
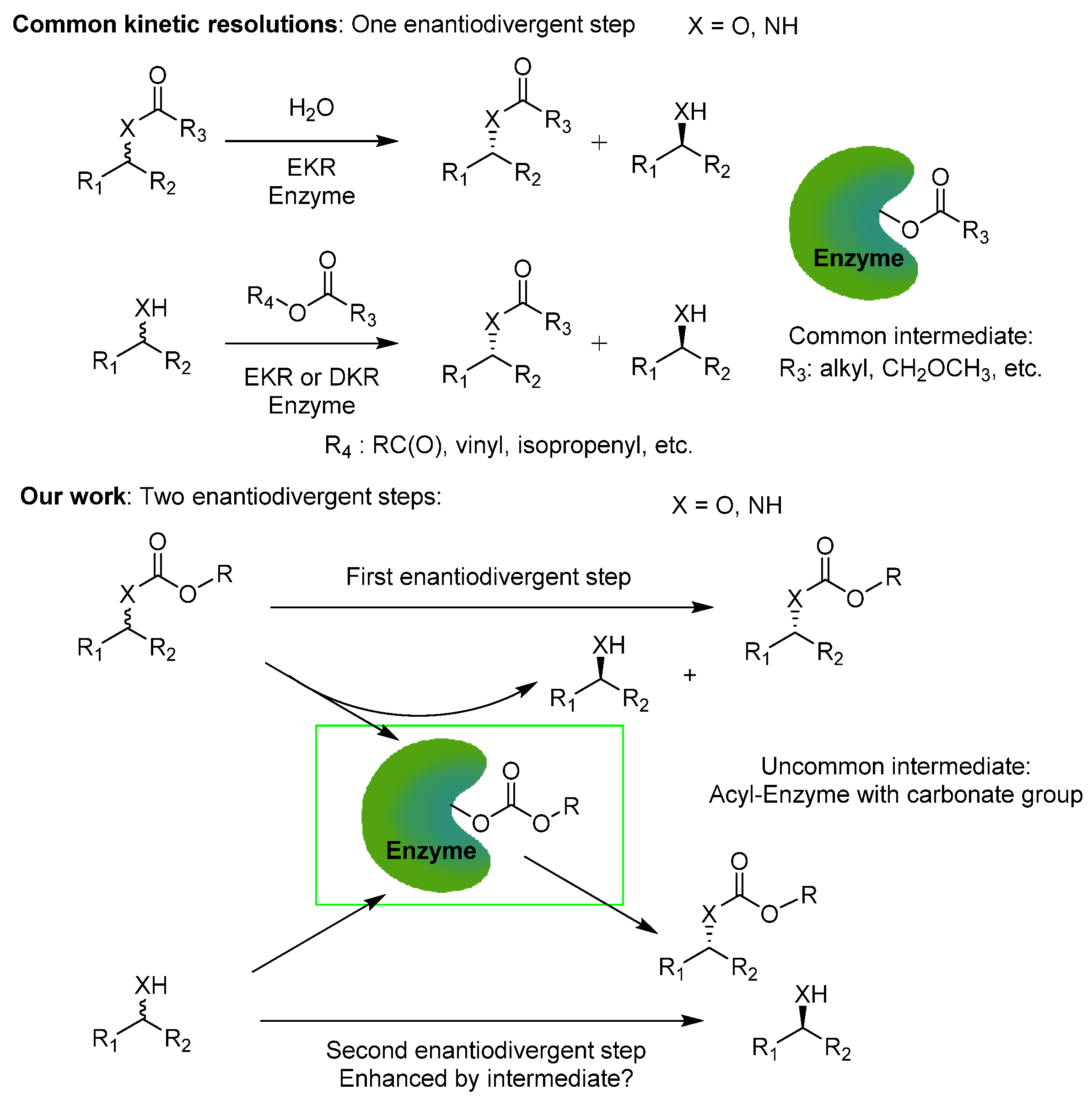
Intensification of Double Kinetic Resolution of Chiral Amines and Alcohols via Chemoselective Formation of a Carbonate–Enzyme Intermediate


Abstract
:
1. Introduction
2. Results and Discussion
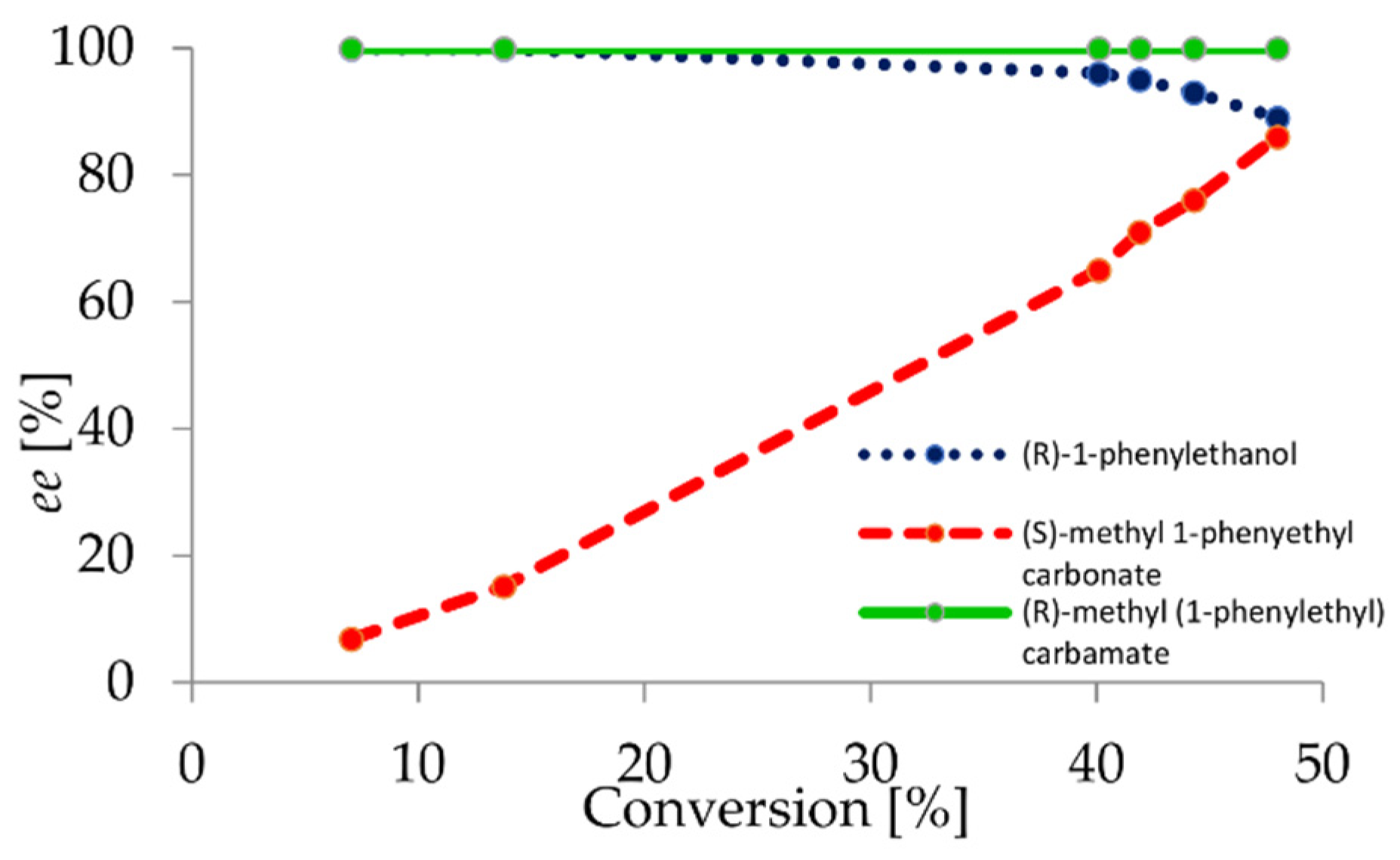
2.1. Chemo- and Enantioselectivity Studies on Carbonate EKR

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | rac-1 2 | Conv. (%) | (S)-1 ee (%) | (R)-2 ee (%) | E 3 |
|---|---|---|---|---|---|
| 1 | 1a | 31 | 40 | >99 | >200 |
| 2 | 1b | 3 | 2 | >99 | >200 |
| 3 | 1c | 13 | 14 | >99 | >200 |
| 4 | 1d | 60 | 6 | 4 | 1 |
| 5 | 1e | 14 | 16 | >99 | >200 |
| 6 | 1f | 37 | 52 | >99 | >200 |
| 7 | 1g | 44 | 45 | 55 | 5 |
| 8 | 1h | 48 | 84 | >99 | >200 |
| 9 | 1i | 35 | 67 | >99 | >200 |
| 10 | 1j | 45 | 81 | >99 | >200 |
| 11 | 1k | 23 | 31 | >99 | >200 |
| 12 | 1l | 55 | 8 | 7 | 1 |
| 13 | 1m | 87 | rac (+/−) | rac (+/−) | nd |
| 14 | 1n | 93 | nd | nd | nd |
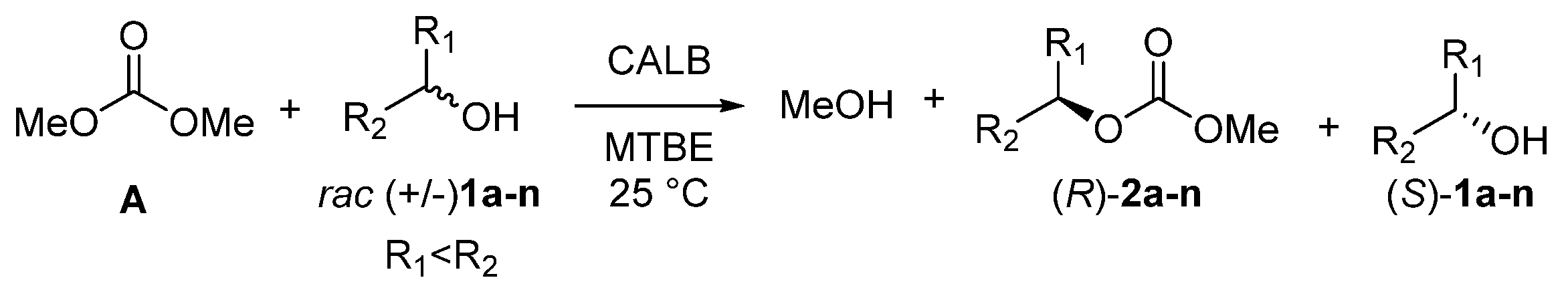
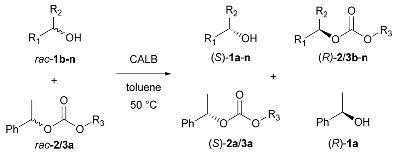
2.2. Carbonate-Mediated DEKR of Alcohols
2.3. Carbonate-Mediated DEKR of Alcohols and Amines
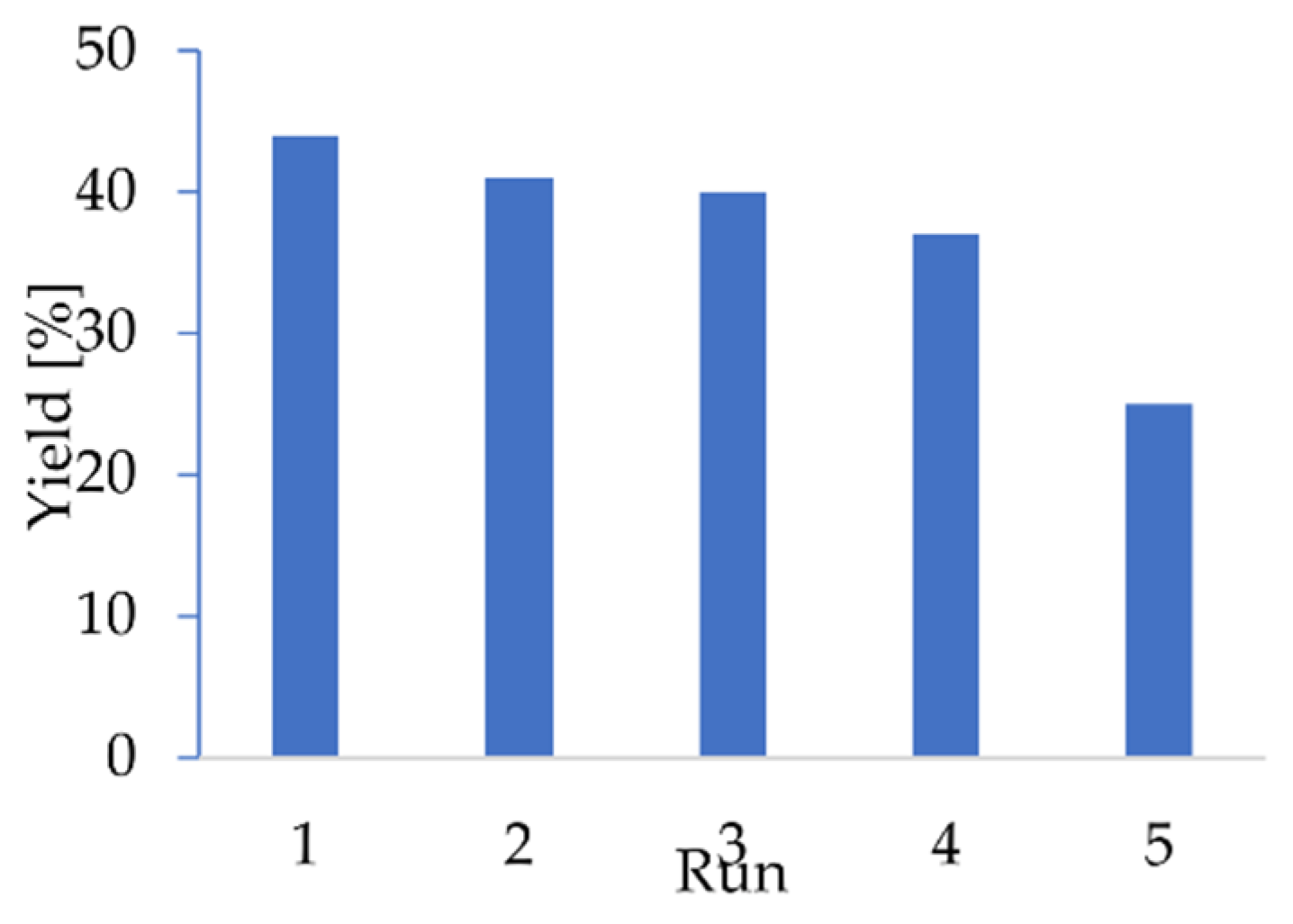
2.4. Upscale Studies
3. Materials and Methods
3.1. Materials and Instrumentation
3.2. Carbonate Synthesis
3.3. Enzymatic Kinetic Resolution of Alcohols
3.4. Double Enzymatic Kinetic Resolution of Alcohols
3.5. Upscale Procedures
3.6. Enantioselectivity Determination
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Forman, S.A.; Warner, D.S. Clinical and Molecular Pharmacology of Etomidate. Anesthesiology 2011, 114, 695–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farlow, M.R.; Cummings, J.L. Effective Pharmacologic Management of Alzheimer’s Disease. Am. J. Med. 2007, 120, 388–397. [Google Scholar] [CrossRef]
- José, C.; Toledi, M.V.; Briand, L.E. Enzymatic kinetic resolution of racemic ibuprofen: Past, present and future. Crit. Rev. Biotechnol. 2016, 36, 891–903. [Google Scholar] [CrossRef] [PubMed]
- Seddigi, Z.S.; Malik, M.S.; Ahmed, S.A.; Babalghith, A.O.; Kamal, A. Lipases in asymmetric transformations: Recent advances in classical kinetic resolution and lipase–metal combinations for dynamic processes. Coord. Chem. Rev. 2017, 348, 54–70. [Google Scholar] [CrossRef]
- Skrobiszewski, A.; Gładkowski, W.; Maciejewska, G.; Wawrzeńczyk, C. Chemoenzymatic Synthesis of trans-β-Aryl-δ-hydroxy-γ-lactones and Enzymatic Kinetic Resolution of Their Racemic Mixtures. Molecules 2016, 21, 1552. [Google Scholar] [CrossRef] [Green Version]
- Xing, X.; Jia, J.-Q.; Zhang, J.-F.; Zhou, Z.-W.; Li, J.; Wang, N.; Yu, X.-Q. CALB Immobilized onto Magnetic Nanoparticles for Efficient Kinetic Resolution of Racemic Secondary Alcohols: Long-Term Stability and Reusability. Molecules 2019, 24, 490. [Google Scholar] [CrossRef] [Green Version]
- Zdun, B.; Cieśla, P.; Kutner, J.; Borowiecki, P. Expanding Access to Optically Active Non-Steroidal Anti-Inflammatory Drugs via Lipase-Catalyzed KR of Racemic Acids Using Trialkyl Orthoesters as Irreversible Alkoxy Group Donors. Catalysts 2022, 12, 546. [Google Scholar] [CrossRef]
- Leśniarek, A.; Chojnacka, A.; Gładkowski, W. Application of Lecitase® Ultra-Catalyzed Hydrolysis to the Kinetic Resolution of (E)-4-phenylbut-3-en-2-yl Esters. Catalysts 2018, 8, 423. [Google Scholar] [CrossRef] [Green Version]
- Puchl’ová, E.; Szolcsányi, P. Scalable Green Approach Toward Fragrant Acetates. Molecules 2020, 25, 3217. [Google Scholar] [CrossRef]
- Koszelewski, D.; Brodzka, A.; Madej, A.; Trzepizur, D.; Ostaszewski, R. Evaluation of gem-Diacetates as Alternative Reagents for Enzymatic Regio- and Stereoselective Acylation of Alcohols. J. Org. Chem. 2021, 86, 6331–6342. [Google Scholar] [CrossRef]
- Wilk, M.; Brodzka, A.; Koszelewski, D.; Samsonowicz-Górski, J.; Ostaszewski, R. Model Studies on the Enzyme-Regulated Stereodivergent Cascade Passerini Reaction. Eur. J. Org. Chem. 2021, 29, 4161–4165. [Google Scholar] [CrossRef]
- Xia, B.; Cheng, G.; Lin, X.; Wu, Q. Dynamic Double Kinetic Resolution of Amines and Alcohols under the Cocatalysis of Raney Nickel/Candida antarctica Lipase B: From Concept to Application. Eur. J. Org. Chem. 2014, 14, 2917–2923. [Google Scholar] [CrossRef]
- Pozo, M.; Pulido, R.; Gotor, V. Vinyl carbonates as novel alkoxycarbonylation reagents in enzymatic synthesis of carbonates. Tetrahedron 1992, 48, 6477–6484. [Google Scholar] [CrossRef]
- Hoben, C.E.; Kanupp, L.; Bäckvall, J.-E. Practical chemoenzymatic dynamic kinetic resolution of primary amines via transfer of a readily removable benzyloxycarbonyl group. Tetrahedron Lett. 2008, 49, 977–979. [Google Scholar] [CrossRef]
- Pozo, M.; Gotor, V. Double enantioselective enzymic synthesis of carbonates and urethanes. Tetrahedron Asymmetry 1995, 6, 2797–2802. [Google Scholar] [CrossRef]
- Pozo, M.; Gotor, V. Kinetic resolution of vinyl carbonates through a lipase-mediated synthesis of their carbonate and carbamate derivatives. Tetrahedron 1993, 49, 10725–10732. [Google Scholar] [CrossRef]
- Paravidino, M.; Hanefeld, U. Enzymatic acylation: Assessing the greenness of different acyl donors. Green Chem. 2011, 13, 2651–2657. [Google Scholar] [CrossRef] [Green Version]
- Faber, K.; Riva, S. Enzyme-catalyzed irreversible acyl transfer. Synthesis 1992, 10, 895. [Google Scholar] [CrossRef]
- Zhou, Y.; Jin, Q.; Gao, Z.; Guo, H.; Zhang, H.; Zhou, X. Asymmetric organic carbonate synthesis catalyzed by an enzyme with dimethyl carbonate: A fruitful sustainable alliance. RSC Adv. 2014, 4, 7013. [Google Scholar] [CrossRef]
- Koszelewski, D.; Ostaszewski, R. The studies on chemoselective promiscuous activity of hydrolases on acylals transformations. Bioorganic Chem. 2019, 93, 102825. [Google Scholar] [CrossRef]
- Hedfors, C.; Hult, K.; Martinelle, M. Lipase chemoselectivity towards alcohol and thiol acyl acceptors in a transacylation reaction. J. Mol. Catal. B Enzym. 2010, 66, 120–123. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, R.; Li, Q.; Zhang, Z.; Feng, Y. Kinetic resolution of rac-alkyl alcohols via lipase-catalyzed enantioselective acylation using succinic anhydride as acylating agent. J. Mol. Catal. B Enzym. 2009, 56, 142–145. [Google Scholar] [CrossRef]
- Parrish, J.P.; Salvatore, R.N.; Jung, K.W. Perspectives on Alkyl Carbonates in Organic Synthesis. Tetrahedron 2000, 56, 8207–8237. [Google Scholar] [CrossRef]
- Gryglewicz, S.; Oko, F.A.; Gryglewicz, G. Synthesis of Modern Synthetic Oils Based on Dialkyl Carbonates. Ind. Eng. Chem. Res. 2003, 42, 5007–5010. [Google Scholar] [CrossRef]
- Ono, Y. Catalysis in the production and reactions of dimethyl carbonate, an environmentally benign building block. Appl. Catal. A Gen. 1997, 155, 133–166. [Google Scholar] [CrossRef]
- Kantam, M.L.; Pal, U.; Sreedhar, B.; Choudary, B.M. An Efficient Synthesis of Organic Carbonates using Nanocrystalline Magnesium Oxide. Adv. Synth. Catal. 2007, 349, 1671–1675. [Google Scholar] [CrossRef]
- Ohkoshi, M.; Michinishi, J.; Hara, S.; Senboku, H. Electrochemical carboxylation of benzylic carbonates: Alternative method for efficient synthesis of arylacetic acids. Tetrahedron 2010, 66, 7732–7737. [Google Scholar] [CrossRef]
- Gao, X.; Xiao, Y.; Wan, X.; Zhang, X. Copper-Catalyzed Highly Stereoselective Trifluoromethylation and Difluoroalkylation of Secondary Propargyl Sulfonates. Angew. Chem. 2018, 57, 3187–3191. [Google Scholar] [CrossRef]
- Whiteoak, C.J.; Kielland, N.; Laserna, V.; Escudero-Adán, E.C.; Martin, E.; Kleij, A.W. A Powerful Aluminum Catalyst for the Synthesis of Highly Functional Organic Carbonates. J. Am. Chem. Soc. 2013, 135, 1228–1231. [Google Scholar] [CrossRef]
- Anastasiadou, D.; Hensen, E.J.M.; Figueiredo, M.C. Electrocatalytic synthesis of organic carbonates. Chem. Commun. 2020, 56, 13082–13092. [Google Scholar] [CrossRef]
- Tundo, P.; Musolino, M.; Aricò, F. The reactions of dimethyl carbonate and its derivatives. Green Chem. 2018, 20, 28–85. [Google Scholar] [CrossRef]
- Bobbink, F.D.; Gruszka, W.; Hulla, M.; Das, S.; Dyson, P.J. Synthesis of cyclic carbonates from diols and CO2 catalyzed by carbenes. Chem. Commun. 2016, 52, 10787–10790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryono, D.E.; Cheng, P.T.W.; Bolton, S.A.; Chen, S.; Shi, Y.; Meng, W.; Tino, J.A.; Sulsky, R.B. Novel Glucokinase Activators and Methods of Using Same. U.S. Patent 2008009465A1, 10 January 2008. [Google Scholar]
- Flynn, B.L.; Baell, J.B.; Harvey, A.J.; Chaplin, J.H.; Paul, D.; Mould, J. Novel Benzofuran Potassium Channel Blockers and Uses Thereof. WO2008040057A1, 10 April 2008. [Google Scholar]
- Molander, G.A.; Harring, L.S. Samarium-promoted cyclopropanation of allylic alcohols. J. Org. Chem. 1989, 54, 3525–3532. [Google Scholar] [CrossRef]
- Zhao, X.; Li, Y.; Lin, X.; Li, J. Substituted Aryl Amine Alcohol Compound and Its Preparation Method and Application. CN108299279A, 20 July 2018. [Google Scholar]
- Tsunoi, S.; Ryu, I.; Okuda, T.; Tanaka, M.; Komatsu, M.; Sonoda, N. New Strategies in Carbonylation Chemistry: The Synthesis of δ-Lactones from Saturated Alcohols and CO. J. Am. Chem. Soc. 1998, 120, 8692–8701. [Google Scholar] [CrossRef]
- Kim, I.-H.; Nishi, K.; Kasagami, T.; Morisseau, C.; Liu, J.-Y.; Tsai, H.-J.; Hammock, B.D. Biologically active ester derivatives as potent inhibitors of the soluble epoxide hydrolase. Bioorganic Med. Chem. Lett. 2012, 22, 5889–5892. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, T.; Kameda, H.; Kamei, T.; Koyanagi, J.; Pichierri, F.; Omata, K.; Ishizaki, M.; Nakamura, H. FICA, a new chiral derivatizing agent for determining the absolute configuration of secondary alcohols by 19F and 1H NMR spectroscopies. Tetrahedron Asymmetry 2013, 24, 1001–1009. [Google Scholar] [CrossRef]
- Wiles, C.; Watts, P.; Haswell, S.J. Clean and selective oxidation of aromatic alcohols using silica-supported Jones’ reagent in a pressure-driven flow reactor. Tetrahedron Lett. 2006, 47, 5261–5264. [Google Scholar] [CrossRef]
- Nakamura, T.; Tateishi, K.; Tsukagoshi, S.; Hashimoto, S.; Watanabe, S.; Soloshonok, V.A.; Aceña, J.L.; Kitagawa, O. Self-disproportionation of enantiomers of non-racemic chiral amine derivatives through achiral chromatography. Tetrahedron 2012, 68, 4013–4017. [Google Scholar] [CrossRef]
- Faber, K. Biotransformations in Organic Chemistry, 7th ed.; Springer: Berlin/Heidelberg, Germany, 2018; pp. 39–43. [Google Scholar]
- Koszelewski, D.; Grischek, B.; Glueck, S.M.; Kroutil, W.; Faber, K. Enzymatic Racemization of Amines Catalyzed by Enantiocomplementary ω-Transaminases. Chem. Eur. J. 2011, 17, 378–383. [Google Scholar] [CrossRef]
- Gu, W.; Martinez, S.; Singh, A.K.; Nguyen, H.; Rozenski, J.; Schols, D.; Hederwijn, P.; Das, K.; De Jonghe, S. Exploring the dNTP -binding site of HIV-1 reverse transcriptase for inhibitor design. Eur. J. Med. Chem. 2021, 225, 113785. [Google Scholar] [CrossRef]
- Plank, T.N.; Drake, J.L.; Kim, D.K.; Funk, T.W. Air-Stable, Nitrile-Ligated (Cyclopentadienone)iron Dicarbonyl Compounds as Transfer Reduction and Oxidation Catalysts. Adv. Synth. Catal. 2012, 354, 597–601. [Google Scholar] [CrossRef]
- Varjosaari, S.E.; Skrypai, V.; Suating, P.; Hurley, J.J.M.; Gilbert, T.M.; Adler, M.J. 1-Hydrosilatrane: A Locomotive for Efficient Ketone Reductions. Eur. J. Org. Chem. 2017, 2, 229–232. [Google Scholar] [CrossRef]
- Margalef, J.; Slagbrand, T.; Tinnis, F.; Adolfsson, H.; Diégueza, M.; Pàmies, O. Third-Generation Amino Acid Furanoside-Based Ligands from d-Mannose for the Asymmetric Transfer Hydrogenation of Ketones: Catalysts with an Exceptionally Wide Substrate Scope. Adv. Synth. Catal. 2016, 358, 4006–4018. [Google Scholar] [CrossRef]
- Tang, S.; Zhang, X.; Tu, H.; You, S. Regio- and Enantioselective Rhodium-Catalyzed Allylic Alkylation of Racemic Allylic Alcohols with 1,3-Diketones. J. Am. Chem. Soc. 2018, 140, 7737–7742. [Google Scholar] [CrossRef]
- Wang, C.; Yamamoto, H. Tungsten-Catalyzed Asymmetric Epoxidation of Allylic and Homoallylic Alcohols with Hydrogen Peroxide. J. Am. Chem. Soc. 2014, 136, 1222–1225. [Google Scholar] [CrossRef]
- Bettoni, L.; Gaillard, S.; Renaud, J.-L. Iron-Catalyzed β-Alkylation of Alcohols. Org. Lett. 2019, 21, 8404–8408. [Google Scholar] [CrossRef]
- Liu, J.; Yang, S.; Tang, W.; Yang, Z.; Xu, J. Iridium-catalyzed efficient reduction of ketones in water with formic acid as a hydride donor at low catalyst loading. Green Chem. 2018, 20, 2118–2124. [Google Scholar] [CrossRef]
- Kanchanadevi, A.; Ramesh, R.; Semeril, D. Efficient and recyclable Ru(II) arene thioamide catalysts for transfer hydrogenation of ketones: Influence of substituent on catalytic outcome. J. Organomet. Chem. 2016, 808, 68–77. [Google Scholar] [CrossRef]
- Amberchan, G.; Snelling, R.A.; Moya, E.; Landi, M.; Lutz, K.; Gatihi, R.; Singaram, B. Reaction of Diisobutylaluminum Borohydride, a Binary Hydride, with Selected Organic Compounds Containing Representative Functional Groups. J. Org. Chem. 2021, 86, 6207–6227. [Google Scholar] [CrossRef]
- Chciuk, T.V.; Anderson, W.R.; Flowers, R.A. Interplay between Substrate and Proton Donor Coordination in Reductions of Carbonyls by SmI2–Water Through Proton-Coupled Electron-Transfer. J. Am. Chem. Soc. 2018, 140, 15342–15352. [Google Scholar] [CrossRef]
- Mizoguchi, H.; Uchida, T.; Katsuki, T. Ruthenium-Catalyzed Oxidative Kinetic Resolution of Unactivated and Activated Secondary Alcohols with Air as the Hydrogen Acceptor at Room Temperature. Angew. Chem. 2014, 126, 3242–3246. [Google Scholar] [CrossRef]
- Huy, P.H.; Motsch, S.; Kappler, S.M. Formamides as Lewis Base Catalysts in SN Reactions—Efficient Transformation of Alcohols into Chlorides, Amines, and Ethers. Angew. Chem. 2016, 55, 10145–10149. [Google Scholar] [CrossRef] [PubMed]
- Houvman, J.A.; Knaus, T.; Costa, M.; Mutti, F.G. Efficient synthesis of enantiopure amines from alcohols using resting E. coli cells and ammonia. Green Chem. 2019, 21, 3846–3857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, B.; Feng, X.; Meng, W.; Du, H. Asymmetric Hydrogenation of Ketones and Enones with Chiral Lewis Base Derived Frustrated Lewis Pairs. Angew. Chem. 2020, 59, 4498–4505. [Google Scholar] [CrossRef] [PubMed]
- Kaithal, A.; van Bonn, P.; Hölscher, M.; Leitner, W. Manganese(I)-Catalyzed β-Methylation of Alcohols Using Methanol as C1 Source. Angew. Chem. 2020, 59, 215–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eleftheriadis, N.; Thee, S.; Biesebeek, J.; der Wouden, P.; Baas, B.-J.; Dekker, F.J. Identification of 6-benzyloxysalicylates as a novel class of inhibitors of 15-lipoxygenase-1. Eur. J. Med. Chem. 2015, 94, 265–275. [Google Scholar] [CrossRef]
- Pacherille, A.; Tuga, B.; Hallooman, D.; Dos Reis, I.; Vermette, M.; Issack, B.B.; Rhyman, L.; Ramasami, P.; Sunasee, R. BiCl3-Facilitated removal of methoxymethyl-ether/ester derivatives and DFT study of –O–C–O– bond cleavage. New J. Chem. 2021, 45, 7109–7116. [Google Scholar] [CrossRef]
- Osumah, A.; Magolan, J.; Waynant, K.V. One-pot carbonyl reduction and carbonate formation using sodium borohydride in dialkyl carbonate solvents. Tetrahedron Lett. 2019, 60, 151203. [Google Scholar] [CrossRef]
- Wu, L.; Tian, S. Immobilization of 1,5,7-Triazabicyclo [4.4.0]dec-5-ene on Magnetic γ-Fe2O3 Nanoparticles: A Highly Recyclable and Efficient Nanocatalyst for the Synthesis of Organic Carbonates. Eur. J. Inorg. Chem. 2014, 12, 2080–2087. [Google Scholar] [CrossRef]
- Takizawa, K.; Sekino, T.; Sato, S.; Yoshino, T.; Kojima, M.; Matsunaga, S. Cobalt-Catalyzed Allylic Alkylation Enabled by Organophotoredox Catalysis. Angew. Chem. 2019, 58, 9199–9203. [Google Scholar] [CrossRef]
- Jin, S.; Hunt, A.J.; Clark, J.H.; McElroy, C.R. Acid-catalysed carboxymethylation, methylation and dehydration of alcohols and phenols with dimethyl carbonate under mild conditions. Green Chem. 2016, 18, 5839–5844. [Google Scholar] [CrossRef] [Green Version]
- Chevella, D.; Macharla, A.K.; Banothu, R.; Sai Gajula, K.; Amrutham, V.; Boosa, M.; Nama, N. Synthesis of non-symmetrical alkyl carbonates from alcohols and DMC over the nanocrystalline ZSM-5 zeolite. Green Chem. 2019, 21, 2938–2945. [Google Scholar] [CrossRef] [Green Version]
- Mutlu, H.; Ruiz, J.; Solleder, S.C.; Meier, M.A.R. TBD catalysis with dimethyl carbonate: A fruitful and sustainable alliance. Green Chem. 2012, 14, 1728–1735. [Google Scholar] [CrossRef]
- Kumara, S.; Jain, S.L. Non-symmetrical dialkyl carbonate synthesis promoted by 1-(3-trimethoxysilylpropyl)-3-methylimidazolium chloride. New J. Chem. 2013, 37, 3057–3061. [Google Scholar] [CrossRef]
- Gu, Q.; Fang, J.; Xu, Z.; Ni, W.; Kong, K.; Hou, Z. CO2 promoted synthesis of unsymmetrical organic carbonate using switchable agents based on DBU and alcohols. New J. Chem. 2018, 42, 13054–13064. [Google Scholar] [CrossRef]
- Hooker, J.M.; Reibel, A.T.; Hill, S.M.; Schueller, M.J.; Fowler, J.S. One-Pot, Direct Incorporation of [11C]CO2 into Carbamates. Angew. Chem. 2009, 48, 3482–3485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, R.; Bao, L.; Sheng, H.; Sun, L.; Chen, M.; Feng, Y.; Zhu, M. Heterobimetallic dinuclear lanthanide alkoxide complexes as acid–base bifunctional catalysts for synthesis of carbamates under solvent-free conditions. RSC Adv. 2016, 6, 78576–78584. [Google Scholar] [CrossRef]
- Leathen, M.L.; Peterson, E.A. Facile preparation of protected benzylic and heteroarylmethyl amines via room temperature Curtius rearrangement. Tetrahedron Lett. 2010, 51, 2888–2891. [Google Scholar] [CrossRef]
- Di Nicola, A.; Arcadi, A.; Rossi, L. BMIm HCO3: An ionic liquid with carboxylating properties. Synthesis of carbamate esters from amines. New J. Chem. 2016, 40, 9895–9898. [Google Scholar] [CrossRef]
- Ji, P.; Manna, K.; Lin, Z.; Feng, X.; Urban, A.; Song, Y.; Lin, W. Single-Site Cobalt Catalysts at New Zr12(μ3-O)8(μ3-OH)8(μ2-OH)6 Metal–Organic Framework Nodes for Highly Active Hydrogenation of Nitroarenes, Nitriles, and Isocyanides. J. Am. Chem. Soc. 2017, 139, 7004–7011. [Google Scholar] [CrossRef]
- Gomm, A.; Lewis, W.; Green, A.P.; O’Reilly, E. A New Generation of Smart Amine Donors for Transaminase-Mediated Biotransformations. Chem. Eur. J. 2016, 22, 12692–12695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martín-Matute, B.; Edin, M.; Bogár, K.; Bäckvall, J.-E. Highly Compatible Metal and Enzyme Catalysts for Efficient Dynamic Kinetic Resolution of Alcohols at Ambient Temperature. Angew. Chem. 2004, 43, 6535–6539. [Google Scholar] [CrossRef] [PubMed]

| Entry | rac-1 2 | Acyl Donors: | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
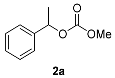 |  | ||||||||||||
| Conv1 3 (%) | (S)-2a 5 ee (%) | (R)-1a 5 ee (%) (E1) | Conv2 4 (%) | (S)-1 5 ee (%) | (R)-2 5 ee (%) (E2) | Conv1 3 (%) | (S)-3a 5 ee (%) | (R)-1a 5 ee (%) (E1) | Conv2 4 (%) | (S)-1 5 ee (%) | (R)-3 5 ee (%) (E2) | ||
| 1 | 1b | 11 | 14 | 87 (14) | 11 | 12 | >99 (>200) | <1 | nd | nd | <1 | nd | nd |
| 2 | 1c | 26 | 45 | 96 (76) | 27 | 41 | 99 (>200) | <1 | nd | nd | <1 | nd | nd |
| 3 | 1d * | 47 | 87 | 95 (111) | 46 | 26 | 31 (2) | 16 | 16 | 81 (11) | 17 | 4 | 20 (2) |
| 4 | 1e | 10 | 11 | 97 (58) | 9 | 10 | >99 (>200) | 10 | 8 | 72 (7) | 12 | 9 | 67 (6) |
| 5 | 1f | 38 | 75 | 95 (86) | 36 | 56 | >99 (>200) | 18 | 11 | 48 (3) | 18 | 13 | 58 (4) |
| 6 | 1g | 42 | 72 | 90 (41) | 42 | 47 | 65 (7) | 38 | 52 | 85 (21) | 36 | 34 | 61 (6) |
| 7 | 1h | 45 | 74 | 93 (61) | 44 | 78 | >99 (>200) | 11 | 8 | 68 (6) | 12 | 14 | >99 (>200) |
| 8 | 1i | 44 | 11 | 15 (1) | 43 | 65 | 83 (21) | <1 | nd | nd | <1 | nd | nd |
| 9 | 1j | 8 | 4 | 92 (25) | 10 | 10 | 98 (72) | <1 | nd | nd | <1 | nd | nd |
| 10 | 1k | 11 | 11 | 98 (55) | 10 | 11 | 97 (73) | 18 | 18 | 81 (11) | 19 | 5 | 21 (2) |
| 11 | 1l | 48 | 88 | 94 (94) | 47 | 48 | 54 (5) | 29 | 38 | 89 (25) | 30 | rac | rac |
| 12 | 1m | 38 | 51 | 83 (18) | 37 | rac | rac | 27 | 29 | 78 (11) | 29 | rac | rac |
| 13 | 1n | 31 | 43 | 96 (75) | nd | nd | nd | 32 | 23 | 51 (4) | 31 | nd | nd |
| 14 | MeOH | nd | nd | nd | nd | nd | nd | 7 | 9 | nd | 8 | nd | 96 (63) |
| 15 | EtOH | 12 | 9 | nd | 13 | nd | 86 (15) | nd | nd | nd | nd | nd | nd |
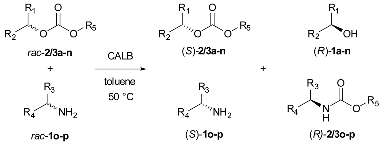
| Entry | rac-2/3 2 | Acyl Acceptors: | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
 |  | ||||||||||||
| Conv1 (%) 3 | (S)-2/3a-n ee (%) | (R) 1a-n ee (%) (E1) 5 | Conv2 (%) 4 | (S)-1o ee (%) | (R)-2/3o ee (%) (E2)5 | Conv1 (%) 3 | (S)-2/3a-n ee (%) | (R) 1a-n ee (%) (E1) 5 | Conv2 (%) 4 | (S)-1p ee (%) | (R)-2/3p ee (%) (E2) 5 | ||
| 1 | 2a | 49 | 87 | 90 (54) | 47 | 89 | >99 (>200) | 51 | >99 | 71 (30) | 51 | 95 | 92 (93) |
| 2 | 3a | 27 | 34 | 82 (14) | 28 | 36 | 85 (18) | 20 | 36 | 85 (15) | 21 | 21 | 52 (4) |
| 3 | 2b | 14 | 15 | 76 (8) | 13 | 15 | >99 (>200) | 29 | 28 | 65 (6) | 30 | 42 | 98 (150) |
| 4 | 3b | 24 | 20 | 62 (5) | 23 | 10 | 33 (2) | <1 | nd | nd | <1 | nd | nd |
| 5 | 2c | 28 | 36 | 89 (25) | 29 | 40 | >99 (>200) | 27 | 34 | 89 (23) | 26 | 30 | >99 (>200) |
| 6 | 2d * | 49 | 24 | 25 (2) * | 48 | 91 | >99 (>200) | 41 | 15 | 22 (2) * | 39 | 65 | 98 (195) |
| 7 | 2e | 36 | 35 | 67 (7) | 36 | 56 | >99 (>200) | 42 | 49 | 57 (6) | 40 | 62 | 93 (52) |
| 8 | 2f | 49 | 89 | 89 (51) | 47 | 89 | >99 (>200) | 47 | 87 | 88 (44) | 47 | 77 | 91 (49) |
| 9 | 2g | 46 | 46 | 56 (8) | 44 | 85 | >99 (>200) | 42 | 37 | 54 (5) | 41 | 62 | 89 (32) |
| 10 | 2h | 47 | 87 | 98 (>200) | 48 | 92 | >99 (>200) | 49 | 92 | >99 (>200) | 47 | 85 | 97 (183) |
| 11 | 2i | 42 | 85 | 98 (>200) | 43 | 76 | >99 (>200) | 49 | >99 | >99 (>200) | 49 | 92 | 97 (>200) |
| 12 | 2j | 46 | 94 | 98 (>200) | 45 | 86 | >99 (>200) | 36 | 55 | >99 (>200) | 35 | 51 | >99 (112) |
| 13 | 2k | 21 | 22 | 83 (13) | 21 | 43 | >99 (>200) | 31 | 40 | 87 (21) | 32 | 44 | 99 (>200) |
| 14 | 2l | 44 | 29 | 44 (3) | 45 | 81 | >99 (>200) | 32 | 22 | 45 (3) | 30 | 45 | >99 (>200) |
| 15 | 2m | 52 | rac | rac | 49 | 95 | >99 (>200) | 45 | rac | rac | 46 | 83 | 98 (>200) |
| 16 | 2n | 48 | nd | nd | 49 | 94 | >99 (>200) | 45 | nd | nd | 44 | 78 | 99 (>200) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Samsonowicz-Górski, J.; Brodzka, A.; Ostaszewski, R.; Koszelewski, D. Intensification of Double Kinetic Resolution of Chiral Amines and Alcohols via Chemoselective Formation of a Carbonate–Enzyme Intermediate. Molecules 2022, 27, 4346. https://doi.org/10.3390/molecules27144346
Samsonowicz-Górski J, Brodzka A, Ostaszewski R, Koszelewski D. Intensification of Double Kinetic Resolution of Chiral Amines and Alcohols via Chemoselective Formation of a Carbonate–Enzyme Intermediate. Molecules. 2022; 27(14):4346. https://doi.org/10.3390/molecules27144346
Chicago/Turabian StyleSamsonowicz-Górski, Jan, Anna Brodzka, Ryszard Ostaszewski, and Dominik Koszelewski. 2022. "Intensification of Double Kinetic Resolution of Chiral Amines and Alcohols via Chemoselective Formation of a Carbonate–Enzyme Intermediate" Molecules 27, no. 14: 4346. https://doi.org/10.3390/molecules27144346
APA StyleSamsonowicz-Górski, J., Brodzka, A., Ostaszewski, R., & Koszelewski, D. (2022). Intensification of Double Kinetic Resolution of Chiral Amines and Alcohols via Chemoselective Formation of a Carbonate–Enzyme Intermediate. Molecules, 27(14), 4346. https://doi.org/10.3390/molecules27144346