
From Repurposing to Redesign: Optimization of Boceprevir to Highly Potent Inhibitors of the SARS-CoV-2 Main Protease †


 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
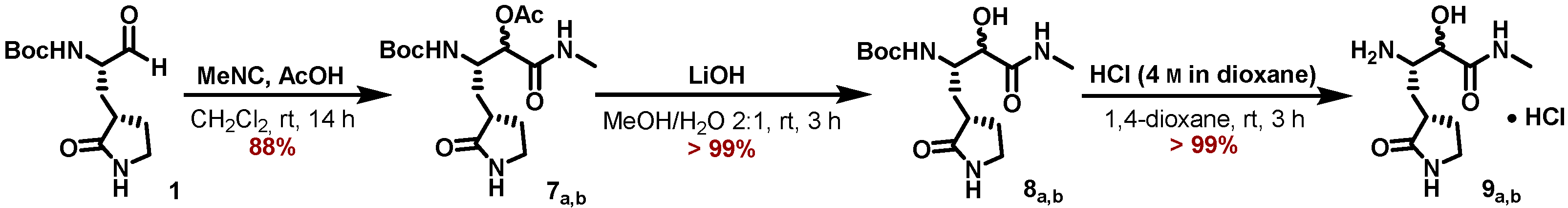{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Inhibition of the SARS-CoV-2 Main Protease by Boceprevir and Telaprevir
2.2. Crystal Structures of SARS-CoV-2 Mpro Complexes with Boceprevir and Telaprevir
2.3. P1′ Moiety
2.4. P1 Substituent
2.5. P2 Position
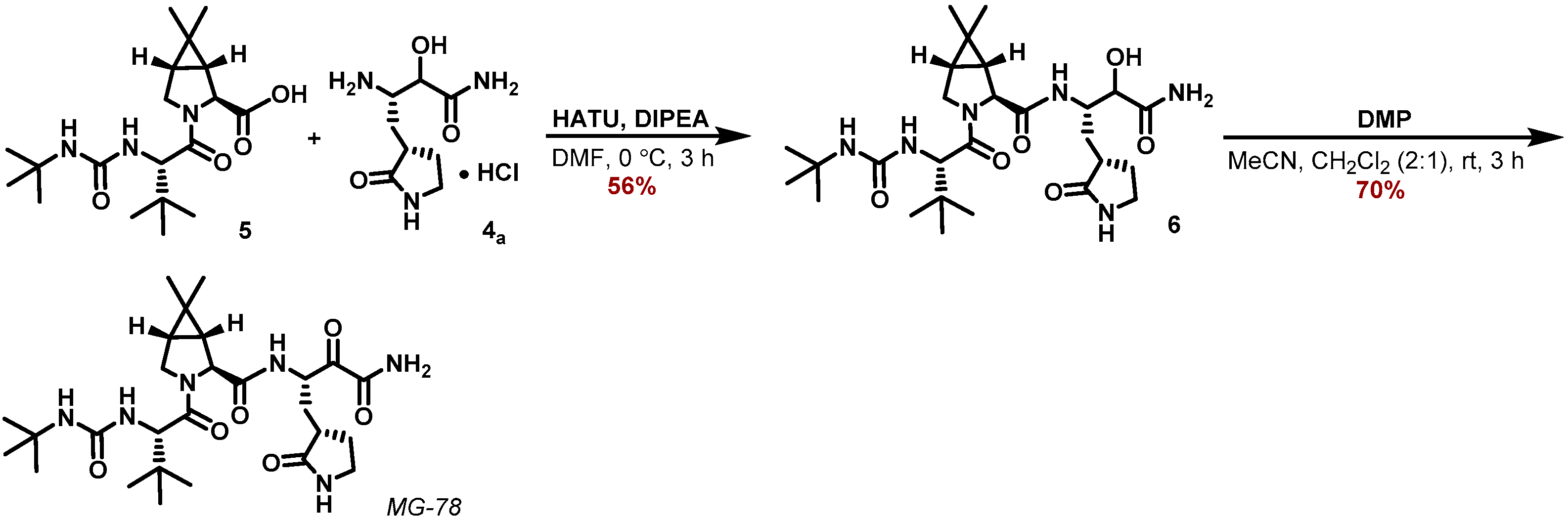
2.6. Design and Synthesis of MG-78
2.7. Crystal Structures of the SARS-CoV Mpro Complexes with MG-78 and MG-131
2.8. P1 Residue
2.9. P2 Residue
2.10. P3 Position
2.11. P4 Residue
2.12. Inhibitory Activity of MG-78 against the Mpro of HCoV NL63
2.13. Is MG-78 Also an Inhibitor of Enterovirus 3C Proteases?
2.14. Binding of MG-78 to the Enterovirus A71 and the Coxsackievirus B3 3C Proteases
2.15. P1 Residue
2.16. P2 Residue
2.17. P3 and P4 Residues
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Inhibitors
5.2. Recombinant Protein Production
5.3. Determination of the Inhibitory Activity of Compounds against Coronavirus Mpros and Enterovirus 3Cpros
5.4. Crystallization and Diffraction Data Collection of SARS-CoV-2 Mpro in Complexes with Boceprevir, Telaprevir, MG-78, or MG-131
5.5. Crystallization and Diffraction Data Collection of the MG-78 Complexes with Enterovirus-A71 3Cpro and Coxsackievirus B3 3Cpro
5.6. Diffraction Data Processing, Structure Solution, and Refinement
5.7. Compound Synthesis
5.7.1. General Experimental Information
5.7.2. Preparation of tert-Butyl ((2S)-1-cyano-1-hydroxy-3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl)carbamate (2a,b)

5.7.3. Preparation of tert-Butyl ((2S)-1-cyano-1-hydroxy-3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl)carbamate (3a,b)

5.7.4. Major Diastereomer 3a
5.7.5. Minor Diastereomer 3b
5.7.6. Preparation of (3S)-3-Amino-2-hydroxy-4-((S)-2-oxopyrrolidin-3-yl)butanamide (4a)

5.7.7. Preparation of (1R,2S,5S)-N-((2S)-4-Amino-3-hydroxy-4-oxo-1-((S)-2-oxopyrrolidin-3-yl) butan-2-yl)-3-((S)-2-(3-(tert-butyl)ureido)-3,3-dimethylbutanoyl)-6,6-dimethyl-3-azabicyclo[3.1.0]hexane-2-carboxamide (6)

5.7.8. Preparation of (1R,2S,5S)-N-((S)-4-Amino-3,4-dioxo-1-((S)-2-oxopyrrolidin-3-yl)butan2-yl)-3-((S)-2-(3-tert-butylureido)-3,3-dimethylbutanoyl)-6,6-dimethyl-3-azabicylo[3.1.0]hexane-2-carboxamide (MG-78)

5.7.9. Preparation of (3S)-3-((tert-Butoxycarbonyl)amino)-1-(methylamino)-1-oxo-4-((S)2-oxopyrrolidin-3-yl)butan-2-yl acetate (7a,b)

5.7.10. Preparation of tert-Butyl ((2S)-3-hydroxy-4-(methylamino)-4-oxo-1-((S)-2-oxo-pyrrolidin-3-yl)butan-2yl)carbamate (8a,b)

5.7.11. Major Diastereomer 8a
5.7.12. Preparation of (3S)-3-Amino-2-hydroxy-N-methyl-4-((S)-2-oxopyrrolidin-3-yl)butanamide (9a,b)

5.7.13. Preparation of (1R,2S,5S)-3-((S)-2-(3-(tert-Butyl)ureido)-3,3-dimethylbutanoyl)-N((2S)3-hydroxy4-(methylamino)-4-oxo-1-((S)-2-oxopyrrolidin-3-yl)butan-2-yl)-6,6-dimethyl -3-azabicyclo[3.1.0]hexane-2-carboxamide (10a,b)

5.7.14. Major Diastereomer 10a
5.7.15. Preparation of (1R,2S,5S)-3-((S)-2-(3-tert-Butylureido)-3,3-dimethylbutanoyl) -6,6-dimethyl-N-((S)-4-(methylamino)-3,4-dioxo-1-((S)-2-oxopyrrolidin-3-yl)butan-2-yl)-3-azabicyclo[3.1.0]hexane-2-carboxamide (MG-131)

Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Dai, W.; Zhang, B.; Jiang, X.-M.; Su, H.; Li, J.; Zhao, Y.; Xie, X.; Jin, Z.; Peng, J.; Liu, F.; et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science 2020, 368, 1331–1335. [Google Scholar] [CrossRef]
- Almaraz-Girón, M.A.; Calderón-Jaimes, E.; Carrillo, A.S.; Díaz-Cervantes, E.; Alonso, E.C.; Islas-Jácome, A.; Domínguez-Ortiz, A.; Castañón-Alonso, S.L. Search for Non-Protein Protease Inhibitors Constituted with an Indole and Acetylene Core. Molecules 2021, 26, 3817. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef]
- Boras, B.; Jones, R.M.; Anson, B.J.; Arenson, D.; Aschenbrenner, L.; Bakowski, M.A.; Beutler, N.; Binder, J.; Chen, E.; Eng, H.; et al. Preclinical characterization of an intravenous coronavirus 3CL protease inhibitor for the potential treatment of COVID19. Nat. Commun. 2021, 12, 6055. [Google Scholar] [CrossRef] [PubMed]
- Zaidman, D.; Gehrtz, P.; Filep, M.; Fearon, D.; Gabizon, R.; Douangamath, A.; Prilusky, J.; Duberstein, S.; Cohen, G.; Owen, C.D.; et al. An automatic pipeline for the design of irreversible derivatives identifies a potent SARS-CoV-2 Mpro inhibitor. Cell Chem. Biol. 2021, 8, 1795–1806. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, N.; Sacco, M.D.; Ma, C.; Hu, Y.; Townsend, J.A.; Meng, X.; Zhang, F.; Zhang, X.; Ba, M.; Szeto, T.; et al. Expedited Approach toward the Rational Design of Noncovalent SARS-CoV-2 Main Protease Inhibitors. J. Med. Chem. 2022, 65, 2848–2865. [Google Scholar] [CrossRef]
- Quan, B.-X.; Shuai, H.; Xia, A.-J.; Hou, Y.; Zeng, R.; Liu, X.-L.; Lin, G.-F.; Qiao, J.-X.; Li, W.-P.; Wang, F.-L.; et al. An orally available Mpro inhibitor is effective against wild-type SARS-CoV-2 and variants including Omicron. Nat. Microbiol. 2022, 7, 716–725. [Google Scholar] [CrossRef] [PubMed]
- Unoh, Y.; Uehara, S.; Nakahara, K.; Nobori, H.; Yamatsu, Y.; Yamamoto, S.; Maruyama, Y.; Taoda, Y.; Kasamatsu, K.; Suto, T.; et al. Discovery of S-217622, a Noncovalent Oral SARS-CoV-2 3CL Protease Inhibitor Clinical Candidate for Treating COVID-19. J. Med. Chem. 2022, 65, 6499–6512. [Google Scholar] [CrossRef]
- Zhang, L.; Lin, D.; Kusov, Y.; Nian, Y.; Ma, Q.; Wang, J.; von Brunn, A.; Leyssen, P.; Lanko, K.; Neyts, J.; et al. α-Ketoamides as broad-spectrum inhibitors of coronavirus and enterovirus replication: Structure-based design, synthesis, and activity assessment. J. Med. Chem. 2020, 63, 4562–4578. [Google Scholar] [CrossRef]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef]
- Oerlemans, R.; Ruiz-Moreno, A.J.; Cong, Y.; Dinesh Kumar, N.; Velasco-Velazquez, M.A.; Neochoritis, C.G.; Smith, J.; Reggiori, F.; Groves, M.R.; Domling, A. Repurposing the HCV NS3-4A protease drug boceprevir as COVID-19 therapeutics. RSC Med. Chem. 2020, 12, 370–379. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Ye, F.; Feng, Y.; Yu, F.; Wang, Q.; Wu, W.; Zhao, C.; Sun, H.; Huang, B.; Niu, P.; et al. Both boceprevir and GC376 efficaciously inhibit SARS-CoV-2 by targeting its main protease. Nat. Commun. 2020, 11, 4417. [Google Scholar] [CrossRef]
- Qiao, J.; Li, Y.S.; Zeng, R.; Liu, F.L.; Luo, R.H.; Huang, C.; Wang, Y.F.; Zhang, J.; Quan, B.; Shen, C.; et al. SARS-CoV-2 Mpro inhibitors with antiviral activity in a transgenic mouse model. Science 2021, 371, 1374–1378. [Google Scholar] [CrossRef]
- Kneller, D.W.; Galanie, S.; Phillips, G.; O’Neill, H.M.; Coates, L.; Kovalevsky, A. Malleability of the SARS-CoV-2 3CL Mpro active-site cavity facilitates binding of clinical antivirals. Structure 2020, 28, 1313–1320. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Prongay, A.J.; Guo, Z.; Yao, N.; Pichardo, J.; Fischmann, T.; Strickland, C.; Myers, J.; Weber, P.C.; Beyer, B.M.; Ingram, R.; et al. Discovery of the HCV NS3/4A Protease Inhibitor (1R,5S)-N-[3-Amino-1-(cyclobutylmethyl)-2,3-dioxopropyl]-3-[2(S)-[[[(1,1-dimethylethyl)amino]carbonyl]amino]-3,3-dimethyl-1-oxobutyl]-6,6-dimenthyl-3-azabicyclo [3.1.0]hexan-2(S)-carboxamide (Sch 503034) II. Key Steps in Structure-Based Optimization. J. Med. Chem. 2007, 50, 2310–2318. [Google Scholar] [CrossRef]
- Romano, K.P.; Ali, A.; Aydin, C.; Soumana, D.; Ozen, A.; Deveau, L.M.; Silver, C.; Cao, H.; Newton, A.; Petropoulos, C.J.; et al. The molecular basis of drug resistance against hepatitis C virus NS3/4A protease inhibitors. PLoS Pathog. 2012, 8, e1002832. [Google Scholar] [CrossRef] [PubMed]
- Brandl, M.; Weiss, M.S.; Jabs, A.; Sühnel, J.; Hilgenfeld, R. C-H···π-interactions in proteins. J. Mol. Biol. 2001, 307, 357–377. [Google Scholar] [CrossRef]
- Weiss, M.S.; Brandl, M.; Sühnel, J.; Pal, D.; Hilgenfeld, R. More hydrogen bonds for the (structural) biologist. Trends Biochem. Sci. 2001, 26, 521–523. [Google Scholar] [CrossRef]
- Anand, K.; Ziebuhr, J.; Wadhwani, P.; Mesters, J.R.; Hilgenfeld, R. Coronavirus main proteinase (3CLpro) structure: Basis for design of anti-SARS drugs. Science 2003, 300, 1763–1767. [Google Scholar] [CrossRef]
- Yang, H.; Xie, W.; Xue, X.; Yang, K.; Ma, J.; Liang, W.; Zhao, Q.; Zhou, Z.; Pei, D.; Ziebuhr, J.; et al. Design of wide-spectrum inhibitors targeting coronavirus main proteases. PLoS Biol. 2005, 3, e324. [Google Scholar] [CrossRef]
- Bhalerao, D.S.; Arkala, A.K.R.; Madhavi, Y.V.; Nagaraju, M.; Gade, S.R.; Kumar, U.K.S.; Bandichhor, R.; Dahanukar, V.H. Synthesis and Process Optimization of Boceprevir: A Protease Inhibitor Drug. Org. Process Res. Dev. 2015, 19, 1559–1567. [Google Scholar] [CrossRef]
- Verschueren, K.H.; Pumpor, K.; Anemüller, S.; Chen, S.; Mesters, J.R.; Hilgenfeld, R. A structural view of the inactivation of the SARS coronavirus main proteinase by benzotriazole esters. Chem. Biol. 2008, 15, 597–606. [Google Scholar] [CrossRef]
- Rut, W.; Groborz, K.; Zhang, L.; Sun, X.; Zmudzinski, M.; Pawlik, B.; Wang, X.; Jochmans, D.; Neyts, J.; Młynarski, W.; et al. SARS-CoV-2 Mpro inhibitors and activity-based probes for patient-sample imaging. Nat. Chem. Biol. 2021, 17, 222–228. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, T.R.; Roqué-Rosell, N.; Birtley, J.R.; Leatherbarrow, R.J.; Curry, S. Structural and Mutagenic Analysis of Foot-and-Mouth Disease Virus 3C Protease Reveals the Role of the β-Ribbon in Proteolysis. J. Virol. 2007, 81, 115–124. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhang, R.; Beyer, B.M.; Durkin, J.; Ingram, R.; Njoroge, F.G.; Windsor, W.T.; Malcolm, B.A. A continuous spectrophotometric assay for the hepatitis C virus serine protease. Anal. Biochem. 1999, 270, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Morrison, J.F.; Walsh, C.T. The behavior and significance of slow-binding enzyme inhibitors. Adv. Enzymol. Relat Areas Mol. Biol. 1988, 61, 201–301. [Google Scholar] [CrossRef] [PubMed]
- Xia, Z.; Sacco, M.; Hu, Y.; Ma, C.; Meng, X.; Zhang, F.; Szeto, T.; Xiang, Y.; Chen, Y.; Wang, J. Rational design of hybrid SARS-CoV-2 main protease inhibitors guided by the superimposed cocrystal structures with the peptidomimetic inhibitors GC-376, telaprevir, and boceprevir. ACS Pharmacol. Transl. Sci. 2021, 4, 1408–1421. [Google Scholar] [CrossRef] [PubMed]
- Dragovich, P.S.; Zhou, R.; Skalitzky, D.J.; Fuhrman, S.A.; Patick, A.K.; Ford, C.E.; Meador, J.W., 3rd; Worland, S.T. Solid-phase synthesis of irreversible human rhinovirus 3C protease inhibitors. Part 1: Optimization of tripeptides incorporating N-terminal amides. Bioorg. Med. Chem. 1999, 7, 589–598. [Google Scholar] [CrossRef]
- Tan, J.; George, S.; Kusov, Y.; Perbandt, M.; Anemüller, S.; Mesters, J.R.; Norder, H.; Coutard, B.; Lacroix, C.; Leyssen, P.; et al. 3C protease of enterovirus 68: Structure-based design of Michael acceptor inhibitors and their broad-spectrum antiviral effects against picornaviruses. J. Virol. 2013, 87, 4339–4351. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; George, S.; Schmidt, M.F.; Al-Gharabli, S.I.; Rademann, J.; Hilgenfeld, R. Peptide aldehyde inhibitors challenge the substrate specificity of the SARS-coronavirus main protease. Antivir. Res. 2011, 92, 204–212. [Google Scholar] [CrossRef]
- Owen, D.R.; Allerton, C.M.N.; Anderson, A.S.; Aschenbrenner, L.; Avery, M.; Berritt, S.; Boras, B.; Cardin, R.D.; Carlo, A.; Coffman, K.J.; et al. An oral SARS-CoV-2 Mpro inhibitor clinical candidate for the treatment of COVID-19. Science 2021, 374, 1586–1593. [Google Scholar] [CrossRef]
- Anand, K.; Palm, G.J.; Mesters, J.R.; Siddell, S.G.; Ziebuhr, J.; Hilgenfeld, R. Structure of coronavirus main protease reveals combination of a chymotrypsin fold with an extra alpha-helical domain. EMBO J. 2002, 21, 3213–3224. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Song, J. The catalysis of the SARS 3C-like protease is under extensive regulation by its extra domain. FEBS J. 2006, 273, 1035–1045. [Google Scholar] [CrossRef] [PubMed]
- Norder, H.; De Palma, A.M.; Selisko, B.; Costenaro, L.; Papageorgiou, N.; Arnan, C.; Coutard, B.; Lantez, V.; De Lamballerie, X.; Baronti, C.; et al. Picornavirus non-structural proteins as targets for new anti-virals with broad activity. Antivir. Res. 2011, 89, 204–218. [Google Scholar] [CrossRef] [PubMed]
- Krug, M.; Weiss, M.S.; Heinemann, U.; Mueller, U. XDSAPP: A graphical user interface for the convenient processing of diffraction data using XDS. J. Appl. Crystallogr. 2012, 45, 568–572. [Google Scholar] [CrossRef]
- Evans, P. Scaling and assessment of data quality. Acta Crystallogr. D Biol. Crystallogr. 2006, 62, 72–82. [Google Scholar] [CrossRef]
- Evans, P.R. An introduction to data reduction: Space-group determination, scaling and intensity statistics. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 282–292. [Google Scholar] [CrossRef]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.W.; McCoy, A.; et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 235–242. [Google Scholar] [CrossRef]
- Vagin, A.; Teplyakov, A. Molecular replacement with MOLREP. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 22–25. [Google Scholar] [CrossRef]
- Lebedev, A.A.; Young, P.; Isupov, M.N.; Moroz, O.V.; Vagin, A.A.; Murshudov, G.N. JLigand: A graphical tool for the CCP4 template-restraint library. Acta Crystallogr. D Biol. Crystallogr. 2012, 68, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [PubMed]
- Murshudov, G.N.; Skubák, P.; Lebedev, A.A.; Pannu, N.S.; Steiner, R.A.; Nicholls, R.A.; Winn, M.D.; Long, F.; Vagin, A.A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 355–367. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Göhl, M.; Zhang, L.; El Kilani, H.; Sun, X.; Zhang, K.; Brönstrup, M.; Hilgenfeld, R. From Repurposing to Redesign: Optimization of Boceprevir to Highly Potent Inhibitors of the SARS-CoV-2 Main Protease. Molecules 2022, 27, 4292. https://doi.org/10.3390/molecules27134292
Göhl M, Zhang L, El Kilani H, Sun X, Zhang K, Brönstrup M, Hilgenfeld R. From Repurposing to Redesign: Optimization of Boceprevir to Highly Potent Inhibitors of the SARS-CoV-2 Main Protease. Molecules. 2022; 27(13):4292. https://doi.org/10.3390/molecules27134292
Chicago/Turabian StyleGöhl, Matthias, Linlin Zhang, Haifa El Kilani, Xinyuanyuan Sun, Kaixuan Zhang, Mark Brönstrup, and Rolf Hilgenfeld. 2022. "From Repurposing to Redesign: Optimization of Boceprevir to Highly Potent Inhibitors of the SARS-CoV-2 Main Protease" Molecules 27, no. 13: 4292. https://doi.org/10.3390/molecules27134292
APA StyleGöhl, M., Zhang, L., El Kilani, H., Sun, X., Zhang, K., Brönstrup, M., & Hilgenfeld, R. (2022). From Repurposing to Redesign: Optimization of Boceprevir to Highly Potent Inhibitors of the SARS-CoV-2 Main Protease. Molecules, 27(13), 4292. https://doi.org/10.3390/molecules27134292