
Synthesis and Stereochemical Characterization of a Novel Chiral α-Tetrazole Binaphthylazepine Organocatalyst

 , , ,
, , ,  , and
, and 
Abstract
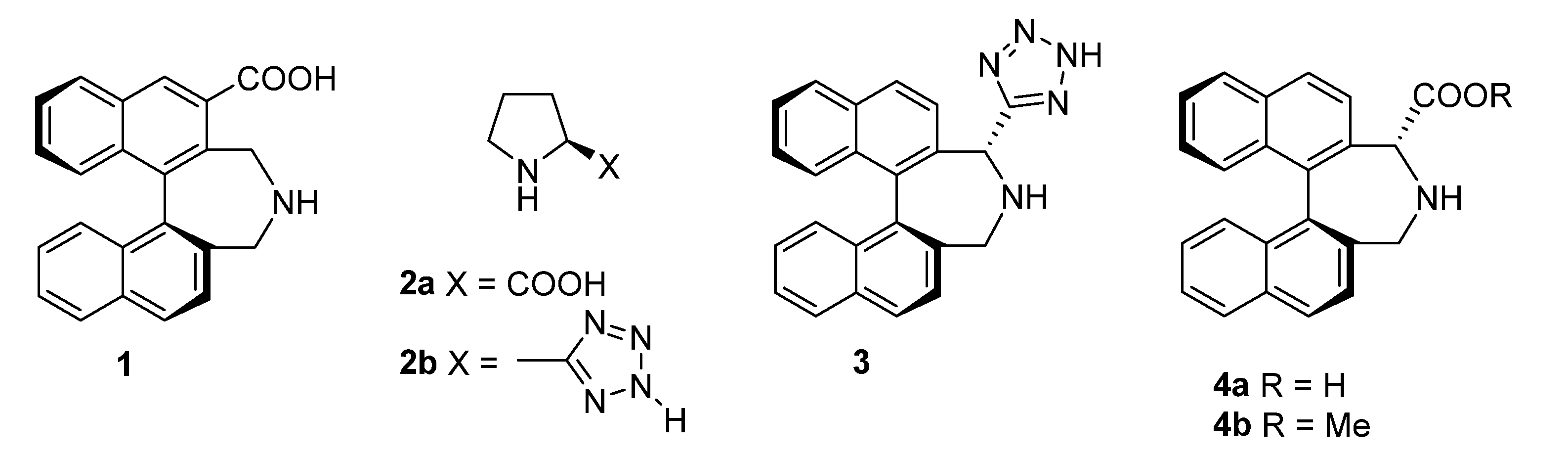
:1. Introduction
2. Results and Discussion
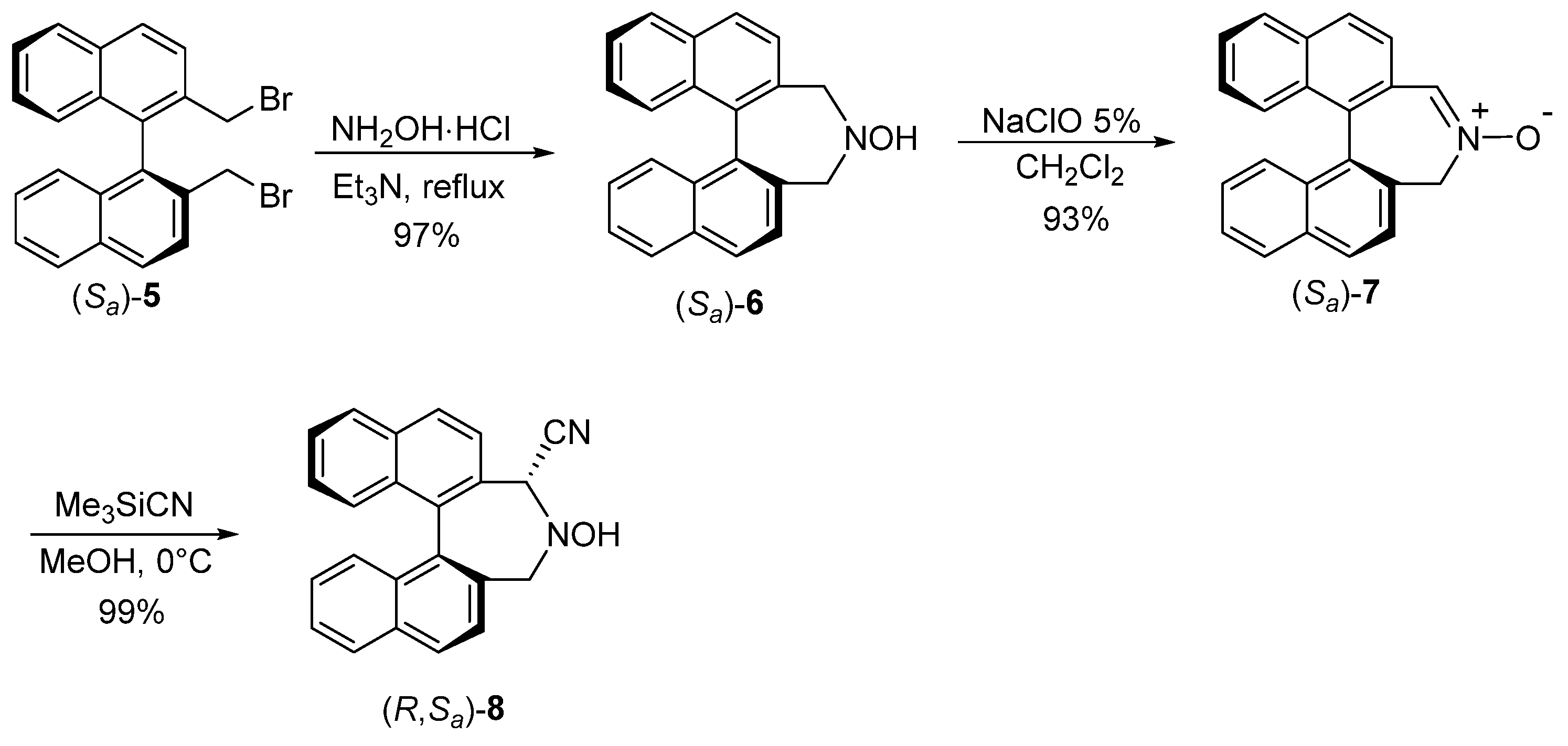
2.1. Synthesis of Binaphthylazepine Catalyst 3
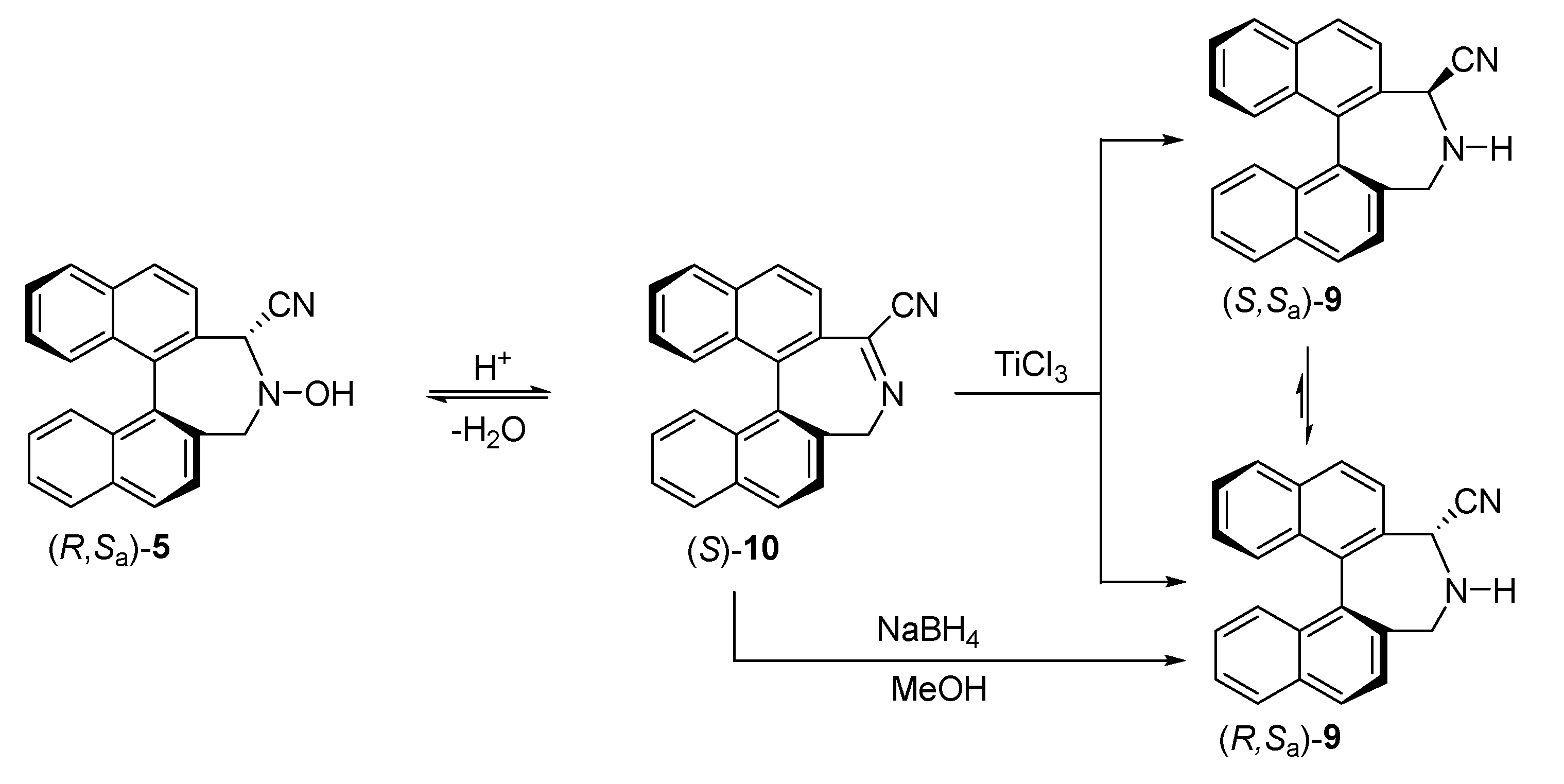
2.1.1. Synthesis of Cyano-Hydroxylamine 8
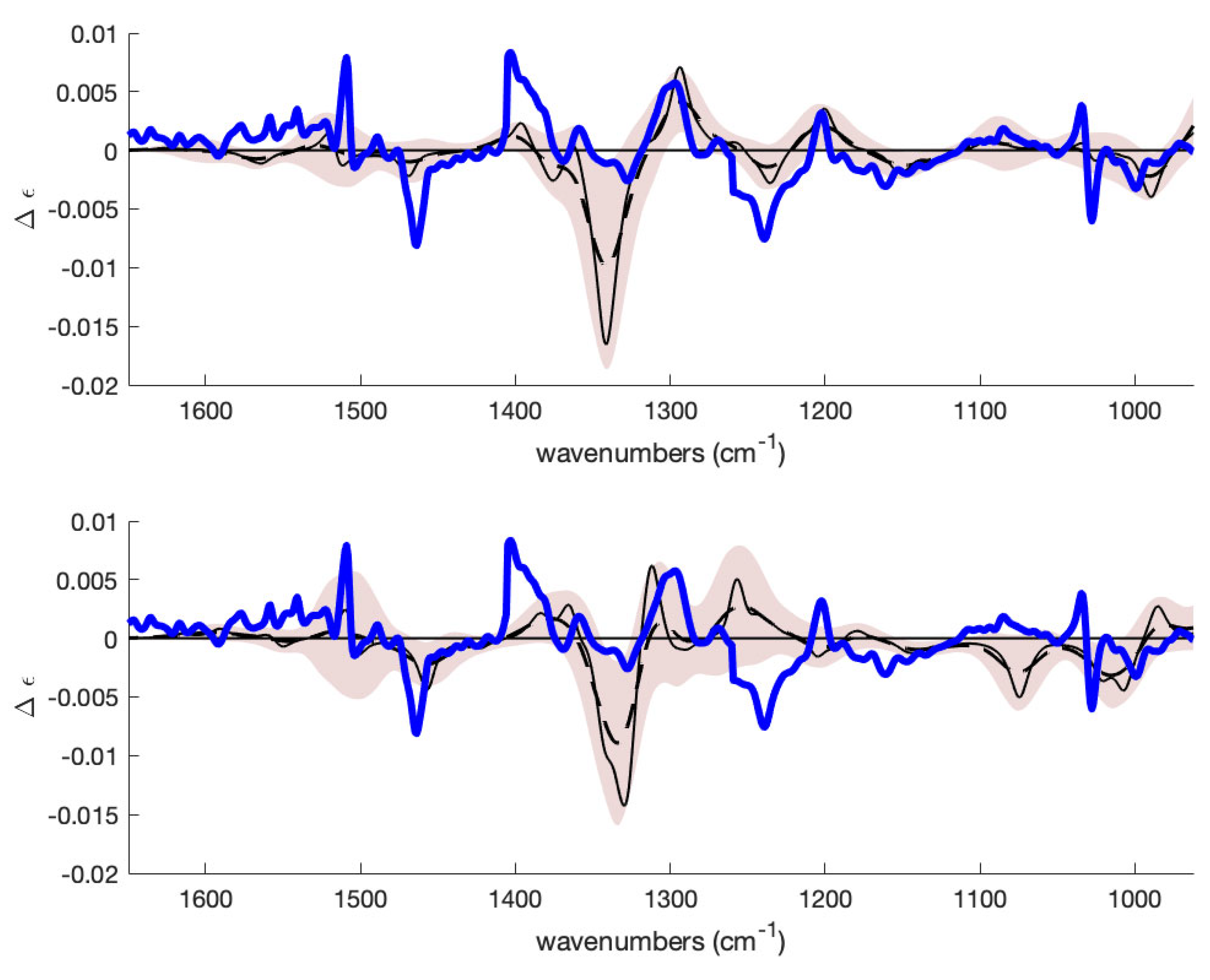
2.1.2. Absolute Configuration Assignment to Cyano-Hydroxylamine 8
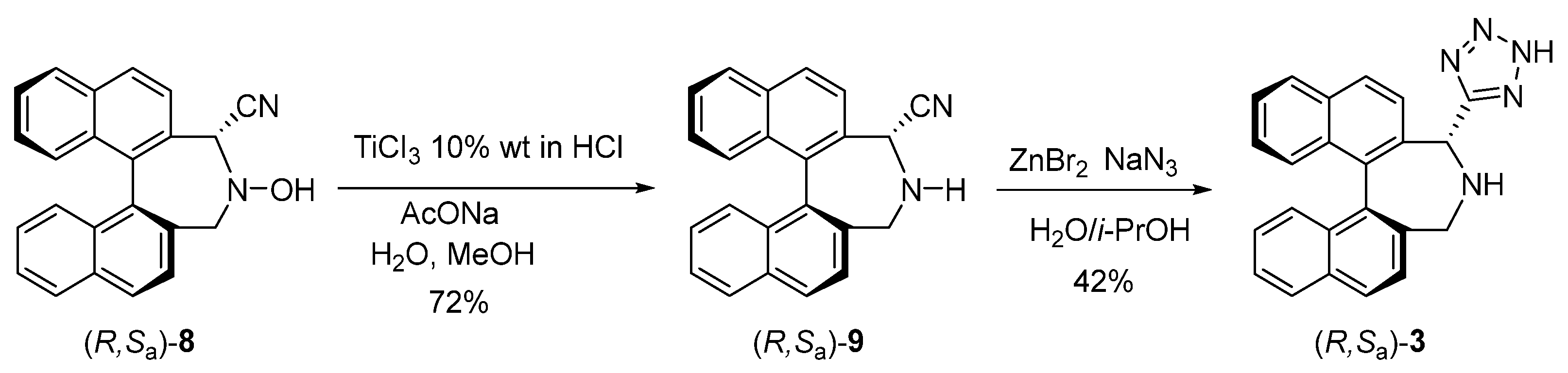
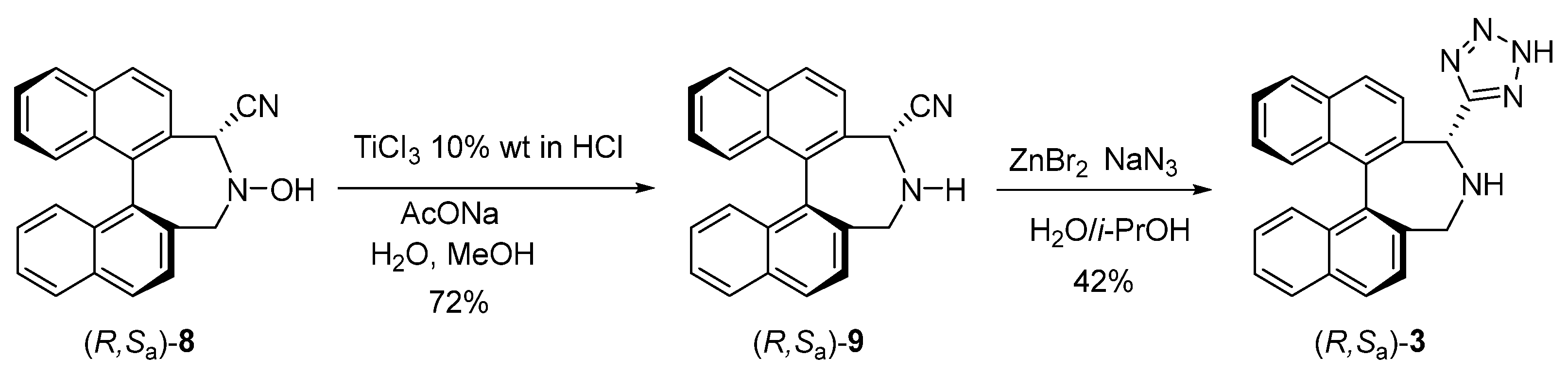
2.2. Synthesis of Tetrazole Binaphthylazepine 3
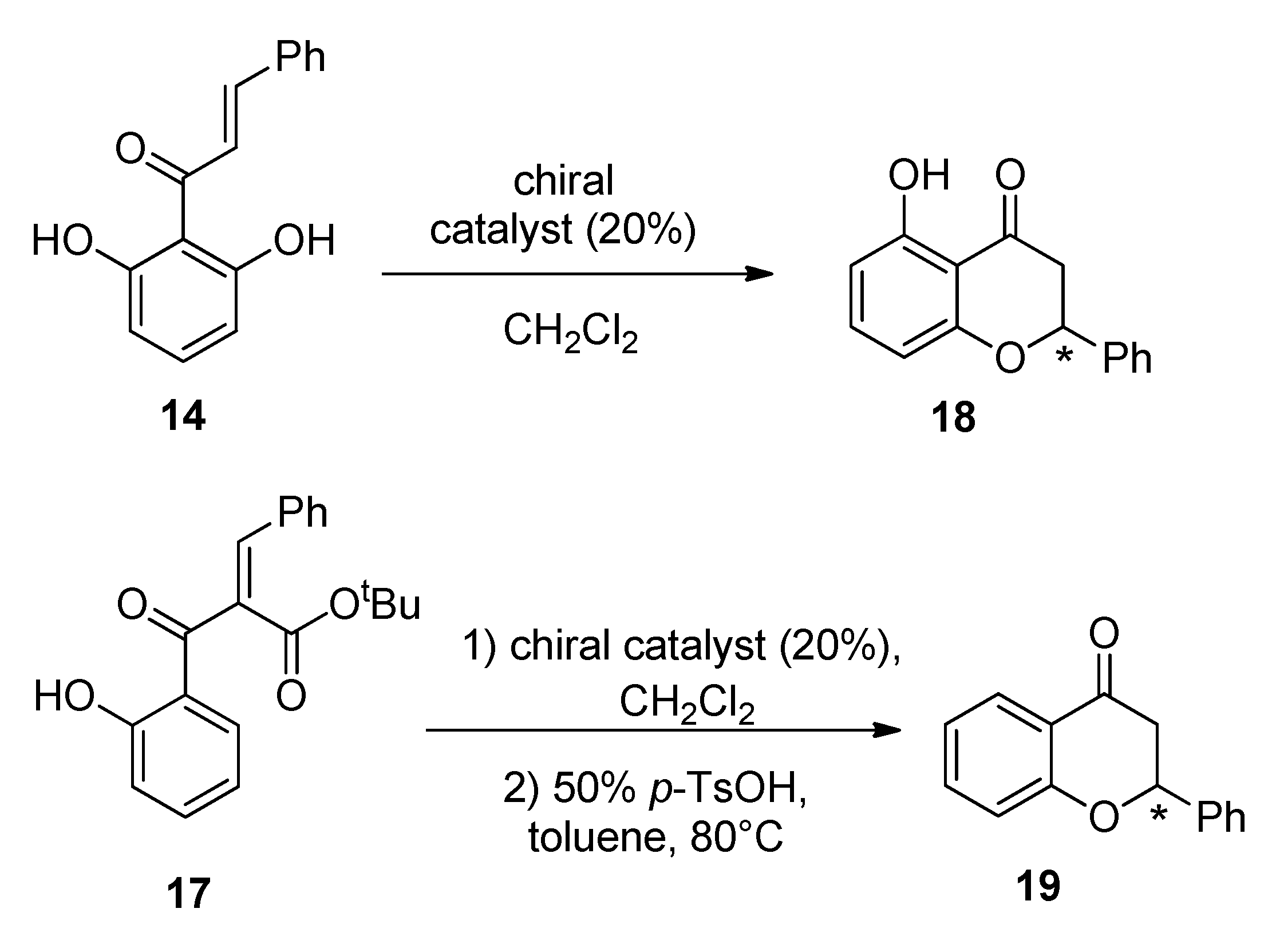
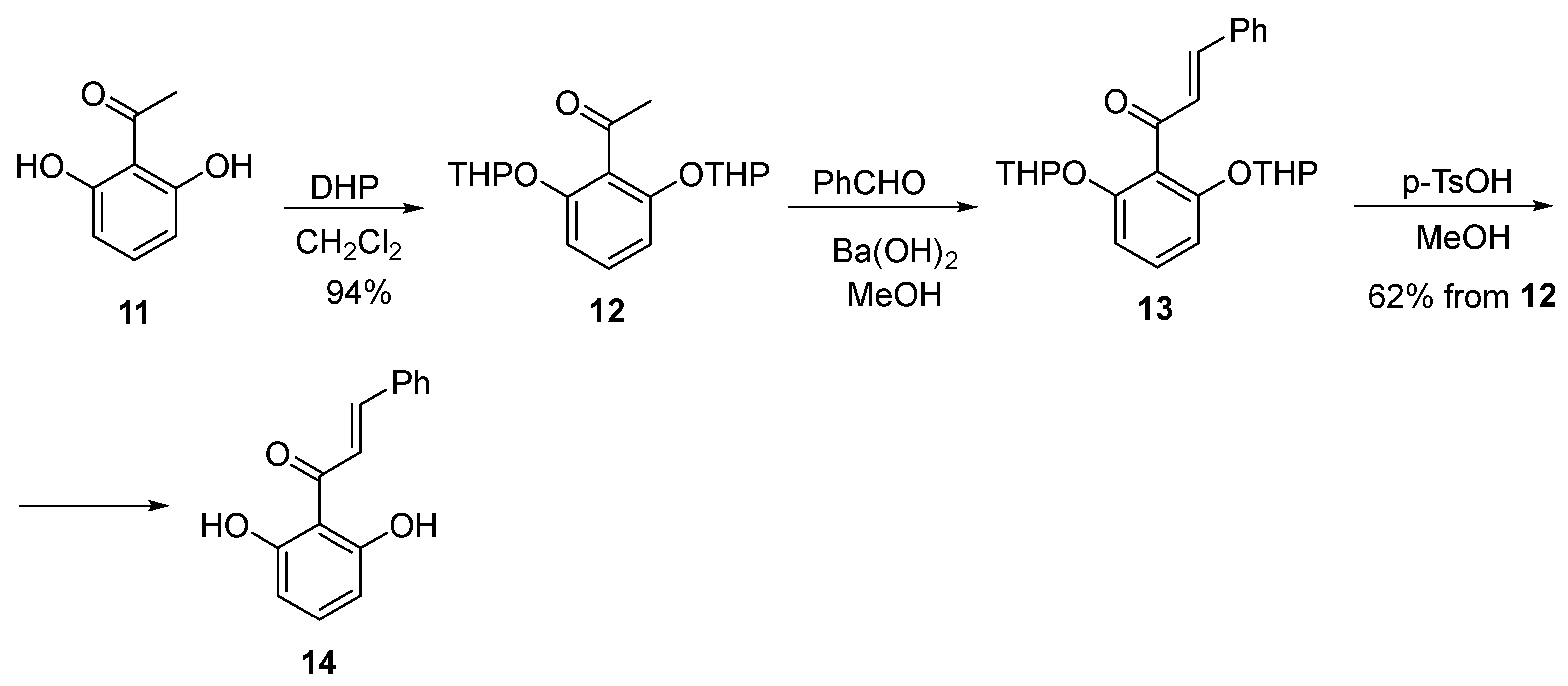
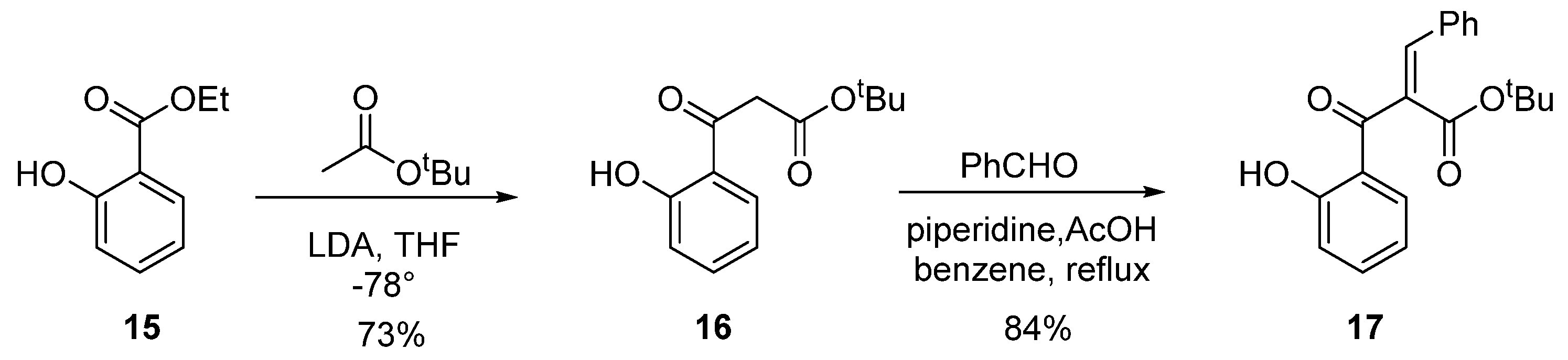
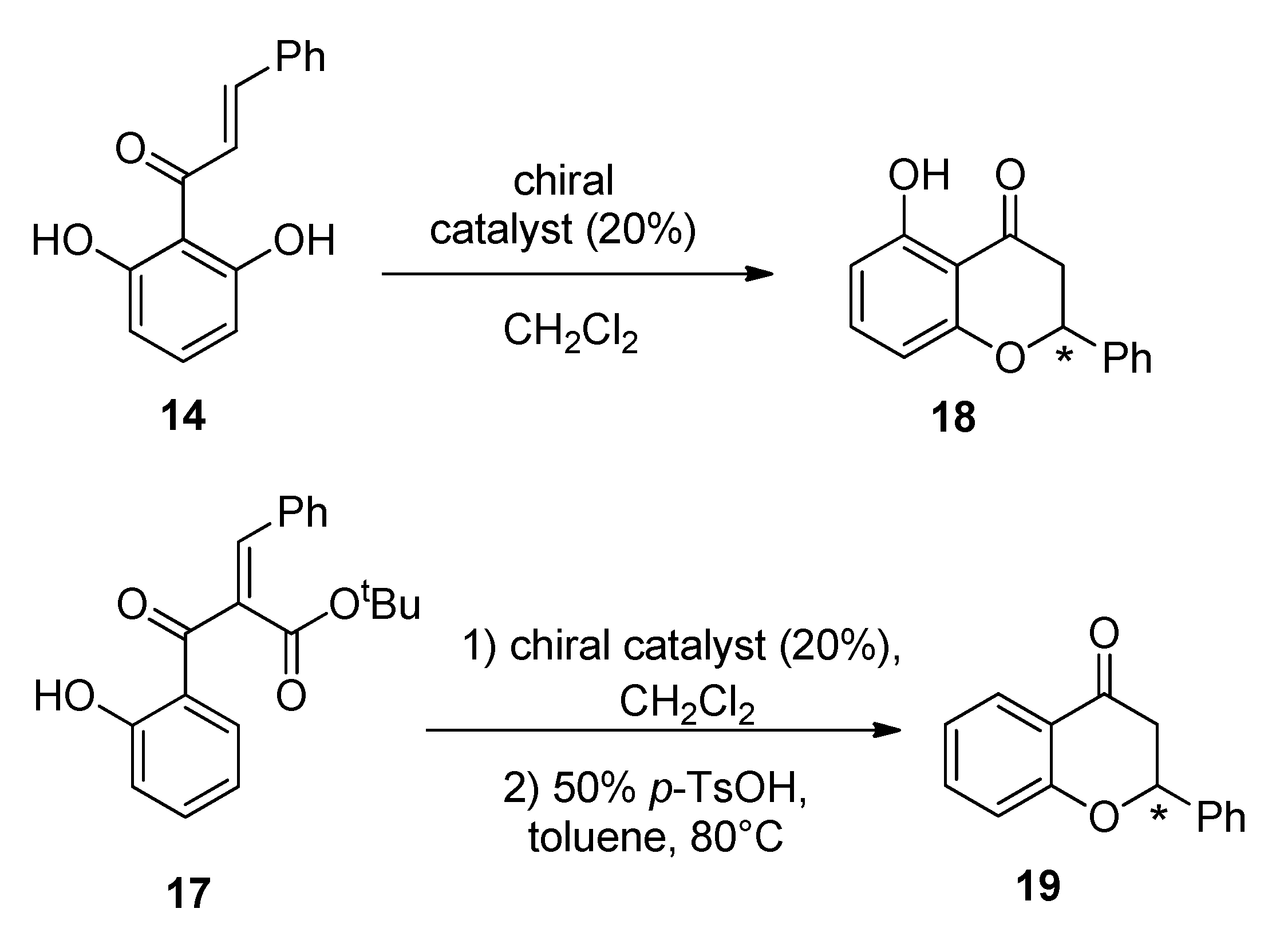
2.3. Asymmetric Catalytic Synthesis of Flavanones
3. Materials and Methods
3.1. General Experimental Procedures
3.2. (S)-(+)-3,5-Dihydro-4H-dinaphth[2,1-c:1′2′-e]azepine-N-hydroxide. [(Sa)-6]
3.3. (S)-(+)-3H-Dinaphth[2,1-c:1′,2′-e]azepine-4-oxide. [(Sa)-7]
3.4. (R,Sa)-(+)-4-Hydroxy-4,5-dihydro-3H-dinaphtho[2,1-c:1’,2’-e]azepine-3-carbonitrile. [(R,Sa)-8]
3.5. (R,Sa)-(+)-4,5-Dihydro-3H-dinaphtho[2,1-c:1’,2’-e]azepine-3-carbonitrile [(R,Sa)-9]
3.6. (R,Sa)-(+)-3-(2H-Tetrazol-5-yl)-4,5-dihydro-3H-dinaphtho[2,1-c:1’,2’-e]azepine [(R,Sa)-3]
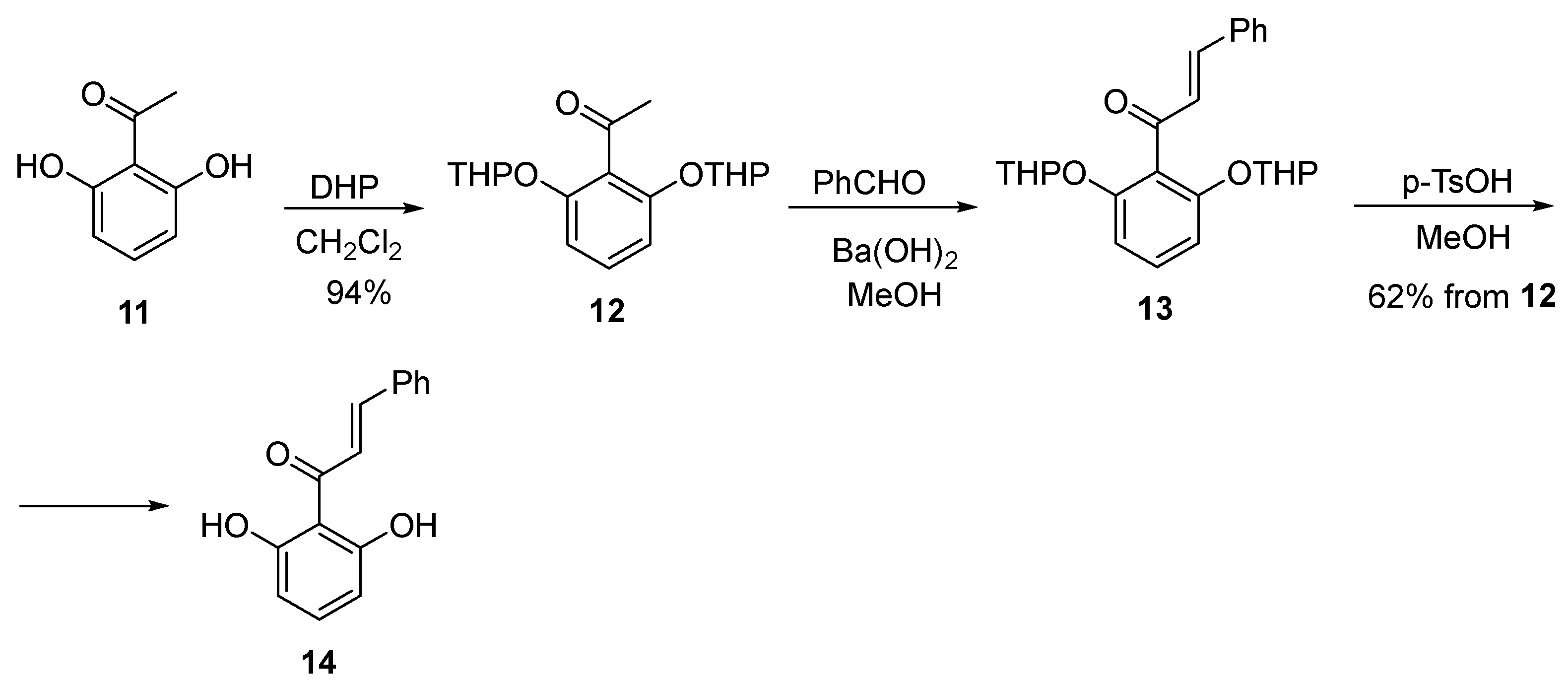
3.7. 1-(2,6-Dihydroxyphenyl)-3-phenylprop-2-en-1-one (14)
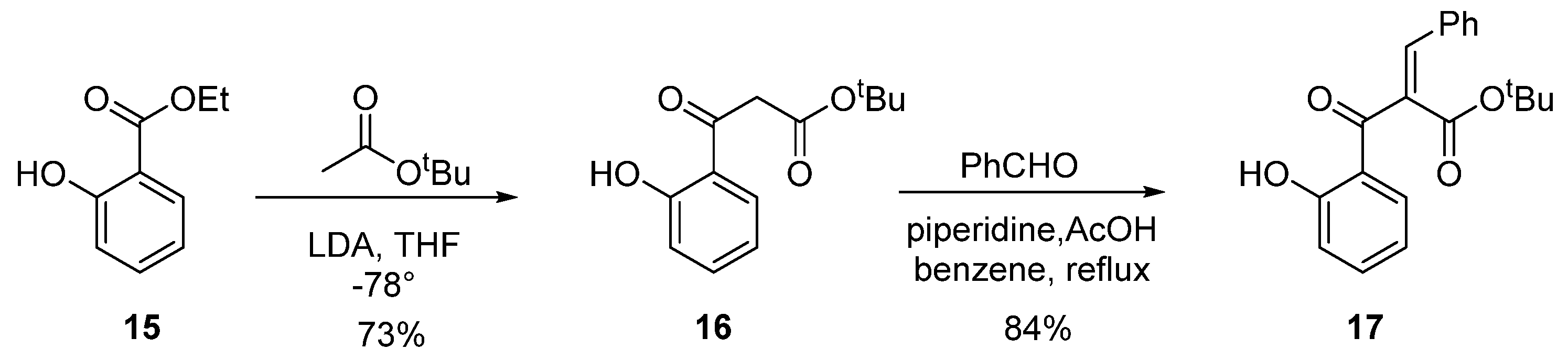
3.8. Tert-Butyl 3-(2-hydroxyphenyl)-3-oxopropanoate (16)
3.9. (E)-Tert-butyl 2-(2-hydroxyphenylcarbonyl)-3-phenylprop-2-enoate (17)
3.10. Asymmetric Oxa-Michael Cyclization of Chalcone 14
3.11. Asymmetric Oxa-Michael Cyclization of Chalcone 17
3.12. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhou, Q.-L. (Ed.) Privileged Chiral Ligands and Catalysts; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2011; ISBN 9783527635207. [Google Scholar]
- Dieéguez, M. (Ed.) Chiral Ligands: Evolution of Ligand Libraries for Asymmetric Catalysis, New directions in organic and biological chemistry, 1st ed.; CRC Press: Boca Raton, FL, USA, 2021; ISBN 9780367428488. [Google Scholar]
- Rosini, C.; Franzini, L.; Raffaelli, A.; Salvadori, P. Synthesis and Applications of Binaphthylic C2-Symmetry Derivatives as Chiral Auxiliaries in Enantioselective Reactions. Synthesis 1992, 1992, 503–517. [Google Scholar] [CrossRef]
- Berthod, M.; Mignani, G.; Woodward, G.; Lemaire, M. Modified BINAP: The How and the Why. Chem. Rev. 2005, 105, 1801–1836. [Google Scholar] [CrossRef] [PubMed]
- Brunel, J.M. BINOL: A Versatile Chiral Reagent. Chem. Rev. 2005, 105, 857–898. [Google Scholar] [CrossRef] [PubMed]
- Mazaleyrat, J.P.; Cram, D.J. Chiral Catalysis of Additions of Alkyllithiums to Aldehydes. J. Am. Chem. Soc. 1981, 103, 4585–4586. [Google Scholar] [CrossRef]
- Superchi, S.; Mecca, T.; Giorgio, E.; Rosini, C. 1,1′-Binaphthylazepine-Based Ligands for Asymmetric Catalysis. Part 2: New Aminoalcohols as Chiral Ligands in the Enantioselective Addition of ZnEt2 to Aromatic Aldehydes. Tetrahedron Asymmetry 2001, 12, 1235–1239. [Google Scholar] [CrossRef]
- Superchi, S.; Giorgio, E.; Scafato, P.; Rosini, C. Rational Design of Chiral 1,1′-Binaphthylazepine-Based Ligands for the Enantioselective Addition of ZnEt2 to Aromatic Aldehydes. Tetrahedron Asymmetry 2002, 13, 1385–1391. [Google Scholar] [CrossRef]
- Pisani, L.; Superchi, S. 1,1′-Binaphthylazepine-Based Ligands for the Enantioselective Dialkylzinc Addition to Aromatic Aldehydes. Tetrahedron Asymmetry 2008, 19, 1784–1789. [Google Scholar] [CrossRef]
- Pisani, L.; Superchi, S.; D’Elia, A.; Scafato, P.; Rosini, C. Synthetic Approach toward Cis-Disubstituted γ- and δ-Lactones through Enantioselective Dialkylzinc Addition to Aldehydes: Application to the Synthesis of Optically Active Flavors and Fragrances. Tetrahedron 2012, 68, 5779–5784. [Google Scholar] [CrossRef]
- Kano, T.; Takai, J.; Tokuda, O.; Maruoka, K. Design of an Axially Chiral Amino Acid with a Binaphthyl Backbone as an Organocatalyst for a Direct Asymmetric Aldol Reaction. Angew. Chem. Int. Ed. 2005, 44, 3055–3057. [Google Scholar] [CrossRef]
- Pisani, L.; Bochicchio, C.; Superchi, S.; Scafato, P. Tropos Amino Alcohol Mediated Enantioselective Aryl Transfer Reactions to Aromatic Aldehydes: Enantioselective Aryl Transfer Reactions to Aromatic Aldehydes. Eur. J. Org. Chem. 2014, 2014, 5939–5945. [Google Scholar] [CrossRef]
- Superchi, S.; Bisaccia, R.; Casarini, D.; Laurita, A.; Rosini, C. Flexible Biphenyl Chromophore as a Circular Dichroism Probe for Assignment of the Absolute Configuration of Carboxylic Acids. J. Am. Chem. Soc. 2006, 128, 6893–6902. [Google Scholar] [CrossRef]
- Vergura, S.; Scafato, P.; Belviso, S.; Superchi, S. Absolute Configuration Assignment from Optical Rotation Data by Means of Biphenyl Chiroptical Probes. Chem. Eur. J. 2019, 25, 5682–5690. [Google Scholar] [CrossRef]
- Santoro, E.; Vergura, S.; Scafato, P.; Belviso, S.; Masi, M.; Evidente, A.; Superchi, S. Absolute configuration assignment to chiral natural products by biphenyl chiroptical probes: The case of the phytotoxins colletochlorin A and agropyrenol. J. Nat. Prod. 2020, 83, 1061–1068. [Google Scholar] [CrossRef]
- Vergura, S.; Orlando, S.; Scafato, P.; Belviso, S.; Superchi, S. Absolute configuration sensing of chiral aryl- and aryloxy-propionic acids by biphenyl chiroptical probes. Chemosensors 2021, 9, 154. [Google Scholar] [CrossRef]
- Vergura, S.; Pisani, L.; Scafato, P.; Casarini, D.; Superchi, S. Central-to-Axial Chirality Induction in Biphenyl Chiroptical Probes for the Stereochemical Characterization of Chiral Primary Amines. Org. Biomol. Chem. 2018, 16, 555–565. [Google Scholar] [CrossRef]
- Hashimoto, T.; Maruoka, K. Recent Development and Application of Chiral Phase-Transfer Catalysts. Chem. Rev. 2007, 107, 5656–5682. [Google Scholar] [CrossRef]
- Kano, T.; Maruoka, K. Unique Properties of Chiral Biaryl-Based Secondary Aminecatalysts for Asymmetric Enamine Catalysis. Chem. Sci. 2013, 4, 907–915. [Google Scholar] [CrossRef]
- Shirakawa, S.; Maruoka, K. Recent Developments in Asymmetric Phase-Transfer Reactions. Angew. Chem. Int. Ed. 2013, 52, 4312–4348. [Google Scholar] [CrossRef]
- Liu, Y.; Arumugam, N.; Almansour, A.I.; Kumar, R.S.; Maruoka, K. Practical Synthesis of Both Enantiomeric Amino Acid, Mannich, and Aldol Derivatives by Asymmetric Organocatalysis. Chem. Rec. 2017, 17, 1059–1069. [Google Scholar] [CrossRef]
- Maruoka, K. Design of High-Performance Chiral Phase-Transfer Catalysts with Privileged Structures. Proc. Jpn. Acad. Ser. B: Phys. Biol. Sci. 2019, 95, 1–16. [Google Scholar] [CrossRef]
- Panday, S.K. Advances in the Chemistry of Proline and Its Derivatives: An Excellent Amino Acid with Versatile Applications in Asymmetric Synthesis. Tetrahedron Asymmetry 2011, 22, 1817–1847. [Google Scholar] [CrossRef]
- Kotsuki, H.; Sasakura, N. Proline-Related Secondary Amine Catalysts and Applications. In Comprehensive Enantioselective Organocatalysis; Dalko, P.I., Ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2013; pp. 1–31. ISBN 9783527658862. [Google Scholar]
- Liu, J.; Wang, L. Recent Advances in Asymmetric Reactions Catalyzed by Proline and Its Derivatives. Synthesis 2016, 49, 960–972. [Google Scholar] [CrossRef]
- Torii, H.; Nakadai, M.; Ishihara, K.; Saito, S.; Yamamoto, H. Asymmetric Direct Aldol Reaction Assisted by Water and a Proline-Derived Tetrazole Catalyst. Angew. Chem. Int. Ed. 2004, 43, 1983–1986. [Google Scholar] [CrossRef]
- Hartikka, A.; Arvidsson, P.I. Rational Design of Asymmetric Organocatalysts––Increased Reactivity and Solvent Scope with a Tetrazolic Acid. Tetrahedron Asymmetry 2004, 15, 1831–1834. [Google Scholar] [CrossRef]
- Cobb, A.J.; Shaw, D.M.; Ley, S.V. 5-Pyrrolidin-2-Yltetrazole: A New, Catalytic, More Soluble Alternative to Proline in an Organocatalytic Asymmetric Mannich-Type Reaction. Synlett 2004, 558–560. [Google Scholar] [CrossRef]
- Bulman Page, P.C.; Kinsey, F.S.; Chan, Y.; Strutt, I.R.; Slawin, A.M.Z.; Jones, G.A. Novel Binaphthyl and Biphenyl α- and β-Amino Acids and Esters: Organocatalysis of Asymmetric Diels–Alder Reactions. A Combined Synthetic and Computational Study. Org. Biomol. Chem. 2018, 16, 7400–7416. [Google Scholar] [CrossRef]
- Lombardo, M.; Trombini, C. Nucleophilic Additions to Nitrones. Synthesis 2000, 2000, 759–774. [Google Scholar] [CrossRef]
- Cicchi, S.; Corsi, M.; Goti, A. Inexpensive and Environmentally Friendly Oxidation of Hydroxylamines to Nitrones with Bleach. J. Org. Chem. 1999, 64, 7243–7245. [Google Scholar] [CrossRef]
- Merino, P.; Lanaspa, A.; Merchan, F.L.; Tejero, T. Diastereoselective Hydrocyanation of Chiral Nitrones. Synthesis of Novel α-(Hydroxyamino) Nitriles. J. Org. Chem. 1996, 61, 9028–9032. [Google Scholar] [CrossRef]
- Autschbach, J. Ab initio Electronic Circular Dichroism and Optical Rotatory Dispersion: From Organic Molecules to Transition Metal Complexes. In Comprehensive Chiroptical Spectroscopy; Berova, N., Polavarapu, P.L., Nakanishi, K., Woody, R.W., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2012; Volume 1, Chapter 21; pp. 593–642. ISBN 9781118012932. [Google Scholar]
- Polavarapu, P.L. Determination of the Structures of Chiral Natural Products Using Vibrational Circular Dichroism. In Comprehensive Chiroptical Spectroscopy; Berova, N., Polavarapu, P.L., Nakanishi, K., Woody, R.W., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2012; Volume 2, Chapter 11; pp. 387–420. ISBN 9781118012925. [Google Scholar]
- Mazzeo, G.; Cimmino, A.; Masi, M.; Longhi, G.; Maddau, L.; Memo, M.; Evidente, A.; Abbate, S. Importance and Difficulties in the Use of Chiroptical Methods to Assign the Absolute Configuration of Natural Products: The Case of Phytotoxic Pyrones and Furanones Produced by Diplodia Corticola. J. Nat. Prod. 2017, 80, 2406–2415. [Google Scholar] [CrossRef]
- Mazzeo, G.; Longhi, G.; Abbate, S.; Buonerba, F.; Ruzziconi, R. Chiroptical Signatures of Planar and Central Chirality in [2]Paracyclo[2](5,8)Quinolinophane Derivatives: Chiroptical Signatures of [2]Paracyclo[2](5,8)Quinolinophanes. Eur. J. Org. Chem. 2014, 2014, 7353–7363. [Google Scholar] [CrossRef]
- Monaco, G.; Aquino, F.; Zanasi, R.; Herrebout, W.; Bultinck, P.; Massa, A. Model-Averaging of Ab Initio Spectra for the Absolute Configuration Assignment via Vibrational Circular Dichroism. Phys. Chem. Chem. Phys. 2017, 19, 28028–28036. [Google Scholar] [CrossRef] [PubMed]
- Monaco, G.; Procida, G.; Di Mola, A.; Herrebout, W.; Massa, A. Error Bounds on Goodness of Fit Indicators in Vibrational Circular Dichroism Spectroscopy. Chem. Phys. Lett. 2020, 739, 137000. [Google Scholar] [CrossRef]
- Efron, B. Bootstrap Methods: Another Look at the Jackknife. Ann. Statist. 1979, 7, 1–26. [Google Scholar] [CrossRef]
- Yamada, K.; Kishikawa, K.; Yamamoto, M. Stereospecificity of the Photorearrangement of Nitronate Anions and Its Utilization for Stereospecific Cleavage of Cyclic Compounds. J. Org. Chem. 1987, 52, 2327–2330. [Google Scholar] [CrossRef]
- Demko, Z.P.; Sharpless, K.B. An Expedient Route to the Tetrazole Analogues of α-Amino Acids. Org. Lett. 2002, 4, 2525–2527. [Google Scholar] [CrossRef]
- Andersen, Ø.M.; Markham, K.R. (Eds.) Flavonoids: Chemistry, Biochemistry, and Applications; CRC, Taylor & Francis: Boca Raton, FL, USA, 2006; ISBN 9780849320217. [Google Scholar]
- Keller, R.B. (Ed.) Flavonoids: Biosynthesis, Biological Effects and Dietary Sources; Nutrition and diet research progress series; Nova Science Publishers: New York, NY, USA, 2009; ISBN 9781607416227. [Google Scholar]
- Zaragozá, C.; Villaescusa, L.; Monserrat, J.; Zaragozá, F.; Álvarez-Mon, M. Potential Therapeutic Anti-Inflammatory and Immunomodulatory Effects of Dihydroflavones, Flavones, and Flavonols. Molecules 2020, 25, 1017. [Google Scholar] [CrossRef]
- Song, M.; Liu, Y.; Li, T.; Liu, X.; Hao, Z.; Ding, S.; Panichayupakaranant, P.; Zhu, K.; Shen, J. Plant Natural Flavonoids Against Multidrug Resistant Pathogens. Adv. Sci. 2021, 8, 2100749. [Google Scholar] [CrossRef]
- Nibbs, A.E.; Scheidt, K.A. Asymmetric Methods for the Synthesis of Flavanones, Chromanones, and Azaflavanones. Eur. J. Org. Chem. 2012, 2012, 449–462. [Google Scholar] [CrossRef]
- Meng, L.; Wang, J. Recent Progress on the Asymmetric Synthesis of Chiral Flavanones. Synlett 2015, 27, 656–663. [Google Scholar] [CrossRef]
- Wang, L.; Gong, X.; Lei, T.; Jiang, S. Research Progress on Asymmetric Synthesis of Flavanones. Chin. J. Org. Chem. 2022, 42, 758. [Google Scholar] [CrossRef]
- Biddle, M.M.; Lin, M.; Scheidt, K.A. Catalytic Enantioselective Synthesis of Flavanones and Chromanones. J. Am. Chem. Soc. 2007, 129, 3830–3831. [Google Scholar] [CrossRef]
- Wang, H.-F.; Xiao, H.; Wang, X.-W.; Zhao, G. Tandem Intramolecular Oxa-Michael Addition/Decarboxylation Reaction Catalyzed by Bifunctional Cinchona Alkaloids: Facile Synthesis of Chiral Flavanone Derivatives. Tetrahedron 2011, 67, 5389–5394. [Google Scholar] [CrossRef]
- Dittmer, C.; Raabe, G.; Hintermann, L. Asymmetric Cyclization of 2′-Hydroxychalcones to Flavanones: Catalysis by Chiral Brønsted Acids and Bases. Eur. J. Org. Chem. 2007, 2007, 5886–5898. [Google Scholar] [CrossRef]
- Hintermann, L.; Dittmer, C. Asymmetric Ion-Pairing Catalysis of the Reversible Cyclization of 2′-Hydroxychalcone to Flavanone: Asymmetric Catalysis of an Equilibrating Reaction. Eur. J. Org. Chem. 2012, 2012, 5573–5584. [Google Scholar] [CrossRef]
- Zhang, Y.-L.; Wang, Y.-Q. Enantioselective Biomimetic Cyclization of 2′-Hydroxychalcones to Flavanones. Tetrahedron Lett. 2014, 55, 3255–3258. [Google Scholar] [CrossRef]
- Zhou, S.; Zhou, Y.; Xing, Y.; Wang, N.; Cao, L. Exploration on Asymmetric Synthesis of Flavanone Catalyzed by (S)-Pyrrolidinyl Tetrazole. Chirality 2011, 23, 504–506. [Google Scholar] [CrossRef]
- Sogawa, S.; Nihro, Y.; Ueda, H.; Izumi, A.; Miki, T.; Matsumoto, H.; Satoh, T. 3,4-Dihydroxychalcones as Potent 5-Lipoxygenase and Cyclooxygenase Inhibitors. J. Med. Chem. 1993, 36, 3904–3909. [Google Scholar] [CrossRef]
- Belviso, C.; Piancastelli, A.; Sturini, M.; Belviso, S. Synthesis of Composite Zeolite-Layered Double Hydroxides Using Ultrasonic Neutralized Red Mud. Microp. Mesop. Mat. 2020, 299, 110108. [Google Scholar] [CrossRef]
- Miles, C.; Main, L.; Nicholson, B. Synthesis of 2′, 6′-Dihydroxychalcones by Using Tetrahydropyran-2-Yl and Trialkylsilyl Protective Groups; the Crystal Structure Determination of 2′,6′-Dihydroxy-2,4,6-Trimethoxychalcone. Aust. J. Chem. 1989, 42, 1103. [Google Scholar] [CrossRef]
- Belviso, S.; Santoro, E.; Penconi, M.; Righetto, S.; Tessore, F. Thioethyl Porphyrazines: Attractive Chromophores for Second-Order Nonlinear Optics and DSSCs. J. Phys. Chem. C 2019, 123, 13074–13082. [Google Scholar] [CrossRef]
- Belviso, S.; Cammarota, F.; Rossano, R.; Lelj, F. Effect of Polyfluorination on Self-Assembling and Electronic Properties of Thioalkyl-Porphyrazines. J. Porphyr. Phthalocyanines 2016, 20, 223–233. [Google Scholar] [CrossRef]
- Wavefunction, Inc. Spartan’02; Wavefunction, Inc.: Irvine, CA, USA, 2019.
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16; Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Singhal, A. Modern Information Retrieval: A Brief Overview. IEEE Data Eng. Bull. 2001, 24, 35–43. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Substrate | Catalyst | Product | Conversion (%) 1 | ee (%) 2 |
|---|---|---|---|---|---|
| 1 | 14 | (R,Sa)-9 | 18 | 40 | 6 |
| 2 | 14 | (R,Sa)-3 | 18 | 60 | 28 |
| 3 | 17 | (R,Sa)-9 | 19 | 60 | 8 |
| 4 | 17 | (R,Sa)-3 | 19 | 70 | 20 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Summa, A.; Scafato, P.; Belviso, S.; Monaco, G.; Zanasi, R.; Longhi, G.; Abbate, S.; Superchi, S. Synthesis and Stereochemical Characterization of a Novel Chiral α-Tetrazole Binaphthylazepine Organocatalyst. Molecules 2022, 27, 5113. https://doi.org/10.3390/molecules27165113
Summa A, Scafato P, Belviso S, Monaco G, Zanasi R, Longhi G, Abbate S, Superchi S. Synthesis and Stereochemical Characterization of a Novel Chiral α-Tetrazole Binaphthylazepine Organocatalyst. Molecules. 2022; 27(16):5113. https://doi.org/10.3390/molecules27165113
Chicago/Turabian StyleSumma, Assunta, Patrizia Scafato, Sandra Belviso, Guglielmo Monaco, Riccardo Zanasi, Giovanna Longhi, Sergio Abbate, and Stefano Superchi. 2022. "Synthesis and Stereochemical Characterization of a Novel Chiral α-Tetrazole Binaphthylazepine Organocatalyst" Molecules 27, no. 16: 5113. https://doi.org/10.3390/molecules27165113
APA StyleSumma, A., Scafato, P., Belviso, S., Monaco, G., Zanasi, R., Longhi, G., Abbate, S., & Superchi, S. (2022). Synthesis and Stereochemical Characterization of a Novel Chiral α-Tetrazole Binaphthylazepine Organocatalyst. Molecules, 27(16), 5113. https://doi.org/10.3390/molecules27165113