- Dochain, S.; Vetica, F.; Puttreddy, R.; Rissanen, K.; Enders, D. Combining Organocatalysis and Lanthanide Catalysis: A Sequential One-Pot Quadruple Reaction Sequence/Hetero-Diels–Alder Asymmetric Synthesis of Functionalized Tricycles. Angew. Chem. Int. Ed. 2016, 55, 16153–16155. [Google Scholar] [CrossRef] [PubMed]
- Vetica, F.; de Figueiredo, R.M.; Cupioli, E.; Gambacorta, A.; Loreto, M.A.; Miceli, M.; Gasperi, T. First asymmetric organocatalyzed domino Friedel–Crafts/lactonization reaction in the enantioselective synthesis of the GABAB receptor modulator (S)-BHFF. Tetrahedron Lett. 2016, 57, 750–753. [Google Scholar] [CrossRef]
- Vetica, F.; Fronert, J.; Puttreddy, R.; Rissanen, K.; Enders, D. Asymmetric organocatalytic synthesis of 4-amino-isochromanones via a direct one-pot intramolecular Mannich reaction. Synthesis 2016, 48, 4451. [Google Scholar]
- Vetica, F.; Bailey, S.; Chauhan, P.; Turberg, M.; Ghaur, A.; Raabe, G.; Enders, D. Desymmetrization of Cyclopentenediones via Organocatalytic Cross-Dehydrogenative Coupling. Adv. Synth. Catal. 2017, 359, 3729. [Google Scholar] [CrossRef]
- Liu, Q.; Chen, X.-Y.; Li, S.; Vetica, F.; Raabe, G.; Enders, D. Two-Step Synthesis of α,β-Unsaturated γ-Amino Acid Esters via N-Heterocyclic Carbene Catalyzed [4 + 2] Cycloaddition of Enals and Nitroso Compounds. Synthesis 2018, 50, 127. [Google Scholar] [CrossRef]
- Vetica, F.; Chauhan, P.; Mahajan, S.; Raabe, G.; Enders, D. Asymmetric Organocatalytic Friedel–Crafts Hydroxyalkylation of Indoles Using Electrophilic Pyrazole-4,5-diones. Synthesis 2018, 50, 1039–1046. [Google Scholar] [CrossRef]
- Bortolami, M.; Magboo, F.J.P.; Petrucci, R.; Vetica, F.; Zollo, G.; Feroci, M. Electrogenerated BF3 From Tetrafluoroborate-Based Ionic Liquids: Theoretical and Experimental Studies Towards Selective Styrene Oxide Isomerization. J. Electrochem. Soc. 2021, 168, 115501. [Google Scholar] [CrossRef]
- Bortolami, M.; Mattiello, L.; Scarano, V.; Vetica, F.; Feroci, M. In Situ Anodically Oxidized BMIm-BF4: A Safe and Recyclable BF3 Source. J. Org. Chem. 2021, 86, 16151–16157. [Google Scholar] [CrossRef]
- Vetica, F.; Bortolami, M.; Petrucci, R.; Rocco, D.; Feroci, M. Electrogenerated NHCs in Organic Synthesis: Ionic Liquids vs Organic Solvents Effects. Chem. Rec. 2021, 21, 2130–2147. [Google Scholar] [CrossRef] [PubMed]
- Pandolfi, F.; Chiarotto, I.; Mattiello, L.; Rocco, D.; Feroci, M. Cathodic Reduction of Caffeine: Synthesis of an Amino-Functionalized Imidazole from a Biobased Reagent. Synlett 2019, 30, 1215–1218. [Google Scholar] [CrossRef]
- Pandolfi, F.; Chiarotto, I.; Rocco, D.; Feroci, M. Electrogenerated superoxide anion induced oxidative amidation of benzoin. Electrochim. Acta 2017, 254, 358–367. [Google Scholar] [CrossRef]
- Scarano, V.; Bortolami, M.; Pandolfi, F.; Petrucci, R.; Rocco, D.; Zollo, G.; Feroci, M. Reaction of Electrogenerated Cyanomethyl Anion with Cyclohexylisocyanate: Synthesis of N-(cyclohexylcarbamoyl)acetamide. An Unexpected Product. J. Electrochem. Soc. 2020, 167, 155514. [Google Scholar] [CrossRef]
- Li, W.; Yu, C.; Tan, X.; Wang, Z.; Qiu, J. Electric-Field-Triggered Graphene Production: From Fundamental Energy Applications to Perspectives. Acc. Mater. Res. 2022, 3, 175–186. [Google Scholar] [CrossRef]
- Deng, J.; Lu, Q.; Mi, N.; Li, H.; Liu, M.; Xu, M.; Tan, L.; Xie, Q.; Zhang, Y.; Yao, S. Electrochemical Synthesis of Carbon Nanodots Directly from Alcohols. Chem. Eur. J. 2014, 20, 4993–4999. [Google Scholar] [CrossRef]
- Miao, P.; Tang, Y.; Han, K.; Wang, B. Facile synthesis of carbon nanodots from ethanol and their application in ferric(iii) ion assay. J. Mater. Chem. A 2015, 3, 15068–15073. [Google Scholar] [CrossRef]
- Canevari, T.C.; Nakamura, M.; Cincotto, F.H.; de Melo, F.M.; Toma, H.E. High performance electrochemical sensors for dopamine and epinephrine using nanocrystalline carbon quantum dots obtained under controlled chronoamperometric conditions. Electrochim. Acta 2016, 209, 464–470. [Google Scholar] [CrossRef]
- Im, H.; Noh, S.; Shim, J.H. Spontaneous formation of core-shell silver-copper oxide by carbon dot-mediated reduction for enhanced oxygen electrocatalysis. Electrochim. Acta 2020, 329, 135172. [Google Scholar] [CrossRef]
- Niu, F.; Xu, Y.; Liu, M.; Sun, J.; Guo, P.; Liu, J. Bottom-up electrochemical preparation of solid-state carbon nanodots directly from nitriles/ionic liquids using carbon-free electrodes and the applications in specific ferric ion detection and cell imaging. Nanoscale 2016, 8, 5470–5477. [Google Scholar] [CrossRef]
- Hou, Y.; Lu, Q.; Deng, J.; Li, H.; Zhang, Y. One-pot electrochemical synthesis of functionalized fluorescent carbon dots and their selective sensing for mercury ion. Anal. Chim. Acta 2015, 866, 69–74. [Google Scholar] [CrossRef] [PubMed]
- An, Q.; Lin, Q.; Huang, X.; Zhou, R.; Guo, X.; Xu, W.; Wang, S.; Xu, D.; Chang, H.-T. Electrochemical synthesis of carbon dots with a Stokes shift of 309 nm for sensing of Fe3+ and ascorbic acid. Dye. Pigment. 2021, 185, 108878. [Google Scholar] [CrossRef]
- Lee, Y.-S.; Hu, C.-C.; Chiu, T.-C. Electrochemical synthesis of fluorescent carbon dots for the selective detection of chlortetracycline. J. Environ. Chem. Eng. 2022, 10, 107413. [Google Scholar] [CrossRef]
- Wang, C.-I.; Wu, W.-C.; Periasamy, A.P.; Chang, H.-T. Electrochemical synthesis of photoluminescent carbon nanodots from glycine for highly sensitive detection of hemoglobin. Green Chem. 2014, 16, 2509–2514. [Google Scholar] [CrossRef]
- Zhang, M.; Wang, H.; Wang, B.; Ma, Y.; Huang, H.; Liu, Y.; Shao, M.; Yao, B.; Kang, Z. Maltase Decorated by Chiral Carbon Dots with Inhibited Enzyme Activity for Glucose Level Control. Small 2019, 15, 1901512. [Google Scholar] [CrossRef]
- Chen, C.-Y.; Tsai, Y.-H.; Chang, C.-W. Evaluation of the dialysis time required for carbon dots by HPLC and the properties of carbon dots after HPLC fractionation. New J. Chem. 2019, 43, 6153–6159. [Google Scholar] [CrossRef]
- Arcudi, F.; Dordevic, L.; Prato, M. Synthesis, Separation, and Characterization of Small and Highly Fluorescent Nitrogen-Doped Carbon NanoDots. Angew. Chem. Int. Ed. 2016, 55, 2107–2112. [Google Scholar] [CrossRef]
- Wang, B.; Song, H.; Tang, Z.; Yang, B.; Lu, S. Ethanol-derived white emissive carbon dots: The formation process investigation and multi-color/white LEDs preparation. Nano Res. 2022, 15, 942–949. [Google Scholar] [CrossRef]
- Penhoat, M.; Barbry, D.; Rolando, C. Direct asymmetric aldol reaction co-catalyzed by l-proline and group 12 elements Lewis acids in the presence of water. Tetrahedron Lett. 2011, 52, 159–162. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| CCDs | Starting Materials | Approach | H2O [mL] | Electrodes +/−/Ref | I/E/ΔE | Time [h] | Q [C] | F/mol | CCDs [mg] |
|---|---|---|---|---|---|---|---|---|---|
| CCD-1(a) | L-Pro (250 mg)/NaOH (200 mg) | Top-down | 25 | C/C/- (b) | 15 mA | 72 | 3888 | 18.5 | 5.0 |
| CCD-2(a) | L-Pro (250 mg)/NaOH (200 mg) | Top-down | 10 | C/C/- (b) | 20 mA | 76 | 5472 | 26.1 | 2.1 |
| CCD-3(c) | L-Pro (200 mg)/NH3 (3 M) | Top-down | 5 | C/Pt/- (d) | 50 mA | 4.6 | 828 | 4.9 | 2.5 |
| CCD-4(a) | L-Pro (575 mg)/NH3 (3 M) | Bottom-up | 10 | Pt/Pt/Ag-AgCl (e) | +3 V | 2 | 864 | 1.8 | 1.6 |
| CCD-5(a) | L-Pro (575 mg)/NH3 (3 M) | Bottom-up | 10 | Pt/Pt/Ag-AgCl (e) | +3 V | 4 | 1555 | 3.2 | 2.0 |
| CCD-6(a) | L-Pro (288 mg)/NH3 (3 M) | Bottom-up | 5 | Pt/Pt/- (e) | 150 mA | 7 | 3780 | 15.7 | 2.4 |
| CCD-7(a) | L-Pro (200 mg)/NH3 (3 M) | Bottom-up | 5 | Pt/Pt/- (e) | 150 mA | 3 | 1620 | 9.6 | 3.8 |
| CCD-8(c) | L-Pro (200 mg)/NH3 (3 M) | Bottom-up | 5 | Pt/Pt/- (e) | 150 mA | 3 | 1620 | 9.6 | 2.6 |
| CCD-9(f) | L-Pro (200 mg)/NH3 (3 M) | Bottom-up | 5 | Pt/Pt/- (e) | 150 mA | 3 | 1620 | 9.6 | 7.2 |
| CCD-10(a) | L-Pro (200 mg)/NH3 (3 M) | Bottom-up | 5 | Pt/Pt/- (e) | ΔE = 12 V | 0.5 | 825 | 4.9 | 1.7 |
| CCD-11(c) | L-Pro (200 mg)/NH3 (3 M) | Bottom-up | 5 | Pt/Pt/- (e) | ΔE = 12 V | 0.5 | 851 | 5.0 | 2.5 |
| CCD-12(c) | L-Pro (200 mg)/NH3 (3 M) | Bottom-up | 5 | Pt/Pt/- (e) | ΔE = 10 V | 0.7 | 992 | 5.9 | 4.7 |
| CCD-13(c) | L-Pro (200 mg)/NH3 (3 M) | Bottom-up | 5 | Pt/Pt/- (e) | ΔE = 8 V | 1.5 | 1704 | 10.1 | 22 |
| CCD-14(c) | L-Pro (200 mg)/NH3 (3 M) | Bottom-up | 5 | Pt/Pt/- (e) | ΔE = 6 V | 5 | 4153 | 24.7 | 46 |
| CD-15(f) | EtOH (10 mL)/NaOH (110 mg) | Bottom-up | 1 | Pt/Pt/Hg2Cl2 (e) | + 3 V | 5 | 300 | 10 |
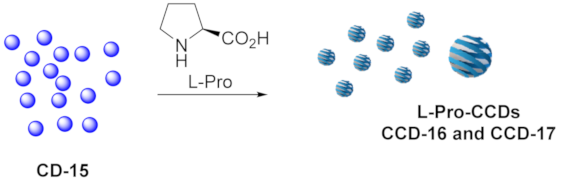
| CCDs | CD-15 [mg] | Approach | H2O [mL] | Reagents | Reaction Conditions |
|---|---|---|---|---|---|
| CCD-16(a) | 5.5 | Adsorption | 1 | L-Pro (55 mg) | 30 min sonication + 2 h stirring at rt |
| CCD-17(a,b) | 5.5 | Functionalization | 5 | L-Pro (55 mg) H2SO4 conc. (35 µL) | 2 h reflux |
| Entry (a) | Catalyst | 1/2 [mmol] | T [°C] (b) | Time [h] | Yield [%] (b) | anti/syn(b) | ee [%] (c) |
|---|---|---|---|---|---|---|---|
| 1 | L-Pro (20 mol%) | 5/0.5 | rt | 24 | 99 | 93/7 | 96 |
| 2 [27] | Solvothermal CCDs (D-proline and citric acid) | 5/0.5 | rt | 24 | 98 | 98/2 | −71 |
| 3 [27] | Solvothermal CCDs (only D-proline) | 5/0.5 | rt | 24 | 58 | 97/3 | −81 |
| 4 | CCD-2 (top-down) | 5/0.5 | rt | 72 | 60 | 51/49 | 0 |
| 5 | CCD-2 (top-down) | 5/0.5 | rt | 162 | 69 | 42/58 | 0 |
| 6 | CCD-4 (potentiostatic +3 V) | 5/0.5 | rt | 144 | traces | - | - |
| 7 (d) | CCD-7 (galvanostatic 150 mA) | 5/0.5 | rt | 24 | traces | 70/30 | 34 |
| 8 (d) | CCD-7 | 5/0.5 | 40 | 24 | 7 | 56/44 | 12 |
| 9 (d) | CCD-7 | 5/0.5 | rt | 168 | 16 | 68/42 | 12 |
| 10 | CCD-10 (12 V) | 5/0.5 | rt | 168 | 15 | 74/26 | 40 |
| 11 (d) | CCD-12 (10 V) | 5/0.5 | rt | 24 | 10 | 70/30 | 48 |
| 12 (d) | CCD-12 | 5/0.5 | rt | 168 | 28 | 70/30 | 50 |
| 13 | CCD-13 (8V) | 5/0.5 | rt | 24 | 15 | 70/30 | 42 |
| 14(d) | CCD-13 (8 V) | 5/0.5 | rt | 72 | 60 | 59/41 | 40 |
| 15(d) | CCD-13 | 5/0.5 | rt | 72 | 35 | 59/41 | 21 |
| 16(d) | CCD-13 | 5/0.5 | rt | 72 | 5 | 59/41 | - |
| 17(e) | CCD-13 + benzoic acid | 5/0.5 | rt | 96 | traces | - | - |
| 18 | CCD-7 (1000 MWCO dialysis) | 5/0.5 | rt | 72 | 22 | 50/50 | 49 |
| 19 | CCD-9 (500 MWCO dialysis) | 5/0.5 | rt | 24 | 85 | 48/52 | 26 |
| 20(d) | CCD-16 (EtOH-derived with L-Pro adsorbed) | 5/0.5 | rt | 24 | 94 | 84/16 | 98 |
| 21(d) | CCD-16 | 5/0.5 | rt | 24 | 78 | 78/22 | 80 |
| 22(d) | CCD-16 | 5/0.5 | rt | 48 | traces | - | - |
| 23(d) | CCD-17 (EtOH-based derivatised with L-Pro) | 5/0.5 | rt | 24 | 83 | 83/17 | 96 |
| 24(d) | CCD-17 | 5/0.5 | rt | 24 | 102 | 84/16 | 98 |
| 25(d) | CCD-17 | 5/0.5 | rt | 24 | 57 | 81/19 | 96 |
| 26(f) | CCD-17 | 5/0.5 | rt | 24 | 0 | - | - |
| 27(f) | L-Pro (20% mol) | 5/0.5 | rt | 24 | 0 | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bortolami, M.; Bogles, I.I.; Bombelli, C.; Pandolfi, F.; Feroci, M.; Vetica, F. Electrochemical Bottom-Up Synthesis of Chiral Carbon Dots from L-Proline and Their Application as Nano-Organocatalysts in a Stereoselective Aldol Reaction. Molecules 2022, 27, 5150. https://doi.org/10.3390/molecules27165150
Bortolami M, Bogles II, Bombelli C, Pandolfi F, Feroci M, Vetica F. Electrochemical Bottom-Up Synthesis of Chiral Carbon Dots from L-Proline and Their Application as Nano-Organocatalysts in a Stereoselective Aldol Reaction. Molecules. 2022; 27(16):5150. https://doi.org/10.3390/molecules27165150
Chicago/Turabian StyleBortolami, Martina, Ingrid Izabela Bogles, Cecilia Bombelli, Fabiana Pandolfi, Marta Feroci, and Fabrizio Vetica. 2022. "Electrochemical Bottom-Up Synthesis of Chiral Carbon Dots from L-Proline and Their Application as Nano-Organocatalysts in a Stereoselective Aldol Reaction" Molecules 27, no. 16: 5150. https://doi.org/10.3390/molecules27165150
APA StyleBortolami, M., Bogles, I. I., Bombelli, C., Pandolfi, F., Feroci, M., & Vetica, F. (2022). Electrochemical Bottom-Up Synthesis of Chiral Carbon Dots from L-Proline and Their Application as Nano-Organocatalysts in a Stereoselective Aldol Reaction. Molecules, 27(16), 5150. https://doi.org/10.3390/molecules27165150