2.1. XRD Analysis
XRD analysis allowed the obtainment of the diffraction patterns of the phases formed during the nanocomposite’s development (PW
0.5, PW
1 and PW
2), as shown in
Figure 1a. After 0.5 h and 1 h of synthesis (PW
0.5 and PW
1, respectively), the aniline polymerization reaction in the presence of O
2, HCl, and ammonium persulfate (APS) resulted in the formation of individual phases assigned to ES–PANI, aniline hydrochloride and WO
3·2H
2O, in addition to residual metallic W (Equation (1)).
The semi-crystalline diffraction pattern of ES–PANI was not clearly observed in the nanocomposite forms. However, its most intense diffraction peaks were observed between 2
θ = 20°–40°. As a semi-crystalline material, the contribution of the ES–PANI phase was observed and characterized by the non-crystalline halo located basically in the same angular region [
19].
The formation of aniline hydrochloride phase was observed in PW
0.5 and PW
1 nanocomposites as a non-polymerized doped monomer. This phase presented diffraction peaks at 2
θ = 10.6°, 21.0°, 22.1°, 22.7° and 28.1° [
20].
Figure 1b shows the disappearance of this phase when the time of synthesis was increased for the formation of the PW
2 nanocomposite. However, only by increasing the concentration of APS, and after 2 h of synthesis, the aniline hydrochloride monomers were fully polymerized to form the ES–PANI phase.
The metallic W phase was easily oxidized to tungsten trioxide (WO
3). However, this phase was not observed in the prepared nanocomposites. Instead, a di-hydrated tungsten oxide (WO
3·2H
2O) resulted from the oxidation process of metallic W during the polymerization of aniline. Different tungsten oxides have been reported in scientific literature. The obtainment of WO
3·2H
2O suggested that the metallic W was oxidized by APS/O
2 and interacted with water molecules from the solution. This phase was clearly identified [
21], presenting diffraction peaks at 2
θ = 12.8°, 23.7°, 24.1°, 27.0° and 27.4°, which corresponded to the planes (010), (001), (200), (011) and (210), respectively. Similarly, the metallic W phase was also identified in the PW
0.5 and PW
1 nanocomposites with diffraction peaks at 2
θ = 40.3°, 58.3°, 73.2° and 87.0° [
22]. After 2 h of synthesis, and using an increased concentration of APS, the metallic W phase was completely converted into WO
3·2H
2O (
Figure 1b).
Changes in diffraction peak intensities were observed as a function of the time of synthesis. A decreased intensity of the diffraction peaks of aniline hydrochloride phase was observed as a function of time, and disappeared completely after 2 h (
Figure 1b). No angular shifts in 2
θ were observed, revealing that the crystalline structure and unit cells of all nanocomposite phases were maintained when the metallic W was converted to WO
3·2H
2O, as well as when the aniline hydrochloride monomers were polymerized. Then, after 2 h, the nanocomposite formed by ES–PANI and WO
3·2H
2O (PW
2) was successfully obtained.
Nanocomposites formed by ES–PANI and tungsten oxides have been applied in several technological applications [
14,
15,
23], and WO
3 is the most reported form. The obtainment of this structure usually applies sodium tungstate di-hydrated (Na
2WO
4·2H
2O), hydrochloric acid, oxalic acid, and deionized water. The system is then heated to remove water molecules [
11,
17,
18]. However, in the methodology of preparation proposed herein, the WO
3·2H
2O phase was simultaneously obtained during the polymerization of aniline using metallic W as precursor. The XRD results showed that the WO
3·2H
2O phase was dependent both on the time of synthesis and APS concentration. This result indicated an alternative synthesis route to obtain PANI–WO
3·2H
2O nanocomposite without using Na
2WO
4·2H
2O as a precursor material.
2.2. Structural Model of the Prepared Nanocomposites
The crystal structure of the aniline tetramer [
24] was used as initial parameters consisting of four monomers located along the
z-direction. Some calculations reported elsewhere [
25] were also considered. The crystal structure of WO
3·2H
2O was based on a previous report [
21] and allowed the obtainment of its atomic coordinates based on the insertion of water molecules. The correction for the Coulombian interaction (DFT+U) for strongly correlated systems was considered and allowed the obtainment of a more accurate model with good representation of the
gap energy.
From the DFT+U calculation, the structure of ES–PANI (doped with Cl
− counter ions) layered on a WO
3·2H
2O plate was obtained after geometric optimization. Two arrangements were chosen to calculate the electronic properties of the nanocomposite. The first one was based on a supercell considering a vacuum region of 15.55 Å between two adjacent layers with constant total energy, allowing the evaluation of the phase interactions along the
xz-plane (
Figure 2a). The second calculation was based on the bulk structural arrangement (
Figure 2b). In addition, the same structures were analyzed as undoped forms, resulting in 4 models, labeled as PWO–ClS (doped system surface), PWO–NS (undoped system surface), PWO–ClB (doped system bulk) and PWO–NB (undoped system bulk).
The bond lengths (
Table 1) of the nanocomposite phases were evaluated in all proposed systems. The bond lengths in polymer and oxide phases are shown in
Figure 3a–d.
The binding energy of the PWO nanocomposite was calculated to access the stability of the phase interactions (Equation (2)):
where
Etc represents the total energy of the nanocomposite, and
Ep and
Eo represent the calculated energy of the individual phases.
The calculated binding energy of the PWO–ClS system was found around –0.109 eV, indicating a stability in the interaction between both phases, which occurred about 0.60 Å from the plane formed by the water molecules. Similarly, the calculated binding energy of the PWO–NS system was found around −0.103 eV, showing less stability in the interaction of phases when compared to the PWO–ClS nanocomposite.
The individual structures composing the PWO–ClB nanocomposite (
b = 14.88 Å (
Figure 2b)) preserved their configurations. No chemical bonds between phases were observed, and a negative binding energy of −0.090 eV was accessed, revealing stability. Similarly, the binding energy of the PWO–NB system was found around −0.237 eV, showing higher stability when compared to the other evaluated nanocomposites. Due to the Coulomb interactions, a difference in the length of the internal bonds of each phase was observed when compared to those of their isolated forms.
The polymer structure was rotated around 28° and attracted by 2 oxide layers upper and lower to the plane of the polymer chain. Hydrogen bonds were observed between the Cl and the H atoms of the upper layer of the water molecules. A plane in the
xz-direction was considered at the threshold separating the phases in the PWO–ClB nanocomposite (two inside the unit cell and another one between two repeated cells along the
y-direction). Then, a distance between the polymer phase and the upper and lower oxide phases was found to be ∆
1 = 1.17 Å and ∆
2 = 1.78 Å, respectively. Similarly, the structure of the undoped systems was obtained based on the same unit cell dimensions for surface area and bulk systems.
Table 2 shows the distances between the WO
3·2H
2O layers and the polymer phase in all systems, as well as the distances between the hydrogen bonds and Cl (
dCl−H), and water molecules (
dH−O).
The distance between the polymer phase and the oxide layer increased when the systems were relaxed to the bulk form. An interaction between the polymeric phase and both upper and bottom oxide layers was observed. Structural rearrangements were also observed by changes in hydrogen bond lengths from 1.36 Å to 1.89 Å, and from 1.36 Å to 2.02 Å in the doped and undoped structures, respectively.
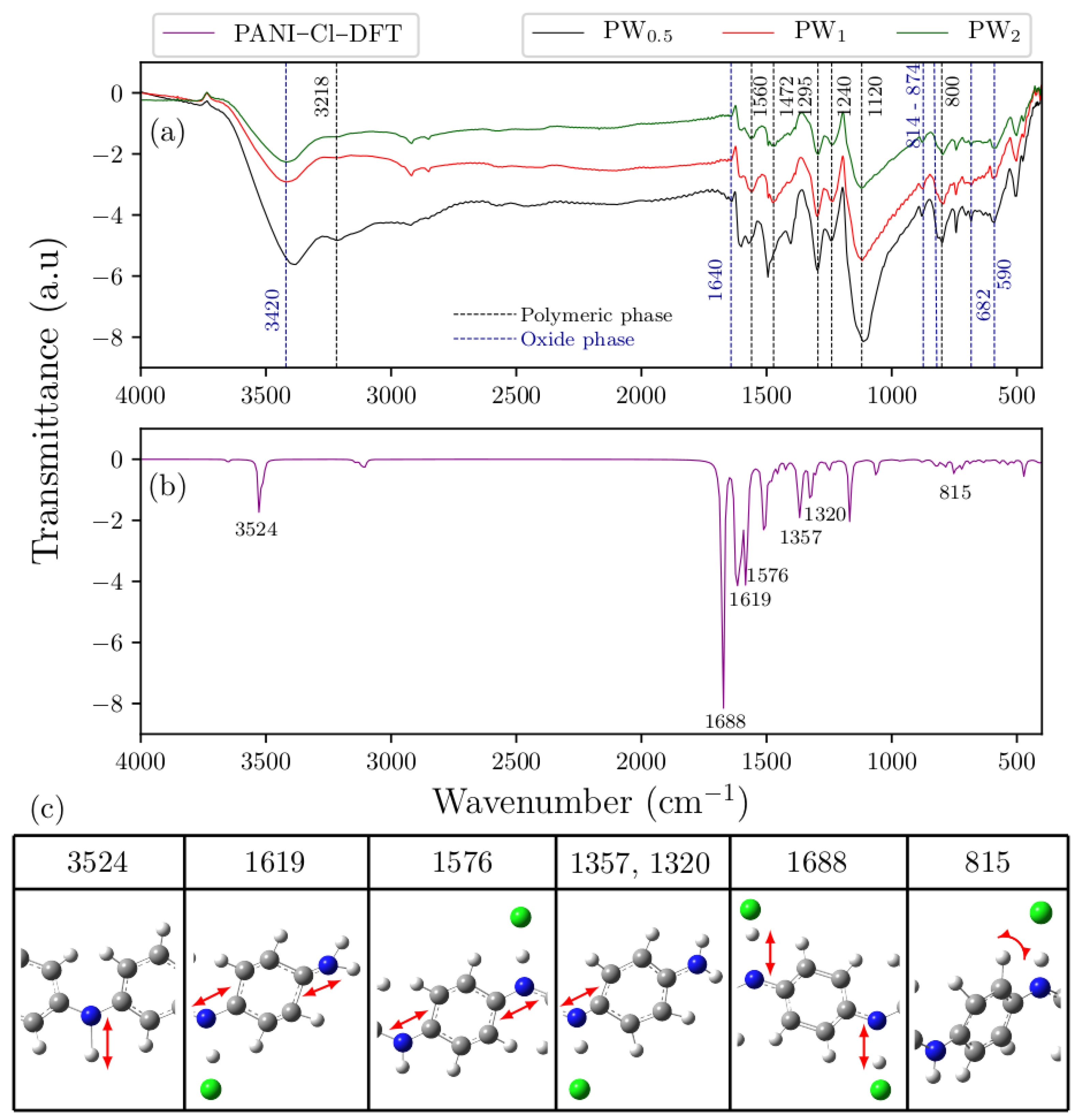
2.3. FTIR Analysis
Figure 4a,b shows the experimental FTIR spectra of PW
0.5, PW
1 and PW
2 nanocomposites, as well as the DFT–based spectrum of ES–PANI polymer phase. The identified vibrational modes are highlighted in
Figure 4c.
The ES–PANI polymer phase spectrum presented 7 main absorption bands at 3218 cm
−1, 1560 cm
−1, 1472 cm
−1, 1295 cm
−1, 1240 cm
−1, 1120 cm
−1 and 800 cm
−1. The band located at 3218 cm
−1 was assigned to the symmetric stretching of the N–H bond [
26]. This band also showed a redshift in the theoretical spectrum to 3524 cm
−1. The stretching of the quinoid and benzenoid structures were observed, respectively, at 1560 cm
−1 and 1472 cm
−1, allowing the characterization of the main molecular structure of ES–PANI. These bands presented a redshift, respectively, to 1619 cm
−1 and 1576 cm
−1 in the theoretical spectrum [
27]. The absorptions at 1295 cm
−1, 1240 cm
−1 and 1120 cm
−1 were assigned to the stretching of the C–N
+ bond of the bipolaronic structure, and to the N–H
+ bond from the delocalized
π–electrons due to the protonation process [
28]. The bands related to the C–N
+ stretching were observed at 1357 cm
−1 and 1320 cm
−1 in the theoretical spectrum. However, the band related to the N–H
+ stretching showed higher intensity at 1688 cm
−1. Interestingly, with increasing the time of synthesis, the intensity of the bands in the range from 1295 cm
−1 to 1120 cm
−1 was decreased. The out–of–plane deformation of the C–H bonds of benzenoid rings [
13] was observed at 800 cm
−1 and at 815 cm
−1, respectively, in the experimental and theoretical spectra.
The ratio between the areas of the quinoid and benzenoid (Q/B) structures (1560 cm
−1 and 1462 cm
−1, respectively) was useful to estimate the doping level of ES–PANI as a function of the time of synthesis [
29]. The Q/B ratio was found to be 0.89 in all nanocomposites, suggesting that the PWO nanocomposite counter ion/chain distance observed in
Table 1 did not result in polymer deprotonation when compared to the distances of the isolated systems.
The doping of polyaniline occurs through the interaction of counter ions and polymer chain, and the doped state occurs in a proportion of quinoid and benzenoid structures (related to the structural defects from these interactions). The distance between polymer and counter ions may reveal a possible deprotonation, returning an insulating state polymer. However, this fact was improbable because the Q/B ratio was greater than 50%, confirming the conductive behavior of the polymer.
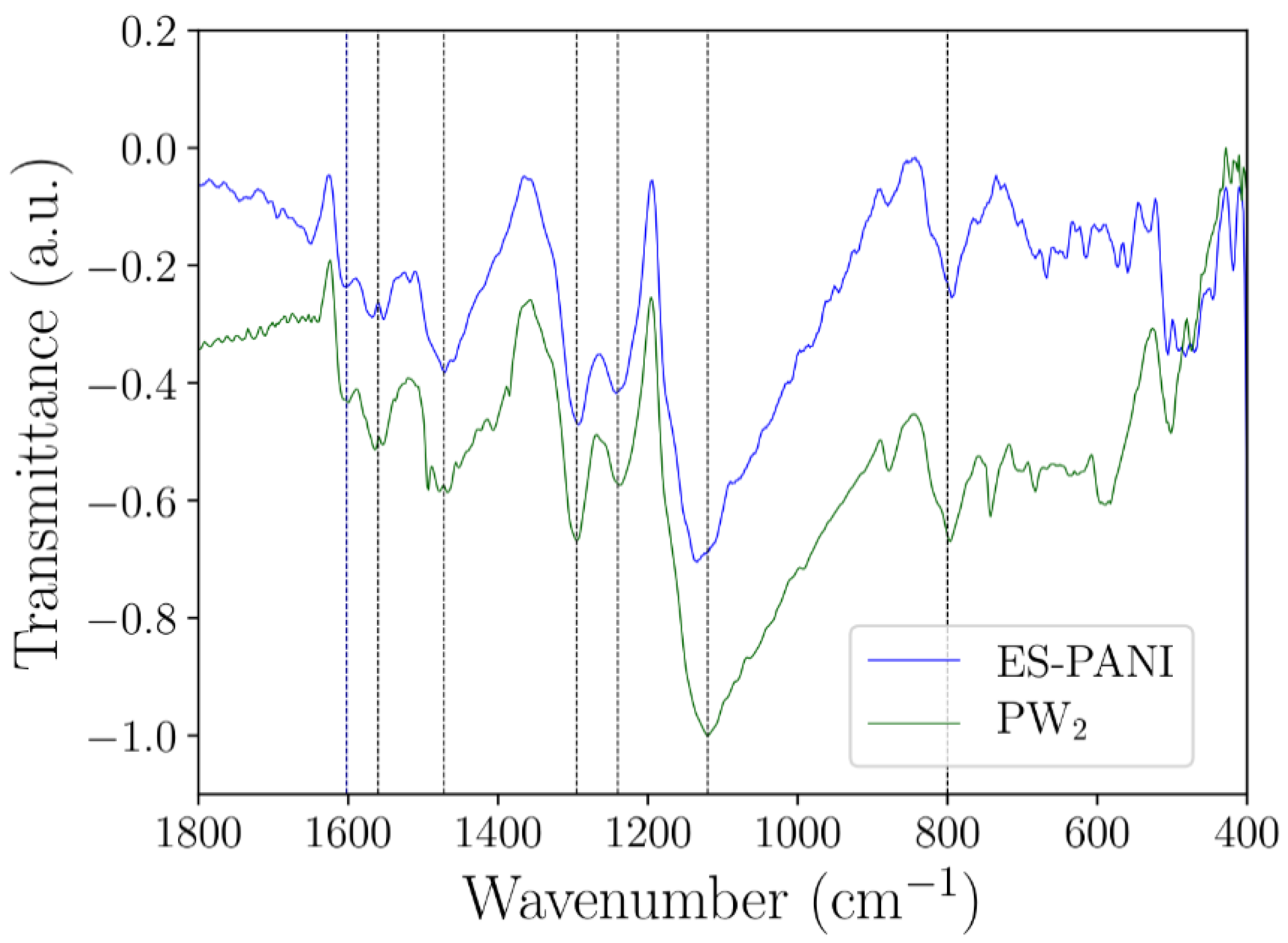
Figure 5 shows the comparison between the ES–PANI and PW
2 spectra. No peak shift (nor new absorption bands) was observed after the nanocomposite formation, pointing to a physical interaction between phases. Similar results were reported in [
13], showing the in-situ incorporation of aluminum oxide into the aniline polymerization medium. The resulting nanocomposite presented enhanced electrical conductivity and electrostatic interaction between phases, with no shift or new absorption FTIR bands.
Despite presenting a physical interaction, some nanocomposites based on polyaniline and inorganic particles can also present chemical interaction. The nanocomposite formed by polyaniline and copper oxide [
12] showed important absorption shifts on the FTIR spectrum. The chemical interaction between phases was also confirmed by the authors through DFT+U calculations.
Our results showed that all identified absorptions (
Table 3) were maintained when the time of synthesis was increased. No intensity variations in the absorption bands related to the protonation of ES–PANI were observed, revealing no deprotonation of the polymer chains in the nanocomposite form.
The absorptions at 3420 cm
−1, 1640 cm
−1, 874 cm
−1, 814 cm
−1, 682 cm
−1 and 590 cm
−1 were observed in the WO
3·2H
2O spectrum. The absorptions at 874 cm
−1 and 814 cm
−1 were assigned to the O–W–O antisymmetric stretching [
18], while the absorptions at 682 cm
−1 and 590 cm
−1 corresponded to the O–W–O symmetric stretching. The band at 1640 cm
−1 was attributed to the in-plane O–H angular deformation [
11], and the absorption at 3420 cm
−1 was assigned to the O–H stretching from both moisture and water molecules of WO
3·2H
2O [
30].
2.4. UV-VIS Analysis
UV-VIS analysis was useful to investigate the main electronic transitions of the nanocomposites and also to evaluate the influence of WO
3·2H
2O in the polymer structure. The UV-VIS spectra of all systems are shown in
Figure 6.
A number of 6 absorption bands resulted from the electronic transitions in all systems. The first absorption at 205 nm observed in the nanocomposite’s spectra was attributed to the W transitions, and a similar transition was also observed in the pure W spectra with a small blueshift.
Characteristic peaks from the polyaniline transitions were observed at 230 nm. The first one was attributed to the
π–π* transitions of the benzenoid structure of the polymer chain, followed by the nonlocal
π–π* transitions at 282 nm [
31]. Absorptions from the transitions of the oxidized W was observed from 350 nm to 445 nm, with contribution from both WO
3·2H
2O and the polymeric phase [
14,
32]. Similar absorption of the polaron
–π* transition was reported [
31]. The absorption at 840 nm was attributed to
π–polaron transitions associated with the polymer doping process [
31].
The π-polaron transitions occurred because, when doped, polymers such as PANI and its derivatives present new energy states located within the gap and close to the lowest energy state (HOMO), which contain a single unpaired electron. The energy level associated with the polaron represents a destabilized bonding orbital and, therefore, presents higher energy than that of HOMO, allowing the π-polaron and polaron-π* transitions, responsible for the conductive behavior of ES–PANI.
All absorptions highlighted in
Figure 6 are listed in
Table 4. The lower energy electronic transitions were associated with the polymer as well as its doped form, which occurred due to the creation of polaronic states.
2.5. Complex Impedance Spectroscopy
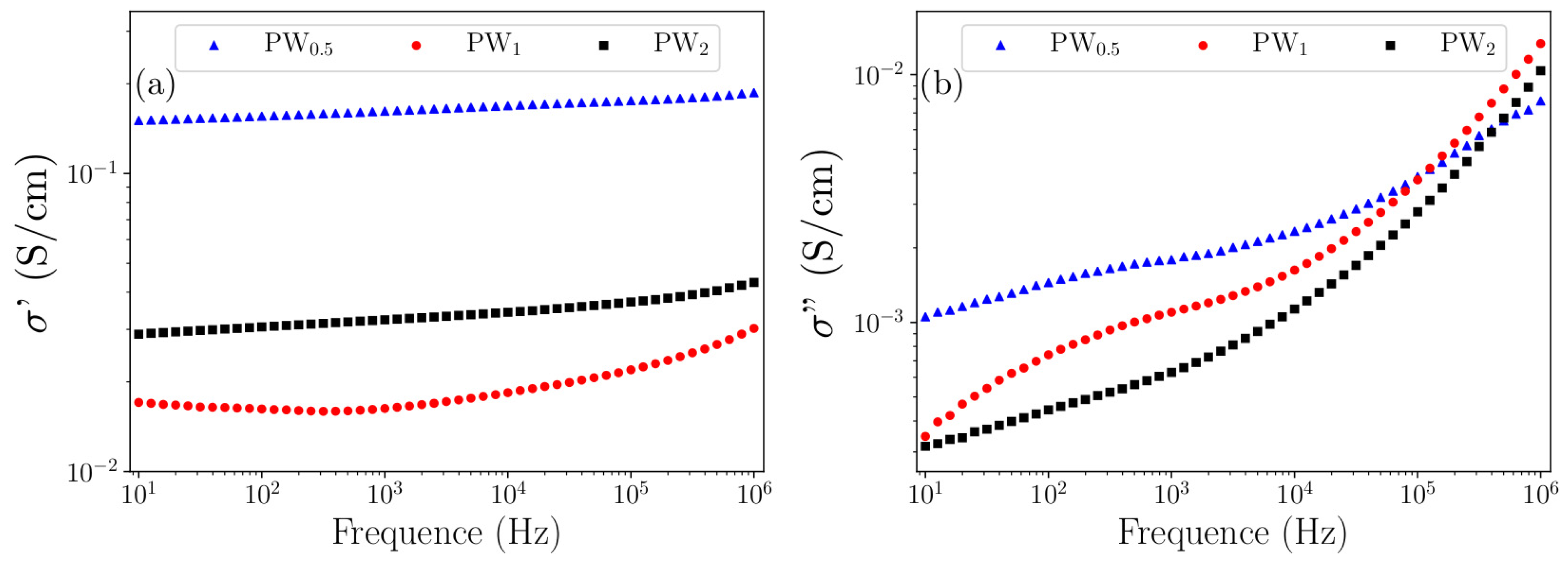
The electrical conductivity of the prepared nanocomposites was analyzed by Complex Impedance Spectroscopy.
Figure 7 shows (a) the dependence of the real and (b) imaginary parts of the complex conductivity as a function of frequency (Equations (3) and (4)):
where
and
represent, respectively, the real and imaginary dielectric permittivity calculated from the complex impedance Z (Equations (5) and (6)):
The real part represents the conduction in phase with the applied electric field, while the imaginary part presents the out-of-phase conduction. It can be observed from the plot of the real part that PW0.5 and PW2 did not present significant changes in the electrical current when the frequency was increased, showing for both nanocomposites a dc conductivity. However, PW1 presented an increase in electrical conductivity at high frequencies. This behavior was assumed to accord with the relation known as Jonscher universal power law, where the electrical conductivity is independent of the frequency when n = 0, and dependent when n > 0. This result is supported by the fact that at low frequencies the disordered regions acted with high resistance, resulting only in a constant conductivity. However, at high frequencies the rate of hopping between the conductive islands and/or between phases increased the conductivity.
It was not possible to assess whether the conductivity maxima showed relaxation peaks resulting from the electrical conduction by hopping due to the applied frequency range in the PW1 imaginary part (around 102 Hz). Thus, we suggested a combined electrical conduction in the nanocomposite form due to the doping characteristics of the polymer, the conduction of the metal phase, the type of ionic structure of the oxide phase, as well as the presence of water in the hydrated structure. This conduction is mostly dc from charge carriers generated through the charge defects of ES–PANI and from the free charge carriers coming from the remaining metallic W in PW0.5 and PW1. However, the electrical conduction is based on hopping between the interfaces when the energy is sufficient to break the potential barrier.
Table 5 presents the values of electrical conductivity of the prepared nanocomposites. PW
0.5 reached 1.4 × 10
−1 S/cm. For PW
1 and PW
2 the electrical conductivity values decreased by one order of magnitude, reaching 1.6 × 10
−2 and 2.9 × 10
−2 S/cm, respectively.
The XRD results showed previously that the metal phase was gradually converted to WO3·2H2O after 2 h of synthesis. As a result, a decreased electronic mobility was verified and assigned to the reduced free charges from the metallic W. This fact was due to its conversion to WO3·2H2O. Despite the interactions between WO3·2H2O and counter ions, the electrical conductivity of the nanocomposites was maintained, suggesting that the distancing of the counter ions observed in the theoretical PWO models in relation to ES–PANI did not result in deprotonation. Compared to the experimental conductivity data of the doped polymer phase, interaction between phases in PW0.5, PW1 and PW2 was observed, resulting in enhanced conductivity values when compared to the pure ES–PANI, as well as showing charge transfer that improved the electronic mobility.
2.6. Band Structure and Density of States (PDOS)
The energy band structures were calculated in the reciprocal space along (Z, Γ, Z) for the polymer system, (Γ, X, U, Z, Γ, S, Z) for the WO3·2H2O system and for all nanocomposites using (Γ, X, U, Z, Γ, S, Z), where (Z, Γ) corresponds to the polymeric chain growth direction of ES–PANI. For all systems, the Fermi energy was adopted as reference for the origin.
Figure 8a–c shows that the PANI–Cl system presented conductive material behavior, where the HOMO band was partially filled. This fact was due to the new energy states created by the addition of counter ions into the undoped PANI, allowing the transfer of electrons to lower energy states [
25]. As a result, the
gap energy was reduced from 2.00 eV (undoped PANI form [
25]) to 0.41 eV (ES–PANI form).
The PDOS projected by atoms facilitated access of the major contribution of Cl atoms, followed by C and N (largest contribution in the LUMO band), as well as the lowest contribution of H atoms. The maximum of the PDOS of the Cl atoms occurred at approximately −0.15 eV below the last occupied state of the HOMO band. Then, the electrons from the higher energy states decayed to the energy states generated by the Cl atoms. For this reason, a charge transfer was allowed between polymer chains and counter ions.
Figure 8d–f shows the band structure and PDOS projected by atoms and orbitals for the WO
3·2H
2O system. The insertion of the Hubbard correction provided an excellent result for the
gap energy of around 2.60 eV. Experimental
gap energy of WO
3·2H
2O and WO
3·H
2O obtained by UV-VIS using the Wood-Tauc method were reported between 2.00 eV and 2.40 eV [
14].
Figure 8e shows the distributed PDOS of the O atoms into 4 groups: O
tot represents the total electron density of oxygen, O
W is the density of oxygens in the
xz–plane, O
H2OW represents the H
2O molecules interacting with the W atoms in the
y-axis, and O
H2O is the water molecules forming the upper and bottom layers. The O atoms in the
xz–plane contributed significantly to the formation of the valence band. Marginal contribution of the O atoms from water, and perpendicular to the
xz–plane, was observed. The PDOS of the O atoms was mainly related to the
p–orbitals, as shown in
Figure 8f. The major contribution in the conduction band was related to the W atoms, followed by O atoms in the
xz–plane (where the
p and
d–orbitals present higher electron density). The electrons from the
d–orbitals of the W atoms migrated to the
p–orbitals of the unoccupied O atoms, so the
d–orbitals did not contribute significantly in the valence band.
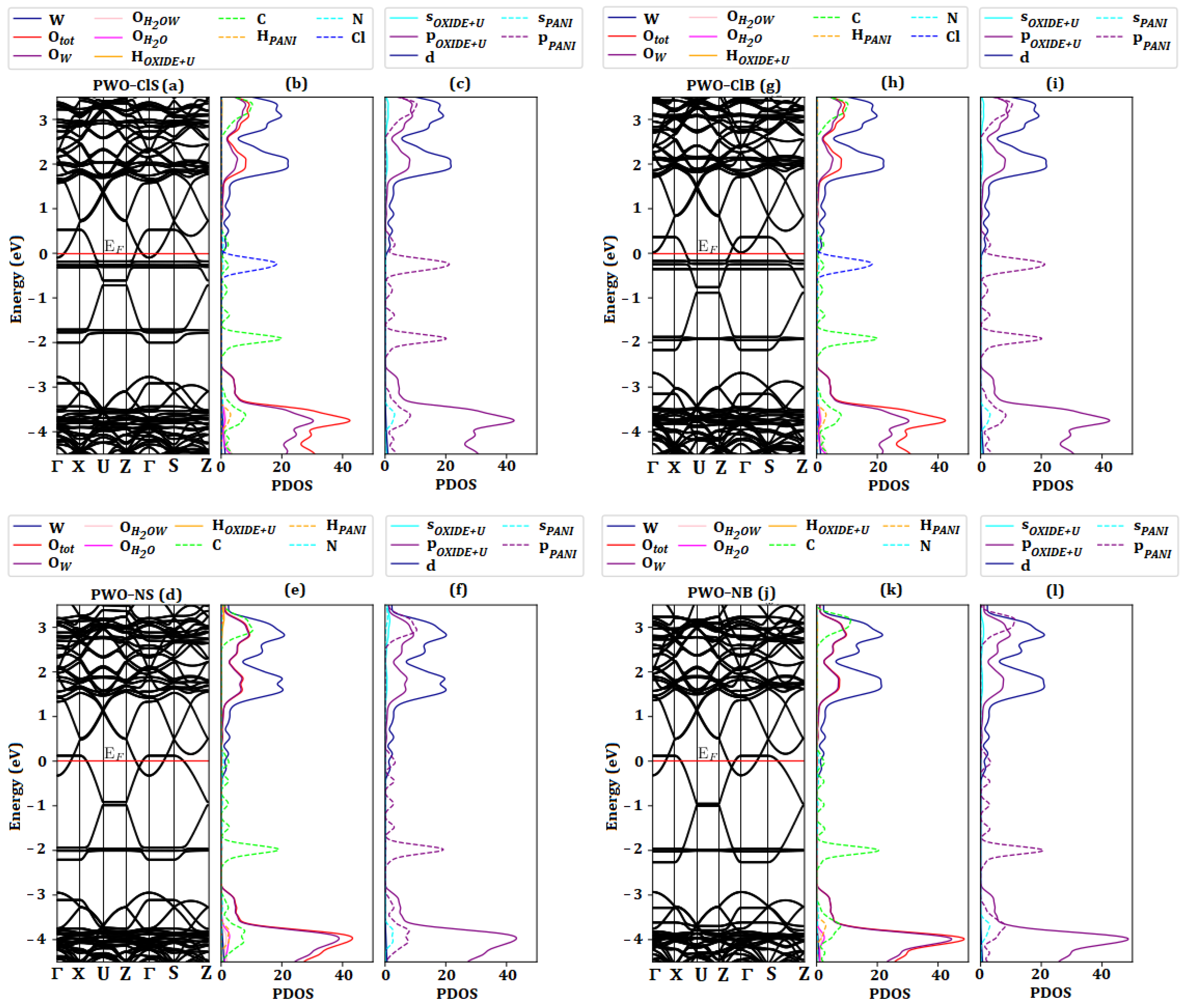
Figure 9a,d,g,j shows the energy band structure of all nanocomposites. The energy bands corresponding to the polymer phase were observed exactly in the region of the
gap energy corresponding to the WO
3·2H
2O phase. All systems were similar, except for the states from the Cl atoms, and besides the difference in the Fermi level caused by the variation of the unit cell. In the doped systems the last occupied valence band state of WO
3·2H
2O was now at the conduction band limit because the polymer phase had higher energy filled levels. Thus, after excitement, the electrons from the polymer phase migrated to the higher unoccupied energy states of WO
3·2H
2O. Comparing the systems of PWO–ClS and PWO–ClB, a small difference in the Fermi energy was observed and related to the unit cell bulk change (∼−0.15 eV). The energy difference in the undoped systems was similar. On the other hand, the doped systems were influenced by counter ions, increasing the energy of the valence band of the WO
3·2H
2O phase.
Figure 9 b,c,e,f,h,i,k and l show the PDOS for atoms and for orbitals of both surface and bulk systems. The O and W atoms presented the major contribution in the valence and conduction bands. However, the largest contribution in the
gap energy of the WO
3·2H
2O phase was related to the C, N and Cl (in doped systems, ES–PANI) atoms. The evaluation of the PDOS for orbitals revealed that the
p–orbitals of the polymer phase were responsible for promoting the electrons to the
d–orbitals of the W atoms (charge transfer between phases). For this reason, the polymer phase acted as a bridge in the
gap energy of the WO
3·2H
2O phase in the nanocomposite forms, behaving as a conducting material. Even with the absence of the Cl atom, the undoped nanocomposite system exhibited the same characteristics as that of the doped one. The interactions between the nanocomposite phases created charge transfers from the
p–orbitals of the polymer to the
d–orbitals of the oxide phase, increasing the electronic mobility. Thus, the contribution of the C and N orbitals of the polymer phase, as well as the W and O orbitals of the oxide phase increased the PDOS of the nanocomposites, improving its electrical conductivity.
2.7. Charge Density
The Lowdin charge was adopted as the charge distribution parameter in order to analyze the charge flow in nanocomposites.
Table 6 shows the Lowdin charge variation for PWO–ClS and PWO–NS systems. We represented Δ
CLowdin (PANI–Cl) here as the charge difference between the PWO–ClS nanocomposite and the PANI–Cl doped polymer, and Δ
CLowdin (WO
3·2H
2O) as the charge difference between the PWO–ClS nanocomposite and the WO
3·2H
2O phase. A decreased charge distribution of PANI–Cl of about 0.2459 was observed in relation to the nanocomposite form, and an equivalent increase of +0.2636 was observed in the WO
3·2H
2O phase. A difference in charge loss/gain between phases was also observed, which infringed the principle of charge conservation. This fact was due to imprecision in the bulk region regarding the position of each atom, resulting in some imprecision in the charge distribution calculation. However, our results revealed a satisfactory understanding of the charge mobility.
Similarly, the ΔCLowdin (PANI) was described as the charge difference between the PWO–NS nanocomposite and the undoped polymer, and the ΔCLowdin (WO3·2H2O) was assigned as the charge difference between the PWO–NS nanocomposite and the WO3·2H2O phase. A loss in charge distribution in the polymer phase of about −0.4961 (relative to the formed nanocomposite) was observed. On the other hand, an equivalent increase of +0.5226 was revealed in the WO3·2H2O phase. These values were twice those of the charge transfer between the doped state and the oxide phase.
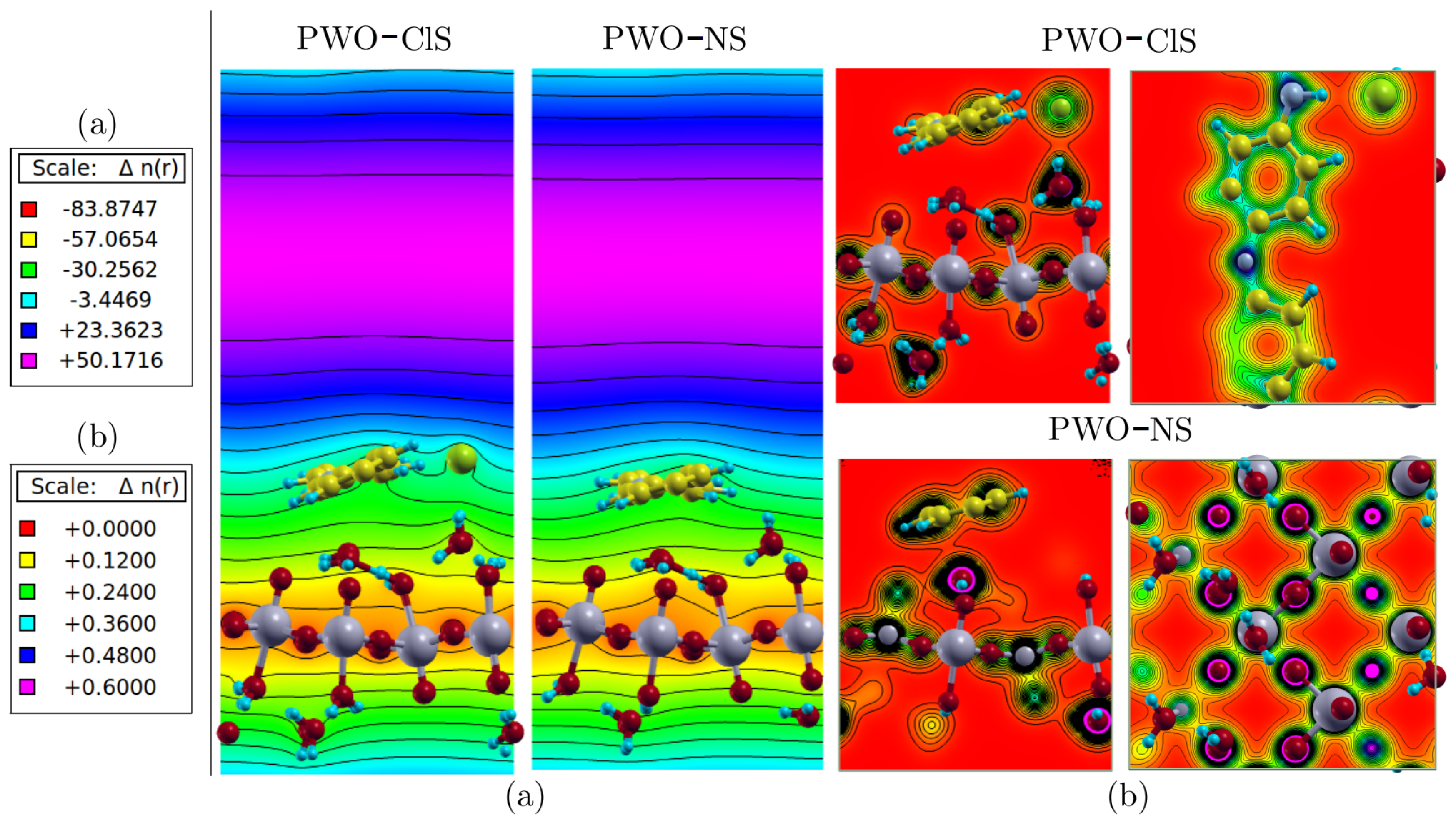
Figure 10 represents (a) the local ion potential map and (b) the charge distribution at the plane normal to the polymer chain growth, showing the electronic interactions between phases. The surface systems showed attractive ionic interactions between polyaniline and the WO
3·2H
2O surface. Higher electronic charge density in the O atom was revealed in the layer where polar W–O bonds along the
xz–plane was observed. The Cl atom caused a marginal distortion in ionic potential distribution when compared to the undoped structure.
The density in both systems showed well-localized behavior, as shown in
Figure 10a, and the highest density around the O atoms, followed by the C atoms along the polymer chain. However, intermediate regions of charge density were observed between the polymer phase and the Cl atom, as well as between the Cl atom and water molecules. In addition, an intermediate region between the water molecules and the oxygen atoms of the WO
3·2H
2O phase was also observed, as well as between the polymer chain and the WO
3·2H
2O phase. Based on color scale, as in
Figure 10b, these regions presented low electron density. In the case of doped conducting polymers by protonation, it is known that the counter ions effectively participate in the electronic conduction, contributing to the electron neutrality of the polymer chain. Then, the electronic conduction occurs by intra- and inter-chain mechanisms, in addition to the hopping between the conducting islands formed by the crystalline regions.
The results observed in
Figure 10 corroborate the experimental data observed by FTIR. The interaction occurring between polymer chains and WO
3·2H
2O phase was clearly electrostatic. The value of the attractive potential around −30.0 (atomic unit) is highlighted in green. This type of interaction resulted in the peak positions of the absorption bands in the FTIR spectra (
Figure 5), since no band shifts/new bands were observed.
2.8. Transmittance and Electrical Current
Quantum transmittance and electric current as a function of voltage was calculated using the
Want package [
33] implemented in the Quantum Espresso software [
34].
Figure 11a shows the plot of the quantum transmittance (2·e
2/h) of the proposed systems as a function of energy (eV), considering the EF = 0.0.
All systems but PANI presented transmittance around the Fermi level, with no gap energy (as seen in the band structure). Thus, PANI–Cl, PWO–ClS and PWO–NS nanocomposites behaved as conductors in terms of electronic charge transport. A region of gap energy of approximately 2.2 eV around the Fermi level emerged in the PANI system, resulting in zero transmittance from −0.2 eV to 2.0 eV. For this reason, we suggest that PANI behaved as an insulating material, since it was the representation of the undoped form (leucoemeraldine).
Similarly, the WO3·2H2O system showed a gap energy of 2.6 eV around the Fermi level, with null transmittance from −1.6 eV to 1.0 eV. The transmittance spectra of the nanocomposites were similar, with a low difference in the Fermi level region resulting from the low influence of the Cl atom on the electronic conduction. The transmittance contributions of each phase were clear as they were quite characteristic when compared to their isolated states. Although PANI and WO3·2H2O present relatively large gap energy, the formation of PWO–NS and PWO–ClS nanocomposites exhibited conducting behavior, showing that the polymeric phase could act as an electronic bridge to reduce the gap energy of the oxide phase.
Figure 11b shows the curves of electrical current (A) as a function of applied voltage (eV) for all proposed systems. The IxV curve of the WO
3·2H
2O system presented non-ohmic behavior from 0.0 eV to 2.0 eV. For higher voltage values the electrical current increased exponentially. The WO
3·2H
2O phase represented an insulating material with no electrical conduction at low voltages. Similarly, the undoped polymer phase presented electrical current from 0.0 eV to 0.37 eV. The electrical current increased linearly from this voltage value. The null energy range of the polymer phase was smaller due to its narrower
gap energy when compared to that of the oxide phase. Thus, it conducted electrical current over a smaller voltage range.
The ES–PANI and nanocomposite systems presented a characteristic curve of conducting materials, where the electrical current changed linearly with the applied voltage (typical ohmic behavior). A good approximation of the electronic properties was observed between the system formed only by PANI and WO3·2H2O (PWO) and the experimental PW system. Then, the final product ES–PANI–WO3·2H2O, which was prepared experimentally, presented enhanced conduction properties, and the PW2 nanocomposite exhibited higher conductivity than that of the individual phases.
2.9. Morphological Analysis
The morphology of the prepared PW
0.5 and PW
2 nanocomposites was evaluated by SEM images. The PW
0.5 nanocomposite (
Figure 12) clearly showed a morphology assigned to the polymeric phase (ES–PANI), constituted mainly of nanofibers [
12,
35]. Microplates of different sizes and thicknesses formed by the WO
3·2H
2O phase were also observed, as shown in
Figure 12a,b [
14]. The regions where ES–PANI was deposited on the WO
3·2H
2O microplates are highlighted in red, revealing a contact surface. The presence of nano-sticks morphology assigned to the aniline hydrochloride [
19] phase is highlighted in blue in
Figure 12a. In addition, the presence of the remaining metallic tungsten was also observed in
Figure 12c, corroborating the results from XRD analysis. The regularity of the morphology of the metallic W was also noted, due to the high symmetry of its crystal structure.
The PW
2 nanocomposite morphology
Figure 12d–f was similar to that of PW
0.5, showing predominantly the polymeric phase. However, the morphology of pure metallic W and aniline hydrochloride was not observed. The morphology assigned to the aniline hydrochloride in the PW
0.5 nanocomposite was not found in the PW
2 sample, corroborating the XRD results. The formation of WO
3·2H
2O [
14] was clearly evidenced, showing that the metallic W was oxidized. The phase interaction between WO
3·2H
2O and polyaniline was clearly observed, corroborating our previous results pointing to a purely electrostatic physical interaction. This observed physical interaction may also be related to the increased electrical conductivity of the PW
2 nanocomposite when compared to the pure ES–PANI, possibly due to new electronic conduction paths created in the nanocomposite material.


 ,
, 










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}