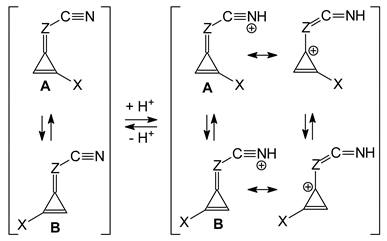
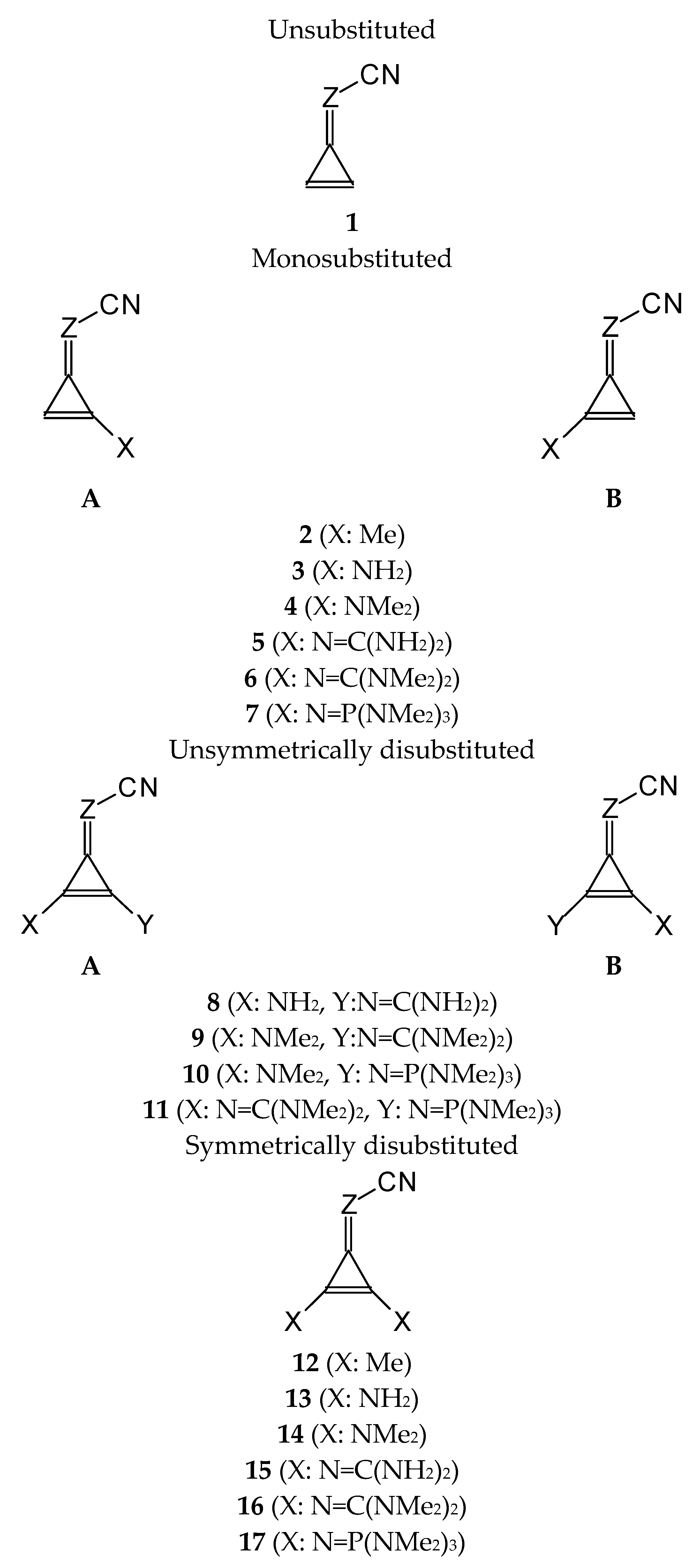
3.1. Unsubstituted Compounds (I.1 and II.1)
The unsubstituted bases
I.1 and
II.1 (
Figure 1) belong to the family of conjugated heterocompounds. In our previous work in this series [
9], we reported the gas-phase structures and acid-base properties of the symmetrically disubstituted derivatives
I.12-I.17 and
II.12-II.17. The non-symmetrical substitution leads to a much larger number of isomers, for which we expect similar properties. In particular, the favored site of protonation in the gas phase, without any doubt, is the cyano N atom for
I.1 and
II.1 as well as for
I.12-I.17 and
II.12-II.17. These nitriles display exceptional basicity in the gas phase when substituents are relatively strong electron donors, X or Y: NR
2, N=C(NR
2)
2, and N=P(NR
2)
3. The imino N-protonated form of
II.1 (ZH
+) and also of
II.12-II.17 can be neglected in the monoprotonation reaction. For
II.1, its Gibbs energy is higher (i.e., less stable) than that of the cyano N-protonated ion (C≡NH
+) by more than 50 kJ mol
−1 (
Table S1 in SM). Hence, the ZH
+ form of
II.1 by its low concentration (<< 1 ppm), can be neglected for our purpose. The same is true for other derivatives (
II.12-II.17), for which the
G-difference is also high (>15 kJ mol
−1).
The next property refers to the acid-base strength measured by gas-phase acidity-basicity parameters. Both the PA and GB of the cyano N atom in
I.1 and
II.1 were estimated in our previous work at the DFT, G2, and G2MP2 levels [
9]. The two parent compounds seem to be stronger bases in the gas phase than Me
2N–C≡N but weaker than Me
2N–CH=CH–C≡N and Me
2N–CH=N–C≡N [
8]. Although the calculated PAs of
I.1 and
II.1 (respectively, 885 and 892 kJ mol
−1,
Table S2 in SM) are lower than the limit for compounds classified as superbases (PA > 1000 kJ mol
−1) [
2,
23], attaching the methylenecyclopropene and cyclopropenimine groups to the nitrile strongly increases the PA of the cyano N atom (by ca. 200 kJ mol
−1), by comparison with the PAs of HC≡N calculated at the same DFT or G2 level.
The imino N atom of the C=N group in
II.1 plays a particular role in this conjugated system. Instead of decreasing the electronic substituent effect of the cyclopropenimine group in comparison to that of the methylenecyclopropene one, it favorably interacts with the electron-accepting (pulling) C≡N function. The electronegative imino N atom in
II.1 withdraws the labile π-electrons present in the cyclopropenimine part, and transmits them to C≡N (
Scheme S1 in SM). For the neutral and cyano N-protonated forms of
II.1, a larger number of resonance structures can be written than for
I.1, in connection with a more delocalized π-electron system [
24,
25]. Hence, the transmission of the electron-donating resonance effect of the cyclopropene cycle through the >C=N– bond to the cyano N-protonation site seems to be stronger in
II.1 than that through the >C=CH– bond in
I.1. Indeed, the DFT-estimated HOMEDs, being geometrical measures of electron delocalization in the conjugated systems, highlight the important differences in the resonance and bond length’s alternation between the neutral forms of
I.1 and
II.1, as well as between their neutral and cyano N-protonated forms [
9].
3.2. Isomerism in Mono- and Disubstituted Derivatives
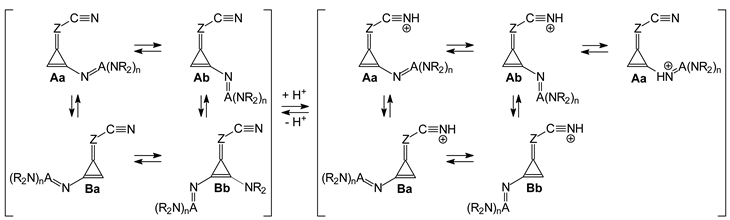
Isomeric phenomena possible in organic heterocompounds frequently influence the acid-base properties of individual isomers and isomeric mixtures. For this reason, they are considered significant for the mono- and disubstituted derivatives of N-bases investigated in this work. First, we considered the geometrical isomerism about the double C=Z bond in all unsymmetrically substituted derivatives, I.2-I.11 and II.2-II.11. Then, we also considered the rotational isomerism about the single C (cyclopropene)–N (substituent) bonds for derivatives I.5-I.11 and II.5-II.11 containing large substituents (N=C(NR2)2 and/or N=P(NR2)3 and abbreviated here as N=A(NR2)n, where A is C or P, and n is 2 or 3, respectively).
Rotation of the NR
2 and Me group(s) only slightly influences the stability of the conjugated system, and was not considered owing to the marginal geometric and thermochemical effects in comparison to those of the large donor substituent(s), N=A(NR
2)
n [
9]. We also neglected the prototropy possible in derivatives containing N=C(NH
2)
2 because the strong electron-withdrawing effect of the –C
2HC=Z–C≡N part in
I.5 and
II.5 strongly favors the imino form (H
2N)
2C=N–C
2HC=Z–C≡N. The same is true for
I.8 and
II.8, for which the analogous effect of the –C
2(NH
2)C=Z–C≡N part also favors the imino form (H
2N)
2C=N–C
2(NH
2)C=Z–C≡N. The replacement of H by Me at the amino N atoms in the guanidino group eliminates prototropy in
I.6,
I.9,
II.6, and
II.9.
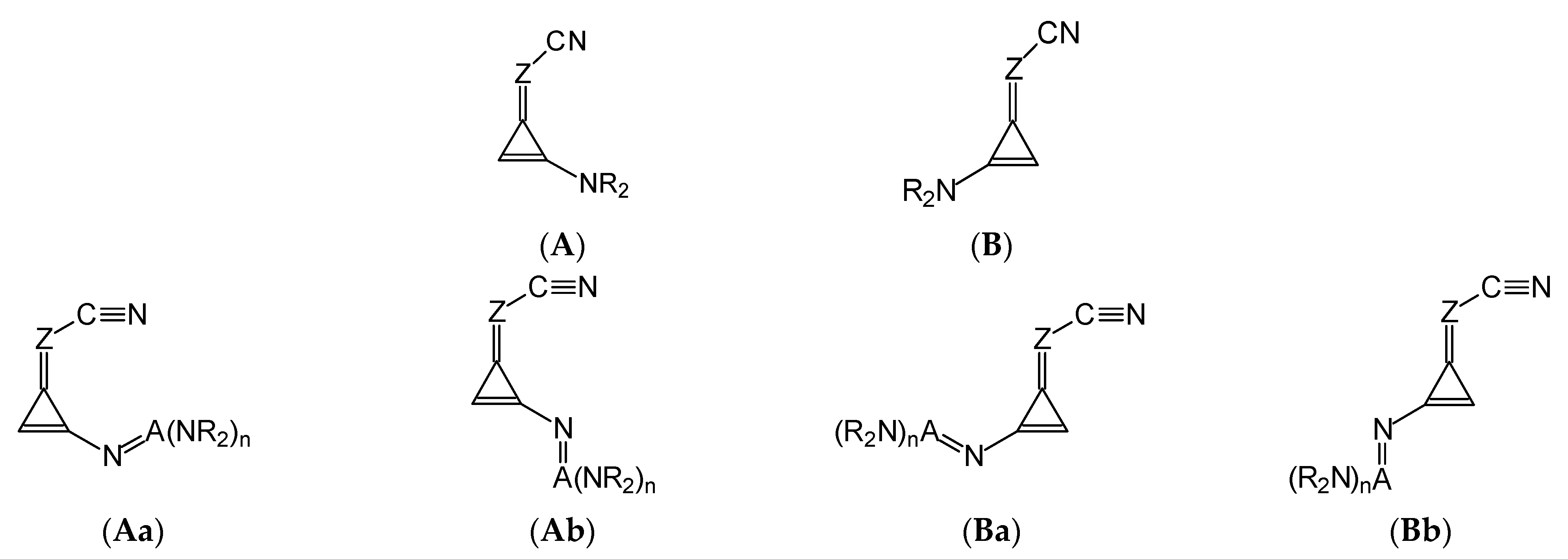
Consequently, for analysis of the geometric, energetic, and basicity parameters of the investigated nitriles, two geometrical isomers were taken into account for derivatives containing one substituent X (
I.2-I.7, and
II.2-II.7): one isomer (
A) with X at the
syn-position and the other one (
B) with X at the
anti-position
vis-à-vis C≡N (
Figure 2). For derivatives containing the large substituents N=A(NR
2)
n, we additionally selected two extreme rotational isomers about the single C (cyclopropene)–N (substituent) bond: one
syn isomer (
a) and the other one
anti (
b)
vis-à-vis C=Z at both
syn- (
A) and
anti-positions (
B) of N=A(NR
2)
n vis-à-vis C≡N. In this way, two isomers (
A and
B) were selected for
I.2-I.4 and
II.2-II.4 with simple substituents (Me, NH
2, and NMe
2), while four isomers (
Aa,
Ab,
Ba, and
Bb) were analyzed for
I.5-I.7 and
II.5-II.7 containing large substituents (N=C(NH
2)
2, N=C(NMe
2)
2, and N=P(NMe
2)
3).
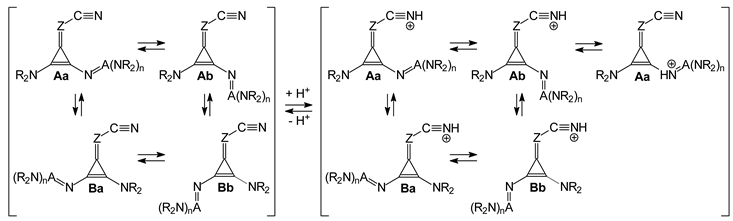
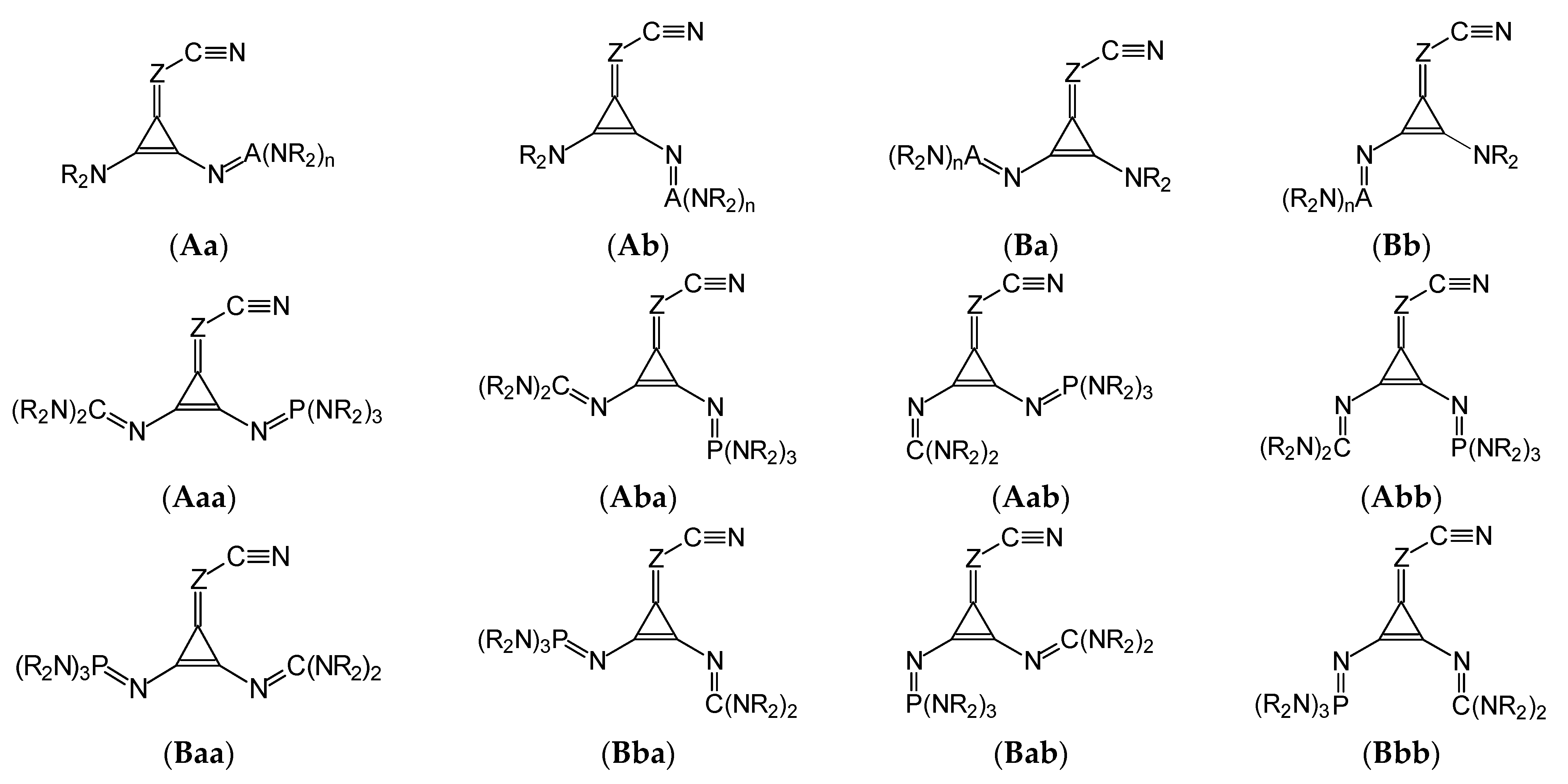
The two types of isomerism (geometrical and rotational) are also possible for derivatives containing two different substituents, NR
2 and N=A(NR
2)
n (
I.8-I.10, and
II.8-II.10). Similar to derivatives with one large substituent (
Figure 2), we examined four isomers (
Aa,
Ab,
Ba, and
Bb) for
I.8-I.10 and
II.8-II.10,
syn (
a) and
anti (
b) rotamers
vis-à-vis C=Z at both
syn- (
A) and
anti-positions (
B)
vis-à-vis C≡N (
Figure 3). A more complicated situation takes place for
I.11 and
II.11, possessing two different large donor substituents, N=C(NR
2)
2 and N=P(NR
2)
3, for which the number of potential isomers is doubled, and eight isomers can be considered,
Aaa,
Aab,
Aba,
Abb, Baa,
Bab,
Bba, and
Bbb included in
Figure 3. For symmetrically disubstituted derivatives with two large N=A(NR
2)
n groups (
I.15-I.17 and
II.15-II.17), only the rotational isomerism about the single bonds C (cyclopropene)–N (X) was investigated, and the four isomers (
aa,
ba,
ab, and
bb) discussed in Part I of this study (as
a–
d, respectively) [
9].
The isomeric phenomena were examined for all neutral as well as cyano and imino N-protonated forms of nitriles containing the methylenecyclopropene (
I.2-I.11) and cyclopropenimine (
II.2-II.11) scaffolds at the DFT1 and/or DFT2 level. Except for the derivatives with two large substituents (
I.11 and
II.11), structures for all isomers considered in this work were found to be stable. For the unsymmetrically disubstituted derivatives
I.11 and
II.11, only six isomers (
Aaa,
Aab,
Aba,
Baa,
Bab, and
Bba) were stable for both the neutral and protonated forms, analogously as for the symmetrically disubstituted derivatives
I.15-I.17 and
II.15-II.17, for which only three isomers (
aa,
ba, and
ab) were previously found to be stable. The conformation
bb for
I.15-I.17 and
II.15-II.17, and also the two conformations
Abb and
Bbb for
I.11 and
II.11 were unstable for both the neutral and imino N-protonated forms. Only structures for the cyano N-protonated forms were found to be stable for these conformations.
Table S1 (SM) summarizes the thermochemical data such as enthalpy (
H) and Gibbs energy (
G) at 298 K and 1 atm that characterize the stability of the isomers studied here for the two series of nitriles,
I.2-I.11 and
II.2-II.11. Note that data for the derivatives already investigated in Part I (
I.1,
I.12-I.17,
II.1, and
II.12-II.17) are also included in this table for easier comparison.
In the following step, we calculated the relative Gibbs energies between these isomers to indicate the most favored structures separately for neutral and monoprotonated forms. Their values are included in
Figures S1–S4 (SM). For neutral derivatives containing one simple substituent (
I.2-I.4 and
II.2-II.4 in
Figure S1), the isomer
A has slightly lower Gibbs energy than
B (Δ
G < 6 kJ mol
−1). A reverse situation takes place for their cyano N-protonated forms. The isomer
B possesses lower Gibbs energy than
A (Δ
G < 6 kJ mol
−1). The isomeric preference and order of substituent effects are analogous in both series (
I and
II).
For neutral mono- and disubstituted derivatives containing one large electron-donor substituent N=A(NR
2)
n (
I.5-I.10 and
II.5-II.9 in
Figures S2 and S3), the isomer
Aa is favored. Important stability also characterizes the isomer
Ba (Δ
G < 10 kJ mol
−1), which predominates for
II.10 (
Figure S3). Isomeric preferences are not the same for nitriles with two large electron-donor substituents (
I.11 and
II.11). The isomer
Bab predominates for
I.11, while
Baa for
II.11 (
Figure S4). However, the isomeric mixtures of
I.11 and
II.11 can also contain other isomers (
Aaa,
Aba,
Aab, and
Bba with Δ
G ≤ 12 kJ mol
−1).
Protonation changes the isomeric preferences, and therefore influences the composition of isomeric mixtures for monocations (
Figures S2–S4 in SM). The cyano N-protonated form of the isomer
Ab is favored for
I.5-I.7 and
II.6-II.7, whereas the cyano N-protonated form of
Ba seems to be more stable for
II.5,
I.8-10, and
II.8-II.10 (
Figures S2 and S3 in SM). Note that the Gibbs energies of some other cyano N-protonated isomers do not differ very much from those of the favored ones (Δ
G ≤ 10 kJ mol
−1), and can also, to various degrees, affect physicochemical properties of the N-protonated forms. For example, the Gibbs energies of the cyano N-protonated isomer
Bb and
Ab for
I.5-I.7, and
Ba and
Ab for
II.5-II.7, differ by no more than 2 kJ mol
−1, and those of the isomers
Aa and
Ba for
I.8-I.10 and
II.9 differ by less than 1 kJ mol
−1. They can predominate in the isomeric mixtures of
I.5-I.10 and
II.5-II.7 and
II.9, therefore determining their properties. In the case of
I.11 and
II.11 (
Figure S4 in SM), the isomer
Aba is favored for the cyano N-protonated form. Nevertheless, other isomers, particularly
Bba and
Bab for
I.11, and
Bba,
Baa,
Bab,
Aab, and
Aaa for
II.11, have very close Gibbs energies to that of
Aba (Δ
G ≤ 12 kJ mol
−1). Note that the Gibbs energy of the guanidino N-imino protonated isomer
Baa of
I.11 is only slightly above that of the favored cyano-protonated derivative (Δ
G ca. 8 kJ mol
−1).
Structural and energetic differences between the geometric and rotational isomers of the investigated nitriles influence the basicity parameters calculated for each potential N-protonation site in the C≡N and C=N (Z, X, and Y) groups. The microscopic (kinetic) basicity parameters (PA
i and GB
i), calculated at the DFT level for these sites according to Equations (S1)–(S3) given in
SM, are listed in
Table S2 (SM). Most estimated PA
is are close to or even higher than 1000 kJ mol
−1. This means that many derivatives investigated here can be classified into the family of strong organic N-bases such as guanidines and phosphazenes [
1,
2,
4].
3.3. Favored Site of Protonation
Similar to the investigated push–pull nitriles containing the unsubstituted methylenecyclopropene and cyclopropenimine groups (
I.1 and
II.1) and symmetrically disubstituted derivatives (
I.12-I.17 and
II.12-II.17) with electron-donor groups, previously investigated in Part I [
9], the cyano N atom appears to be the favored site of protonation also in the unsymmetrically substituted derivatives
I.2-I.11 and
II.2-II.11 examined here (
Figure 1). The Gibbs energies estimated for isomers protonated at the imino N atom in C=Z, X, or Y (N=A(NR
2)
n) groups are higher than those protonated at the cyano N atom by ca. 10–120 kJ mol
−1 (
Figures S1–S4 in SM). In our previous articles on the basicity of amino, guanidino, and phosphazeno nitriles [
7,
8,
9], we also established computationally that protonation at the amino N atom is negligible. The favored site of protonation (cyano N atom) in compounds of series
I and
II is a consequence of the strong conjugation between the electron-acceptor (C≡N) and electron-donor (XYC
2C=Z) groups in which the C
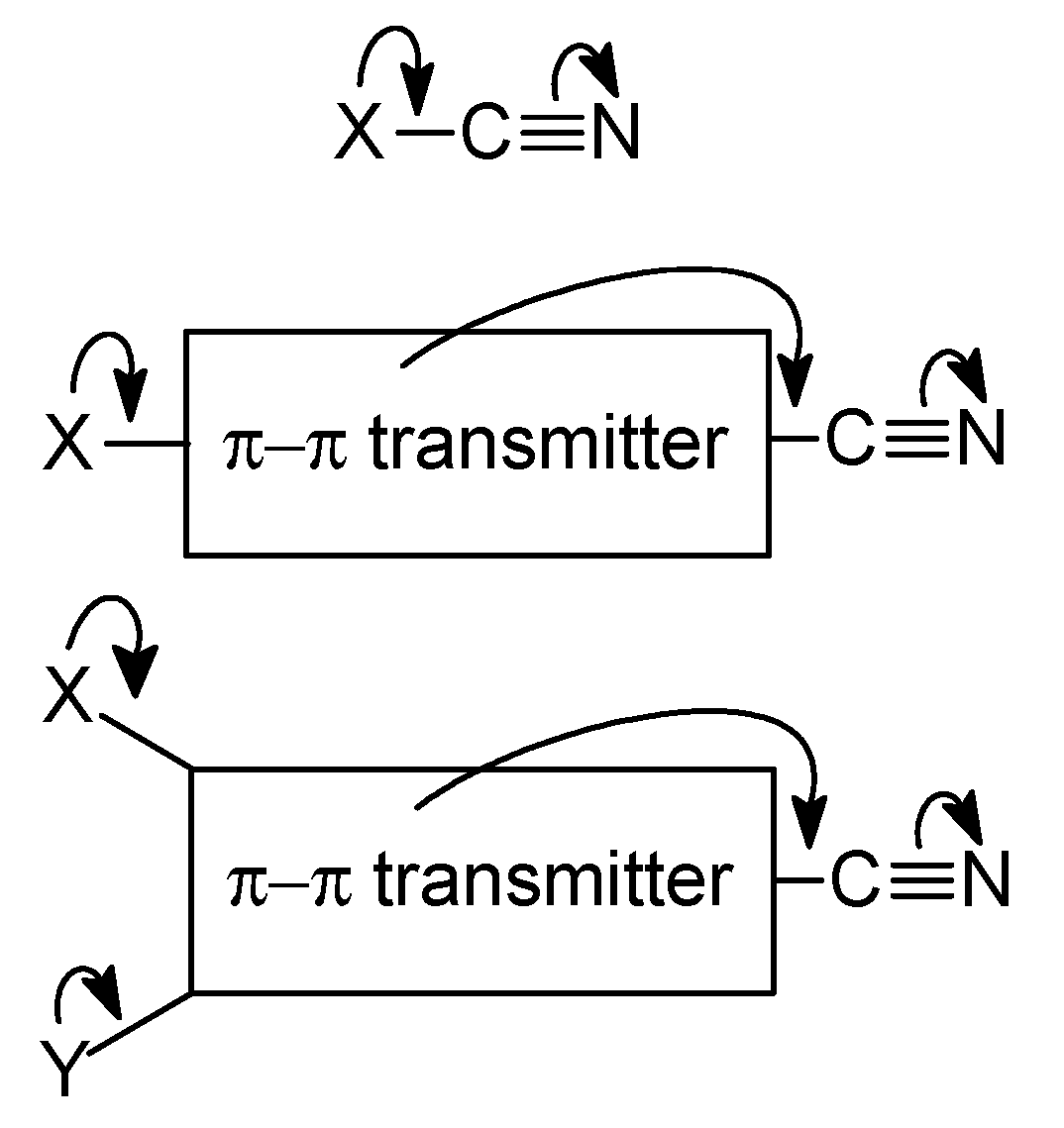
2C=Z scaffolds act as good transmitters for the electronic substituent effects of X and Y.
A perusal of the C (cyclopropene)=Z and Z–C (cyano) bond lengths in the mono- and disubstituted push–pull nitriles
I.2-I.11 and
II.2-II.11 (see
Table S3 in SM) suggests that the energy barriers for geometrical isomerism can be considerably lower than those in the case of unconjugated substituted alkenes and imines. The cyano N-protonation shortens the Z–C bond (by 0.07–0.08 and 0.08–0.09 Å for series
I and
II, respectively) and lengthens the C=Z bond (by 0.05–0.06 and 0.03–0.05 Å for series
I and
II, respectively). These effects can facilitate the rotation of the cyano group about the C=Z bond (
Figure S5 and Table S5 in SM), lengthened by stronger electron delocalization in the cyano N-protonated forms. Moreover, the C=Z bond in the ionic forms (1.40–1.43 Å for
I and 1.32–1.36 Å for
II) and even in neutral isomers (1.35–1.38 Å for
I and 1.29–1.32 Å for
II) are longer than those of the double C=C and C=N bonds, calculated at the same DFT level for H
2C=CH
2 (1.33 Å) and H
2C=NH (1.27 Å). In many protonated forms of nitriles, they are also longer than those for the aromatic molecules: benzene (1.39 Å) and
s-1,3,5-triazine (1.33 Å), in which π-electrons are completely delocalized and there are no typical double C=C and C=N bonds. This observation indicates that the rotation of the cyano group about only partially double C=Z bonds in nitriles (
I and
II) can be less restricted than those in alkenes and imines containing unconjugated groups [
26,
27,
28,
29,
30].
Smaller variations in bond lengths when proceeding from the neutral to cyano N-protonated forms are found for the C–X and C–Y bonds (all C–N) about which substituents X and Y can rotate. For large electron-donor substituents (N=A(NR
2)
n), these bonds shorten by ca. 0.02–0.05 Å (
Table S4 in SM). In the neutral forms, bond lengths (1.33–1.37 Å) are close to or even longer than that for aromatic
s-1,3,5-triazine, whereas they are slightly shorter for the monocations (1.30–1.33 Å). These variations indicate that rotation about the C–X and C–Y bonds can be slightly more restricted for the protonated forms for which n–π conjugation appears stronger than for the neutral ones. However, energy barriers can be of the same (or even lower) order of magnitude than those for isomerism about the C=Z bonds.
Based on our theoretical analyses on the relative Gibbs energies for the neutral and N-protonated (cyano and imino) derivatives, and additionally on the C=Z, C–X, and C–Y bonds lengths for investigated isomers, we can conclude with a high probability that in the gas phase (i) unsymmetrically substituted nitriles (I.2-I.11 and II.2-II.11) can be in the form of mixtures of geometric (A and B) and rotational (a and b) isomers, (ii) the cyano N atom is preferentially protonated, and (iii) protonation of the imino N atom may be considered only in a few cases: ZH+ for II.11, and X(or Y)H+ for I.6, I.7, I.9, I.10, and I.11. However, from a physicochemical point of view, their participation in the isomeric mixture of monocations does not appear to be very significant (8 ≤ ΔG ≤ 20 kJ mol−1).
3.4. Electron Delocalization in Mono- and Disubstituted Derivatives
The addition of one or two substituents such as Me, NR
2, and N=A(NR
2)
n at the C atoms of the cyclopropene ring in the parent systems
I.1 and
II.1 (
Figure 1), influences the distribution of all labile n- and π-electrons in monosubstituted (
I.2-I.7 and
II.2-II.7) and unsymmetrically disubstituted derivatives (
I.8-I.11 and
II.8-II.11) investigated in this work, as in the symmetrically disubstituted derivatives (
I.12-I.17 and
II.12-II.17) already studied in Part I [
9]. The pushing effects of the amino, guanidino, and phosphazeno groups have been documented for other π-electron N-bases, their structures and thermochemical properties (particularly for imines) [
1,
2,
4,
5,
31,
32,
33]. In the case of nitriles with the methylenecyclopropene and cyclopropenimine transmitters, the pushing effects of NR
2 and N=A(NR
2)
n can be qualitatively illustrated by various resonance structures that can be written for their neutral and protonated forms (
Schemes S2 and S3 in SM). Using the appropriate quantitative descriptors of electron delocalization, we can distinguish significant changes when proceeding from the neutral to protonated forms, particularly in the transmitter parts that are common for all derivatives of series
I and
II, studied here and previously [
9].
To evaluate the electron delocalization quantitatively in the neutral and cyano N-protonated forms of new mono- and disubstituted derivatives, we applied the HOMED procedure [
20,
21]. We calculated the HOMED indices (see Computational details in
SM) for the three-bonds transmitter (cyclopropene ring, HOMED3) in series
I and
II, and for two four-bonds transmitters (methylenecyclopropene in
I, cyclopropenimine in
II, HOMED4) according to Equation (S9). These values were compared with the calculated HOMED values found for the parent compounds and for the symmetrically disubstituted derivatives already reported in Part I [
9]. The HOMA procedure [
22] has also been applied, and the HOMA values estimated for the cyclopropene ring in the neutral and protonated nitriles. For selected derivatives, the estimated HOMED3s and HOMED4s are listed in
Table S6, and HOMAs in
Table S7 (SM).
For the Me derivatives
I.2 and
II.2, the values of HOMED3 and HOMED4 do not vary very much for isomers
A and
B for the neutral and cyano N-protonated forms (
Table S6 in SM). Generally, the HOMED differences are not larger than 0.007 HOMED units. HOMEDs are also higher for the protonated than for the neutral forms of
I.2 and
II.2 by 0.10–0.14 and 0.05–0.09 HOMED units, respectively. This HOMED trend is analogous to that for the parent compounds,
I.1 and
II.1, for which HOMEDs increase (by 0.09–0.14 and 0.05–0.09, respectively) when going from the neutral to protonated forms (
Scheme S1 in SM). When compared to the parent compounds
I.1 and
II.1, the HOMED values for the neutral and cyano N-protonated forms of
I.2 and
II.2 are slightly higher by 0.01–0.03 HOMED units, confirming some electron-donor (polarizability and σ-π hyperconjugation) effects of Me. Stronger effects (shift by 0.03–0.06 HOMED units) occur for derivatives
I.12 and
II.12 containing two Me groups [
9]. The HOMED trend for the neutral and ionic forms of
I.12 and
II.12 is analogous to that for
I.2 and
II.2. The HOMED values increase when going from the neutral to ionic forms by 0.10–0.13 and 0.05–0.09, respectively.
Variations in HOMED3 and HOMED4 values in the amino (NH
2 and NMe
2) derivatives, monosubstituted and symmetrically disubstituted, can be observed in
Scheme 1. Except for the HOMED4 values for isomers
A and
B of the neutral NMe
2 nitrile (
I.4), isomerization effects are not significant for these compounds. HOMEDs for isomers
A and
B of monosubstituted nitriles do not differ by more than 0.02 HOMED units. Some differences take place between mono- and disubstituted compounds of series
I and
II when proceeding from the neutral to protonated forms. The HOMED values are higher for the protonated (0.96–0.99) than for neutral forms (0.85–0.95) in the NH
2 and NMe
2 derivatives of series
I, as for the parent compound
I.1. Nevertheless, only for the neutral forms, the NMe
2 group(s) cause a slight increase in HOMEDs in comparison to the NH
2 one(s). For the ionic forms, the effects of the NH
2 and NMe
2 groups are almost the same, indicating some kind of saturation of electron delocalization in the π-electron systems or some additional favorable interactions between the substituent and other functions. A slightly different situation takes place for the NH
2 and NMe
2 derivatives of series
II. The HOMED values do not vary so much when proceeding from the neutral to protonated forms as for series
I. For the NMe
2 derivatives, the HOMED values even decrease for monocations. This can be explained by some kind of unfavorable steric interaction (repulsion) of the larger NMe
2 group (their CH parts) with a positively charged system. Only for the neutral forms, the trend of substituent effects is similar to that for series
I, i.e., HOMEDs estimated for the NMe
2 derivatives are slightly higher than those for the NH
2 ones.
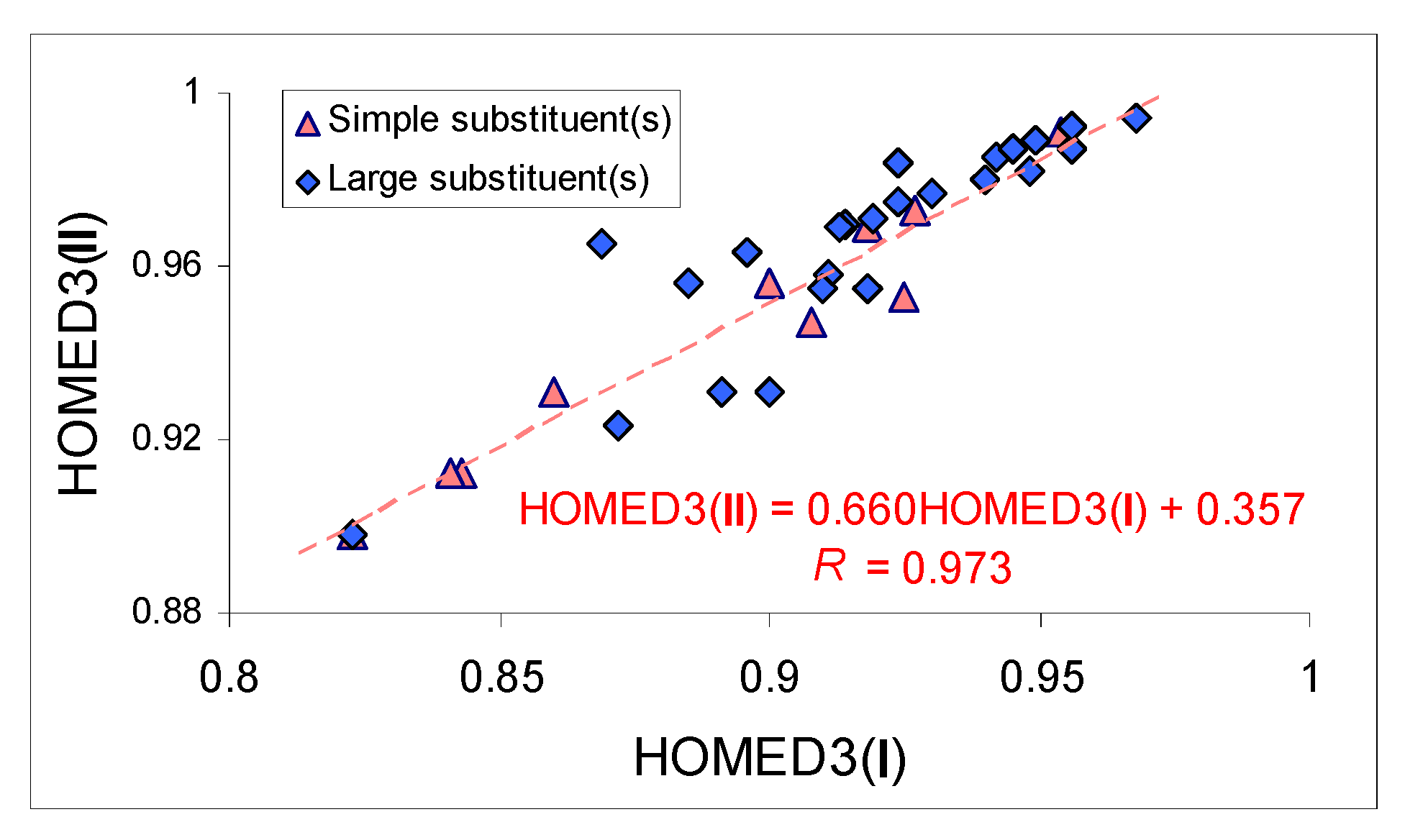
Taking into account the small differences between the HOMED values of isomers
A and
B, and almost analogous HOMED orders in neutral monosubstituted and symmetrically disubstituted derivatives containing simple groups (H < Me < NH
2 < NMe
2), we plotted the HOMED3s estimated for nitriles of series
II against those of series
I. Indeed, some parallelism exists between the corresponding HOMED values, and a quite good linear relationship is found with a correlation coefficient
R equal to 0.973 (
Figure 4). Note that points corresponding to the isomers
A of the monosubstituted derivatives containing NH
2 and NMe
2 slightly deviate from this line, plotted for a subfamily of nitriles with simple substituent (s). A different situation in HOMED variations and more significant deviations from the linear trend occur in the other subfamily containing large substituents.
A comparison of the HOMED values estimated for neutral nitriles containing N=C(NR
2)
2 and N=P(NR
2)
3 (
Table S6 in SM) shows that owing to their strong pushing character, the HOMED values increase for the selected structural transmitters to a higher degree for disubstituted than for monosubstituted compounds, and generally, in parallel to the increase in the substituent pushing effects. However, HOMED differences between the geometrical and rotational isomers are considerably higher for these derivatives than those for nitriles with simple substituent(s). For example, the highest isomeric effects (0.03–0.05 HOMED units) occur for
I.5 and
II.5 and the smallest ones (≤0.02 HOMED units) for
I.8-I.10 and
II.8-II.10. Hence, deviations of points from the linear trend given in
Figure 4 are considerably greater for derivatives with large substituent(s) than for those with simple group(s). They are greater for
I.5-I.7 and
II.5-II.7 than for
I.8-I.10 and
II.8-II.10.
Moreover, the HOMED indices estimated for compounds of series
I with large substituent(s) increase when proceeding from the neutral to cyano N-protonated forms. In series
II, the HOMED differences are considerably smaller, and in some cases, even the HOMED values for monocations are lower than those for neutrals. Some saturation of electron delocalization for monocations, already signaled for strong electron donor groups in the symmetrically disubstituted derivatives reported in Part I [
9], is also observed for the monosubstituted and unsymmetrically disubstituted nitriles studied here. Consequently, the HOMED3 indices referring to the cyclopropene ring are close to unity for most of the protonated nitriles as for aromatic systems (benzene), indicating a strong electron delocalization. Owing to the significant differences between the isomers and the relatively small variations in the HOMED3 values (0.9–1.0) for monocations, a comparison of their values for the nitriles of series
I and
II is instead presented in a scatter plot (
Figure S6 in SM).
Since the HOMA index is based on the already delocalized reference molecule (butadiene) [
22], values of this descriptor (
Table S7 in SM) are considerably smaller than those of HOMED3 based on the almost non-delocalized reference molecules (ethane and ethene) [
20,
21], which are also applied in the original HOMA procedure [
34,
35]. The use of different reference molecules for the C–C and C=C bond lengths in the hypothetical structure of cyclohexatriene leads to a different “zero” in the HOMA and HOMED scales. They correspond to moderately and non-delocalized cyclohexatriene, respectively. Only unity in the HOMA and HOMED scales refers to the same completely delocalized benzene molecule. However, the general HOMA increase trend for the investigated nitriles is analogous to that of HOMED. The HOMA values are higher for the cyano N-protonated than for the neutral forms. They also increase when the pushing effects of substituent(s) are enhanced.
3.5. Microscopic Basicities and Substituent Effects
All nitriles studied here and previously, and containing the methylenecyclopropene and cyclopropenimine intercalated scaffolds unsymmetrically and symmetrically substituted by electron-donor group(s),
I.2-I.17 and
II.2-II.17, can be classified into the family of nitriles (
I) and iminonitriles (
II), respectively. They can be considered as homolog families of conjugated (push–pull) nitriles. Their basicity parameters as well as substituent and push–pull effects can be quantitatively compared according to the methods of LFER (Linear Free Energy Relationship) analysis, similar to those reported in the literature for a series of homolog π-electron systems having the same site of protonation/deprotonation and different substituents, or, in other words, for series of acids and bases for which the mechanism of protonation/deprotonation is the same [
36,
37,
38,
39].
Taking into account the microscopic gas-phase basicity parameters (PA
is and GB
is) given in
Table S2 (SM) for compounds of series
I and
II, we can make the following observations. Generally, the PA
is and GB
is of monosubstituted nitriles
I.2-I.7 and
II.2-II.7 are enhanced in parallel to a greater size than substituent X and its stronger electron-donor effect, which increases as follows: Me < NH
2 < NMe
2 < N=C(NH
2)
2 < N=C(NMe
2)
2 < N=P(NMe
2)
3. This general order is analogous to that reported previously for series of nitriles X–C≡N containing substituent X directly bonded to the C atom of the C≡N group [
7,
8]. For nitriles studied here, PA
is and GB
is depend additionally on possible geometrical and/or rotational isomerism. For derivatives
I.2-I.4 and
II.2-II.4 with simple substituents (Me, NH
2, and NMe
2), the geometrical isomerism differentiates PA
is of the isomers
A and
B by 2–10 kJ mol
−1. Geometrical and rotational isomerisms, possible in
I.5-I.7 and
II.5-II.7 containing one large substituent (N=C(NH
2)
2, N=C(NMe
2)
2, and N=P(NMe
2)
3), cause considerably stronger effects; PA
is for isomers
Aa,
Ab,
Ba, and
Bb vary by 10–50 kJ mol
−1.
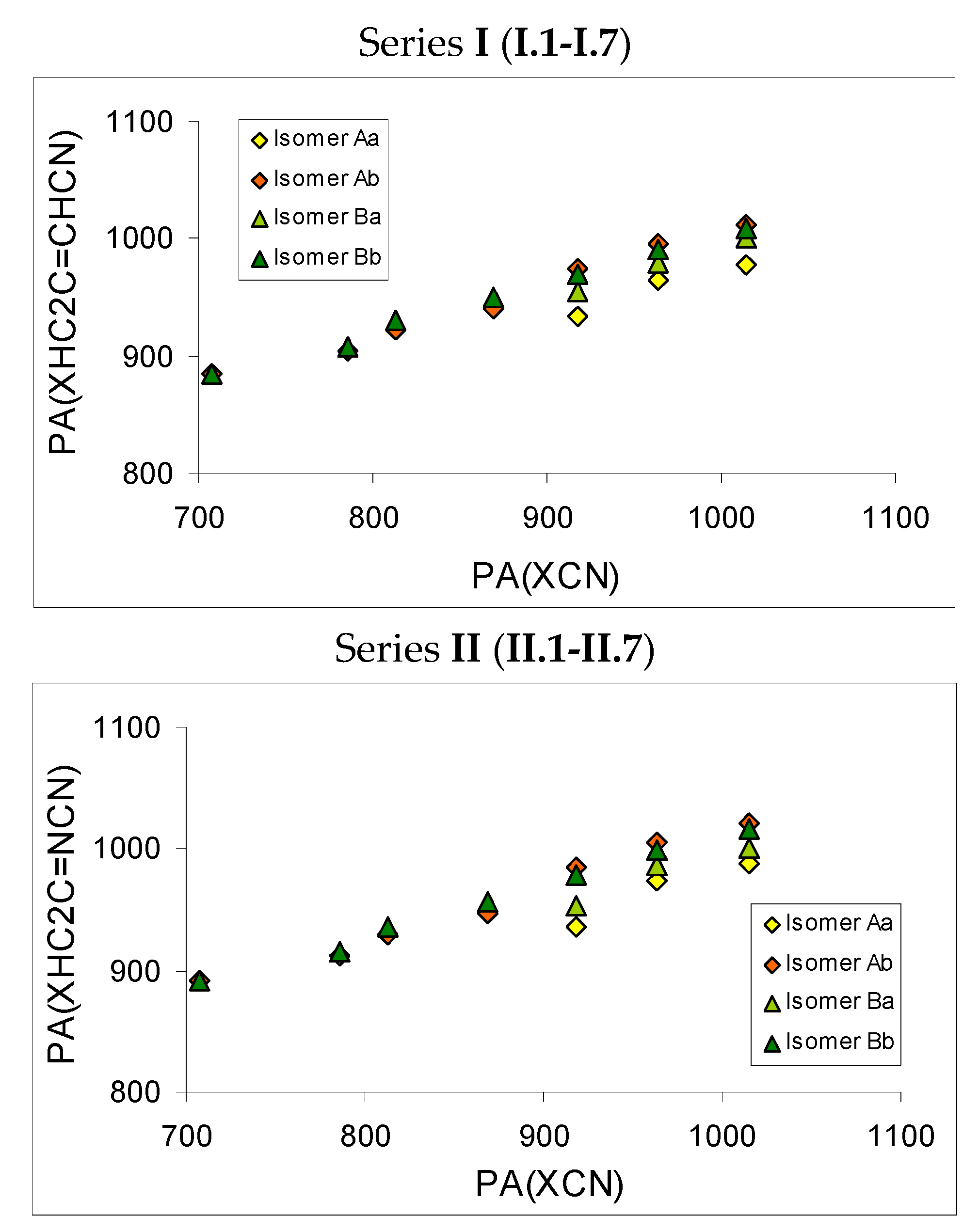
Figure 5 illustrates these substituent and isomeric effects on PA
is for monosubstituted derivatives of series
I (
I.2-I.7) and
II (
II.2-II.7). The point corresponding to PAs of the parent compounds
I.1 and
II.1 is also included in this figure. Although PA
is for geometrical and rotational isomers vary in a different way, linear trends exist between substituent isomeric effects for the monosubstituted derivatives studied here (
I.1-I.7 and
II.1-II.7) and previously (X–C≡N) [
8]. In our previous report, analogous linear trends were described for the symmetrically disubstituted derivatives
I.12-I.17 and
II.12-II.17 [
9].
The gas-phase total substituent effect for each isomer of
I.2-I.17 and
II.2-II.17 can be estimated as a difference between the DFT-calculated PA
i (or GB
i) of substituted derivatives and that of the corresponding parent system,
I.1 and
II.1, respectively. This difference is usually abbreviated in the literature as δPA
i, and, in the case of the nitriles studied here, corresponds to the sum of the electronic substituent effects, intramolecular interactions between the substituent and N-protonation or other N sites, and steric effects. For δPA
i estimation, we considered all isomers of the monosubstituted nitriles (
I.2-I.7 and
II.2-II.7) and unsymmetrically disubstituted derivatives (
I.8-I.11 and
II.8-II.11) investigated in this work, and also the symmetrically disubstituted compounds (
I.12-I.17 and
II.12-II.17) previously reported in Part I [
9].
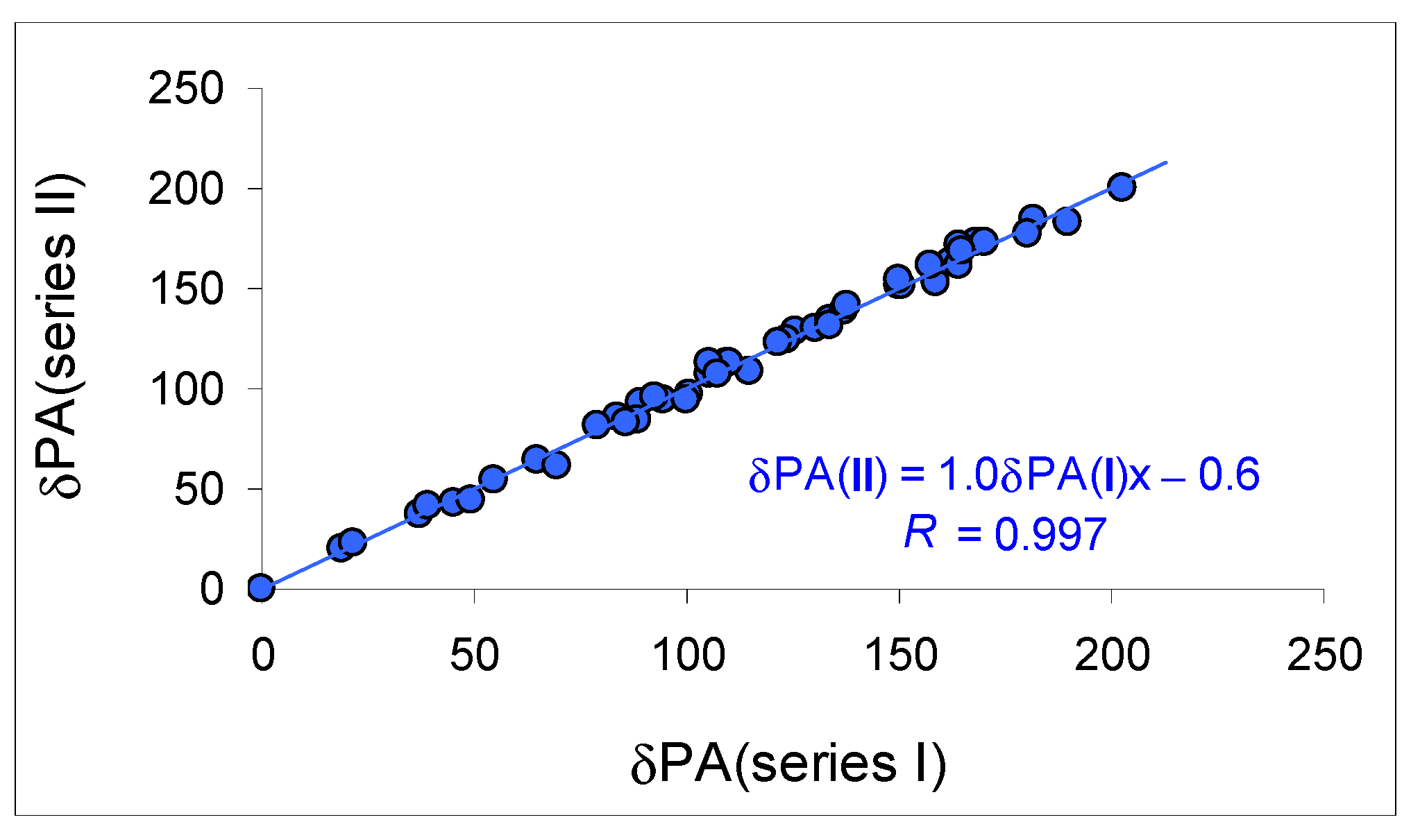
Figure 6 presents an excellent linear relationship between δPA
is determined for all isomers of series
I (
I.2-I.17) and
II (
II.2-II.17). This relationship (with a correlation coefficient
R equal to 0.997) indicates that generally the structural replacement of the CH group in series
I by the imino N atom in series
II does not destroy π–π and n–π conjugation in the π-electron system nor transmission of the pushing substituent effects to the pulling C≡N group (protonation center). Some small deviations of points for derivatives containing the N=C(NH
2)
2 (
5Aa,
5Ba,
8Ba) or N=P(NR
2)
3 group (
7Ba,
10Ba,
11Bba) can be attributed to different favorable and unfavorable intramolecular interactions or steric effects between various functional groups, e.g., amino, imino, and cyano, as signaled previously for symmetrically disubstituted derivatives [
9].
Figure S7 (SM) shows some intramolecular interactions for derivatives with one or two N=C(NH
2)
2 groups.
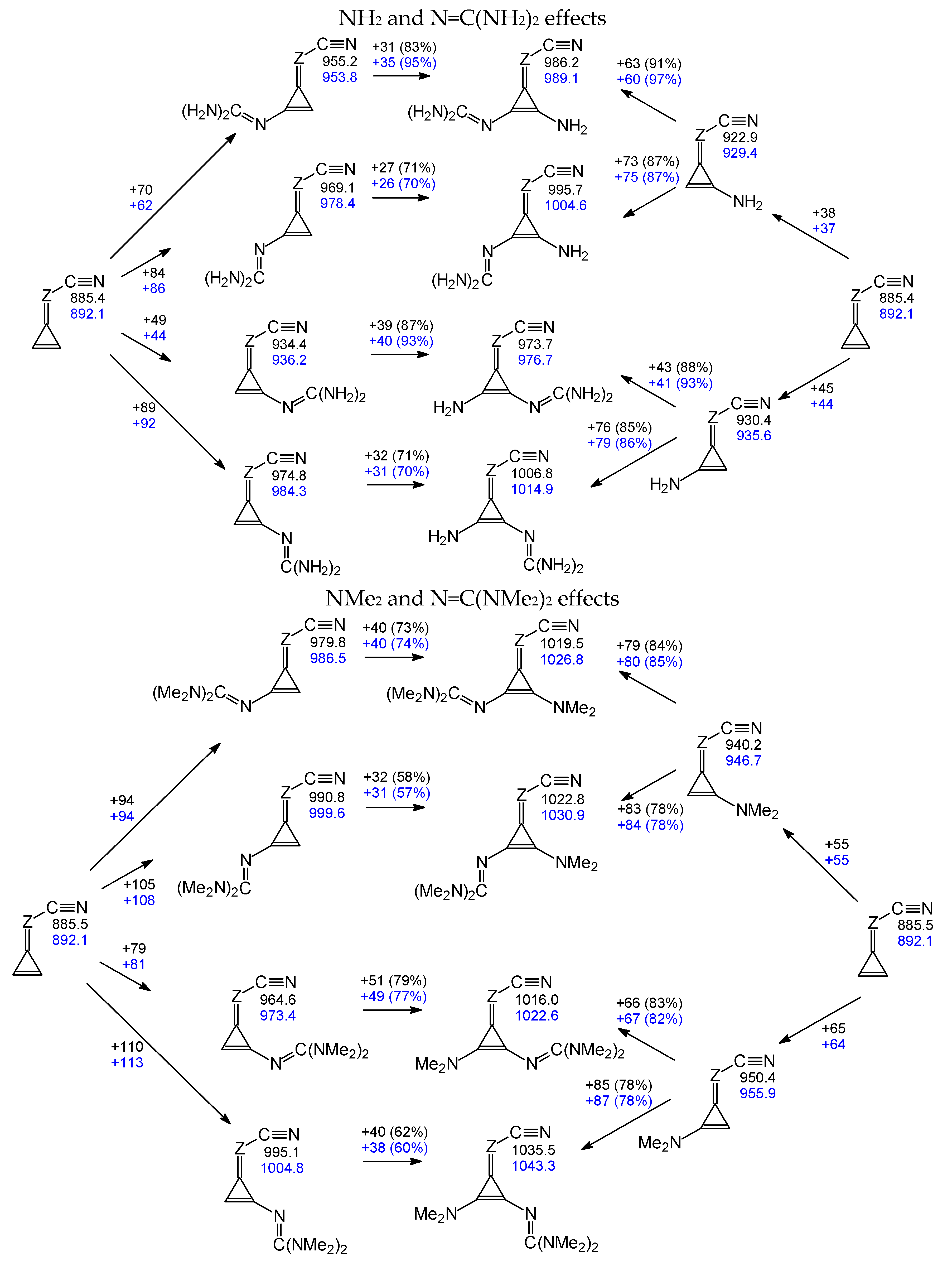
3.6. Partial Substituent Effect and Additivity
All substituents chosen in this work are electron donors. Me is σ–π hyperconjugated as in toluene, NR2 is n–π conjugated, N=C(NR2)2 (two NR2 and C=N as transmitter) is n–π cross-conjugated (double-conjugated or Y-conjugated), and N=P(NR2)3 (three NR2 and P=N as transmitter) is n–π cross(triple)-conjugated. We consider them as “simple conjugated” (Me, NR2) and “cross-conjugated” (N=C(NR2)2 and N=P(NR2)3) substituents, or “simple” and “large” (or “more complex”) groups.
Although possessing a high predictive value, the linear relationship (presented in
Figure 6) gives no information about the partial gas-phase substituent effects of the investigated simple and large groups for mono- and disubstituted derivatives. Without additional analysis, it is not possible to answer the question of the additivity or non-additivity of the substituent effects. For this reason, substituent effects were separately estimated and analyzed for each derivative. First, nitriles containing simple substituents X (Me, NH
2, and NMe
2) were examined. Starting from PA
is of the parent compounds (
I.1 and
II.1) and proceeding to the isomers
A and
B of
I.2-I.4 and
II.2-II.4, and next, going from
A and
B of
I.2-I.4 and
II.2-II.4 to symmetrically disubstituted nitriles
I.12-I.14 and
II.12-II.14, the partial substituent effects on PA
is of the cyano N atom were calculated and are given in
Scheme S4 (SM). This analysis shows that transmission of the substituent effects from the
syn-position
vis-à-vis C≡N in the isomer
A of
I.2-I.4 and
II.2-II.4 is slightly weaker than that from the
anti-position in the isomer
B. Due to the small distance between
syn-X and C≡N, some additional unfavorable intramolecular effects can also take place. Furthermore, introduction of the second substituent Me and NMe
2 in the cyclopropene ring of
I.2, II.2,
I.4, and
II.4 causes weaker effects on PA
is than that of the first substituent (
ca. 90% and 70–90%, respectively). This suggests that substituent effects are not additive in the cross-conjugated disubstituted derivatives
I.12,
I.14,
II.12, and
II.14. A slightly different trend is observed for the NH
2 derivatives
I.13 and
II.13. Effects of the second group are slightly stronger (≤110%) than those of the first one for both isomers,
A and
B.
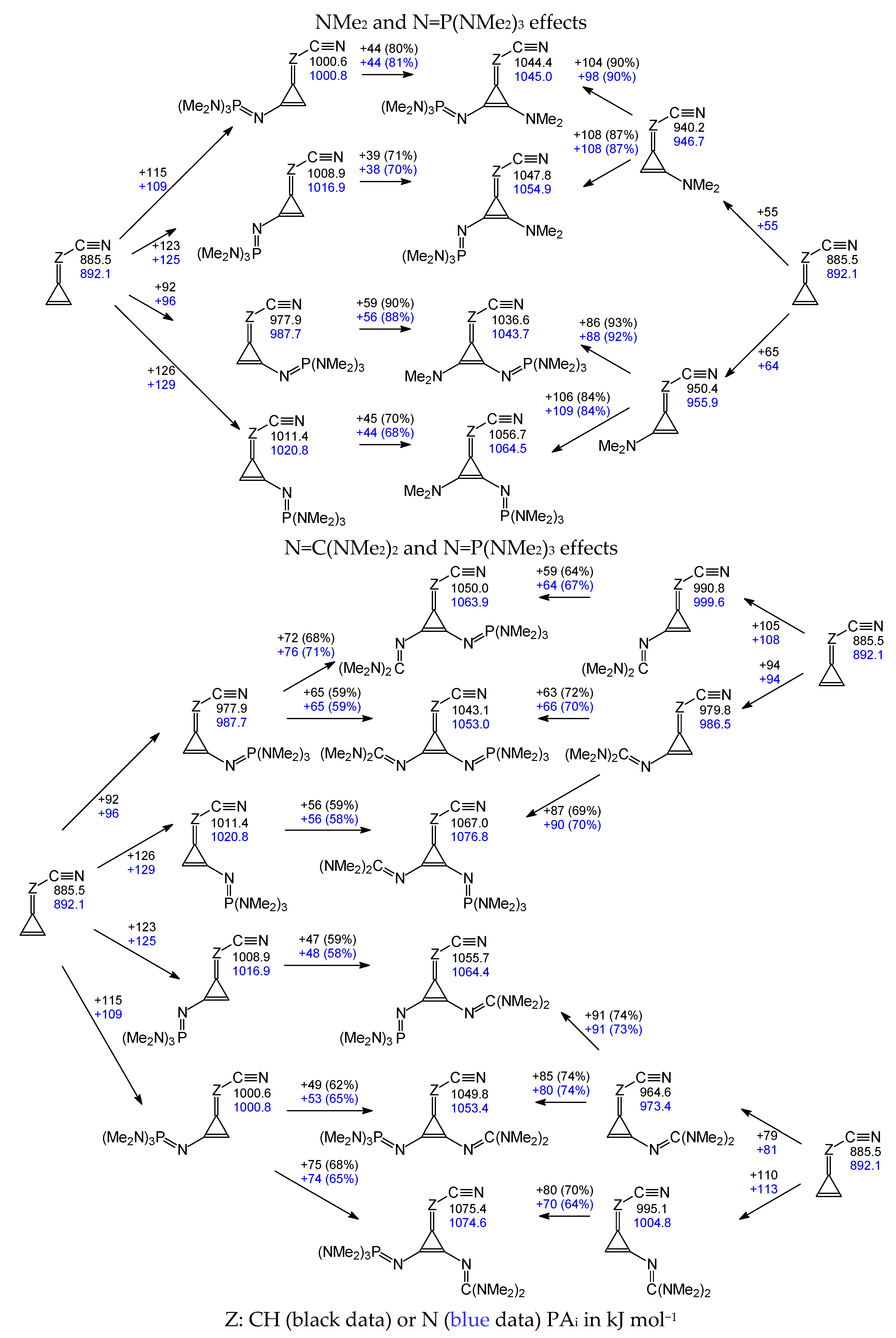
Similarly, the partial electronic substituent effects on PA
is of the cyano N atom can be examined for mono- and disubstituted derivatives containing large substituents (N=C(NH
2)
2, N=C(NMe
2)
2, and N=P(NMe
2)
3). Proceeding from the parent compounds (
I.1 and
II.1), the effects of the first substituent in the isomers
Aa,
Ab,
Ba, and
Bb of the monosubstituted derivatives (
I.5-I.7 and
II.5-II.7), then those of the second substituent in the unsymmetrically and symmetrically disubstituted nitriles (
I.8-I.17 and
II.8-II.17) were calculated.
Scheme S5 (SM) illustrates the partial substituent effects estimated in this way for the monosubstituted (
I.5-I.7 and
II.5-II.7) and symmetrically disubstituted derivatives containing N=C(NH
2)
2, N=C(NMe
2)
2, and N=P(NMe
2)
3 (
I.15-I.17 and
II.15-II.17). On the other hand, the partial substituent effects estimated for the unsymmetrically disubstituted derivatives with NH
2 and N=C(NH
2)
2, NMe
2 and N=C(NMe
2)
2, NMe
2 and N=P(NMe
2)
3, and N=C(NMe
2)
2 and N=P(NMe
2)
3 (
I.8-I.11 and
II.8-II.11) are compared in
Scheme 2. Generally, no additivity of the substituent effects takes place for the derivatives with large substituent(s). However, some parallelism of partial substituent effects is observed between the series of mono- and disubstituted nitriles containing the same type of substituent, X in monosubstituted nitriles and Y in disubstituted derivatives. Some examples are included in
Figure S8 (SM), showing the degree of non-additivity that can be expected when comparing mono- and disubstituted series.
Adding donor substituents on the transmitter should increase the basicity of the nitrile. Note that cross conjugation of substituents can depend on various factors such as the structure of substituents, their position
vis-à-vis C≡N, and conformation
vis-à-vis C=Z. The additivity of the effects of two substituents can be tested formally by comparing, in an arbitrary order, the effect observed when introducing a substituent on the unsubstituted compound with the effect observed when introducing this substituent at the same position and, when possible, in the same conformation, on the compound already bearing another substituent. Therefore, deviation from additivity is calculated here as the ratio of the latter to the former expressed as a percentage, given in
Scheme 2 (see also
Schemes S4 and S5 in SM). Perfect additivity would correspond to a 100% ratio. Actually, these percentages are in almost all cases lower than 100%, highlighting a deviation from the additivity of the donor effects of each substituent. These observations are discussed in the following on the grounds of partial saturation of the resonance effect.
If the two substituents are identical, the difference in their respective effects comes only from the initial position of the first group relative to C≡N (on the same side or opposite). The nitriles of series
I and
II bearing two identical substituents were studied earlier [
9] and their basicities can now be compared with those of the monosubstituted systems (
Schemes S4 and S5 in SM). For the methyl substitutions, the attenuation of the effect of the second Me is about 90% for both series, and the order of introduction has a relatively small effect. For the NMe
2 substitution, a significant difference is observed between series
I and
II, with a respective attenuation of about 75% and 90%. For the NH
2 substitution, we obtain an unexpected deviation from additivity, with values in the range 106–110%, meaning that there is a synergy, instead of a competing donor effect, between the two groups. This can be partially explained by differences in geometries (consequently, in conjugations) of neutral monosubstituted and disubstituted derivatives. Note that NH
2 is planar in the DFT structures of monosubstituted neutral compounds, while in disubstituted ones two amino groups take the tetrahedral conformation. In cyano-protonated derivatives, the amino groups are planar in both types of derivatives.
For the larger substituents, the problem is complicated by their different conformations and their combinations (
Scheme S5 in SM) when testing the additivity. The comparison was made between groups in the same position and conformation. Additionally, intramolecular interactions can also affect the substituent effects and, consequently, basicity of the N-cyano site. Some examples of intramolecular interactions for guanidino (N=C(NH
2)
2) derivatives are shown in
Figure S7 (SM). Targeting the superbases, we consider the electron-donor groups in positions and conformations leading to the largest basicity increases. Within this category of effects, the addition of the second substituent is about 60–70% for N=C(NMe
2)
2 and N=P(NMe
2)
3, with little difference between series
I and
II. Notably, the basicity enhancement by the second substituent is reduced by only 75–90% by the weak donors (Me, N(Me
2)) but by 60–70% for the strong donors (N=C(NMe
2)
2 and N=P(NMe
2)
3), indicating the partial saturation effect mentioned earlier. The strength of electron donors, deduced from the addition of the first substituent to
1, is as follows: Me < NH
2 < NMe
2 < N=C(NH
2)
2 < N=C(NMe
2)
2 < or ≈ N=P(NMe
2)
3.
We expect to gain more information from the partial substituent effects on the basicity of unsymmetrically disubstituted derivatives
I.8-I.11 and
II.8-II.11. In these cases, the order, in which substitution is introduced, changes the attenuation of the effects of the second substitution, as observed in
Scheme 2. For the discussion, we consider only the electron-donor groups in the position and conformation leading to the largest basicity increases.
When going from derivatives bearing the simple substituent NH2 or NMe2, attenuations for the effect of the second substituent N=C(NH2)2, N=C(NMe2)2, and N=P(NMe2)3 vary from 78 to 97% in series I and II. On the other hand, proceeding from a nitrile already containing the large substituents N=C(NH2)2, N=C(NMe2)2, and N=P(NMe2)3, attenuations for the second simple substituent NH2 and NMe2 are from 57 to 95% for series I and II. In many cases, considering only the largest effects on basicity, we see that first introducing a strong electron-donating group attenuates the effect of a weak donating group more than for the inverse order of introduction. We interpret this observation in terms of a saturation effect of the electron donation, i.e., a large electron donation to the conjugated system hinders the power of a weaker donating group.
The groups N=C(NMe2)2 and N=P(NMe2)3 have similar electron-donation strengths, so the order of introduction in the conjugated system brings slight differences. If we still consider the strongest effects on basicity, introducing the first N=C(NMe2)2 leads to an attenuation of 70% and 64% for series I and II, respectively, and 68% and 65% when starting with N=P(NMe2)3 as the first substitution. These similar attenuation factors show that the two groups experience a comparable mutual interaction. These observations are consistent with our interpretation in terms of the partial saturation effect of electron donation.
The partial substituent effects in our series of nitriles involve a blend of different contributions: principally, an electronic donor effect, modified by the group position relative to the nitrile functional group and its conformation relative to the cyclopropene plane. When we consider the strongest increases in the first and second substitution in each cycle of
Scheme 2,
Schemes S4 and S5 (SM), leading to the strongest bases, the substituent effects appear fairly regular and can be interpreted in terms of the partial saturation of the pushing effect in the push–pull system. All detailed partial substituent effects estimated for derivatives studied here and previously are compiled in
Table S8 (SM).
3.7. Macroscopic Basicities
The discussed above microscopic PA
is and GB
is (
Table S2 in SM) give information on the basicity properties of potential sites of protonation (cyano and imino N atoms) in individual isomers. They are also employed in this work for analysis of the total and partial substituent effects in mono- and disubstituted derivatives containing simple (Me, NH
2, and NMe
2) and large groups (N=C(NH
2)
2, N=C(NMe
2)
2, and N=P(NMe
2)
3). These effects encompass the pushing effect transmitted through the methylenecyclopropene and cyclopropenimine parts to the favored site of protonation as well as all intramolecular interactions between the substituents and other groups in the π-electron systems. On the other hand, the macroscopic PA
ms and GB
ms can inform about the observable gas-phase basicity properties of the isomeric mixture at the temperature of 298 K, and can be compared with those of other N-bases. For their estimations, we considered only those isomers of the neutral and protonated forms that can significantly contribute in the isomeric mixture, i.e., for which the relative Gibbs energies are not higher than 20 kJ mol
−1.
For nitriles containing one simple substituent Me, NH
2, and NMe
2, the relative Gibbs energies (Δ
G < 6 kJ mol
−1) of individual isomers for both the neutral and C≡NH
+ forms are not very high (
Figure S1 in SM). Assuming that geometrical isomerism has no high energy barrier in the conjugated system, particularly for cyano-N-protonated forms (
Figure S5 and Table S5 in SM), we considered the isomers
A and
B in the isomeric mixtures of neutral and protonated derivatives
I.2-I.4 and
II.2-II.4. The imino N(Z)-protonated forms of
II.2-II.4 are neglected since they have considerably higher
Gs than the corresponding C≡NH
+ forms (Δ
G 40–50 kJ mol
−1). The same is true for the protonation of the amino N atom. We showed previously that the
syn- and
anti-amino N-protonation in
I.13 and
II.13 require exceptionally more energy (
ca. 200 kJ mol
−1) than the cyano one [
9]. Taking into account the relative Gibbs energies between the isomers
A and
B, we calculated first the isomeric equilibrium constants and percentage contents of the two isomers in the neutral and protonated isomeric mixtures, and next the macroscopic basicity parameters PA
m and GB
m for each derivative with a simple substituent according to Equations (S4)–(S8) given in the
SM. The estimated percentage contents of
A and
B, PA
ms and GB
ms for the isomeric mixtures are summarized in
Table 1. The scheme of the favored acid-base equilibria is also included in this table. The estimated values of the macroscopic basicity parameters are intermediate between those found for individual isomers (
Table S2 in SM).
Although four isomers are possible in the isomeric mixtures of the neutral forms of
I.5-I.10 and
II.5-II.10 containing one large substituent (N=C(NH
2)
2, N=C(NMe
2)
2, and N=P(NMe
2)
3), not all of them contribute significantly to the basicity properties. The relative Gibbs energies for several isomers are higher than 20 kJ mol
−1 (
Figure S2), and their contribution to the isomeric mixtures can be neglected. On the other hand, all four isomers have to be considered in the isomeric mixtures of the protonated forms. Their Δ
Gs are smaller than 20 kJ mol
−1. The isomer
Aa of the monocation protonated at the imino N atom in the N=C(NMe
2)
2 and N=P(NMe
2)
3 group of nitriles in series
I (Δ
Gs ≤ 20 kJ mol
−1) has also to be taken into account. The other monoprotonated isomers (Δ
Gs > 20 kJ mol
−1) can be neglected in their isomeric mixtures. Calculations, performed according to Equations (S4)–(S8) included in the
SM for the neutral and protonated isomeric mixtures of
I.5-I.10 and
II.5-II.10, give the possibility to estimate the percentage contents of all considered neutral and ionic forms, and next, to calculate the macroscopic basicity parameters PA
m and GB
m for these nitriles. The estimated parameters together with the scheme of acid-base equilibria are included in
Table 2. Isomers that slightly contribute to the isomeric mixture (<0.5%) marginally influence the macroscopic basicity parameters. Similarly to the simple derivatives
I.2-I.4 and
II.2-II.4, the calculated PA
m and GB
m values are situated between those of the microscopic values found for individual isomers (
Table S2 in SM).
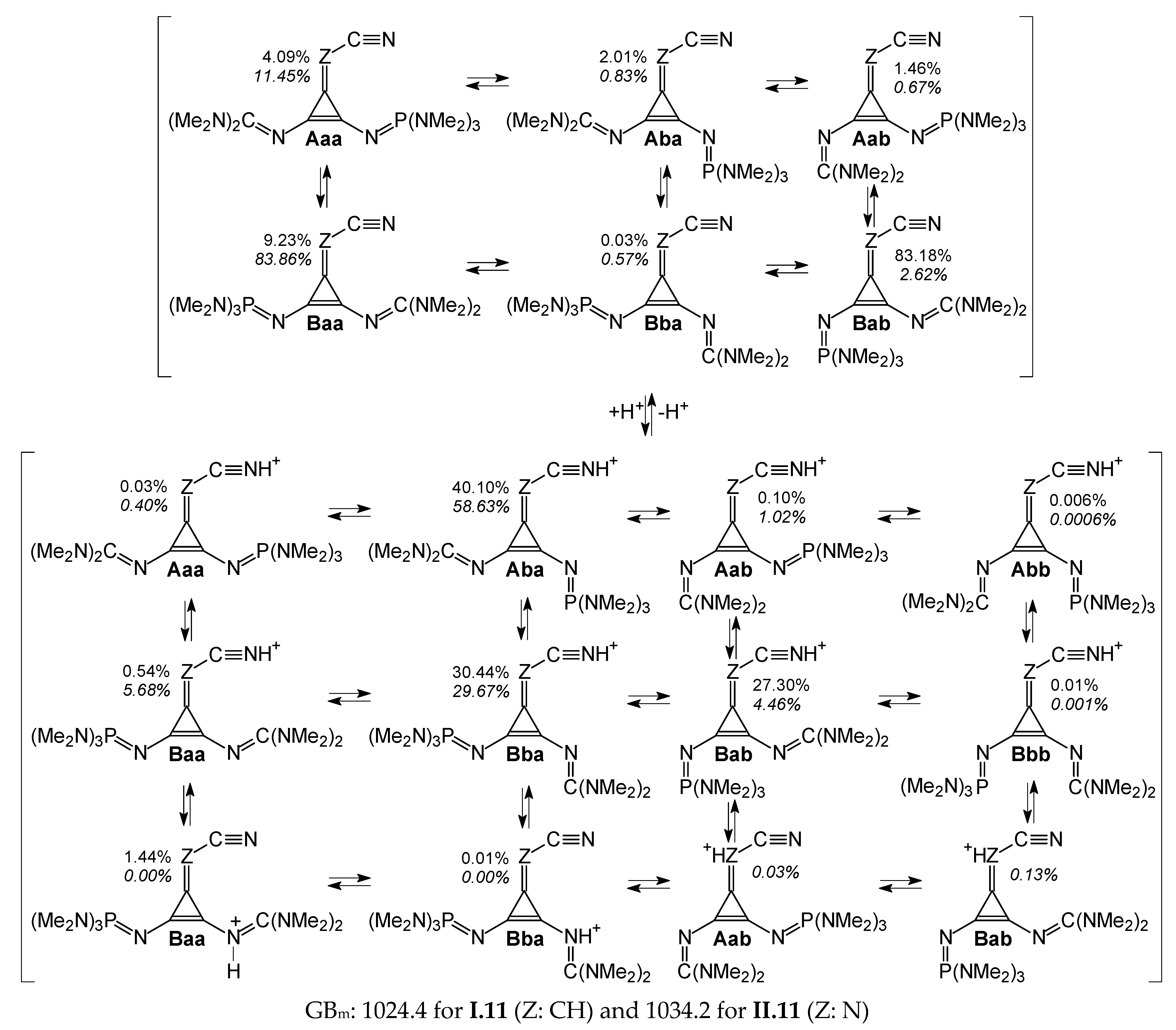
Nitriles unsymmetrically substituted by two different large substituents (
I.11 and
II.11) are specific cases, for which eight isomers are possible for the neutral and protonated forms (
Figure 3). Two structures only (
Abb and
Bbb) are found to be stable for the cyano N-protonated forms (
Table S1 in SM). For the neutral and other protonated forms, only six isomers (
Aaa,
Aba,
Aab,
Baa,
Bba, and
Bab) are stable and can be present in the isomeric mixtures. Regarding their relative Gibbs energies (
Figure S4 in SM), all of them have to be considered in the isomeric mixture of the neutral nitriles. Using Equation (S5) given in
SM, we estimated their percentage contents for neutral
I.11 and
II.11 and these are included in
Table S9 (SM). For protonated nitriles, the relative Gibbs energies vary from 0 to 72 kJ mol
−1. Most of the imino N-protonated isomers in the guanidino or phosphazeno substituent and in the C=Z group (with Δ
Gs > 20 kJ mol
−1) can be neglected in the ionic isomeric mixtures. According to Equation (S6), their percentage contents are very low (see
Table S9 in SM). Only the guanidino-protonated isomers
I.11Baa and
I.11Bba, and imino N(Z)-protonated isomers
II.11Aab and
II.11Bab slightly contribute to the ionic isomeric mixtures. Among the cyano N-protonated isomers, only
I.11Abb,
II.11Abb, and
II.11Bbb can be neglected in these mixtures. Estimating the macroscopic basicity parameters for
I.11 and
II.11, according to Equations (S7) and (S8) included in the
SM, leads to the following PA
m and GB
m values: 1055.1 and 1024.4, and 1061.7 and 1034.2 kJ mol
−1, respectively. The favored acid-base equilibria for these two nitriles are summarized in
Scheme 3.
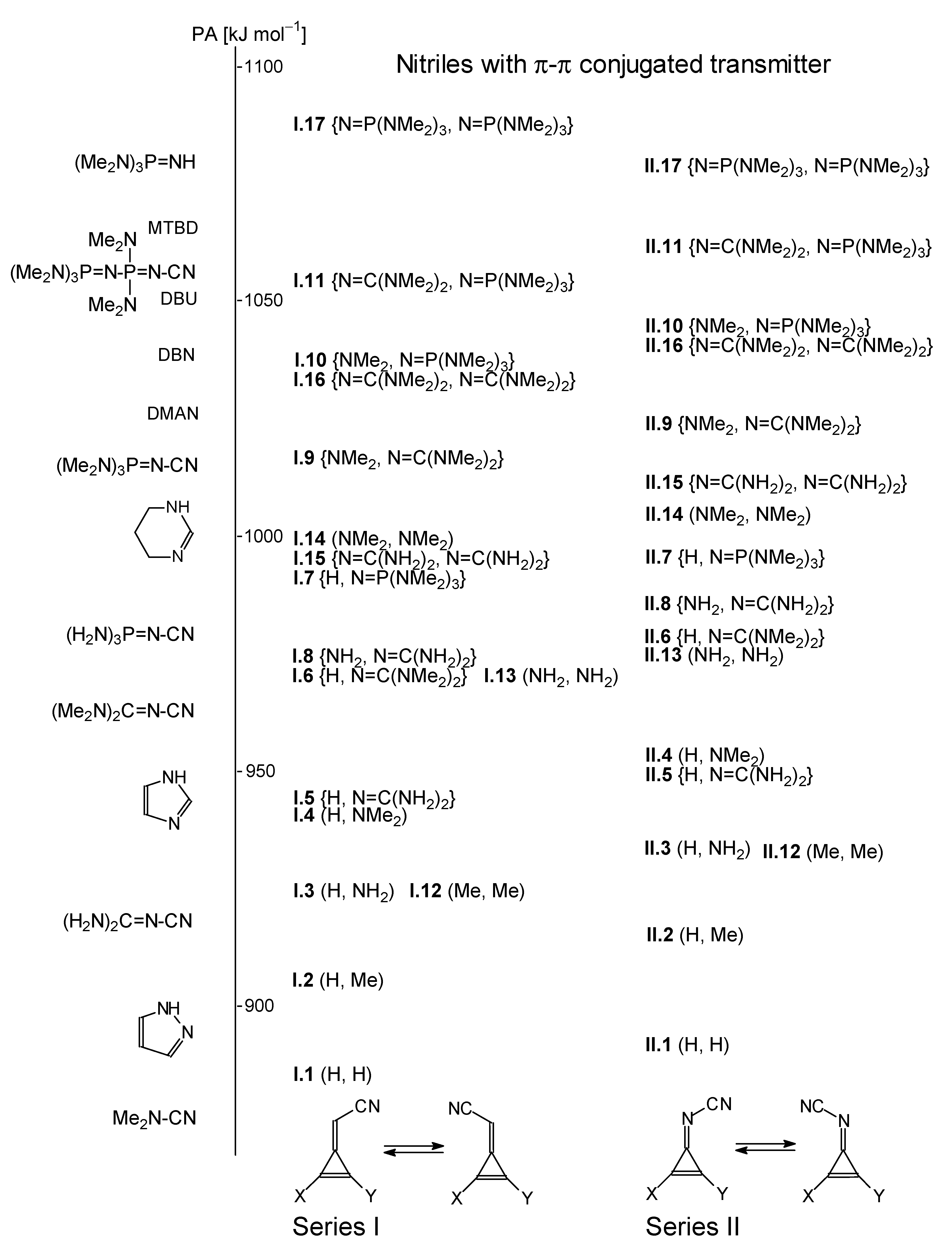
Taking into account all macroscopic basicities estimated for series
I and
II of the isomeric nitriles studied in this work, as well as in part I, [
9], we can see that unsymmetrically and symmetrically substituted derivatives with two strong electron donor substituents such as guanidino and phosphazeno groups ((N=CNMe
2)
2 and N=P(NMe
2)
3, respectively} possess PAs higher than that of DMAN. Hence, they can be classified as strong bases, also called superbases. Their basicities are close to those of bicyclic N-bases (amidines DBN and DBU, and guanidine MTBD) and of the simplest phosphazene ((Me
2N)
3P=NH) [
2].
Scheme 4 shows the position of all new nitriles containing the methylenecyclopropene (
I.1-I.17) and cyclopropenimine (
II.1-II.17) scaffolds in the general PA scale of superbases.



 ,
, 



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}