Ligand Binding Prediction Using Protein Structure Graphs and Residual Graph Attention Networks

 , ,
, ,
Abstract
:1. Introduction
- (i)
- We propose an effective graph-Siamese-like network, PSG-BAR, to simultaneously operate on structural graphs of proteins and molecular graphs of drugs for binding affinity prediction.
- (ii)
- We introduce an attention-based readout strategy to generate graph-level embedding for proteins. These attention scores are shown to correlate with some known physical properties of binding sites. We call this learning component the interaction attention module.
2. Methods
2.1. Dataset
2.2. Protein and Ligand Graph Construction
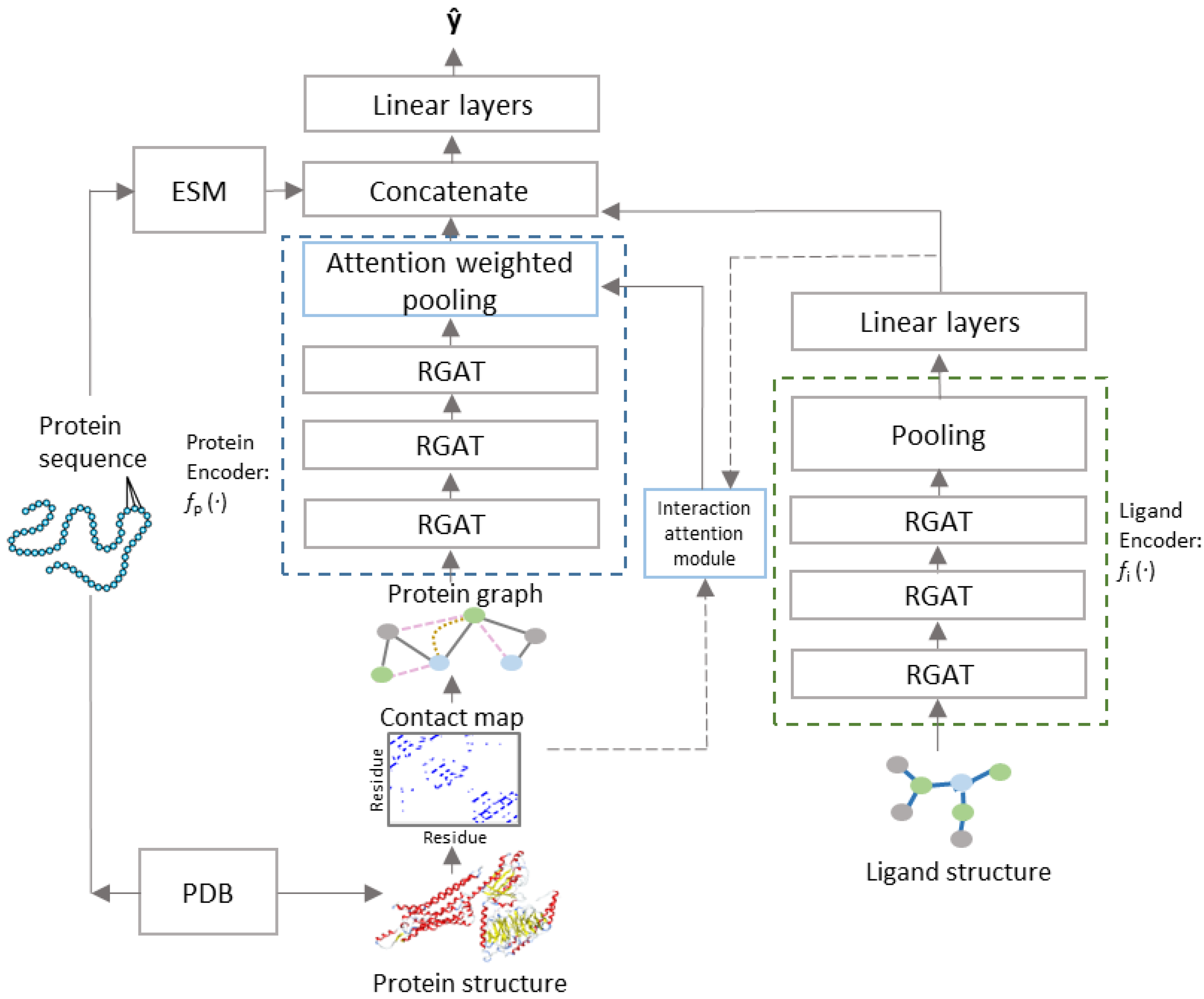
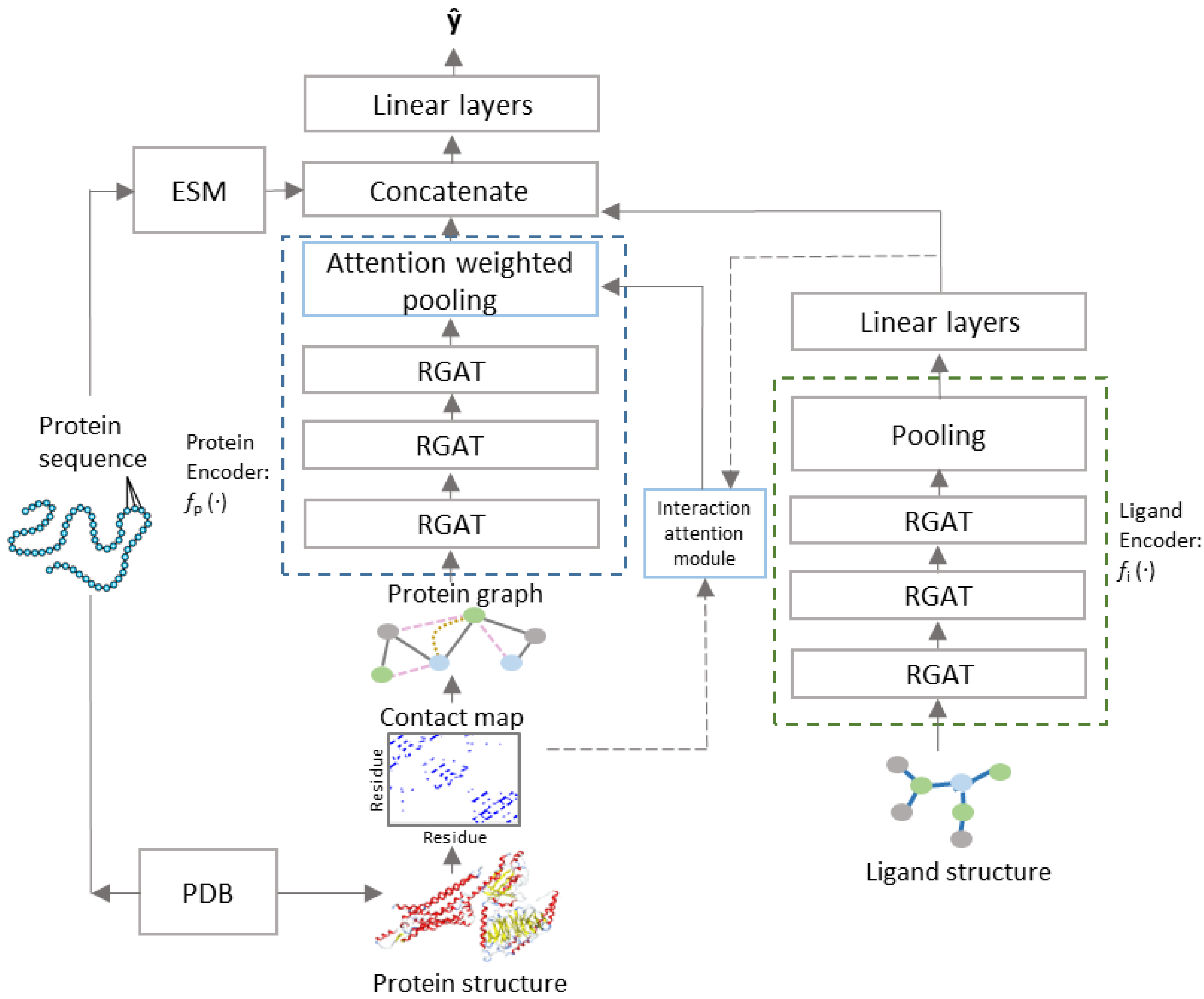
2.3. Model Architecture
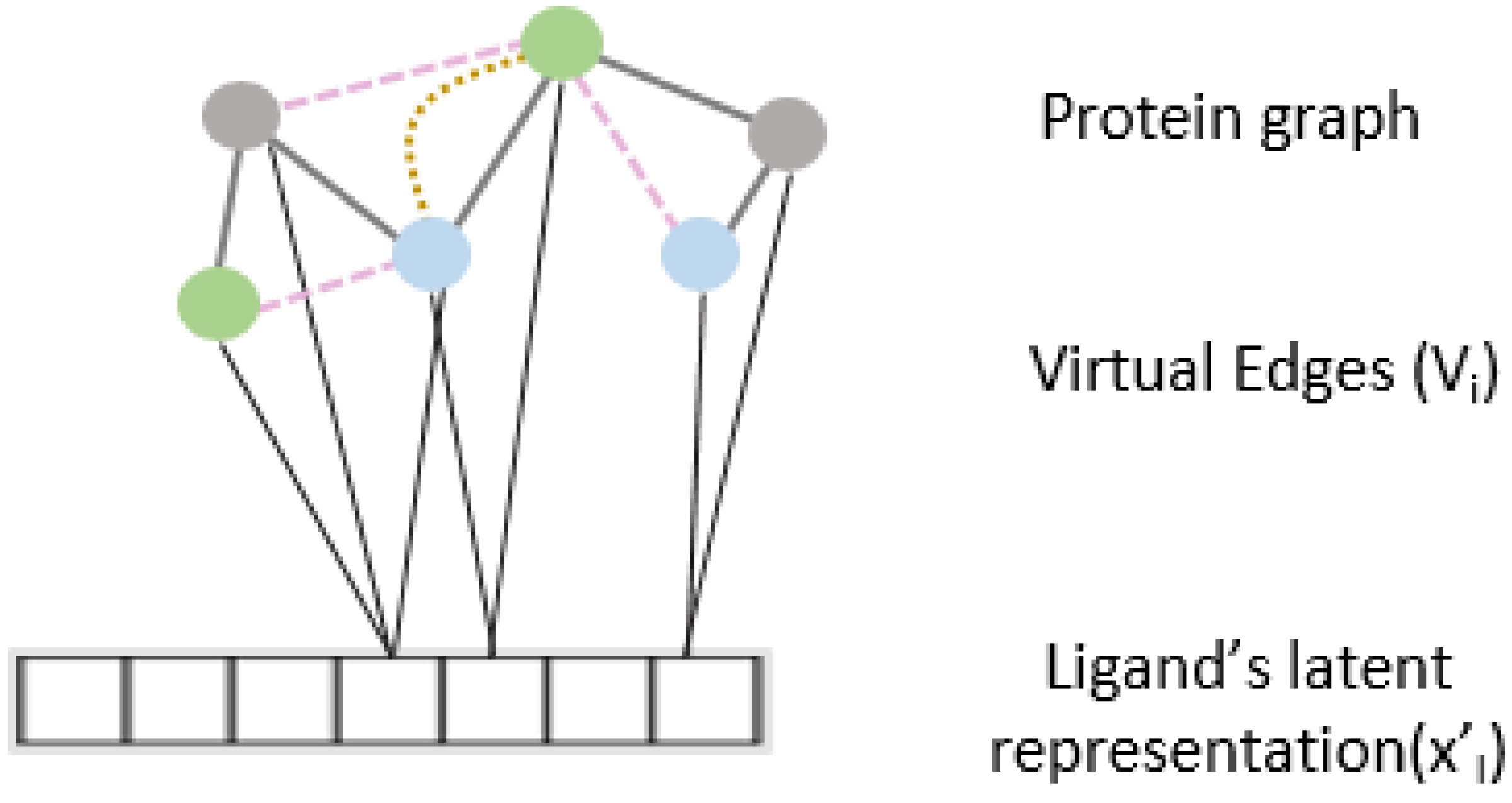
2.4. Interaction Attention Module
2.5. Training and Hyperparameter Tuning
3. Results
3.1. Binding Affinity Prediction Results
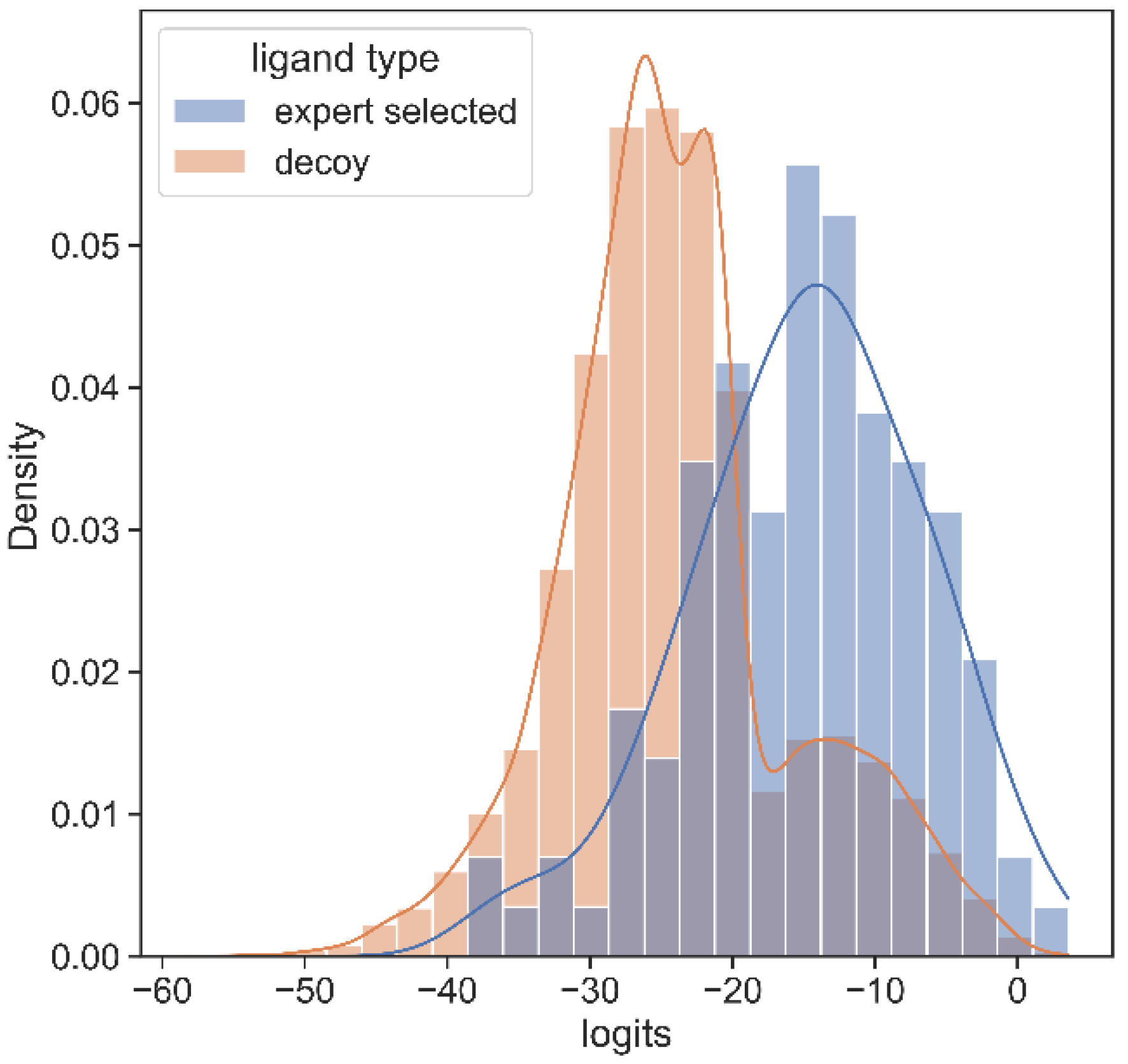
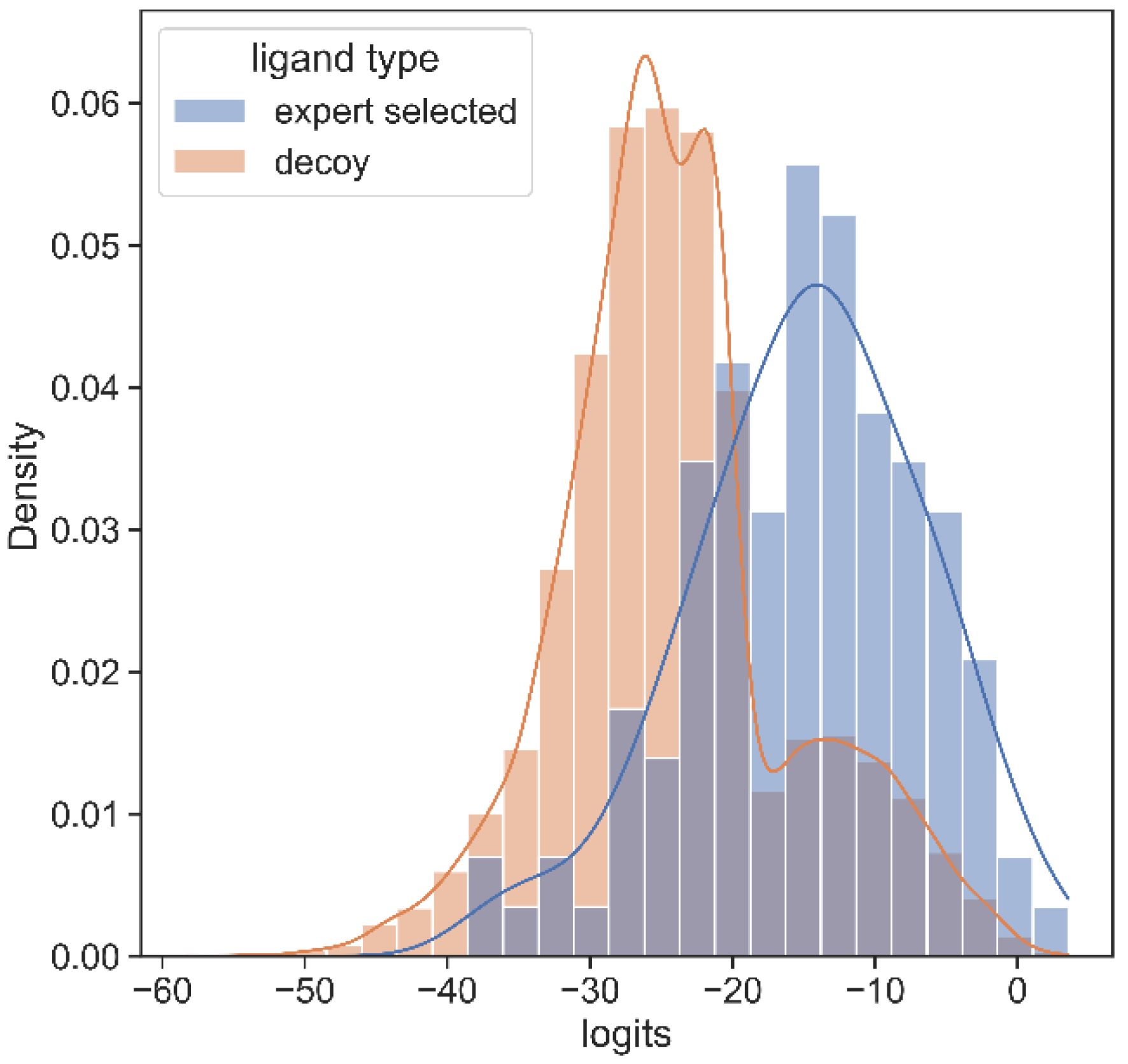
3.2. SARS Inhibitor Prediction
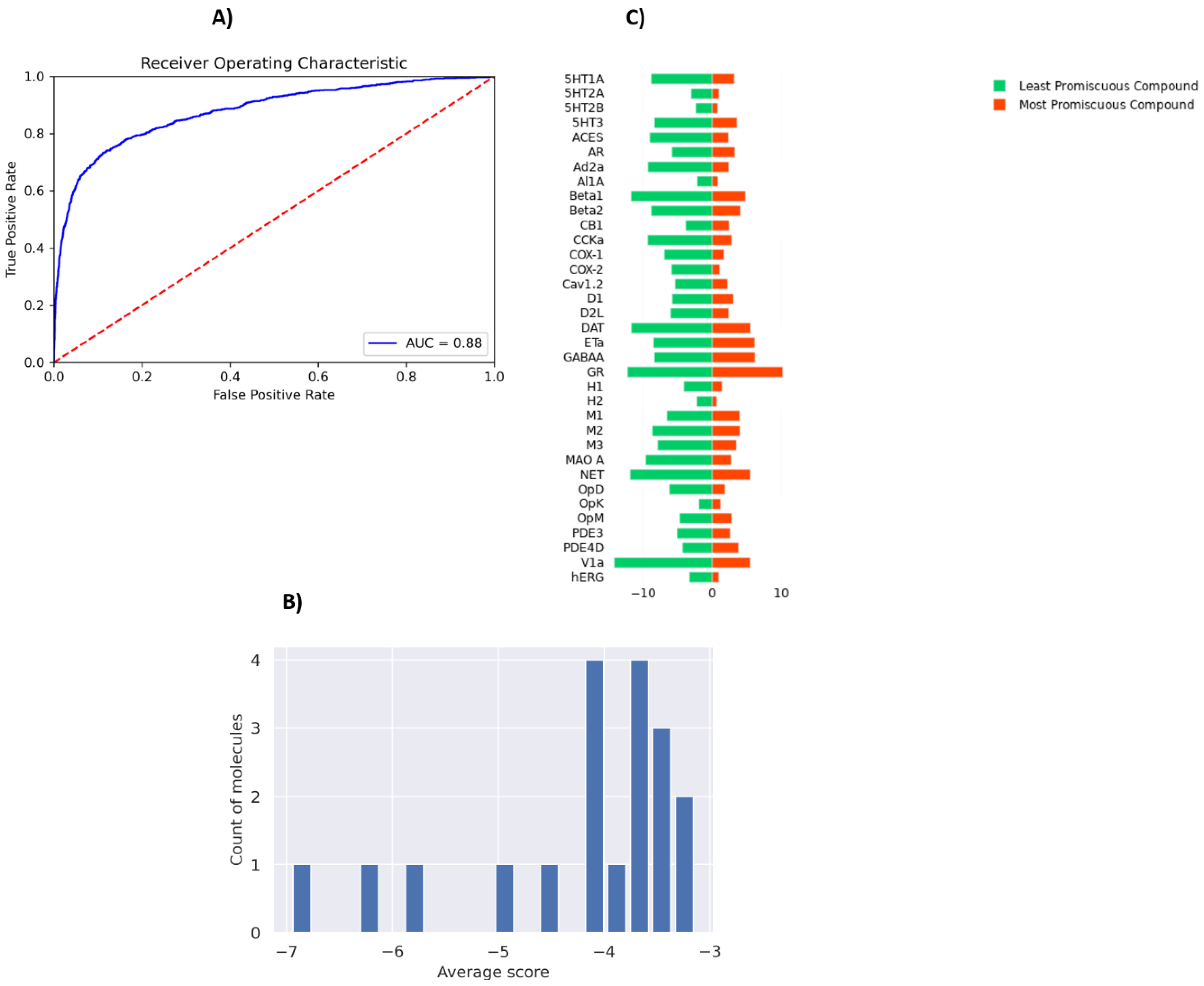
3.3. SARS CoV-2 MPro Experimental Validation
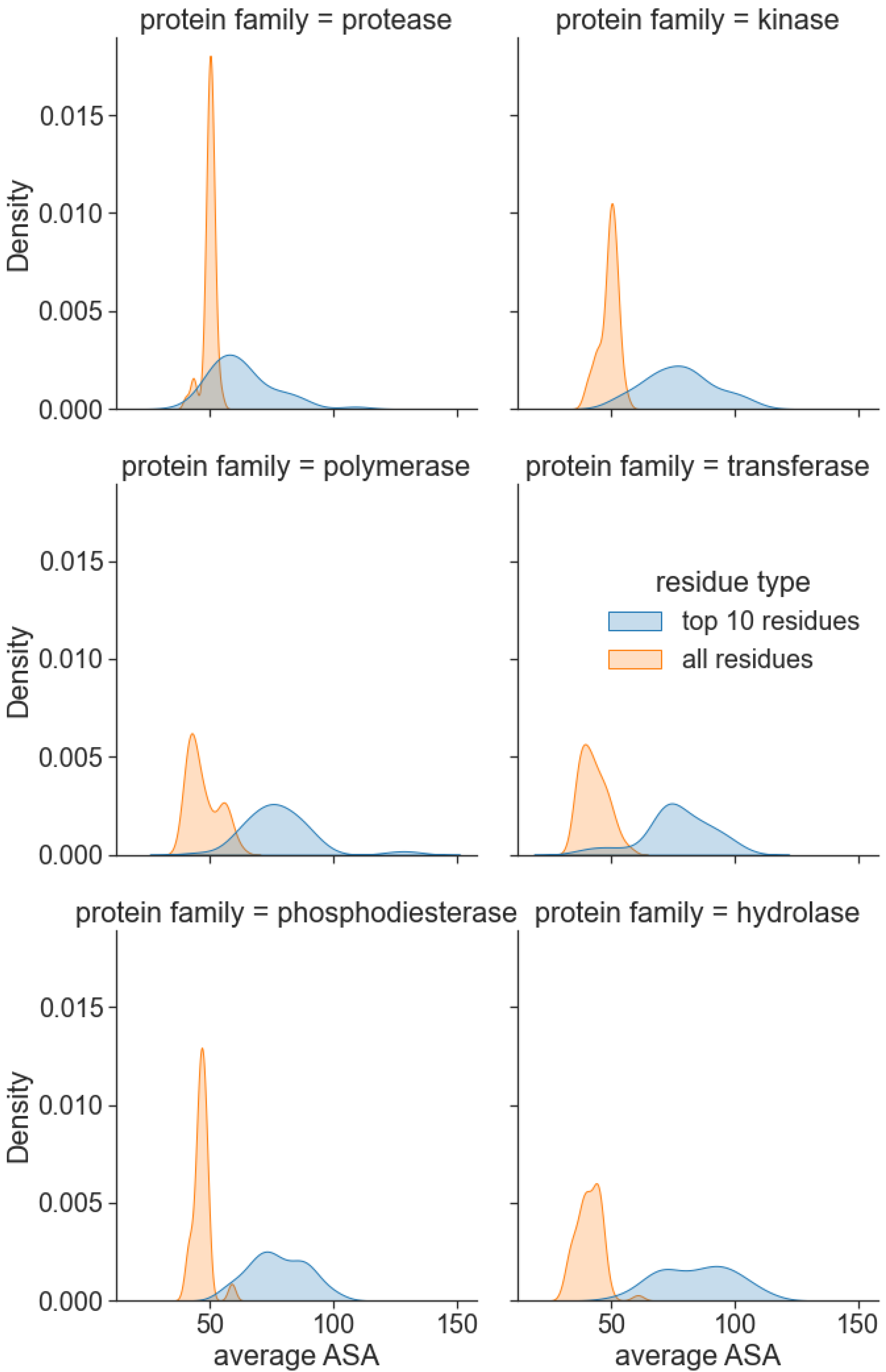
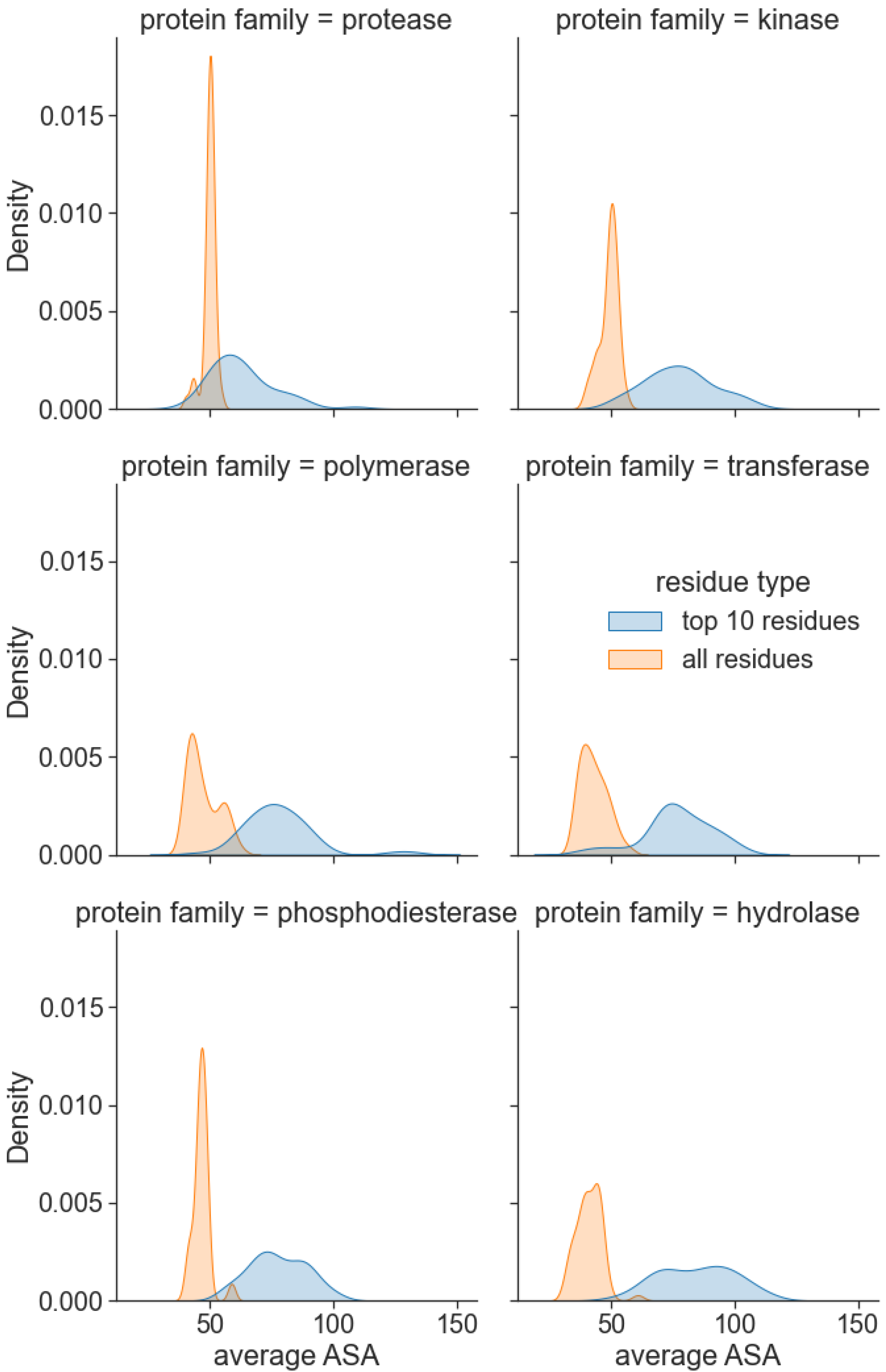
3.4. Attention Centrality
3.5. Drug Promiscuity
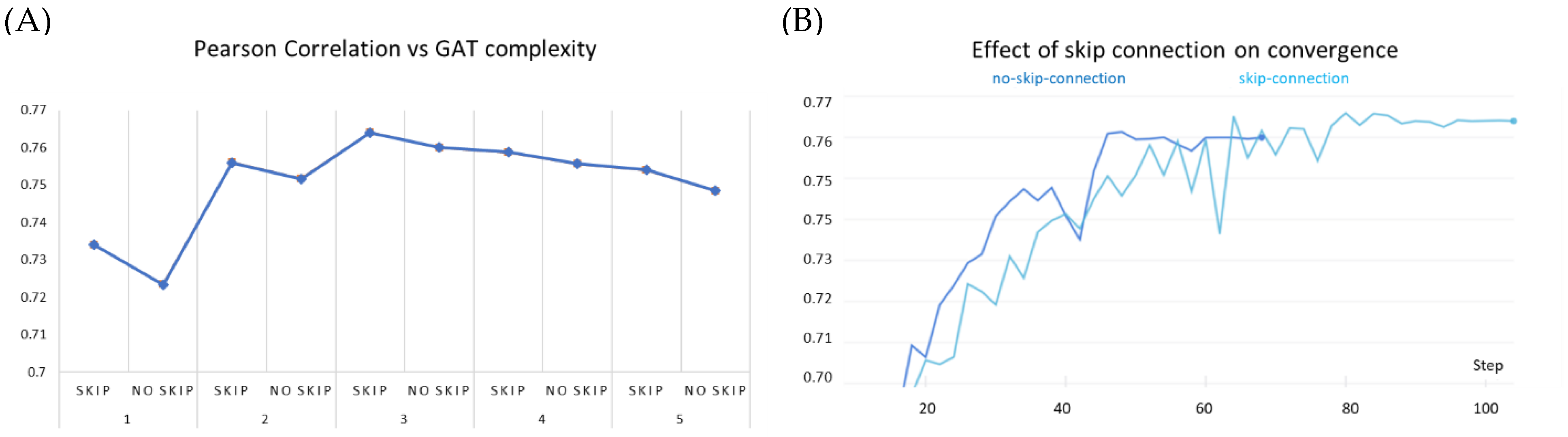
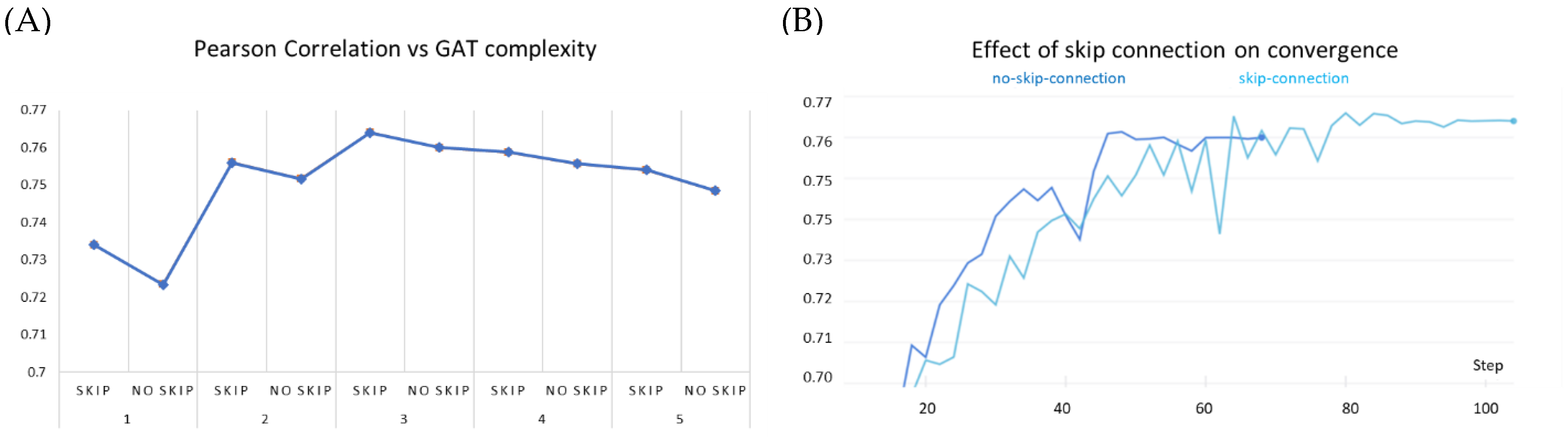
3.6. Ablation Studies
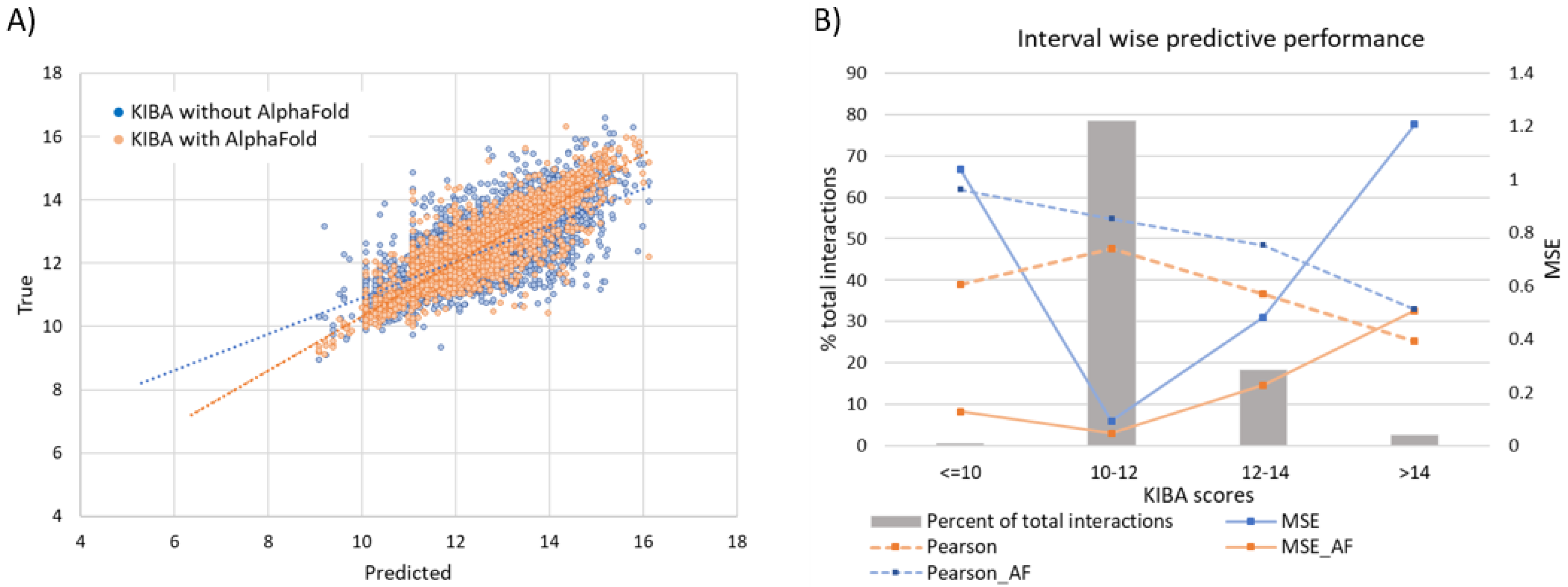
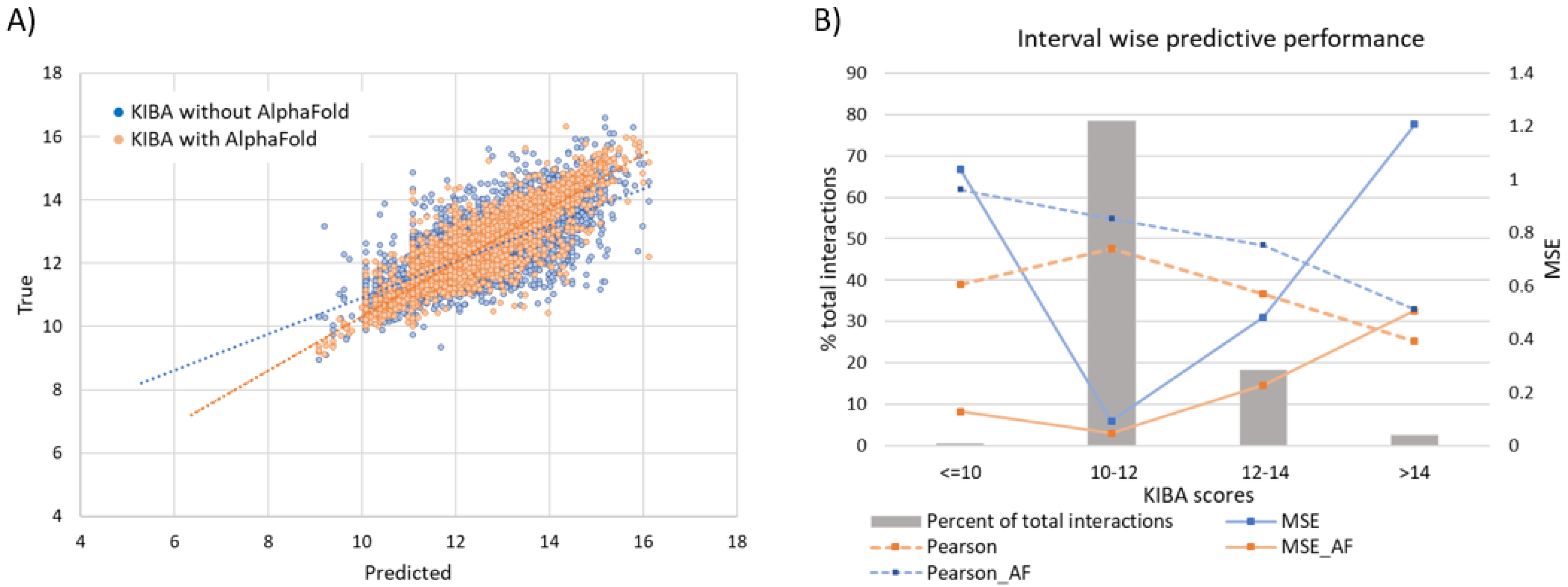
3.6.1. Effect of Augmenting KIBA Dataset with AlphaFold Structures
3.6.2. Effect of Secondary Structure Features of Proteins
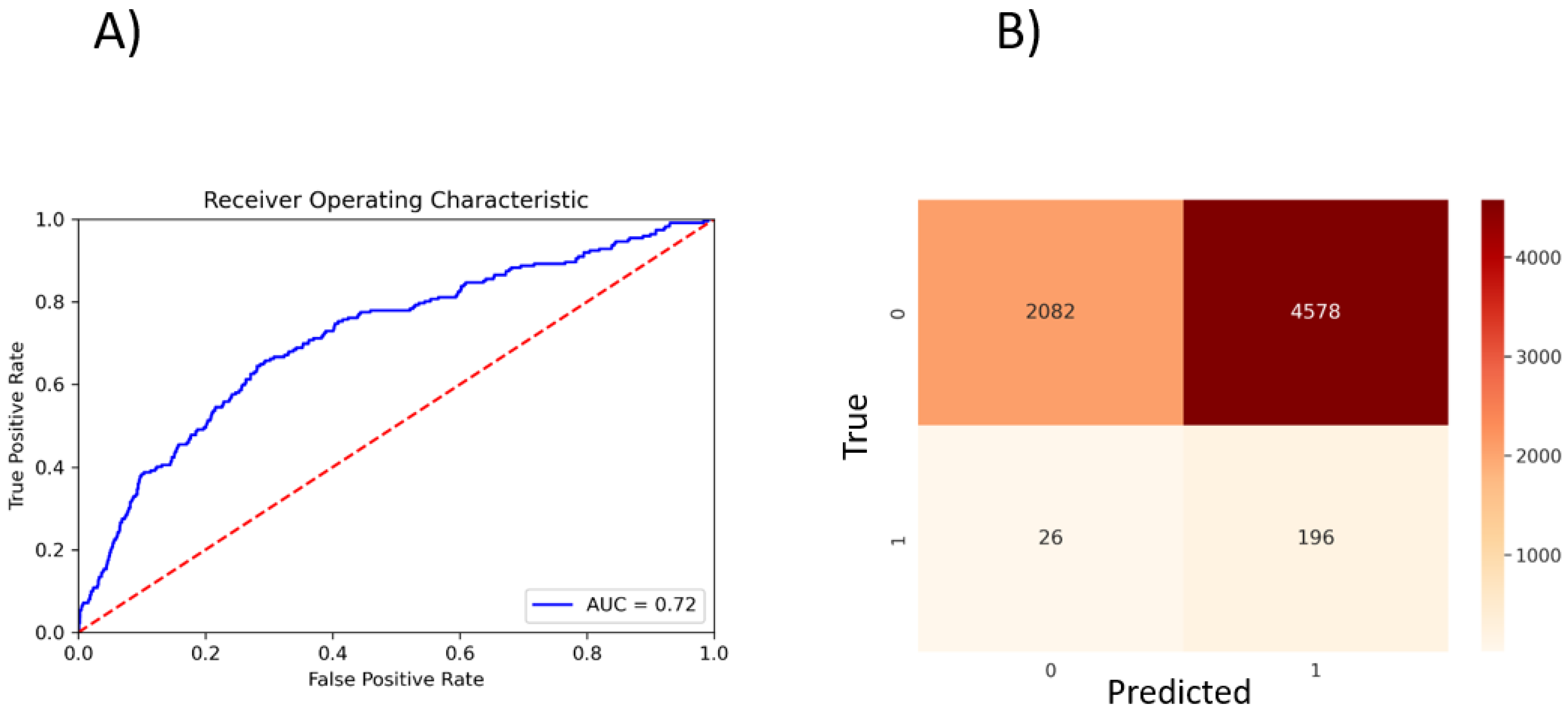
3.7. Error Analysis of Prediction of Effective Binders
4. Discussion and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Yang, X.; Kui, L.; Tang, M.; Li, D.; Wei, K.; Chen, W.; Miao, J.; Dong, Y. High-throughput transcriptome profiling in drug and biomarker discovery. Front. Genet. 2020, 11, 19. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.M.; Mytelka, D.S.; Dunwiddie, C.T.; Persinger, C.C.; Munos, B.H.; Lindborg, S.R.; Schacht, A.L. How to improve R&D productivity: The pharmaceutical industry’s grand challenge. Nat. Rev. Drug Discov. 2010, 9, 203–214. [Google Scholar] [PubMed]
- Dara, S.; Dhamercherla, S.; Jadav, S.S.; Babu, C.M.; Ahsan, M.J. Machine Learning in Drug Discovery: A Review. Artif. Intell. Rev. 2022, 55, 1947–1999. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.; Cao, S.; Su, P.-C.; Patel, R.; Shah, D.; Chokshi, H.B.; Szukala, R.; Johnson, M.E.; Hevener, K.E. Hit identification and optimization in virtual screening: Practical recommendations based on a critical literature analysis: Miniperspective. J. Med. Chem. 2013, 56, 6560–6572. [Google Scholar] [CrossRef]
- Medina-Franco, J. Grand challenges of computer-aided drug design: The road ahead. Front. Drug Discov. 2021, 1, 728551. [Google Scholar]
- Maia, E.H.B.; Assis, L.C.; De Oliveira, T.A.; Da Silva, A.M.; Taranto, A.G. Structure-based virtual screening: From classical to artificial intelligence. Front. Chem. 2020, 8, 343. [Google Scholar] [CrossRef]
- Gentile, F.; Agrawal, V.; Hsing, M.; Ton, A.-T.; Ban, F.; Norinder, U.; Gleave, M.E.; Cherkasov, A. Deep Docking: A Deep Learning Platform for Augmentation of Structure Based Drug Discovery. ACS Cent. Sci. 2020, 6, 939–949. [Google Scholar] [CrossRef]
- Bender, B.J.; Gahbauer, S.; Luttens, A.; Lyu, J.; Webb, C.M.; Stein, R.M.; Fink, E.A.; Balius, T.E.; Carlsson, J.; Irwin, J.J.; et al. A practical guide to large-scale docking. Nat. Protoc. 2021, 16, 4799–4832. [Google Scholar] [CrossRef]
- Wójcikowski, M.; Ballester, P.J.; Siedlecki, P. Performance of machine-learning scoring functions in structure-based virtual screening. Sci. Rep. 2017, 7, 46710. [Google Scholar] [CrossRef]
- Irwin, J.J.; Tang, K.G.; Young, J.; Dandarchuluun, C.; Wong, B.R.; Khurelbaatar, M.; Moroz, Y.S.; Mayfield, J.; Sayle, R.A. ZINC20—A free ultralarge-scale chemical database for ligand discovery. J. Chem. Inf. Model. 2020, 60, 6065–6073. [Google Scholar] [CrossRef]
- Jones, D.; Kim, H.; Zhang, X.; Zemla, A.; Stevenson, G.; Bennett, W.F.D.; Kirshner, D.; Wong, S.E.; Lightstone, F.C.; Allen, J.E. Improved Protein–Ligand Binding Affinity Prediction with Structure-Based Deep Fusion Inference. J. Chem. Inf. Model. 2021, 61, 1583–1592. [Google Scholar] [CrossRef] [PubMed]
- Pandey, M.; Fernandez, M.; Gentile, F.; Isayev, O.; Tropsha, A.; Stern, A.C.; Cherkasov, A. The transformational role of GPU computing and deep learning in drug discovery. Nat. Mach. Intell. 2022, 4, 211–221. [Google Scholar] [CrossRef]
- Kim, J.; Park, S.; Min, D.; Kim, W. Comprehensive Survey of Recent Drug Discovery Using Deep Learning. Int. J. Mol. Sci. 2021, 22, 9983. [Google Scholar] [CrossRef] [PubMed]
- Cortés-Ciriano, I.; Ain, Q.U.; Subramanian, V.; Lenselink, E.B.; Méndez-Lucio, O.; IJzerman, A.P.; Wohlfahrt, G.; Prusis, P.; Malliavin, T.E.; van Westen, G.J. Polypharmacology modelling using proteochemometrics (PCM): Recent methodological developments, applications to target families, and future prospects. MedChemComm 2015, 6, 24–50. [Google Scholar] [CrossRef]
- Gao, K.; Nguyen, D.D.; Sresht, V.; Mathiowetz, A.M.; Tu, M.; Wei, G.-W. Are 2D fingerprints still valuable for drug discovery? Phys. Chem. Chem. Phys. 2020, 22, 8373–8390. [Google Scholar] [CrossRef]
- Tetko, I.V.; Karpov, P.; Van Deursen, R.; Godin, G. State-of-the-art augmented NLP transformer models for direct and single-step retrosynthesis. Nat. Commun. 2020, 11, 5575. [Google Scholar] [CrossRef]
- Wen, M.; Zhang, Z.; Niu, S.; Sha, H.; Yang, R.; Yun, Y.; Lu, H. Deep-learning-based drug–target interaction prediction. J. Proteome Res. 2017, 16, 1401–1409. [Google Scholar] [CrossRef]
- Tian, Q.; Bilgic, B.; Fan, Q.; Liao, C.; Ngamsombat, C.; Hu, Y.; Witzel, T.; Setsompop, K.; Polimeni, J.R.; Huang, S.Y. DeepDTI: High-fidelity six-direction diffusion tensor imaging using deep learning. NeuroImage 2020, 219, 117017. [Google Scholar] [CrossRef]
- Öztürk, H.; Özgür, A.; Ozkirimli, E. DeepDTA: Deep drug–target binding affinity prediction. Bioinformatics 2018, 34, i821–i829. [Google Scholar] [CrossRef]
- Lee, I.; Keum, J.; Nam, H. DeepConv-DTI: Prediction of drug-target interactions via deep learning with convolution on protein sequences. PLoS Comput. Biol. 2019, 15, e1007129. [Google Scholar] [CrossRef]
- Cherkasov, A.; Muratov, E.N.; Fourches, D.; Varnek, A.; Baskin, I.I.; Cronin, M.; Dearden, J.; Gramatica, P.; Martin, Y.C.; Todeschini, R. QSAR modeling: Where have you been? Where are you going to? J. Med. Chem. 2014, 57, 4977–5010. [Google Scholar] [CrossRef] [PubMed]
- Hilpert, K.; Fjell, C.D.; Cherkasov, A. Short linear cationic antimicrobial peptides: Screening, optimizing, and prediction. In Peptide-Based Drug Design; Springer: Berlin/Heidelberg, Germany, 2008; pp. 127–159. [Google Scholar]
- Cherkasov, A.; Shi, Z.; Fallahi, M.; Hammond, G.L. Successful in silico discovery of novel nonsteroidal ligands for human sex hormone binding globulin. J. Med. Chem. 2005, 48, 3203–3213. [Google Scholar] [CrossRef] [PubMed]
- Cherkasov, A. Inductive QSAR descriptors. Distinguishing compounds with antibacterial activity by artificial neural networks. Int. J. Mol. Sci. 2005, 6, 63–86. [Google Scholar] [CrossRef]
- Liu, Z.; Li, Y.; Han, L.; Li, J.; Liu, J.; Zhao, Z.; Nie, W.; Liu, Y.; Wang, R. PDB-wide collection of binding data: Current status of the PDBbind database. Bioinformatics 2015, 31, 405–412. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Jiménez, J.; Skalic, M.; Martinez-Rosell, G.; De Fabritiis, G. K deep: Protein–ligand absolute binding affinity prediction via 3d-convolutional neural networks. J. Chem. Inf. Model. 2018, 58, 287–296. [Google Scholar] [CrossRef]
- Stepniewska-Dziubinska, M.M.; Zielenkiewicz, P.; Siedlecki, P. Development and evaluation of a deep learning model for protein–ligand binding affinity prediction. Bioinformatics 2018, 34, 3666–3674. [Google Scholar] [CrossRef]
- Lim, J.; Ryu, S.; Park, K.; Choe, Y.J.; Ham, J.; Kim, W.Y. Predicting Drug–Target Interaction Using a Novel Graph Neural Network with 3D Structure-Embedded Graph Representation. J. Chem. Inf. Model. 2019, 59, 3981–3988. [Google Scholar] [CrossRef]
- Nguyen, T.M.; Nguyen, T.; Le, T.M.; Tran, T. GEFA: Early fusion approach in drug-target affinity prediction. IEEE/ACM Trans. Comput. Biol. Bioinform. 2022, 19, 718–728. [Google Scholar] [CrossRef]
- Son, J.; Kim, D. Development of a graph convolutional neural network model for efficient prediction of protein-ligand binding affinities. PLoS ONE 2021, 16, e0249404. [Google Scholar] [CrossRef]
- Jiang, M.; Li, Z.; Zhang, S.; Wang, S.; Wang, X.; Yuan, Q.; Wei, Z. Drug–target affinity prediction using graph neural network and contact maps. RSC Adv. 2020, 10, 20701–20712. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Szwajda, A.; Shakyawar, S.; Xu, T.; Hintsanen, P.; Wennerberg, K.; Aittokallio, T. Making sense of large-scale kinase inhibitor bioactivity data sets: A comparative and integrative analysis. J. Chem. Inf. Model. 2014, 54, 735–743. [Google Scholar] [CrossRef] [PubMed]
- Leckband, D.; Israelachvili, J.; Schmitt, F.; Knoll, W. Long-range attraction and molecular rearrangements in receptor-ligand interactions. Science 1992, 255, 1419–1421. [Google Scholar] [CrossRef] [PubMed]
- Dunbar Jr, J.B.; Smith, R.D.; Yang, C.-Y.; Ung, P.M.-U.; Lexa, K.W.; Khazanov, N.A.; Stuckey, J.A.; Wang, S.; Carlson, H.A. CSAR benchmark exercise of 2010: Selection of the protein–ligand complexes. J. Chem. Inf. Model. 2011, 51, 2036–2046. [Google Scholar] [CrossRef]
- Veličković, P.; Cucurull, G.; Casanova, A.; Romero, A.; Lio, P.; Bengio, Y. Graph attention networks. arXiv 2017, arXiv:1710.10903. [Google Scholar]
- Davis, M.I.; Hunt, J.P.; Herrgard, S.; Ciceri, P.; Wodicka, L.M.; Pallares, G.; Hocker, M.; Treiber, D.K.; Zarrinkar, P.P. Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2011, 29, 1046–1051. [Google Scholar] [CrossRef]
- Sussman, J.L.; Lin, D.; Jiang, J.; Manning, N.O.; Prilusky, J.; Ritter, O.; Abola, E.E. Protein Data Bank (PDB): Database of three-dimensional structural information of biological macromolecules. Acta Crystallogr. Sect. D Biol. Crystallogr. 1998, 54, 1078–1084. [Google Scholar] [CrossRef]
- Gilson, M.K.; Liu, T.; Baitaluk, M.; Nicola, G.; Hwang, L.; Chong, J. BindingDB in 2015: A public database for medicinal chemistry, computational chemistry and systems pharmacology. Nucleic Acids Res. 2016, 44, D1045–D1053. [Google Scholar] [CrossRef]
- Wang, R.; Fang, X.; Lu, Y.; Yang, C.-Y.; Wang, S. The PDBbind database: Methodologies and updates. J. Med. Chem. 2005, 48, 4111–4119. [Google Scholar] [CrossRef]
- Shim, J.; Hong, Z.-Y.; Sohn, I.; Hwang, C. Prediction of drug–target binding affinity using similarity-based convolutional neural network. Sci. Rep. 2021, 11, 4416. [Google Scholar] [CrossRef]
- He, T.; Heidemeyer, M.; Ban, F.; Cherkasov, A.; Ester, M. SimBoost: A read-across approach for predicting drug–target binding affinities using gradient boosting machines. J. Cheminform. 2017, 9, 24. [Google Scholar] [CrossRef] [PubMed]
- Kouranov, A.; Xie, L.; de la Cruz, J.; Chen, L.; Westbrook, J.; Bourne, P.E.; Berman, H.M. The RCSB PDB information portal for structural genomics. Nucleic Acids Res. 2006, 34 (Suppl. 1), D302–D305. [Google Scholar] [CrossRef] [PubMed]
- Tokars, V.; Mesecar, A. QFRET-Based Primary Biochemical High Throughput Screening Assay to Identify Inhibitors of the SARS Coronavirus 3C-Like Protease (3CLPro). Available online: https://pubchem.ncbi.nlm.nih.gov/bioassay/1706 (accessed on 1 July 2022).
- Yang, H.; Xie, W.; Xue, X.; Yang, K.; Ma, J.; Liang, W.; Zhao, Q.; Zhou, Z.; Pei, D.; Ziebuhr, J. Design of wide-spectrum inhibitors targeting coronavirus main proteases. PLoS Biol. 2005, 3, e324. [Google Scholar]
- Yang, K.K.; Wu, Z.; Bedbrook, C.N.; Arnold, F.H. Learned protein embeddings for machine learning. Bioinformatics 2018, 34, 2642–2648. [Google Scholar] [CrossRef] [PubMed]
- Duvaud, S.; Gabella, C.; Lisacek, F.; Stockinger, H.; Ioannidis, V.; Durinx, C. Expasy, the Swiss Bioinformatics Resource Portal, as designed by its users. Nucleic Acids Res. 2021, 49, W216–W227. [Google Scholar] [CrossRef] [PubMed]
- Jamasb, A.R.; Lió, P.; Blundell, T.L. Graphein-a python library for geometric deep learning and network analysis on protein structures. bioRxiv 2020. [Google Scholar] [CrossRef]
- Kearnes, S.; McCloskey, K.; Berndl, M.; Pande, V.; Riley, P. Molecular graph convolutions: Moving beyond fingerprints. J. Comput. -Aided Mol. Des. 2016, 30, 595–608. [Google Scholar] [CrossRef]
- Rives, A.; Meier, J.; Sercu, T.; Goyal, S.; Lin, Z.; Liu, J.; Guo, D.; Ott, M.; Zitnick, C.L.; Ma, J. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. Proc. Natl. Acad. Sci. USA 2021, 118, e2016239118. [Google Scholar] [CrossRef]
- Nguyen, T.; Le, H.; Venkatesh, S. GraphDTA: Prediction of drug–target binding affinity using graph convolutional networks. bioRxiv 2019, 684662. [Google Scholar] [CrossRef]
- Nascimento, A.C.; Prudêncio, R.B.; Costa, I.G. A multiple kernel learning algorithm for drug-target interaction prediction. BMC Bioinform. 2016, 17, 46. [Google Scholar] [CrossRef]
- Zhao, L.; Wang, J.; Pang, L.; Liu, Y.; Zhang, J. GANsDTA: Predicting drug-target binding affinity using GANs. Front. Genet. 2020, 10, 1243. [Google Scholar] [CrossRef] [PubMed]
- Karimi, M.; Wu, D.; Wang, Z.; Shen, Y. DeepAffinity: Interpretable deep learning of compound–protein affinity through unified recurrent and convolutional neural networks. Bioinformatics 2019, 35, 3329–3338. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Fu, T.; Glass, L.M.; Zitnik, M.; Xiao, C.; Sun, J. DeepPurpose: A deep learning library for drug–target interaction prediction. Bioinformatics 2020, 36, 5545–5547. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Zhou, J.; Xu, T.; Huang, L.; Wang, F.; Xiong, H.; Huang, W.; Dou, D.; Xiong, H. Structure-aware interactive graph neural networks for the prediction of protein-ligand binding affinity. In Proceedings of the 27th ACM SIGKDD Conference on Knowledge Discovery & Data Mining, Singapore, 14–18 August 2021; pp. 975–985. [Google Scholar]
- Danel, T.; Spurek, P.; Tabor, J.; Śmieja, M.; Struski, Ł.; Słowik, A.; Maziarka, Ł. Spatial graph convolutional networks. In International Conference on Neural Information Processing; Springer: Berlin/Heidelberg, Germany, 2020; pp. 668–675. [Google Scholar]
- Gentile, F.; Fernandez, M.; Ban, F.; Ton, A.-T.; Mslati, H.; Perez, C.F.; Leblanc, E.; Yaacoub, J.C.; Gleave, J.; Stern, A. Automated discovery of noncovalent inhibitors of SARS-CoV-2 main protease by consensus Deep Docking of 40 billion small molecules. Chem. Sci. 2021, 12, 15960–15974. [Google Scholar] [CrossRef]
- Mesecar, A. A taxonomically-driven approach to development of potent, broad-spectrum inhibitors of coronavirus main protease including SARS-CoV-2 (COVID-19). Be Publ. 2020. [Google Scholar]
- Schrödinger Release 2020–4: Glide; Schrödinger, LLC: New York, NY, USA, 2020.
- Ietswaart, R.; Arat, S.; Chen, A.X.; Farahmand, S.; Kim, B.; DuMouchel, W.; Armstrong, D.; Fekete, A.; Sutherland, J.J.; Urban, L. Machine learning guided association of adverse drug reactions with in vitro target-based pharmacology. EBioMedicine 2020, 57, 102837. [Google Scholar] [CrossRef]
- Stank, A.; Kokh, D.B.; Fuller, J.C.; Wade, R.C. Protein binding pocket dynamics. Acc. Chem. Res. 2016, 49, 809–815. [Google Scholar] [CrossRef]
- Liu, T.; Altman, R. Identifying druggable targets by protein microenvironments matching: Application to transcription factors. CPT Pharmacomet. Syst. Pharmacol. 2014, 3, 1–9. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dataset | Unique Targets | Unavailable PDBs | Unique Ligands | Unique Interactions |
|---|---|---|---|---|
| PDBBind | 9619 | 0 | 7981 | 9777 |
| KIBA | 467 | 181 | 2356 | 124,374 |
| DAVIS | 442 | 139 | 68 | 20,604 |
| BindingDB | 1038 | 100 | 13,222 | 41,142 |
| AID 1706 | 1 | 0 | 290,765 | 290,765 |
| Dataset | Architecture | MSE (↓) | Pearson (↑) |
|---|---|---|---|
| DAVIS | GCNConvNet [51] | 0.284 | 0.804 |
| GINConvNet [51] | 0.257 | 0.824 | |
| DGraphDTA [32] | 0.241 | 0.837 | |
| GEFA [30] | 0.228 | 0.846 | |
| PSG-BAR | 0.237 | 0.856 | |
| KIBA | KronRLS [52] | 0.261 | 0.752 |
| GANsDTA [53] | 0.387 | 0.662 | |
| SimCNN-DTA [41] | 0.257 | 0.757 | |
| SimBoost [42] | 0.204 | - | |
| PSG-BAR | 0.200 | 0.850 | |
| PSG-BAR w/AF | 0.177 | 0.865 | |
| BindingDB | DeepAffinity [54] | 1.212 | 0.700 |
| DeepPurpose [55] | 0.765 | 0.836 | |
| PSG-BAR | 0.651 | 0.864 | |
| PDBBind | GAT [56] | 3.115 | 0.601 |
| SGCN [57] | 2.505 | 0.686 | |
| SIGN [56] | 1.731 | 0.797 | |
| KDeep [27] | 1.612 | 0.820 | |
| PSG-BAR | 1.660 | 0.762 |
| Dataset | Architecture | MSE (↓) | Pearson (↑) |
|---|---|---|---|
| DAVIS | Warm | 0.237 | 0.856 |
| Cold Drug | 0.902 | 0.456 | |
| Cold Protein | 0.436 | 0.612 | |
| Cold Protein–Ligand | 0.910 | 0.357 | |
| KIBA | Warm | 0.200 | 0.850 |
| Cold Drug | 0.362 | 0.601 | |
| Cold Protein | 0.298 | 0.756 | |
| Cold Protein–Ligand | 0.415 | 0.360 | |
| BindingDB | Warm | 0.651 | 0.864 |
| Cold Drug | 1.353 | 0.720 | |
| Cold Protein | 1.811 | 0.540 | |
| Cold Protein–Ligand | 2.102 | 0.515 | |
| PDBBind | Warm | 1.660 | 0.762 |
| Cold Drug | 1.895 | 0.694 | |
| Cold Protein | 2.011 | 0.602 | |
| Cold Protein–Ligand | 2.100 | 0.599 |
| With Surface Features | Without Surface Features | |||
|---|---|---|---|---|
| Dataset | MSE | Pearson | MSE | Pearson |
| BindingDB | 0.651 | 0.864 | 0.678 | 0.851 |
| PDBBind | 1.660 | 0.762 | 1.744 | 0.749 |
| KIBA | 0.200 | 0.850 | 0.209 | 0.837 |
| DAVIS | 0.237 | 0.856 | 0.249 | 0.845 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pandey, M.; Radaeva, M.; Mslati, H.; Garland, O.; Fernandez, M.; Ester, M.; Cherkasov, A. Ligand Binding Prediction Using Protein Structure Graphs and Residual Graph Attention Networks. Molecules 2022, 27, 5114. https://doi.org/10.3390/molecules27165114
Pandey M, Radaeva M, Mslati H, Garland O, Fernandez M, Ester M, Cherkasov A. Ligand Binding Prediction Using Protein Structure Graphs and Residual Graph Attention Networks. Molecules. 2022; 27(16):5114. https://doi.org/10.3390/molecules27165114
Chicago/Turabian StylePandey, Mohit, Mariia Radaeva, Hazem Mslati, Olivia Garland, Michael Fernandez, Martin Ester, and Artem Cherkasov. 2022. "Ligand Binding Prediction Using Protein Structure Graphs and Residual Graph Attention Networks" Molecules 27, no. 16: 5114. https://doi.org/10.3390/molecules27165114
APA StylePandey, M., Radaeva, M., Mslati, H., Garland, O., Fernandez, M., Ester, M., & Cherkasov, A. (2022). Ligand Binding Prediction Using Protein Structure Graphs and Residual Graph Attention Networks. Molecules, 27(16), 5114. https://doi.org/10.3390/molecules27165114