
Targeting Glutaminase by Natural Compounds: Structure-Based Virtual Screening and Molecular Dynamics Simulation Approach to Suppress Cancer Progression

 , , , , and
, , , , and
Abstract
1. Introduction
2. Methodology
2.1. Protein Preparation
2.2. Compound Library Preparation
2.3. Virtual Screening (VS) and Molecular Docking
2.4. Physiochemical and Drug-Likeness Properties Prediction
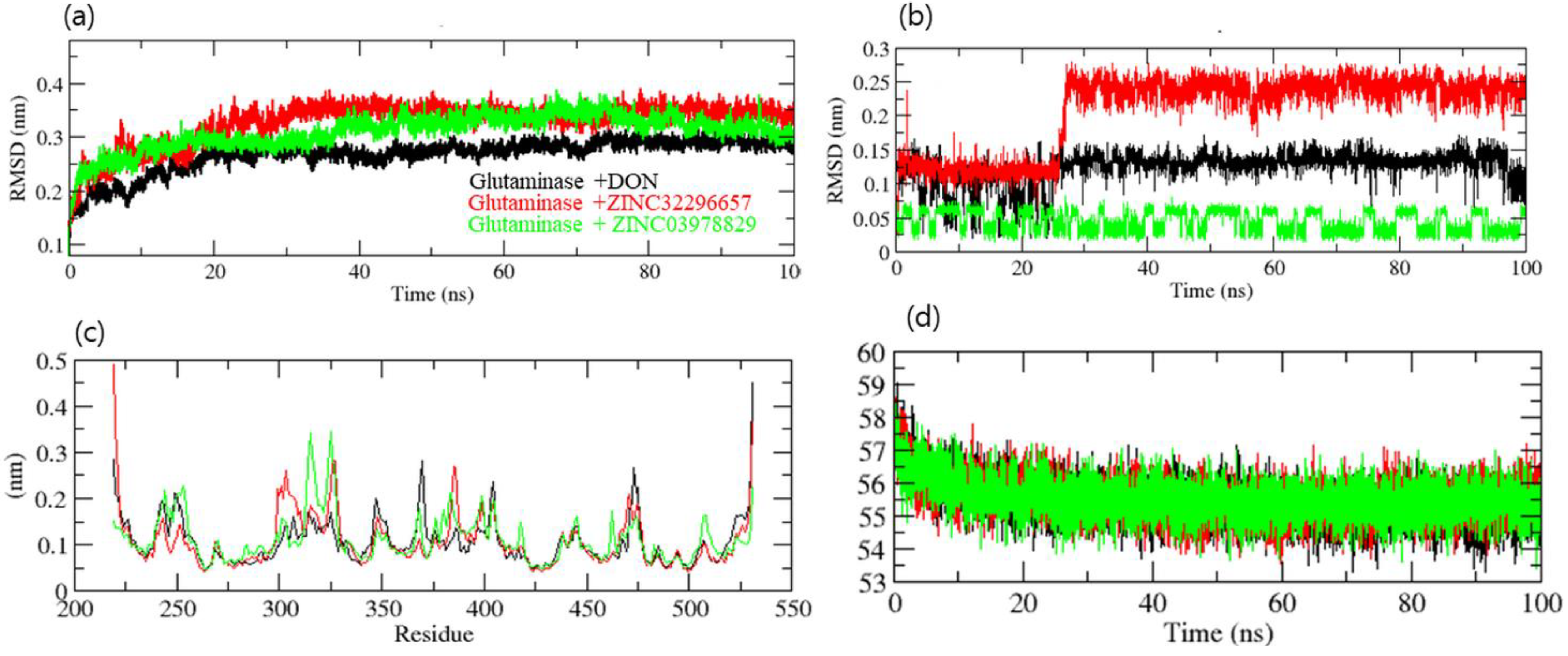
2.5. Molecular Dynamics Simulation
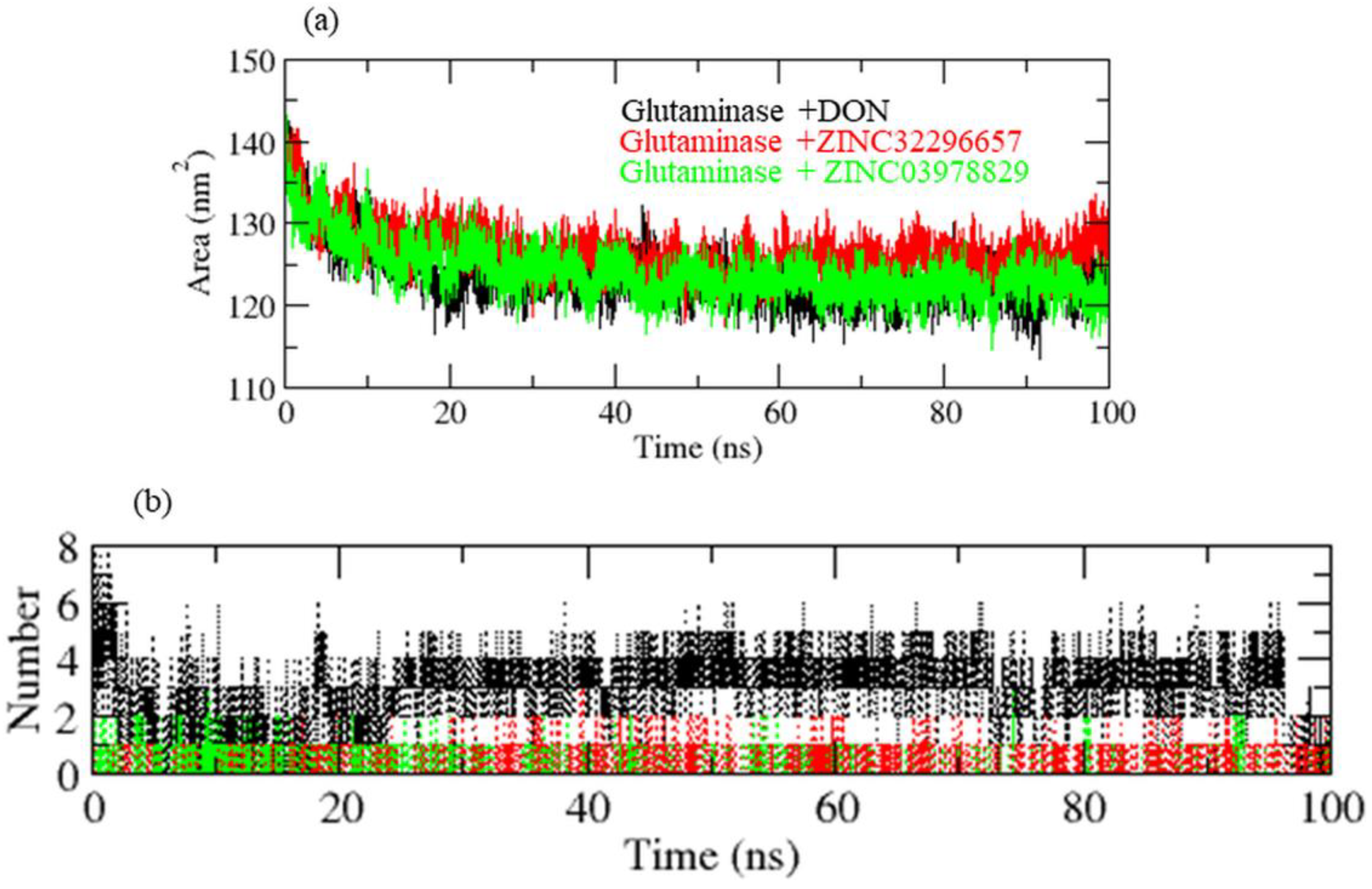
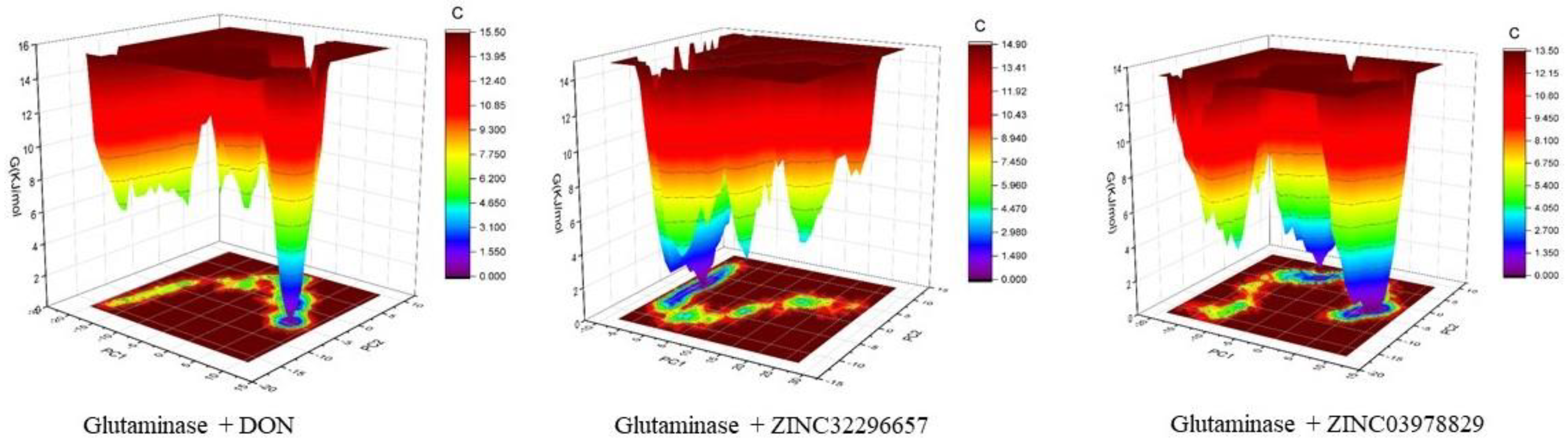
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| GLU | Glutamine |
| GLS | Glutaminase |
| TCM | Traditional Chinese medicine |
| BE | Binding energy |
| MD | Molecular dynamics |
| CADD | Computer-assisted drug design |
| VS | Virtual screening |
| DON | 6-Diazo-5-Oxo-L-Norleucine |
| RMSD | Root mean square deviation |
| RMSF | Root mean square fluctuation |
| SASA | Solvent-accessible surface area |
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Boroughs, L.K.; DeBerardinis, R.J. Metabolic pathways promoting cancer cell survival and growth. Nat. Cell Biol. 2015, 17, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Masisi, B.K.; El Ansari, R.; Alfarsi, L.; Rakha, E.A.; Green, A.R.; Craze, M.L. The role of glutaminase in cancer. Histopathology 2020, 76, 498–508. [Google Scholar] [CrossRef]
- Cheng, T.; Sudderth, J.; Yang, C.; Mullen, A.R.; Jin, E.S.; Mates, J.M.; DeBerardinis, R.J. Pyruvate carboxylase is required for glutamine-independent growth of tumor cells. Proc. Natl. Acad. Sci. USA 2011, 108, 8674–8679. [Google Scholar] [CrossRef]
- Szeliga, M.; Bogacinska-Karas, M.; Rozycka, A.; Hilgier, W.; Marquez, J.; Albrecht, J. Silencing of GLS and overexpression of GLS2 genes cooperate in decreasing the proliferation and viability of glioblastoma cells. Tumour Biol. 2014, 35, 1855–1862. [Google Scholar] [CrossRef]
- van den Heuvel, A.P.; Jing, J.; Wooster, R.F.; Bachman, K.E. Analysis of glutamine dependency in non-small cell lung cancer: GLS1 splice variant GAC is essential for cancer cell growth. Cancer Biol. Ther. 2012, 13, 1185–1194. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, J.; Zhao, Y.; Yue, X.; Zhu, Y.; Wang, X.; Wu, H.; Blanco, F.; Li, S.; Bhanot, G.; et al. Glutaminase 2 is a novel negative regulator of small GTPase Rac1 and mediates p53 function in suppressing metastasis. eLife 2016, 5, e10727. [Google Scholar] [CrossRef]
- Wang, J.B.; Erickson, J.W.; Fuji, R.; Ramachandran, S.; Gao, P.; Dinavahi, R.; Wilson, K.F.; Ambrosio, A.L.; Dias, S.M.; Dang, C.V.; et al. Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell 2010, 18, 207–219. [Google Scholar] [CrossRef]
- Le, A.; Lane, A.N.; Hamaker, M.; Bose, S.; Gouw, A.; Barbi, J.; Tsukamoto, T.; Rojas, C.J.; Slusher, B.S.; Zhang, H.; et al. Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab. 2012, 15, 110–121. [Google Scholar] [CrossRef]
- Wicker, C.A.; Hunt, B.G.; Krishnan, S.; Aziz, K.; Parajuli, S.; Palackdharry, S.; Elaban, W.R.; Wise-Draper, T.M.; Mills, G.B.; Waltz, S.E.; et al. Glutaminase inhibition with telaglenastat (CB-839) improves treatment response in combination with ionizing radiation in head and neck squamous cell carcinoma models. Cancer Lett. 2021, 502, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.Y.; Zhang, L.L.; Ding, J.; Lu, J.J. Anticancer drug discovery from Chinese medicinal herbs. Chin. Med. 2018, 13, 35. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Lu, J.J.; Ding, J. Natural products in cancer therapy: Past, present and future. Nat. Prod. Bioprospect. 2021, 11, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; MacKerell, A.D., Jr. Computer-aided drug design methods. Methods Mol. Biol. 2017, 1520, 85–106. [Google Scholar] [CrossRef]
- Macalino, S.J.; Gosu, V.; Hong, S.; Choi, S. Role of computer-aided drug design in modern drug discovery. Arch. Pharm. Res. 2015, 38, 1686–1701. [Google Scholar] [CrossRef]
- Ehrman, T.M.; Barlow, D.J.; Hylands, P.J. Virtual screening of Chinese herbs with Random Forest. J. Chem. Inf. Model. 2007, 47, 264–278. [Google Scholar] [CrossRef]
- Lionta, E.; Spyrou, G.; Vassilatis, D.K.; Cournia, Z. Structure-based virtual screening for drug discovery: Principles, applications and recent advances. Curr. Top. Med. Chem. 2014, 14, 1923–1938. [Google Scholar] [CrossRef]
- Huey, R.; Morris, G.M.; Forli, S. Using AutoDock 4 and AutoDock vina with AutoDockTools: A tutorial. Scripps Res. Inst. Mol. Graph. Lab. 2012, 10550, 92037. [Google Scholar]
- Sander, T.; Freyss, J.; von Korff, M.; Rufener, C. DataWarrior: An open-source program for chemistry aware data visualization and analysis. J. Chem. Inf. Model. 2015, 55, 460–473. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Pol-Fachin, L.; Fernandes, C.L.; Verli, H. GROMOS96 43a1 performance on the characterization of glycoprotein conformational ensembles through molecular dynamics simulations. Carbohydr. Res. 2009, 344, 491–500. [Google Scholar] [CrossRef] [PubMed]
- Schuttelkopf, A.W.; van Aalten, D.M. PRODRG: A tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 1355–1363. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Thangavelu, K.; Chong, Q.Y.; Low, B.C.; Sivaraman, J. Structural basis for the active site inhibition mechanism of human kidney-type glutaminase (KGA). Sci. Rep. 2014, 4, 3827. [Google Scholar] [CrossRef]
- Copeland, R.A. Conformational adaptation in drug-target interactions and residence time. Future Med. Chem. 2011, 3, 1491–1501. [Google Scholar] [CrossRef]
- Wang, Y.; Xiao, J.; Suzek, T.O.; Zhang, J.; Wang, J.; Bryant, S.H. PubChem: A public information system for analyzing bioactivities of small molecules. Nucleic Acids Res. 2009, 37, W623–W633. [Google Scholar] [CrossRef]
- Li, X.J.; Kong, D.X.; Zhang, H.Y. Chemoinformatics approaches for traditional Chinese medicine research and case application in anticancer drug discovery. Curr. Drug Discov. Technol. 2010, 7, 22–31. [Google Scholar] [CrossRef]
- Jamal, S.; Scaria, V.; Open Source Drug Discovery Consortium. Data-mining of potential antitubercular activities from molecular ingredients of traditional Chinese medicines. PeerJ 2014, 2, e476. [Google Scholar] [CrossRef][Green Version]
- Dai, S.X.; Li, W.X.; Han, F.F.; Guo, Y.C.; Zheng, J.J.; Liu, J.Q.; Wang, Q.; Gao, Y.D.; Li, G.H.; Huang, J.F. In silico identification of anti-cancer compounds and plants from traditional Chinese medicine database. Sci. Rep. 2016, 6, 25462. [Google Scholar] [CrossRef]
- Ling, C.Q. Traditional Chinese medicine is a resource for drug discovery against 2019 novel coronavirus (SARS-CoV-2). J. Integr. Med. 2020, 18, 87–88. [Google Scholar] [CrossRef] [PubMed]
- Aier, I.; Varadwaj, P.K.; Raj, U. Structural insights into conformational stability of both wild-type and mutant EZH2 receptor. Sci. Rep. 2016, 6, 34984. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, R.E.; Haider, M.K. Hydrogen bonds in proteins: Role and strength. In eLS; John Wiley & Sons, Ltd.: Chichester, UK, 2001. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Name | Mol. wt | cLogP | cLogS | H-Acceptors | H-Donors | Drug-Likeness | Mutagenic | Tumorigenic | Reproductive Effective | Irritant | Drug Score |
|---|---|---|---|---|---|---|---|---|---|---|---|
| ZINC42835355 | 608.733 | 6.1432 | −6.513 | 8 | 2 | 4.6261 | none | none | none | none | 0.221249 |
| ZINC17465983 | 546.526 | 4.8554 | −7.85 | 10 | 6 | 1.0735 | none | high | low | none | 0.115695 |
| ZINC05823171 | 436.59 | 4.4475 | −4.742 | 4 | 2 | 0.4587 | none | none | none | none | 0.433376 |
| ZINC05618656 | 470.476 | 4.4782 | −4.586 | 7 | 6 | −0.47845 | none | none | high | none | 0.214637 |
| ZINC38800324 | 436.59 | 5.3389 | −6.055 | 4 | 2 | −0.61958 | none | none | none | none | 0.252781 |
| ZINC28642721 | 538.463 | 2.9812 | −6.422 | 10 | 6 | −0.73675 | none | none | none | none | 0.257962 |
| ZINC13384046 | 484.459 | 4.3143 | −5.366 | 8 | 7 | −0.75118 | high | high | none | none | 0.107838 |
| ZINC33830992 | 442.725 | 6.499 | −6.347 | 2 | 2 | −0.95276 | none | none | none | none | 0.189819 |
| ZINC32296657 | 438.606 | 4.4297 | −5.029 | 4 | 1 | −1.0323 | none | none | none | none | 0.323614 |
| ZINC05520028 | 538.463 | 4.68 | −8.842 | 10 | 6 | −1.1275 | none | none | none | high | 0.104357 |
| ZINC34124041 | 470.779 | 7.9297 | −7.327 | 2 | 0 | −3.2521 | none | none | none | none | 0.117694 |
| ZINC04097720 | 426.726 | 7.5888 | −6.968 | 1 | 0 | −3.3053 | none | none | none | none | 0.132645 |
| ZINC04722964 | 426.726 | 7.5888 | −6.968 | 1 | 0 | −3.3053 | none | none | none | none | 0.132645 |
| ZINC08214547 | 576.768 | 3.0443 | −5.279 | 8 | 4 | −3.6238 | none | none | none | none | 0.221118 |
| ZINC03978829 | 456.708 | 6.0021 | −6.111 | 3 | 2 | −3.658 | none | none | none | none | 0.164992 |
| ZINC33831792 | 422.694 | 7.3798 | −6.44 | 1 | 0 | −4.1072 | none | none | none | none | 0.140914 |
| ZINC31165761 | 424.71 | 7.4843 | −6.704 | 1 | 0 | −4.5197 | none | none | none | none | 0.134382 |
| ZINC06041333 | 632.879 | 7.6697 | −7.838 | 6 | 2 | −6.334 | none | none | none | none | 0.082226 |
| ZINC33831775 | 424.71 | 7.4221 | −6.687 | 1 | 0 | −6.4 | none | none | none | none | 0.133989 |
| ZINC05884271 | 514.441 | 4.4706 | −7.734 | 10 | 6 | −6.6779 | none | high | none | none | 0.094558 |
| DON | 145.157 | −2.6809 | −0.868 | 4 | 2 | −19.893 | none | none | none | high | 0.295486 |
| Name | Mol. Wt. | ALogP | Rotatable Bonds | Molecular_Polar Surface Area |
|---|---|---|---|---|
| ZINC31165761 | 424.702 | 7.404 | 0 | 17.07 |
| ZINC32296657 | 438.599 | 5.328 | 2 | 63.6 |
| ZINC33830992 | 442.717 | 6.451 | 0 | 40.46 |
| ZINC33831775 | 424.702 | 7.449 | 0 | 17.07 |
| ZINC33831792 | 424.702 | 7.598 | 0 | 17.07 |
| ZINC34124041 | 470.77 | 7.865 | 2 | 26.3 |
| ZINC38800324 | 436.583 | 5.522 | 0 | 74.6 |
| ZINC03978829 | 455.692 | 5.018 | 1 | 60.36 |
| ZINC04097720 | 426.717 | 7.443 | 0 | 17.07 |
| ZINC04722964 | 426.717 | 7.443 | 0 | 17.07 |
| ZINC05823171 | 436.583 | 4.261 | 0 | 74.6 |
| ZINC08214547 | 576.761 | 2.887 | 3 | 117.84 |
| DON | 144.149 | −2.354 | 4 | 83.22 |
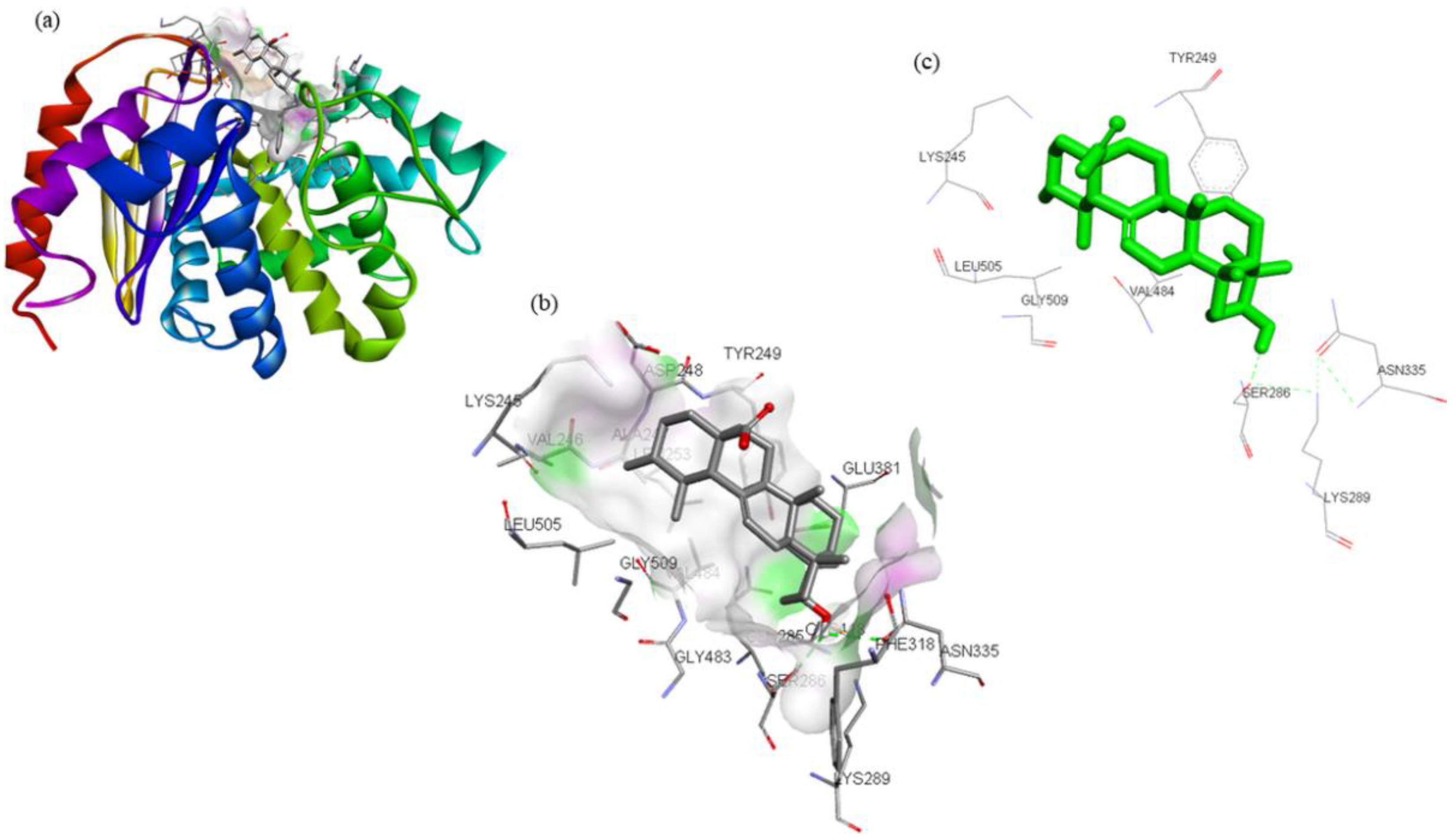
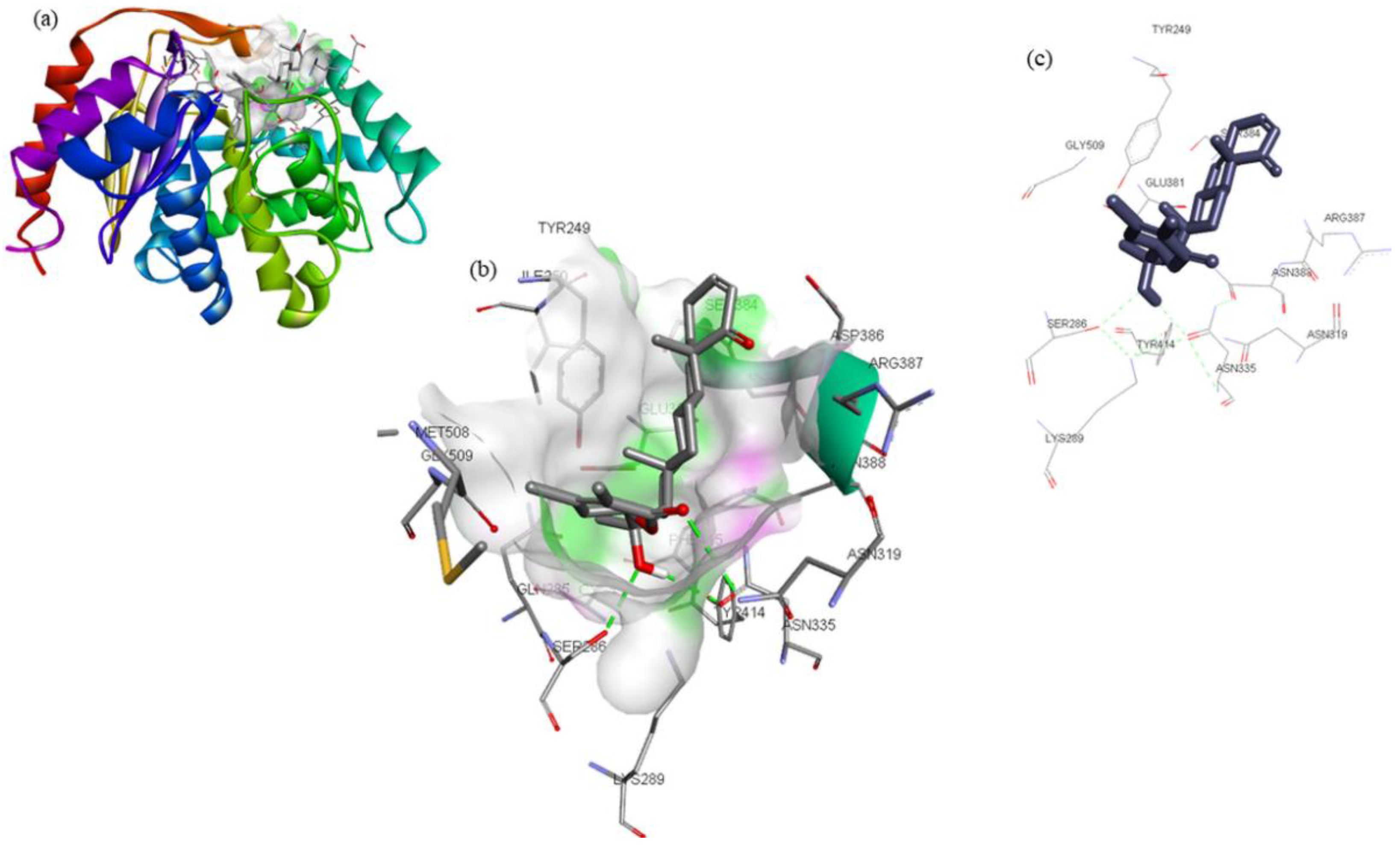
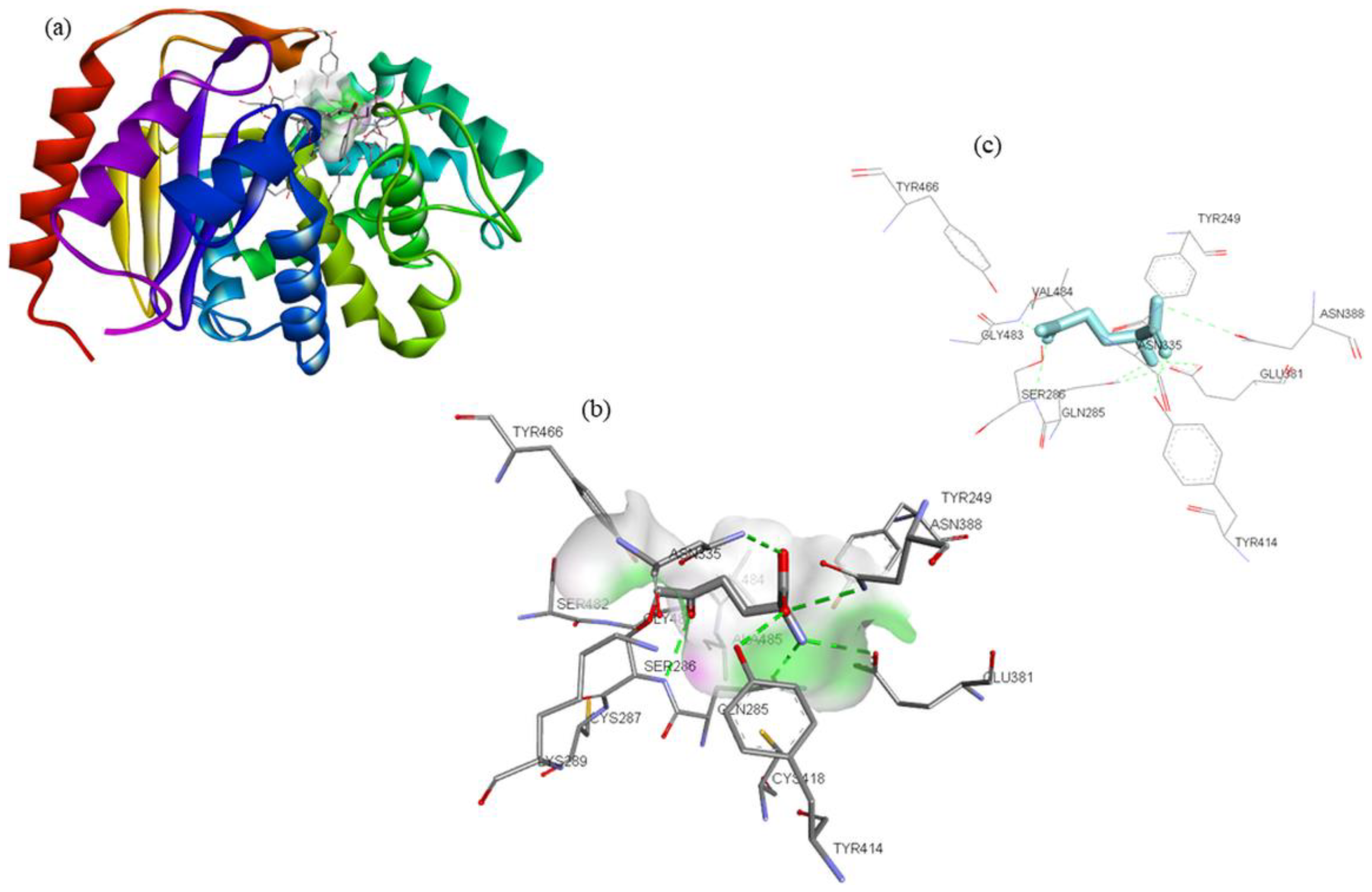
| Compounds | Structure | Target | Binding Energy (kcal/mol) | Interacting Residues |
|---|---|---|---|---|
| ZINC03978829 |  | GLS | −9.3 | Lys245, Tyr249, Ser286, Lys289, Asn335, Val484, Leu505, and Gly509 |
| ZINC32296657 |  | −9.7 | Tyr249, Ser286, Lys289, Asn319, Asn335, Glu381, Ser384, Arg387, Asn388, Tyr414, and Gly509 | |
| DON * |  | −4.7 | Tyr249, Gln285, Ser286, Asn335, Glu381, Asn388, Tyr414, Tyr466, Gly483, and Val484 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tabrez, S.; Zughaibi, T.A.; Hoque, M.; Suhail, M.; Khan, M.I.; Khan, A.U. Targeting Glutaminase by Natural Compounds: Structure-Based Virtual Screening and Molecular Dynamics Simulation Approach to Suppress Cancer Progression. Molecules 2022, 27, 5042. https://doi.org/10.3390/molecules27155042
Tabrez S, Zughaibi TA, Hoque M, Suhail M, Khan MI, Khan AU. Targeting Glutaminase by Natural Compounds: Structure-Based Virtual Screening and Molecular Dynamics Simulation Approach to Suppress Cancer Progression. Molecules. 2022; 27(15):5042. https://doi.org/10.3390/molecules27155042
Chicago/Turabian StyleTabrez, Shams, Torki A. Zughaibi, Mehboob Hoque, Mohd Suhail, Mohammad Imran Khan, and Azhar U. Khan. 2022. "Targeting Glutaminase by Natural Compounds: Structure-Based Virtual Screening and Molecular Dynamics Simulation Approach to Suppress Cancer Progression" Molecules 27, no. 15: 5042. https://doi.org/10.3390/molecules27155042
APA StyleTabrez, S., Zughaibi, T. A., Hoque, M., Suhail, M., Khan, M. I., & Khan, A. U. (2022). Targeting Glutaminase by Natural Compounds: Structure-Based Virtual Screening and Molecular Dynamics Simulation Approach to Suppress Cancer Progression. Molecules, 27(15), 5042. https://doi.org/10.3390/molecules27155042