
Structural, Electronic, Reactivity, and Conformational Features of 2,5,5-Trimethyl-1,3,2-diheterophosphinane-2-sulfide, and Its Derivatives: DFT, MEP, and NBO Calculations


 ,
,  and
and
Abstract
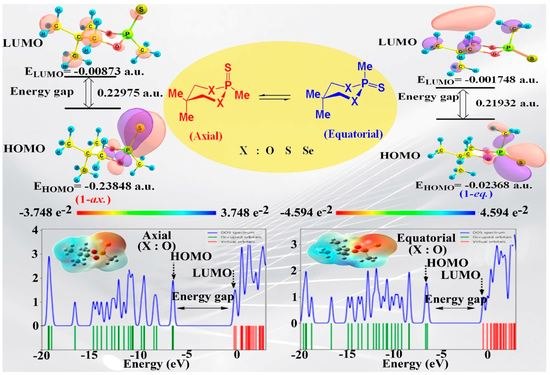
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Computational Models
3. Results and Discussion
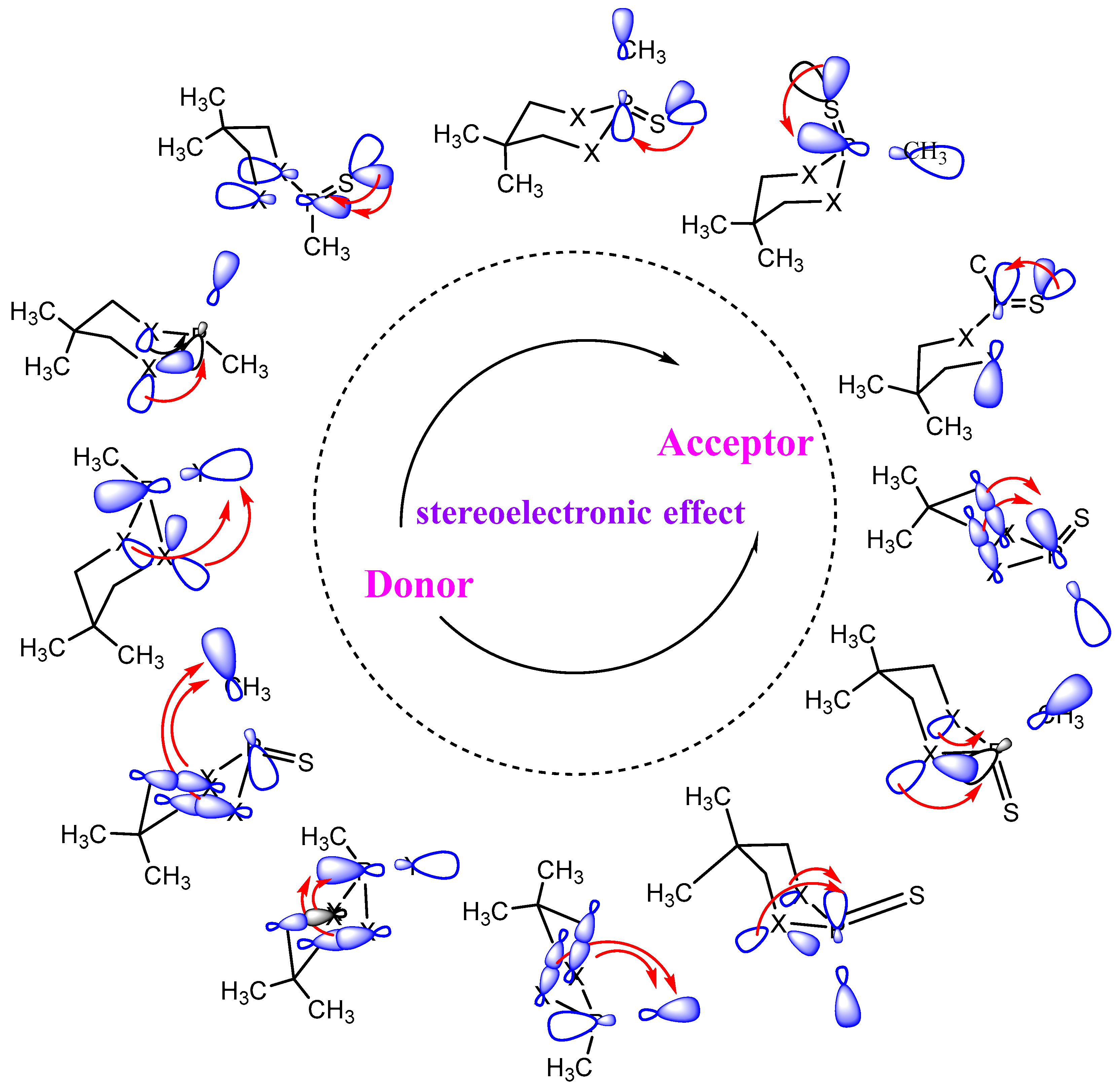
3.1. Conformational Preferences
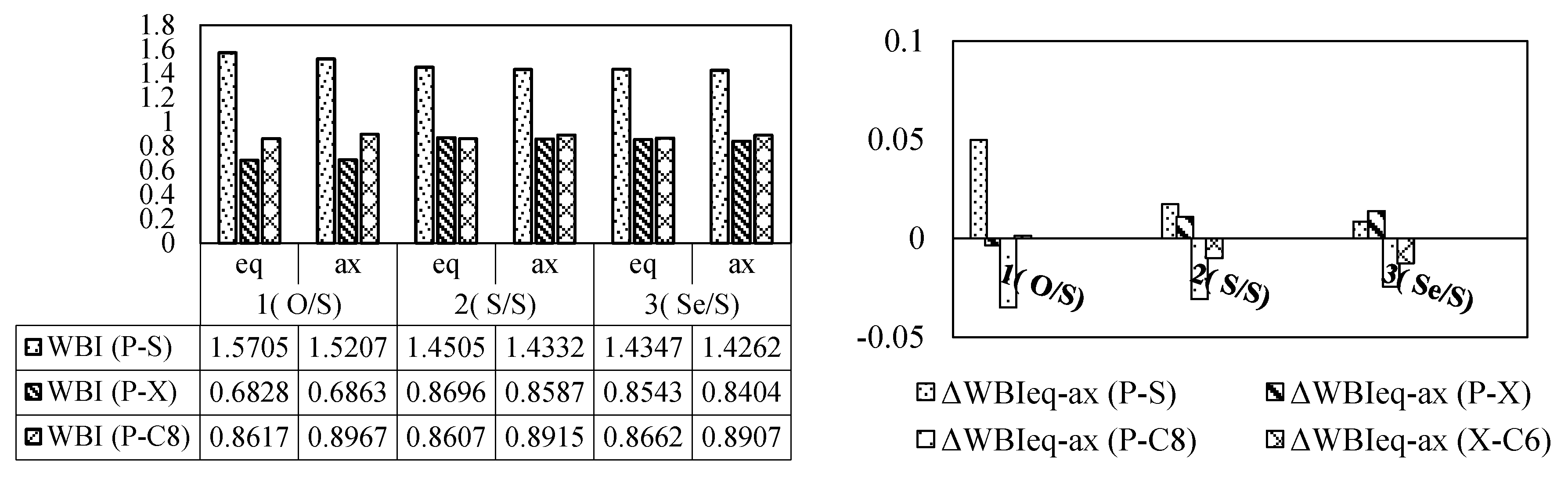
3.2. Bond Order and Structural Parameters
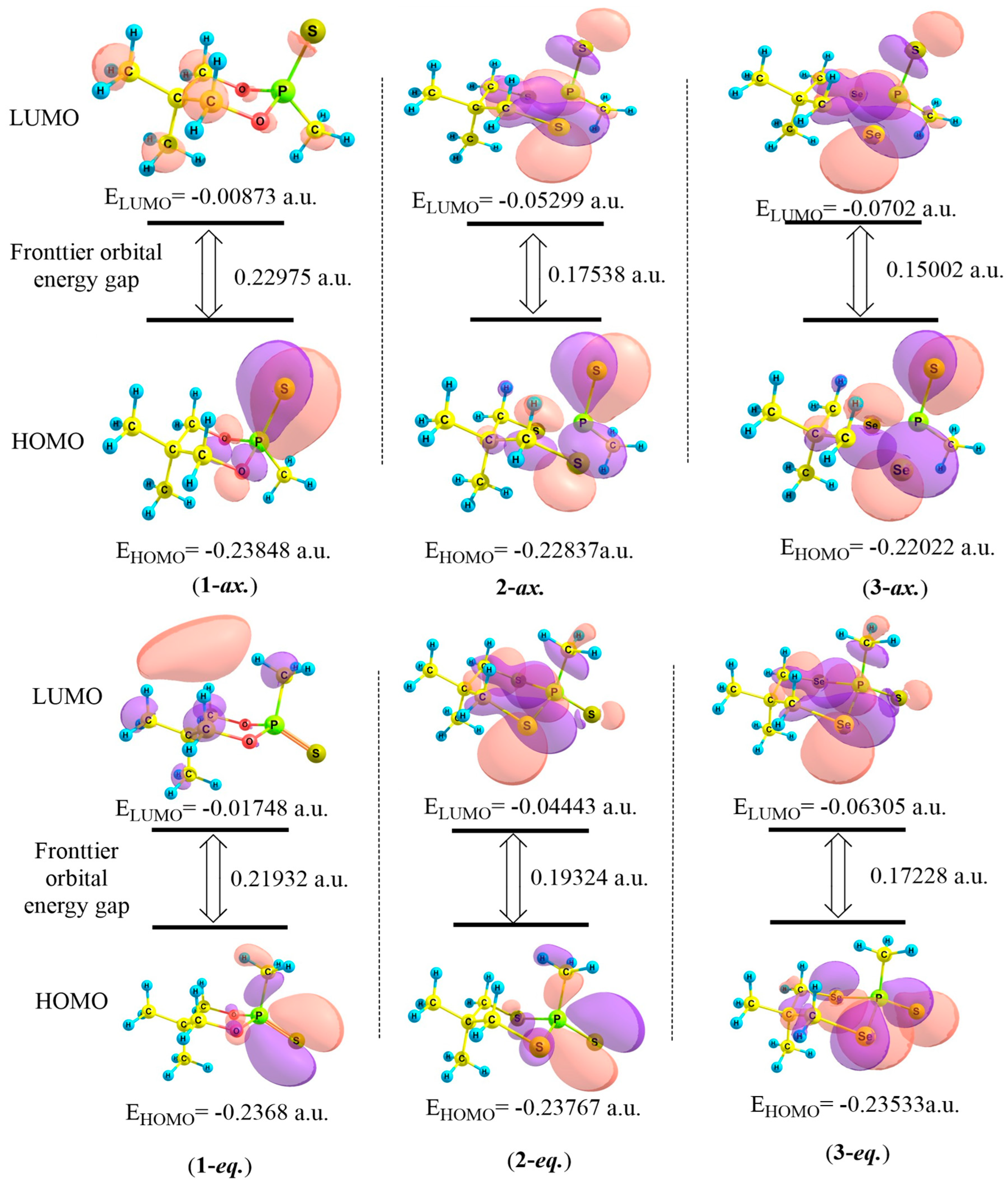
3.3. HOMO–LUMO Analysis
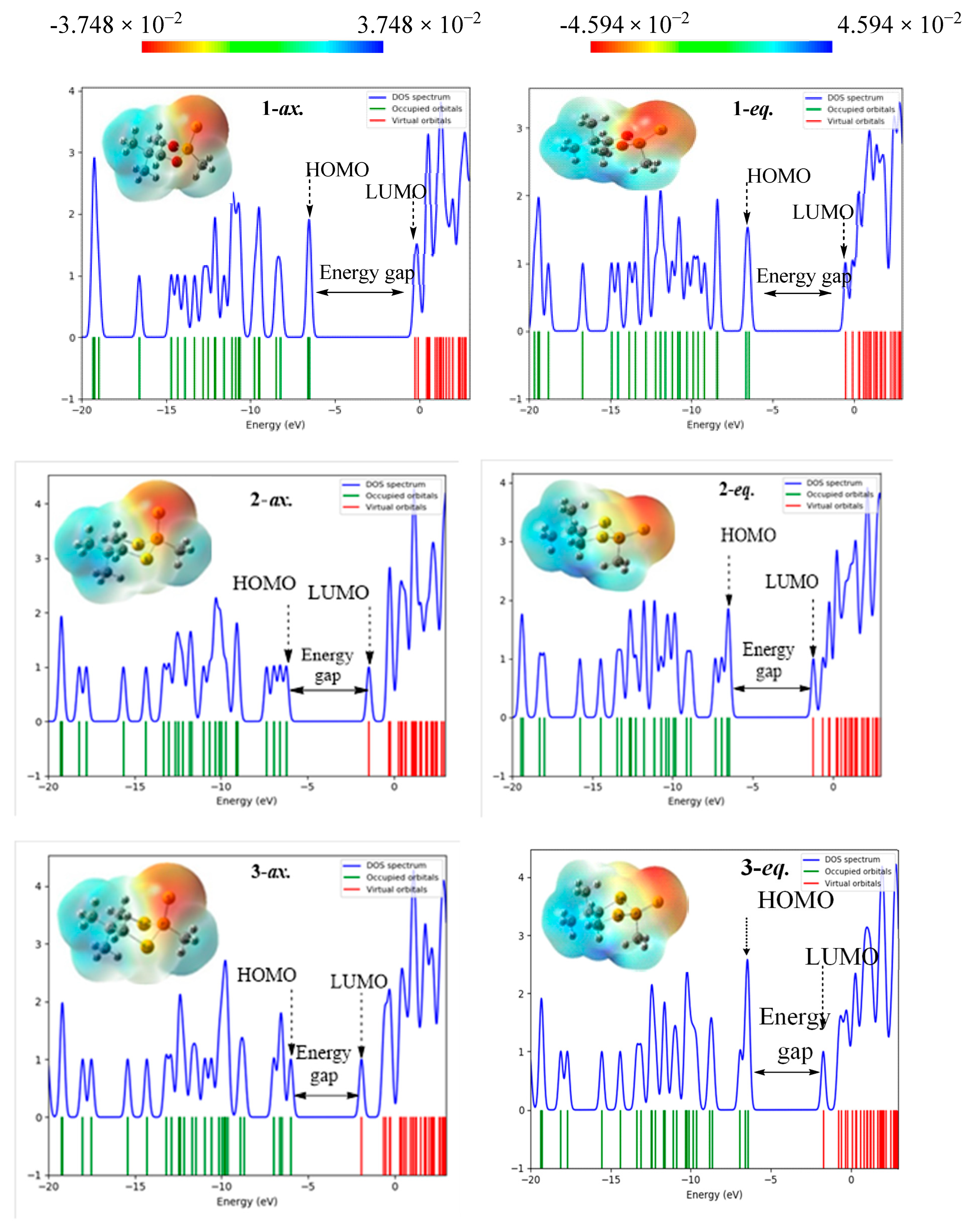
3.4. Molecular Electrostatic Potential (MEP) and Density of States (DOS) Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yao, J.; Wang, Z.; Guo, L.; Xu, X.; Liu, L.; Xu, L.; Song, S.; Xu, C.; Kuang, H. Advances in immunoassays for organophosphorus and pyrethroid pesticides. TrAC Trends Anal. Chem. 2020, 131, 116022. [Google Scholar] [CrossRef]
- Sidhu, G.K.; Singh, S.; Kumar, V.; Dhanjal, D.S.; Datta, S.; Singh, J. Toxicity, monitoring and biodegradation of organophosphate pesticides: A review. Crit. Rev. Environ. Sci. Technol. 2019, 49, 1135–1187. [Google Scholar] [CrossRef]
- Faiz, Y.; Siddique, N.; He, H.; Sun, C.; Waheed, S. Occurrence and profile of organophosphorus compounds in fine and coarse particulate matter from two urban areas of China and Pakistan. Environ. Pollut. 2018, 233, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Demkowicz, S.; Rachon, J.; Daśko, M.; Kozak, W. Selected organophosphorus compounds with biological activity. Applications in medicine. RSC Adv. 2016, 6, 7101–7112. [Google Scholar] [CrossRef]
- Songa, E.A.; Okonkwo, J.O. Recent approaches to improving selectivity and sensitivity of enzyme-based biosensors for organophosphorus pesticides: A review. Talanta 2016, 155, 289–304. [Google Scholar] [CrossRef]
- Oganov, A.R.; Pickard, C.J.; Zhu, Q.; Needs, R.J. Structure prediction drives materials discovery. Nat. Rev. Mater. 2019, 4, 331–348. [Google Scholar] [CrossRef]
- Muratov, E.N.; Amaro, R.; Andrade, C.H.; Brown, N.; Ekins, S.; Fourches, D.; Isayev, O.; Kozakov, D.; Medina-Franco, J.L.; Merz, K.M.; et al. A critical overview of computational approaches employed for COVID-19 drug discovery. Chem. Soc. Rev. 2021, 50, 9121–9151. [Google Scholar] [CrossRef]
- Liu, D.; Xu, S.; Pei, G.; Xu, J.; Zhao, X.; Kong, C.; Yang, Z.; Yang, T. Geometries, electronic structures, and bonding properties of endohedral Group-14 Zintl clusters TM@E10 (TM = Fe, Co, Ni; E = Ge, Sn, Pb). J. Comput. Chem. 2022, 43, 828–838. [Google Scholar] [CrossRef]
- Khalaf, M.M.; Tantawy, A.H.; Soliman, K.A.; Abd El-Lateef, H.M. Cationic gemini-surfactants based on waste cooking oil as new ‘green’ inhibitors for N80-steel corrosion in sulphuric acid: A combined empirical and theoretical approaches. J. Mol. Struct. 2020, 1203, 127442. [Google Scholar] [CrossRef]
- Sayadian, M.; Sadegh, H.; Ali, G.A.M. NMR spectra of Azobenzene-bridged calyx [8] arene complexes by ab initio hartree-fock calculations as nanostructure compound. Int. J. Nano Dimens. 2018, 9, 228–237. [Google Scholar]
- Hasanzadeh, N.; Nori-Shargh, D.; Yahyaei, H.; Mousavi, S.N.; Kamrava, S. Exploring the Origin of the Generalized Anomeric Effects in the Acyclic Nonplanar Systems. J. Phys. Chem. A 2017, 121, 5548–5560. [Google Scholar] [CrossRef] [PubMed]
- Alabugin, I.V.; Kuhn, L.; Krivoshchapov, N.V.; Mehaffy, P.; Medvedev, M.G. Anomeric effect, hyperconjugation and electrostatics: Lessons from complexity in a classic stereoelectronic phenomenon. Chem. Soc. Rev. 2021, 50, 10212–10252. [Google Scholar] [CrossRef] [PubMed]
- Praly, J.P.; Lemieux, R.U. Influence of solvent on the magnitude of the anomeric effect. Can. J. Chem. 1987, 65, 213–223. [Google Scholar] [CrossRef]
- Ortega, P.G.R.; Montejo, M.; Valera, M.S.; González, J.J.L. Anomeric effect in pyranose-ring derivatives containing carbon, silicon, and germanium as anomeric centers: An ab initio systematic study. Struct. Chem. 2019, 30, 2245–2255. [Google Scholar] [CrossRef]
- Ichikawa, Y.; Kaneno, D.; Saeki, N.; Minami, T.; Masuda, T.; Yoshida, K.; Kondo, T.; Ochi, R. Protecting group-free method for synthesis of N-glycosyl carbamates and an assessment of the anomeric effect of nitrogen in the carbamate group. Carbohydr. Res. 2021, 505, 108280. [Google Scholar] [CrossRef]
- Tsipis, C.A.; Bakalbassis, E.G.; Zisopoulou, S.A.; Gallos, J.K. Probing the anomeric effect and mechanism of isomerization of oxazinane rings by DFT methods. Org. Biomol. Chem. 2021, 19, 1066–1082. [Google Scholar] [CrossRef]
- Zhu, F.; Walczak, M.A. Stereochemistry of transition metal complexes controlled by the metallo-anomeric effect. J. Am. Chem. Soc. 2020, 142, 15127–15136. [Google Scholar] [CrossRef]
- Lesarri, A.; Vega-Toribio, A.; Suenram, R.D.; Brugh, D.J.; Nori-Shargh, D.; Boggs, J.E.; Grabow, J.U. Structural evidence of anomeric effects in the anesthetic isoflurane. Phys. Chem. Chem. Phys. 2011, 13, 6610–6618. [Google Scholar] [CrossRef] [Green Version]
- Jameh-Bozorghi, S.; Nori-Shargh, D.; Mousavi, S.N.; Rezaei, A. Hybrid-DFT study and NBO interpretation of the configurational behavior of 2-halotetrahydrothiopyran S-oxides. Phosphorus Sulfur Silicon Relat. Elem. 2013, 188, 839–849. [Google Scholar] [CrossRef]
- Huang, Y.; Zhong, A.-G.; Yang, Q.; Liu, S. Origin of anomeric effect: A density functional steric analysis. J. Chem. Phys. 2011, 134, 084103. [Google Scholar] [CrossRef]
- Wiberg, K.B.; Bailey, W.F.; Lambert, K.M.; Stempel, Z.D. The Anomeric Effect: It’s Complicated. J. Org. Chem. 2018, 83, 5242–5255. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Ying, F.; Wu, W.; Mo, Y. How Solvent Influences the Anomeric Effect: Roles of Hyperconjugative versus Steric Interactions on the Conformational Preference. J. Org. Chem. 2014, 79, 1571–1581. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Chen, Z.; Wu, W.; Mo, Y. How the Generalized Anomeric Effect Influences the Conformational Preference. Chem. A Eur. J. 2013, 19, 1436–1444. [Google Scholar] [CrossRef] [PubMed]
- Reed, A.E.; Rague Schleyer, P.V. The Anomeric Effect with Central Atoms Other Than Carbon. 1. Strong Interactions between Nonbonded Substituents in Polyfluorinated First- and Second-Row Hydrides. J. Am. Chem. Soc. 1987, 109, 7362–7373. [Google Scholar] [CrossRef]
- Mikolajczyk, M.; Graczyk, P.P. Synthesis and Conformational Behavior of 2-Phosphonio-and 2-Phosphinyl-1,3-dithianes. Operation of the Generalized Anomeric Effect in the S-C-P+ System. J. Org. Chem. 1995, 60, 5190–5208. [Google Scholar] [CrossRef]
- Alabugin, I.V.; Manoharan, M.; Zeidan, T.A. Homoanomeric Effects in Six-Membered Heterocycles. J. Am. Chem. Soc. 2003, 125, 14014–14031. [Google Scholar] [CrossRef]
- Balabanovich, A.I.; Prokopovich, V.P. Thermal decomposition study of 5,5,5′,5′,5″,5″- hexamethyltris(1,3,2-dioxaphosphorinanemethan)amine 2,2′,2″-trioxide and its mixtures with melamine phosphate. J. Anal. Appl. Pyrolysis 2006, 76, 169–177. [Google Scholar] [CrossRef]
- Kruszynski, R.; Czubacka, E.; Trzesowska-Kruszynska, A.; Bartczak, T.J.; Bruzik, K.S.; Knopik, P.; Kudzin, Z.; Stec, W.J.; Wolf, W.M. Conformation of sterically hindered 4-methyl-2-oxo-2-trityl-1,3,2- dioxaphosphorinane in the solid state and the solution. J. Chem. Crystallogr. 2011, 41, 908–918. [Google Scholar] [CrossRef] [Green Version]
- Dutasta, J.P.; Grand, A.; Robert, J.B.; Taieb, M. Conformational analysis of 2-thiono-1,3,2-dioxaphosphorinanes. Tetrahedron Lett. 1974, 15, 2659–2662. [Google Scholar] [CrossRef]
- Patois, C.; Ricard, L.; Savignac, P. 2-Alkyl-5,5-dimethyl-1,3,2-dioxaphosphorinan-2-ones a- Lithiated Carbanions. Synthesis, Stability, and Conformation. J. Chem. Soc. Perkin Trans. 1990, 6, 1577–1581. [Google Scholar] [CrossRef]
- Sartillo-Piscil, F.; Sánchez, M.; Cruz-Gregorio, S.; Quintero, L. Conformational and configurational analysis of 2-phenoxy-2-oxo-1,3,2- dioxaphosphorinanes. Conformational and configurational dependence upon conformation of the diol precursor. Tetrahedron 2004, 60, 3001–3008. [Google Scholar] [CrossRef]
- Wölfling, J.; Kovács-Pénzes, P.; Zupkó, I.; Schneider, G.; Frank, É. Synthesis, stereochemistry and cytotoxic activity of novel steroidal 16-spiro-1,3,2-dioxaphosphorinanes. J. Mol. Struct. 2012, 1013, 39–44. [Google Scholar] [CrossRef]
- Masnabadi, N.; Manesh, A.T.; Azarakhshi, F. Ab Initio Calculations of the Conformational Preferences of 1,3-Oxathiane S-Oxide and its Analogs Containing S and SE Atoms—Evidence for Stereoelectronic Interactions Associated with the Anomeric Effects. Phosphorus Sulfur Silicon Relat. Elem. 2013, 188, 1053–1063. [Google Scholar] [CrossRef]
- Wang, X.; Li, S.; Zhao, L.; Xu, C.; Gao, J. A DFT and TD-DFT study on electronic structures and UV-spectra properties of octaethyl-porphyrin with different central metals (Ni, V, Cu, Co). Chin. J. Chem. Eng. 2020, 28, 532–540. [Google Scholar] [CrossRef]
- Masoumeh, V.-N.; Ghiasi, R.; Shafiei, F. Conformational Analysis of 2-Methoxy-2-oxo-1,3,2-dioxaphosphorinane and Its Methylthio and Methylselenyl Analogues. Russ. J. Phys. Chem. A 2020, 94, 772–777. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Into a Functional of the Electron Density F F. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.; Trucks, G.; Schlegel, H.E.A.; Scuseria, G.W.; Robb, M.; Cheeseman, J.; Montgomery, J., Jr.; Vreven, T.; Kudin, K.; Burant, J. Gaussian 03, Revision C. 02; Gaussian, Inc.: Wallingford, CT, USA, 2004. [Google Scholar]
- Cotton, F.A.; Rice, C.E.; Rice, G.W. The crystal and molecular structures of bis(2,4-pentanedionato)chromium. Inorg. Chim. Acta 1977, 24, 231–234. [Google Scholar] [CrossRef]
- Port, V.C.; Cormanich, R.A. There and back again: The role of hyperconjugation in the fluorine: Gauche effect. Phys. Chem. Chem. Phys. 2021, 23, 17329–17337. [Google Scholar] [CrossRef]
- Vermeeren, P.; Doppert, M.T.; Bickelhaupt, F.M.; Hamlin, T.A. How metallylenes activate small molecules. Chem. Sci. 2021, 12, 4526–4535. [Google Scholar] [CrossRef]
- Vermeeren, P.; Hamlin, T.A.; Bickelhaupt, F.M. Chemical reactivity from an activation strain perspective. Chem. Commun. 2021, 57, 5880–5896. [Google Scholar] [CrossRef] [PubMed]
- Alabugin, I.V.; Zeidan, T.A. Stereoelectronic effects and general trends in hyperconjugative acceptor ability of σ bonds. J. Am. Chem. Soc. 2002, 124, 3175–3185. [Google Scholar] [CrossRef] [PubMed]
- Mattelaer, C.A.; Mattelaer, H.P.; Rihon, J.; Froeyen, M.; Lescrinier, E. Efficient and Accurate Potential Energy Surfaces of Puckering in Sugar-Modified Nucleosides. J. Chem. Theory Comput. 2021, 17, 3814–3823. [Google Scholar] [CrossRef]
- Alabugin, I.V.; Bresch, S.; dos Passos Gomes, G. Orbital hybridization: A key electronic factor in control of structure and reactivity. J. Phys. Org. Chem. 2015, 28, 147–162. [Google Scholar] [CrossRef]
- Zhang, J.X.; Sheong, F.K.; Lin, Z. Unravelling Chemical Interactions with Principal Interacting Orbital Analysis. Chem. A Eur. J. 2018, 24, 9639–9650. [Google Scholar] [CrossRef]
- Saputri, W.D.; Pranowo, H.D.; Hofer, T.S. Can’t we negotiate the importance of electron correlation? HF vs RIMP2 in ab initio quantum mechanical charge field molecular dynamics simulations of Cu+ in pure liquid ammonia. J. Mol. Liq. 2022, 347, 118286. [Google Scholar] [CrossRef]
- Doust Mohammadi, M.; Abdullah, H.Y. The Adsorption of Chlorofluoromethane on Pristine, Al-, Ga-, P-, and As-doped Boron Nitride Nanotubes: A PBC-DFT, NBO, and QTAIM Study. ChemistrySelect 2020, 5, 12115–12124. [Google Scholar] [CrossRef]
- Mihalovits, L.M.; Ferenczy, G.G.; Keserű, G.M. The role of quantum chemistry in covalent inhibitor design. Int. J. Quantum Chem. 2021, 122, e26768. [Google Scholar] [CrossRef]
- Mahross, M.H.; Taher, M.A.; Mostfa, M.A.; Chong, K.F.; Ali, G.A.M. Experimental and quantum investigations of novel corrosion inhibitors based triazene derivatives for mild steel. J. Mol. Struct. 2021, 1242, 130831. [Google Scholar] [CrossRef]
- Ou, Q.; Subotnik, J.E. Comparison between GW and Wave-Function-Based Approaches: Calculating the Ionization Potential and Electron Affinity for 1D Hubbard Chains. J. Phys. Chem. A 2016, 120, 4514–4525. [Google Scholar] [CrossRef]
- Mostfa, M.A.; Gomaa, H.; Othman, I.M.M.; Ali, G.A.M. Experimental and theoretical studies of a novel synthesized azopyrazole-benzenesulfonamide derivative as an efficient corrosion inhibitor for mild steel. J. Iran. Chem. Soc. 2020, 18, 1231–1241. [Google Scholar] [CrossRef]
- Aihara, J.I. Reduced HOMO-LUMO Gap as an Index of Kinetic Stability for Polycyclic Aromatic Hydrocarbons. J. Phys. Chem. A 1999, 103, 7487–7495. [Google Scholar] [CrossRef]
- Abraham, C.S.; Prasana, J.C.; Muthu, S. Quantum mechanical, spectroscopic and docking studies of 2-Amino-3-bromo-5-nitropyridine by Density Functional Method. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2017, 181, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Madkour, L.H.; Kaya, S.; Guo, L.; Kaya, C. Quantum chemical calculations, molecular dynamic (MD) simulations and experimental studies of using some azo dyes as corrosion inhibitors for iron. Part 2: Bis–azo dye derivatives. J. Mol. Struct. 2018, 1163, 397–417. [Google Scholar] [CrossRef]
- Parthasarathi, R.; Padmanabhan, J.; Elango, M.; Subramanian, V.; Chattaraj, P.K. Intermolecular reactivity through the generalized philicity concept. Chem. Phys. Lett. 2004, 394, 225–230. [Google Scholar] [CrossRef]
- Rouhani, M. Evaluation of structural properties and antioxidant capacity of Proxison: A DFT investigation. Comput. Theor. Chem. 2021, 1195, 113096. [Google Scholar] [CrossRef]
- Sheikhi, M.; Azarakhshi, F.; Tafreshi, E.S.; Kaviani, S.; Shahab, S.; Ahmadianarog, M. Theoretical Study of the Resveratrol Adsorption on B12N12 and Mg-Decoration B12N12 Fullerenes. Bull. Korean Chem. Soc. 2021, 42, 878–888. [Google Scholar] [CrossRef]
- Karthick, T.; Tandon, P. Computational approaches to find the active binding sites of biological targets against busulfan. J. Mol. Modeling 2016, 22, 142. [Google Scholar] [CrossRef]
- Lipin, R.; Dhanabalan, A.K.; Gunasekaran, K.; Solomon, R.V. Piperazine-substituted derivatives of favipiravir for Nipah virus inhibition: What do in silico studies unravel? SN Appl. Sci. 2021, 3, 110. [Google Scholar] [CrossRef]
- Basri, R.; Khalid, M.; Shafiq, Z.; Tahir, M.S.; Khan, M.U.; Tahir, M.N.; Naseer, M.M.; Braga, A.A.C. Exploration of chromone-based thiosemicarbazone derivatives: SC-XRD/DFT, spectral (IR, UV−Vis) characterization, and quantum chemical analysis. ACS Omega 2020, 5, 30176–30188. [Google Scholar] [CrossRef]
- Chaudhary, T.; Chaudhary, M.K.; Joshi, B.D.; de Santana, M.S.A.; Ayala, A.P. Spectroscopic (FT-IR, Raman) analysis and computational study on conformational geometry, AIM and biological activity of cephalexin from DFT and molecular docking approach. J. Mol. Struct. 2021, 1240, 130594. [Google Scholar] [CrossRef]
- Noureddine, O.; Issaoui, N.; Medimagh, M.; Al-Dossary, O.; Marouani, H. Quantum chemical studies on molecular structure, AIM, ELF, RDG and antiviral activities of hybrid hydroxychloroquine in the treatment of COVID-19: Molecular docking and DFT calculations. J. King Saud Univ. Sci. 2021, 33, 101334. [Google Scholar] [CrossRef] [PubMed]
- Motlagh, N.M.; Rouhani, M.; Mirjafary, Z. Aminated C20 fullerene as a promising nanosensor for detection of A-234 nerve agent. Comput. Theor. Chem. 2020, 1186, 112907. [Google Scholar] [CrossRef]
- Aboulouard, A.; Mtougui, S.; Demir, N.; Moubarik, A.; Idrissi, M.E.; Can, M. New non-fullerene electron acceptors-based on quinoxaline derivatives for organic photovoltaic cells: DFT computational study. Synth. Met. 2021, 279, 116846. [Google Scholar] [CrossRef]
- Masnabadi, N. DFT Study and NBO Analysis of Conformation Properties of 2, 5, 5-Trimethyl-1, 3, 2-Dioxaphosphinane 2-Selenide and Their Dithia and Diselena Analogous. J. Sci. Islamic Repub. Iran 2020, 31, 137–146. [Google Scholar]
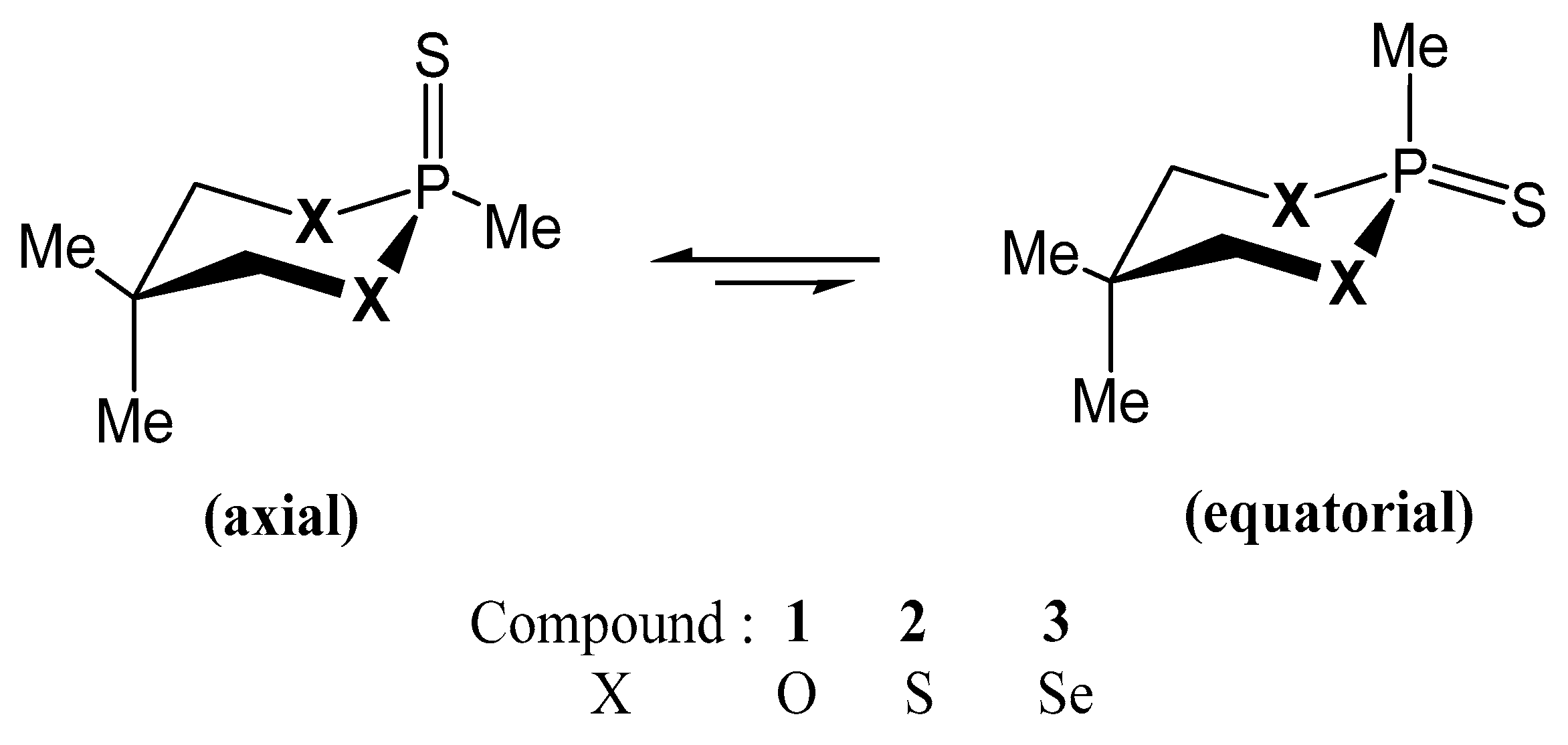


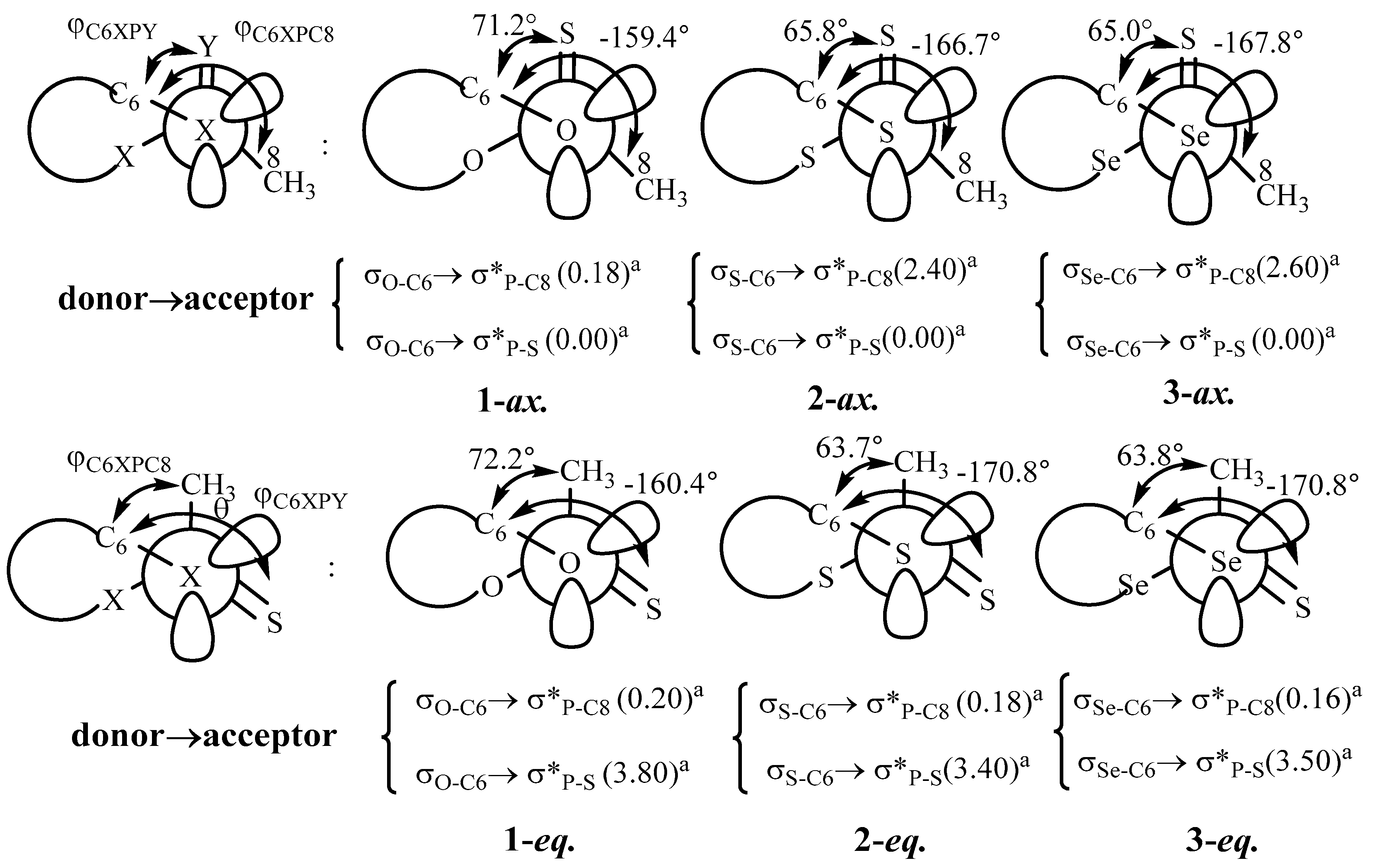

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Masnabadi, N.; Thalji, M.R.; Alhasan, H.S.; Mahmoodi, Z.; Soldatov, A.V.; Ali, G.A.M. Structural, Electronic, Reactivity, and Conformational Features of 2,5,5-Trimethyl-1,3,2-diheterophosphinane-2-sulfide, and Its Derivatives: DFT, MEP, and NBO Calculations. Molecules 2022, 27, 4011. https://doi.org/10.3390/molecules27134011
Masnabadi N, Thalji MR, Alhasan HS, Mahmoodi Z, Soldatov AV, Ali GAM. Structural, Electronic, Reactivity, and Conformational Features of 2,5,5-Trimethyl-1,3,2-diheterophosphinane-2-sulfide, and Its Derivatives: DFT, MEP, and NBO Calculations. Molecules. 2022; 27(13):4011. https://doi.org/10.3390/molecules27134011
Chicago/Turabian StyleMasnabadi, Nasrin, Mohammad R. Thalji, Huda S. Alhasan, Zahra Mahmoodi, Alexander V. Soldatov, and Gomaa A. M. Ali. 2022. "Structural, Electronic, Reactivity, and Conformational Features of 2,5,5-Trimethyl-1,3,2-diheterophosphinane-2-sulfide, and Its Derivatives: DFT, MEP, and NBO Calculations" Molecules 27, no. 13: 4011. https://doi.org/10.3390/molecules27134011
APA StyleMasnabadi, N., Thalji, M. R., Alhasan, H. S., Mahmoodi, Z., Soldatov, A. V., & Ali, G. A. M. (2022). Structural, Electronic, Reactivity, and Conformational Features of 2,5,5-Trimethyl-1,3,2-diheterophosphinane-2-sulfide, and Its Derivatives: DFT, MEP, and NBO Calculations. Molecules, 27(13), 4011. https://doi.org/10.3390/molecules27134011