
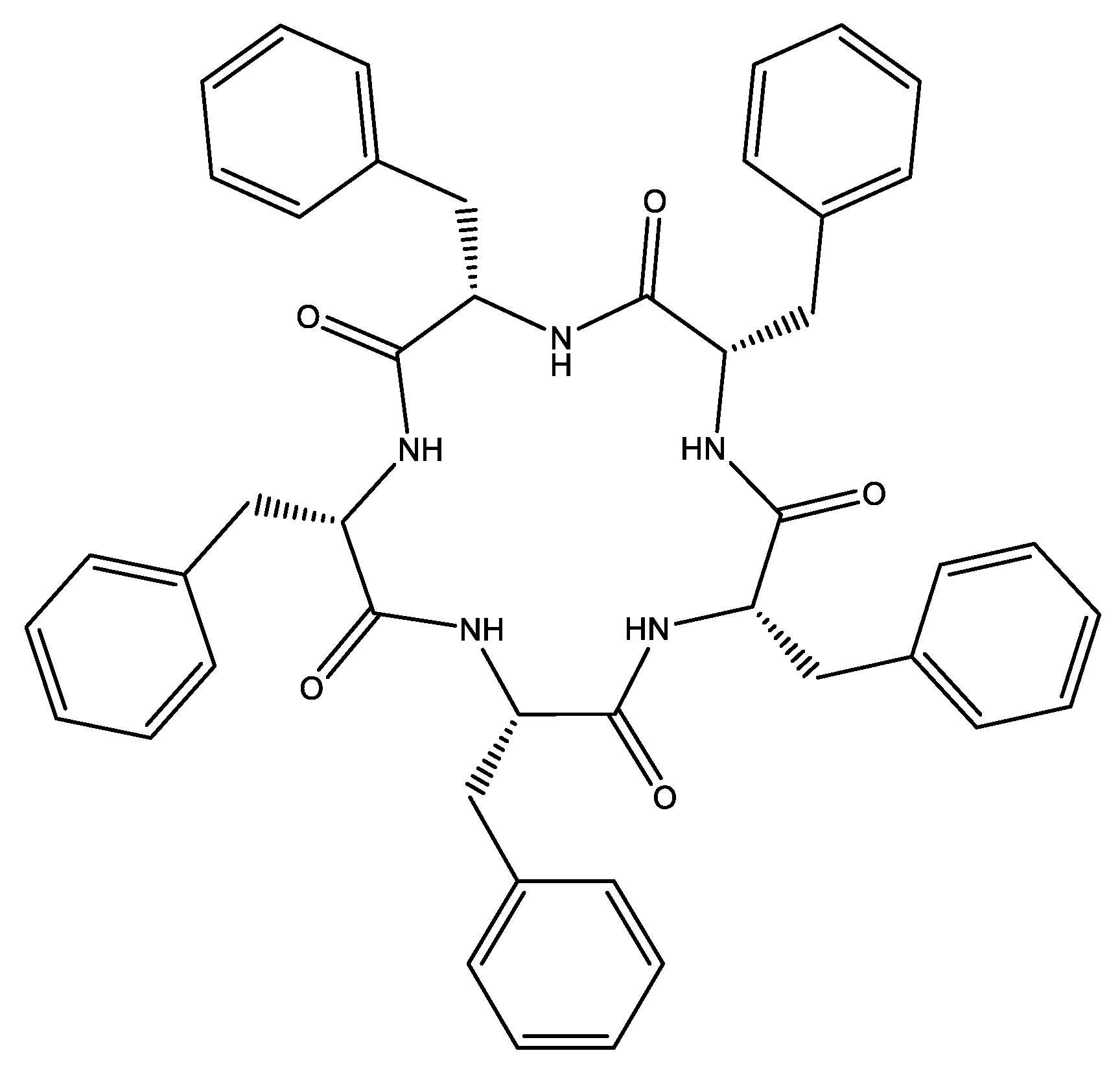
Anion-Sensing Properties of Cyclopentaphenylalanine

 , ,
, ,  ,
,  ,
, 
 and
and 
Abstract
:1. Introduction
2. Experimental Section
2.1. Materials
2.2. Physicochemical Measurements
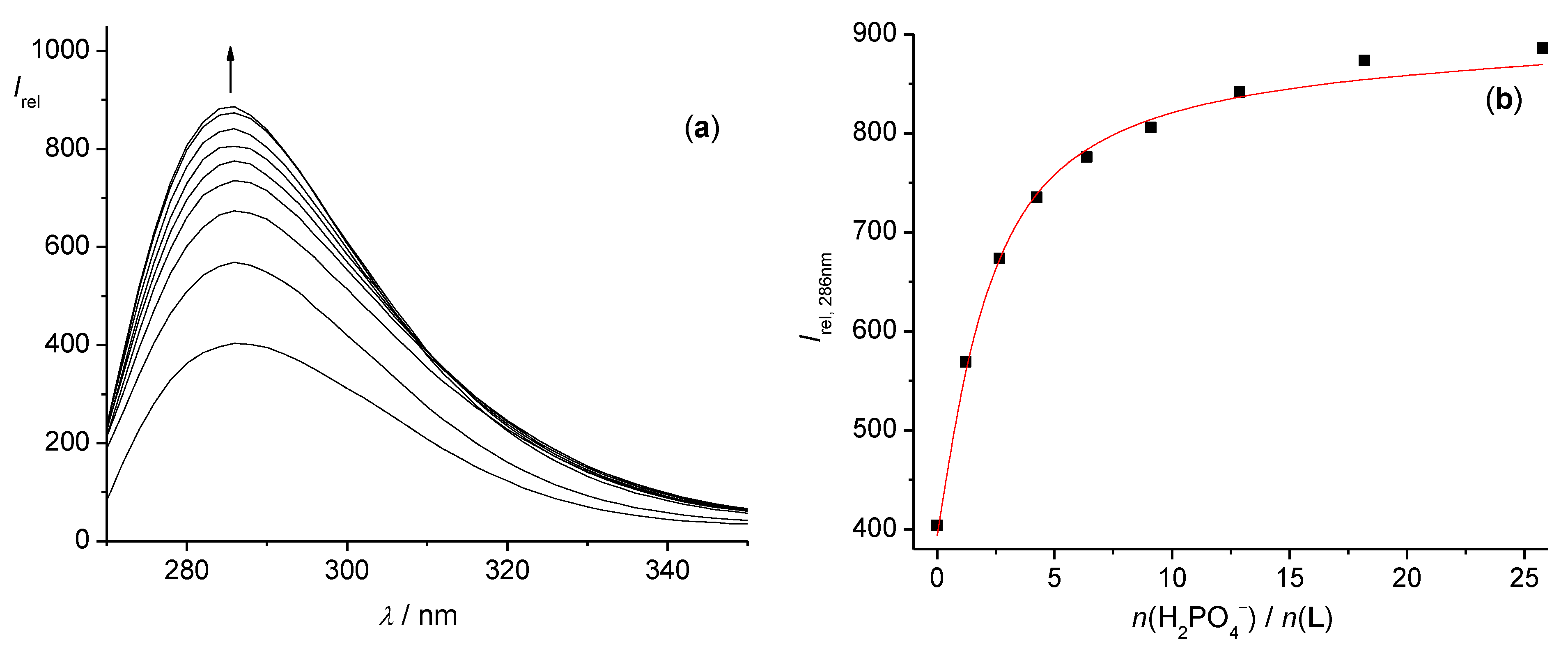
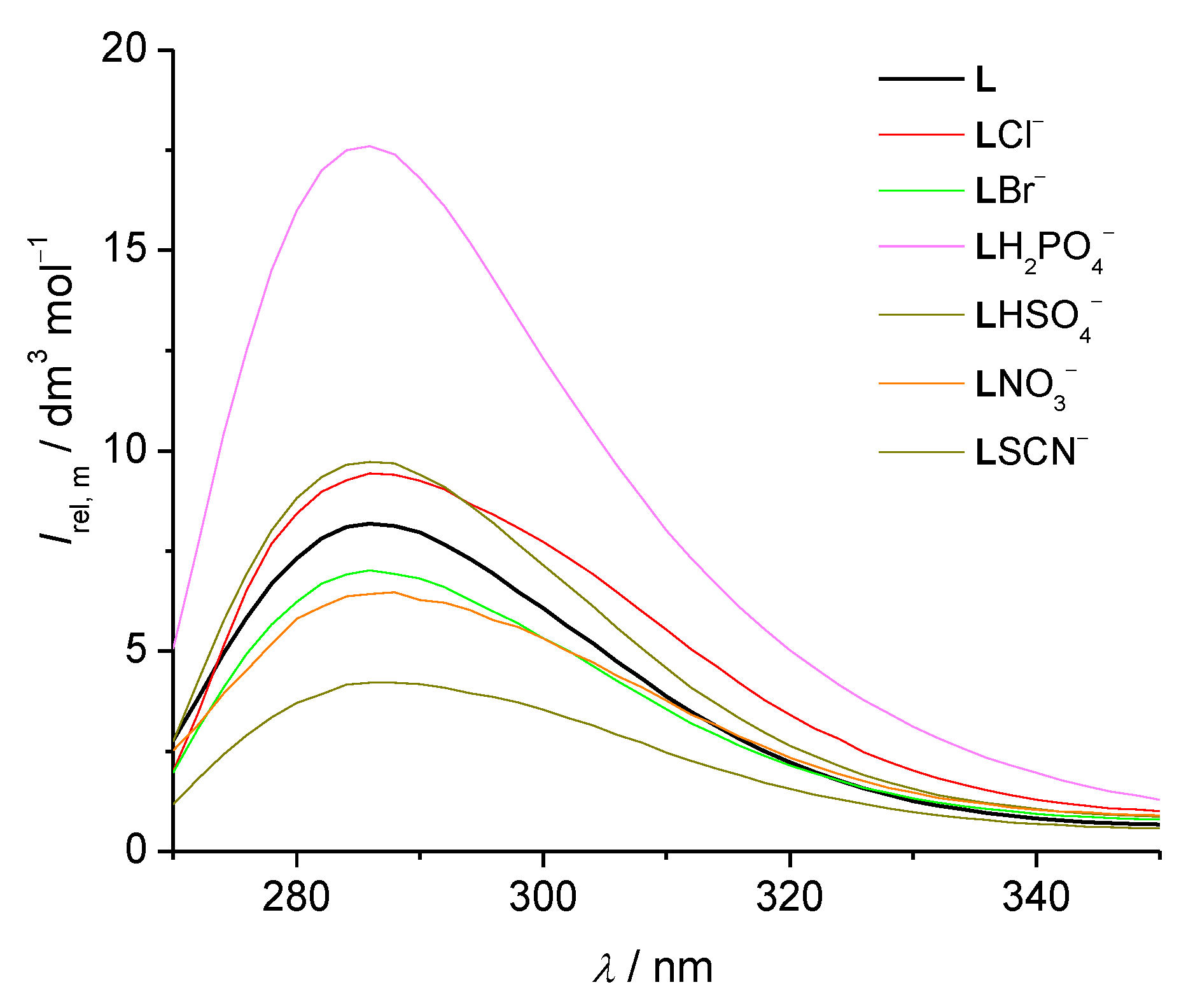
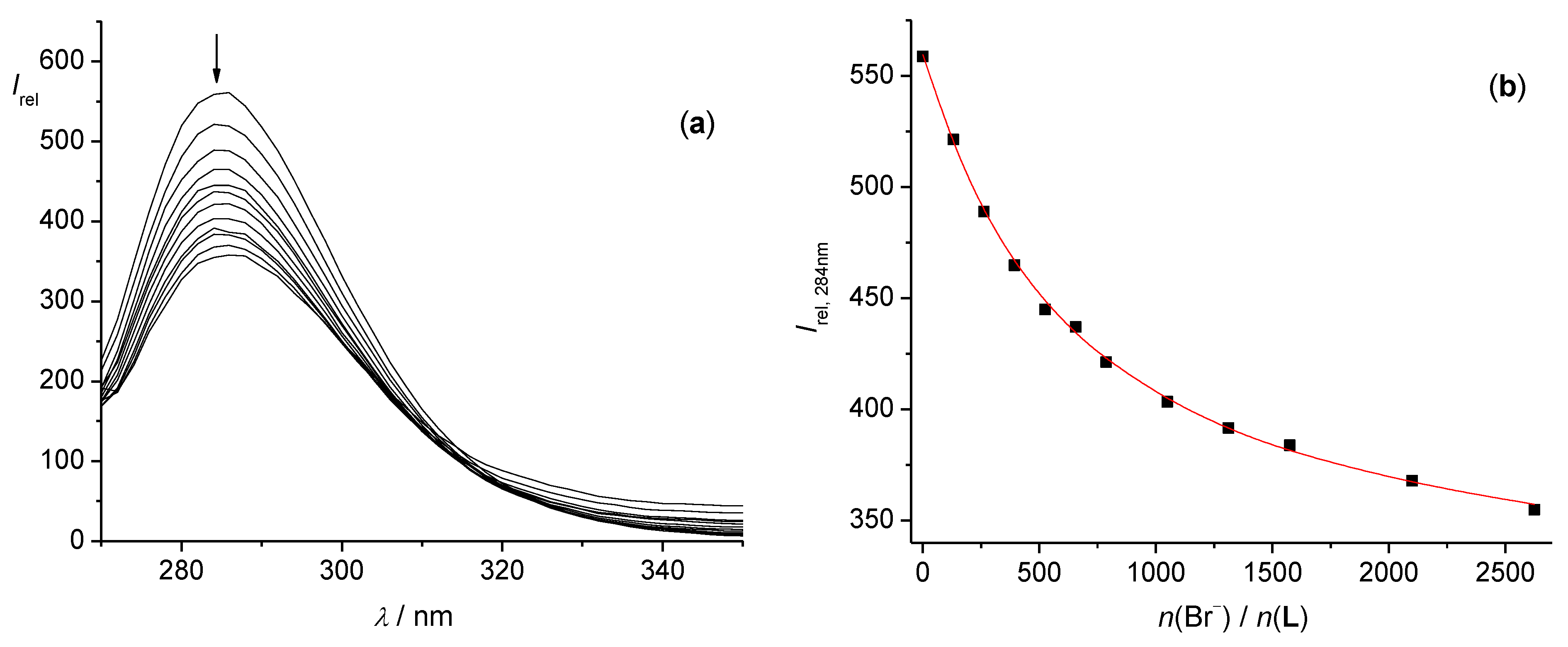
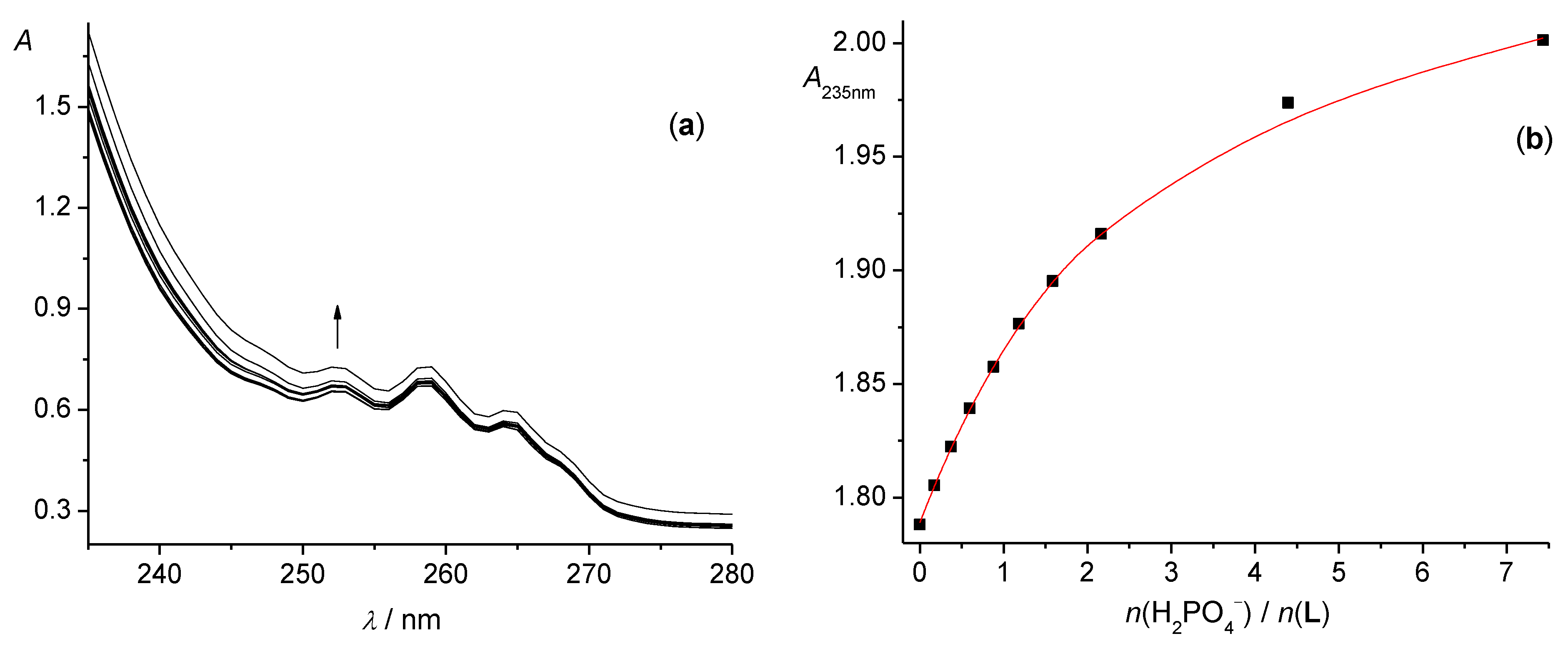
2.2.1. Fluorimetric, Spectrophotometric, and Circular Dichroism Titrations
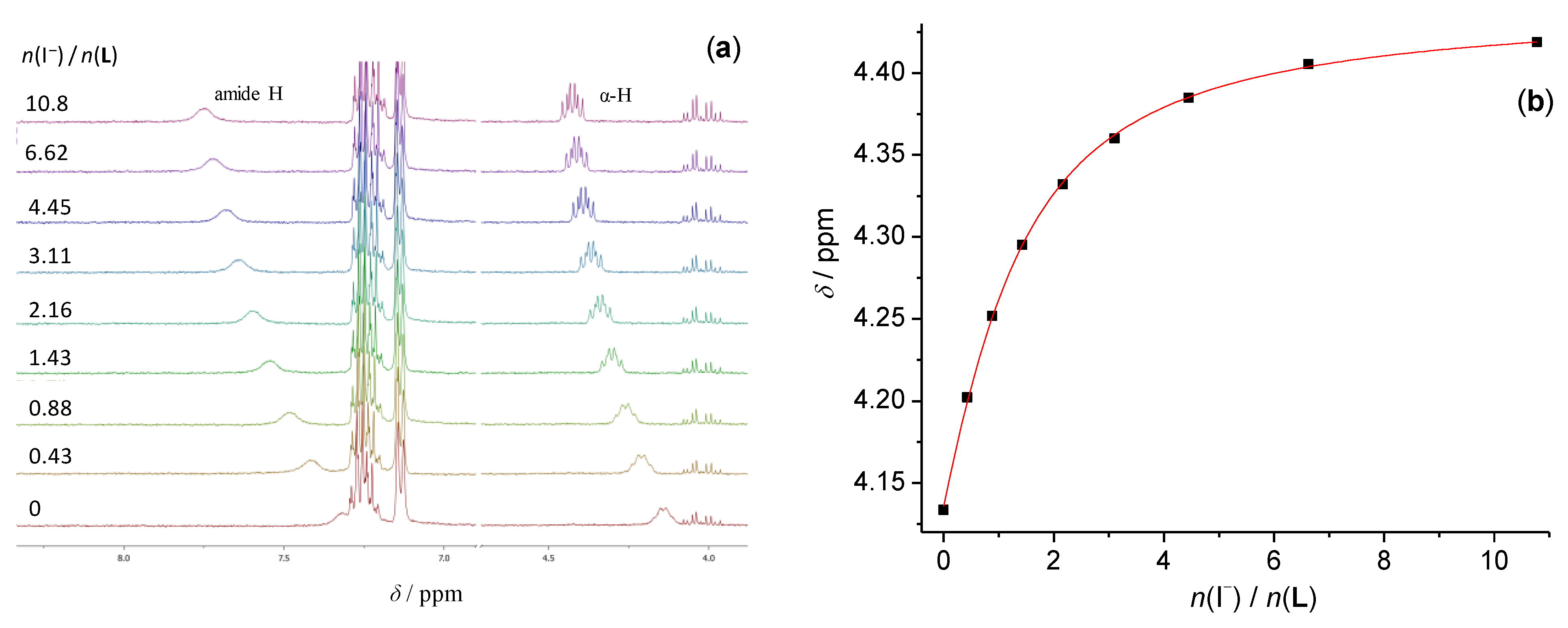
2.2.2. 1H NMR Titrations
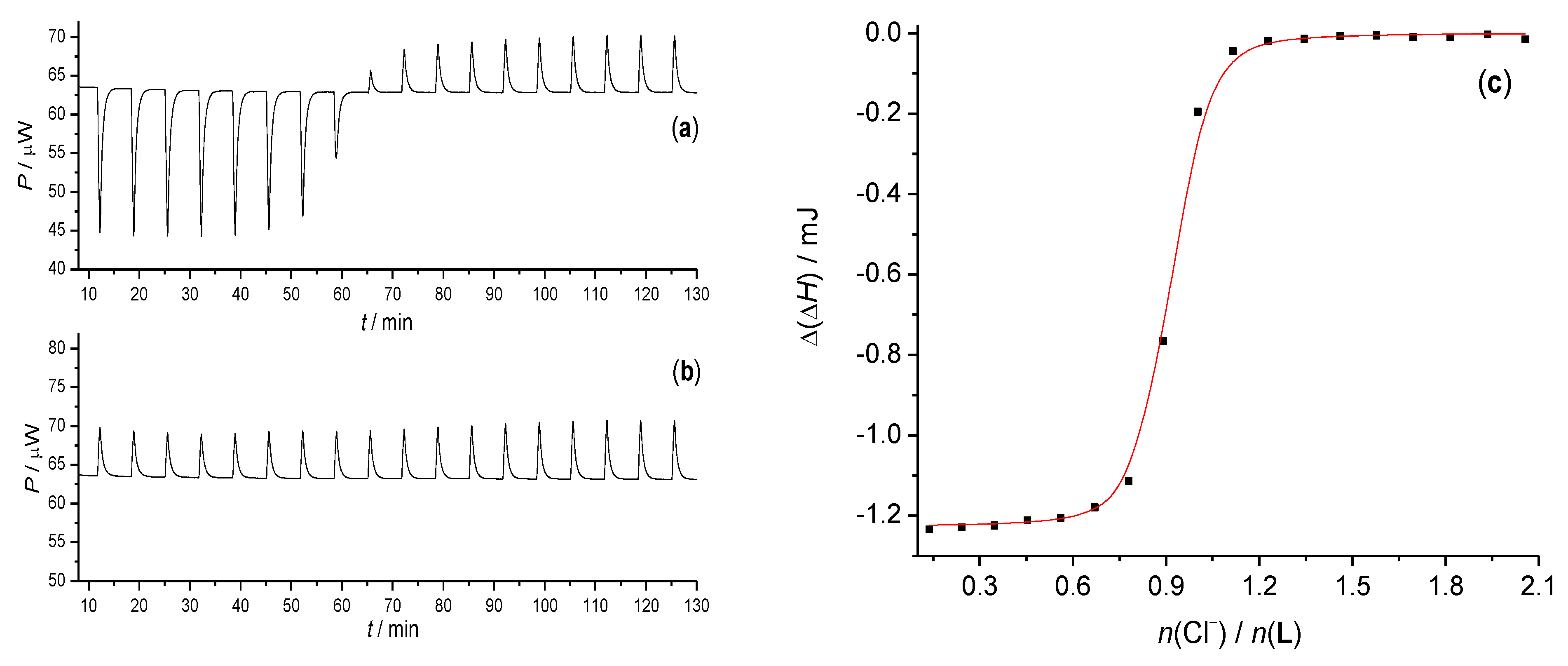
2.2.3. Isothermal Titration Calorimetry
2.2.4. Solubility Determinations
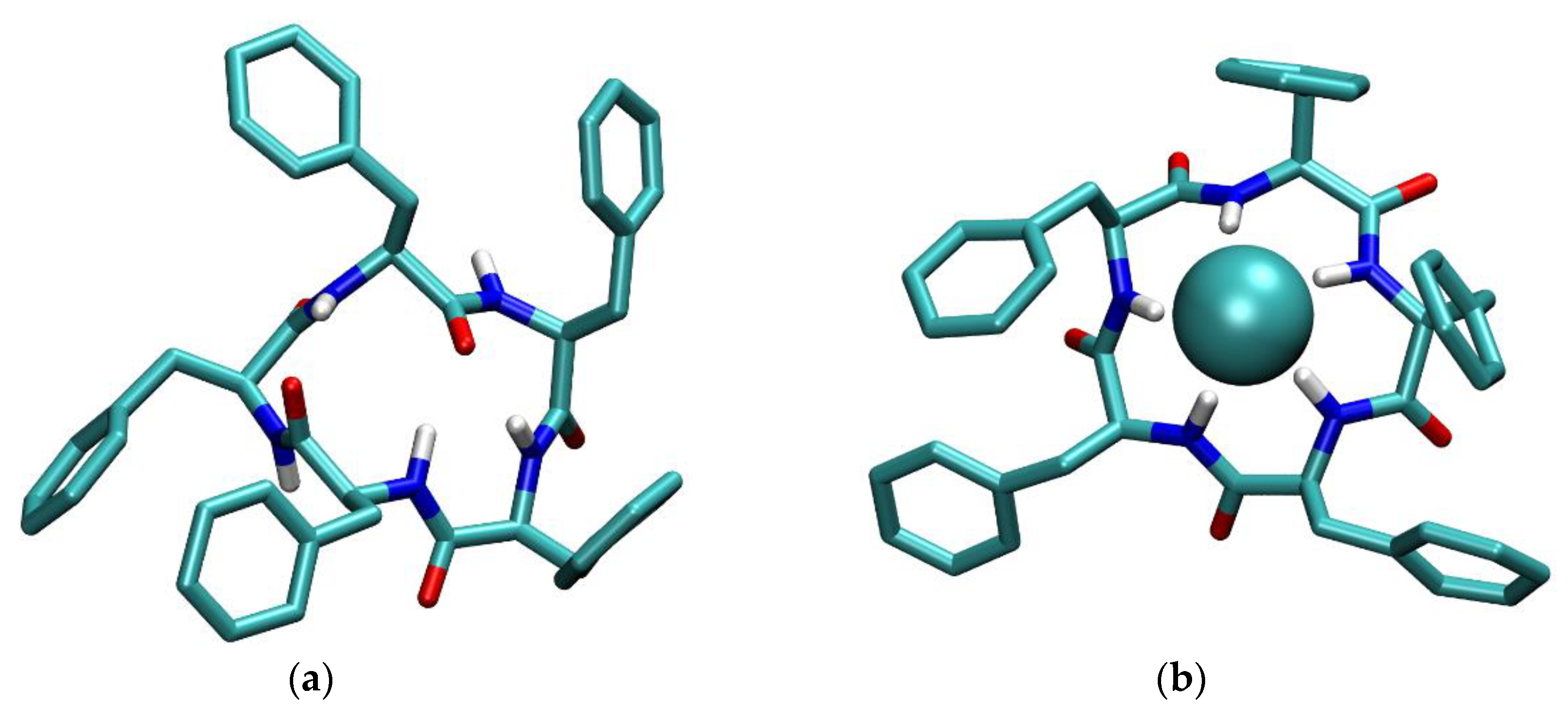
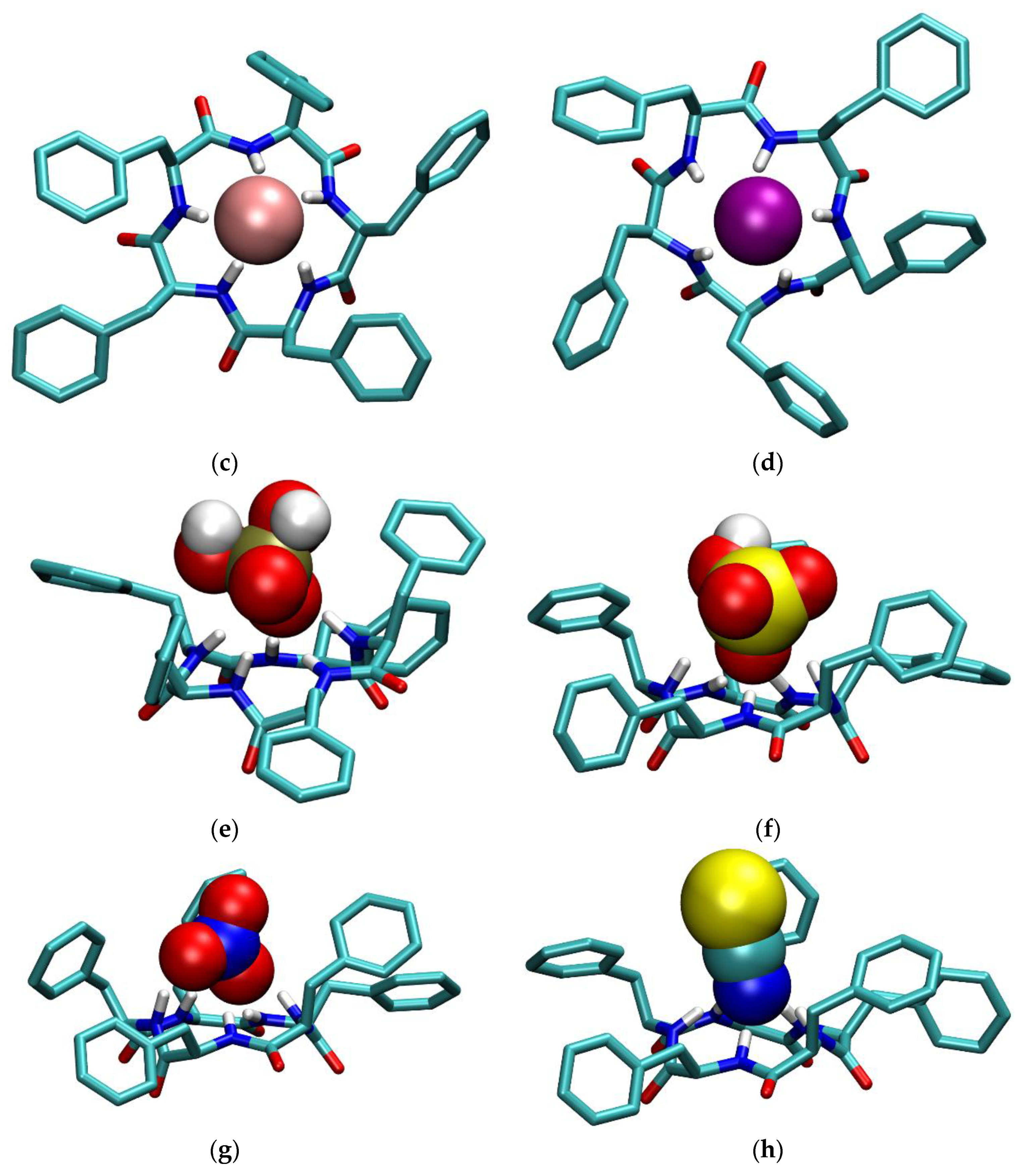
2.3. Molecular Dynamics Simulations
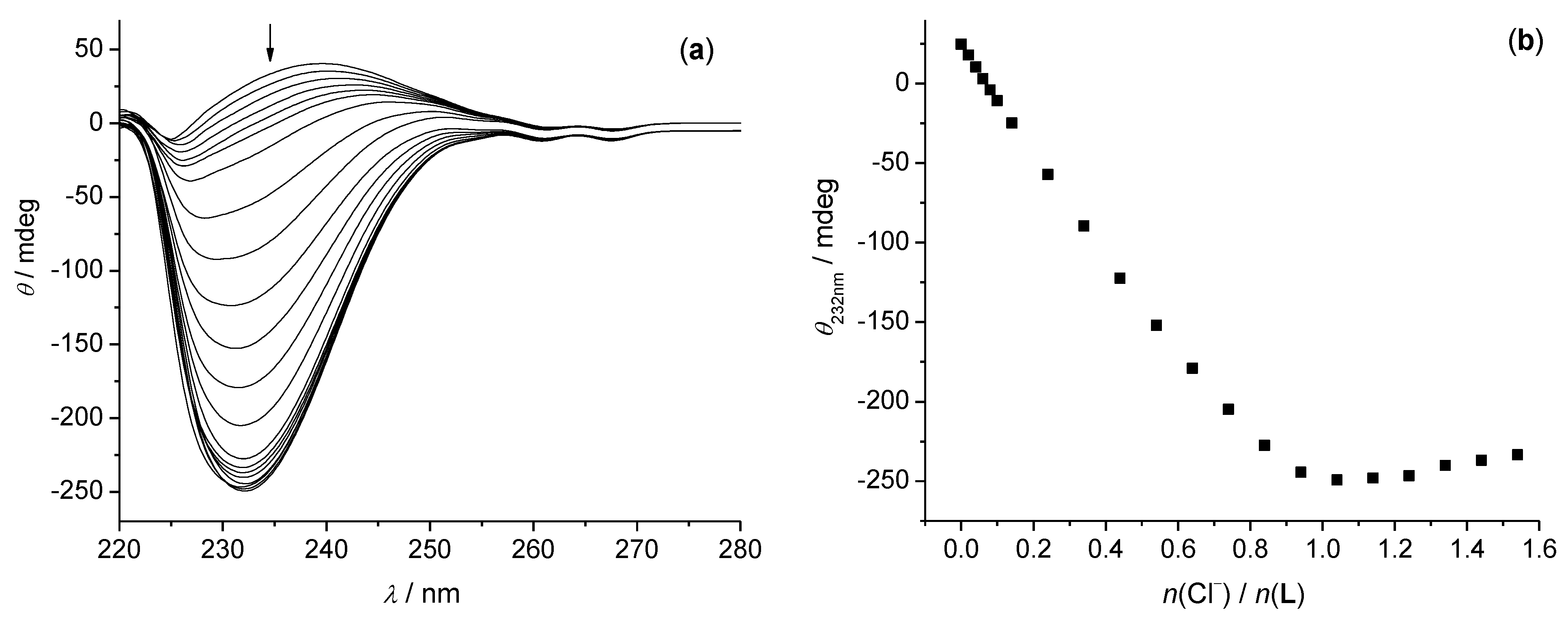
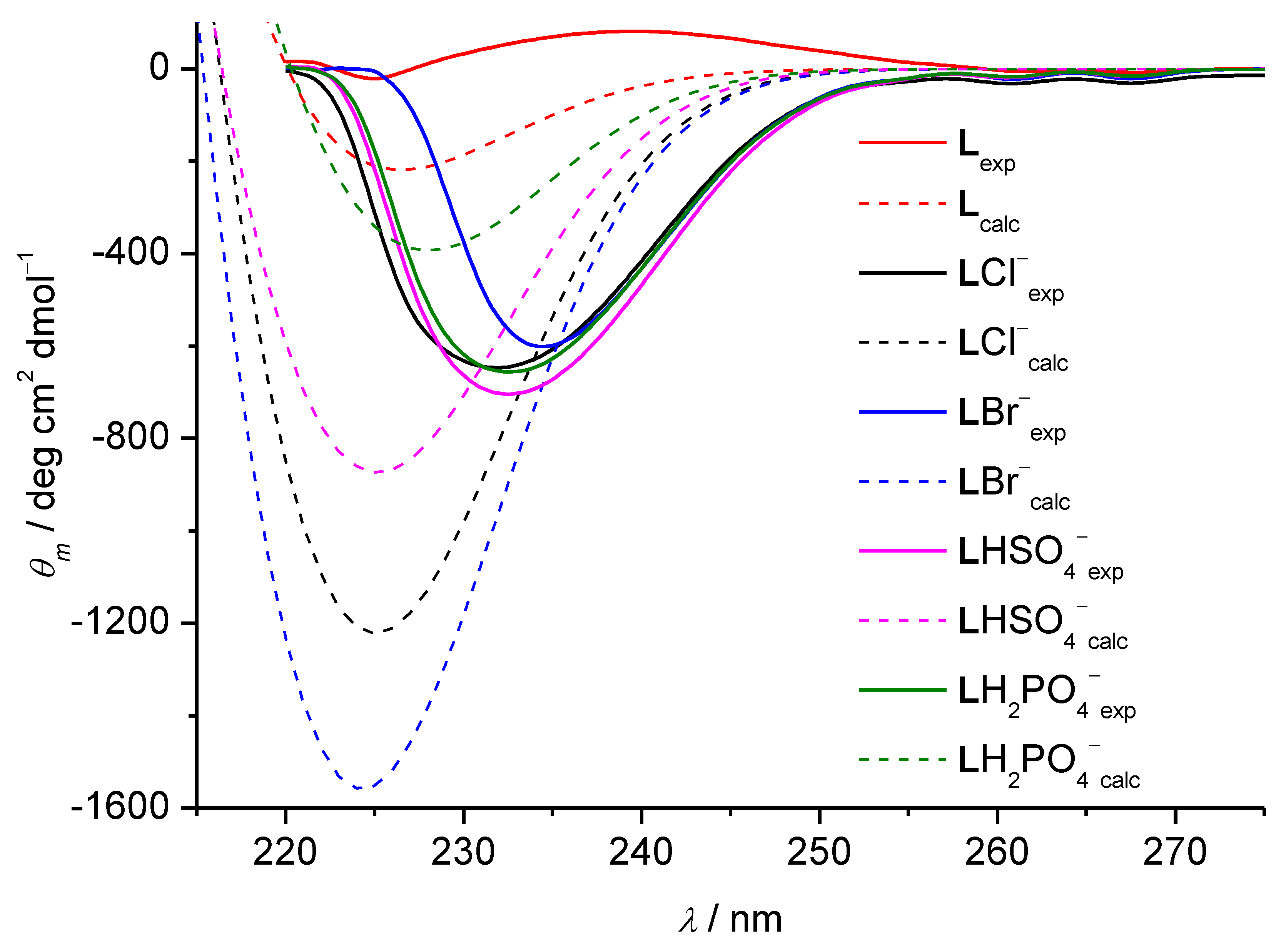
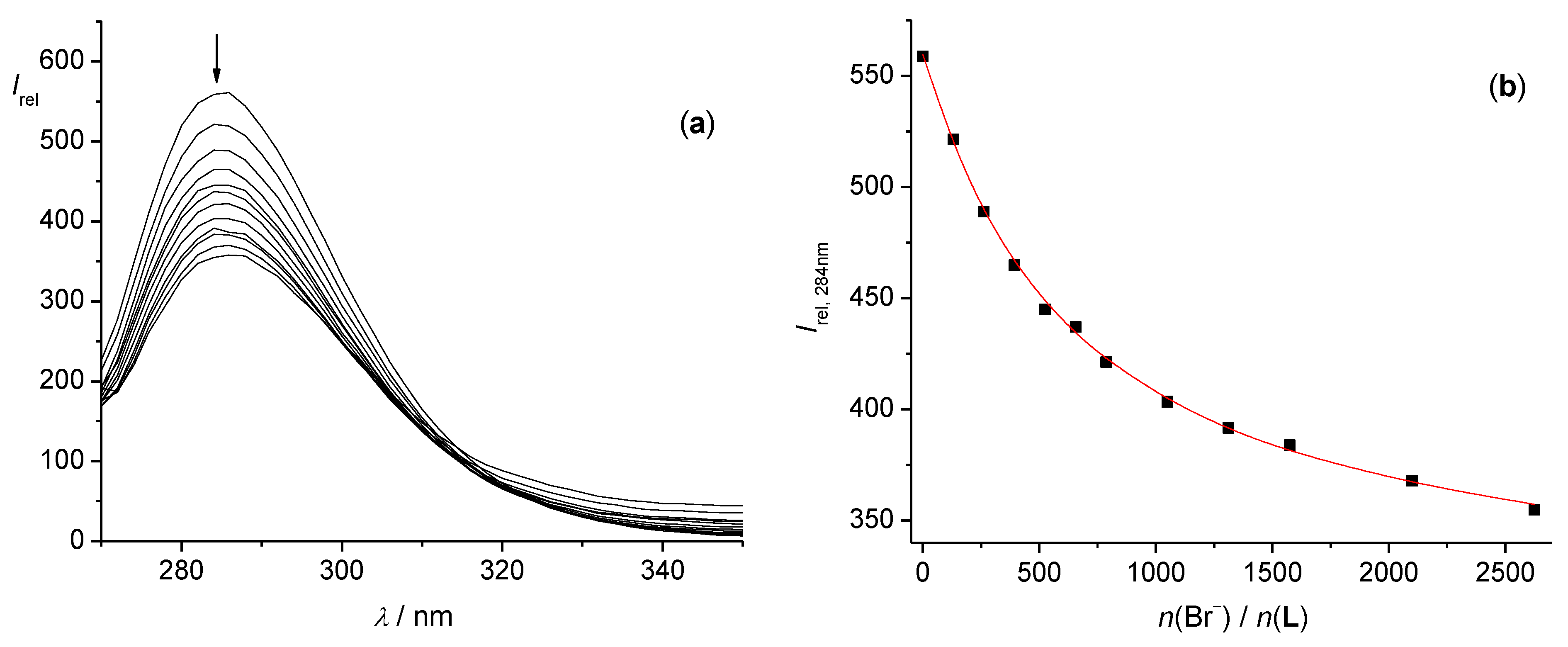
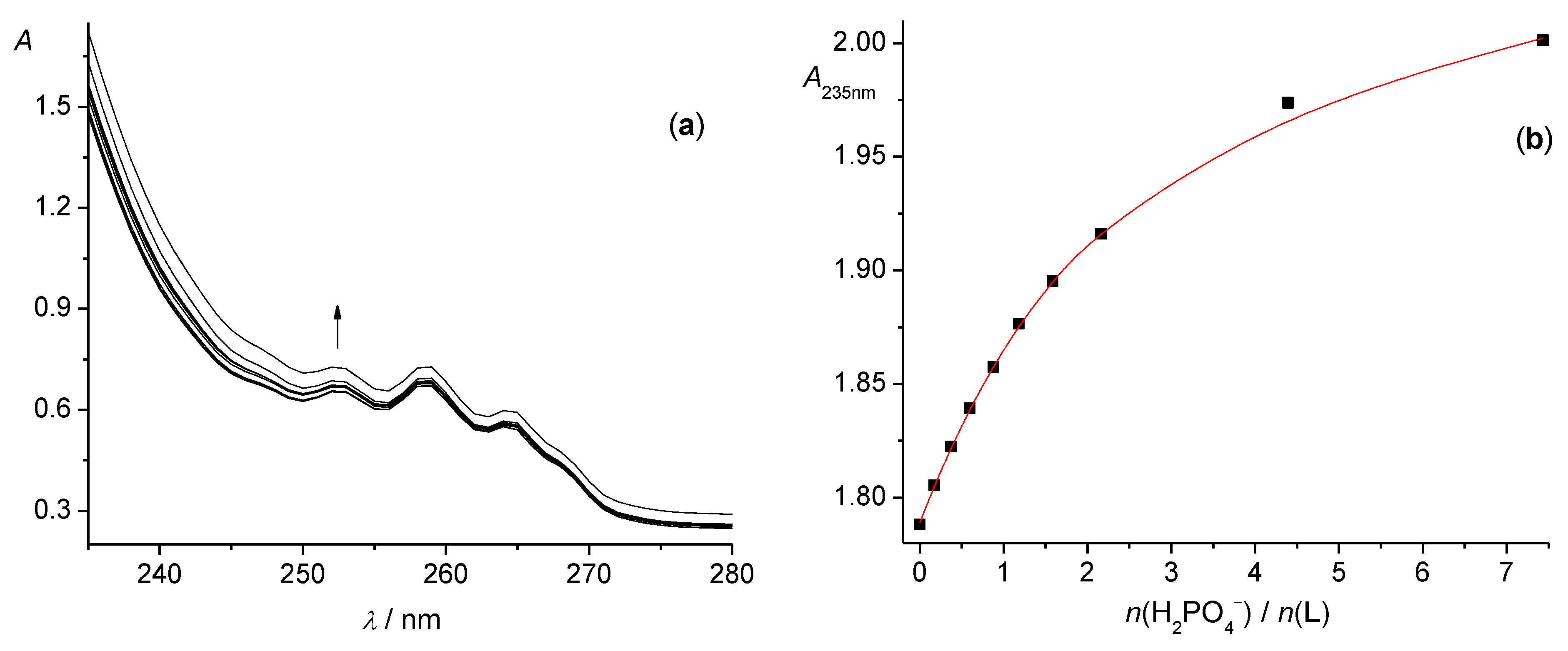
3. Results and Discussion
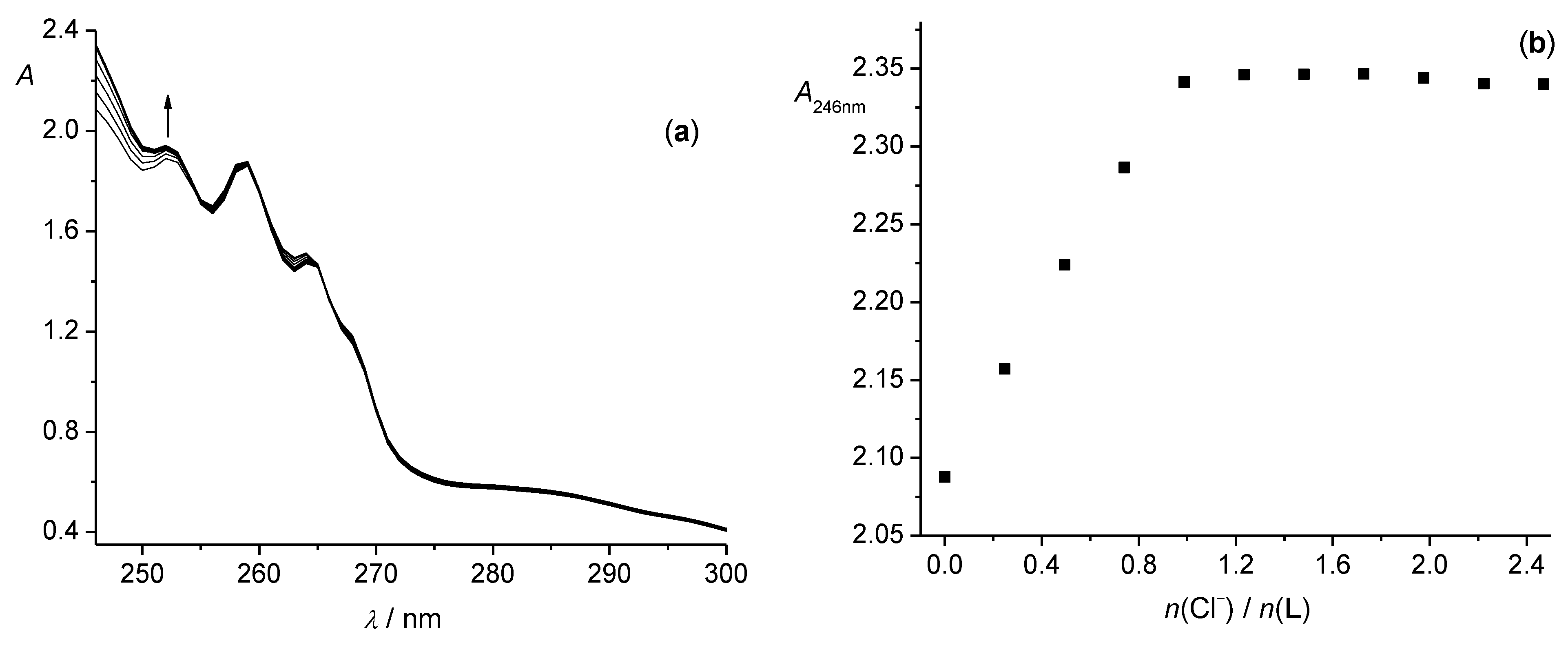
3.1. Anion Complexes of L in Acetonitrile
3.2. Anion Complexes of L in Methanol
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Amendola, V.; Fabbrizzi, L.; Mosca, L. Anion recognition by hydrogen bonding: Urea-based receptors. Chem. Soc. Rev. 2010, 39, 3889–3915. [Google Scholar] [CrossRef]
- Bregović, N.; Cindro, N.; Bertoša, B.; Barišić, D.; Frkanec, L.; Užarević, K.; Tomišić, V. Dehydroacetic Acid Derivatives Bearing Amide or Urea Moieties as Effective Anion Receptors. Chem. A Eur. J. 2017, 23, 10396–10406. [Google Scholar] [CrossRef] [PubMed]
- Barišić, D.; Cindro, N.; Kulcsár, M.J.; Tireli, M.; Užarević, K.; Bregović, N.; Tomišić, V. Protonation and Anion Binding Properties of Aromatic Bis-Urea Derivatives—Comprehending the Proton Transfer. Chem. Eur. J. 2019, 25, 4695–4706. [Google Scholar] [CrossRef] [PubMed]
- Barišić, D.; Cindro, N.; Vidović, N.; Bregović, N.; Tomišić, V. Protonation and anion-binding properties of aromatic sulfonylurea derivatives. RSC Adv. 2021, 11, 23992–24000. [Google Scholar] [CrossRef] [PubMed]
- Rather, I.A.; Wagay, S.A.; Ali, R. Emergence of anion-π interactions: The land of opportunity in supramolecular chemistry and beyond. Coord. Chem. Rev. 2020, 415, 213327. [Google Scholar] [CrossRef]
- Busschaert, N.; Caltagirone, C.; Van Rossom, W.; Gale, P.A. Applications of Supramolecular Anion Recognition. Chem. Rev. 2015, 115, 8038–8155. [Google Scholar] [CrossRef]
- Gale, P.A. Anion receptor chemistry. Chem. Commun. 2010, 47, 82–86. [Google Scholar] [CrossRef] [Green Version]
- Mirski, V.M.; Yatsimirsky, A.K. Artificial Receptors for Chemical Sensors; WILEY-VCH Verlag & Co. KGaA, Boschstr: Weinheim, Germany, 2011; Volume 12, p. 69469. [Google Scholar]
- Matsumoto, A. Anion Sensing, Topics in Current Chemistry 255; Springe: Berlin/Heidelberg, Germany, 2005. [Google Scholar]
- Horvat, G.; Tarana, S.; Vidović, N.; Cindro, N.; Speranza, G.; Tomišić, V. Thermodynamic and MD studies of anion complexation by cyclopentaleucine in acetonitrile and dimethyl sulfoxide. J. Mol. Liq. 2021, 340, 116848. [Google Scholar] [CrossRef]
- Sergeant, G.E.; Jolliffe, K.A. Anion recognition using a simple cyclic peptide. Supramol. Chem. 2020, 32, 233–237. [Google Scholar] [CrossRef]
- Bartl, J.; Kubik, S. Anion Binding of a Cyclopeptide-Derived Molecular Cage in Aqueous Solvent Mixtures. Chempluschem 2020, 85, 963–969. [Google Scholar] [CrossRef]
- Kubik, S. Anion Recognition in Aqueous Media by Cyclopeptides and Other Synthetic Receptors. Acc. Chem. Res. 2017, 50, 2870–2878. [Google Scholar] [CrossRef] [PubMed]
- Elmes, R.B.P.; Jolliffe, K.A. Anion recognition by cyclic peptides. Chem. Commun. 2015, 51, 4951–4968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tay, H.M.; Beer, P. Optical sensing of anions by macrocyclic and interlocked hosts. Org. Biomol. Chem. 2021, 19, 4652–4677. [Google Scholar] [CrossRef] [PubMed]
- Bregović, N.; Cindro, N.; Frkanec, L.; Tomišić, V. Complexation of fluoride anion and its ion pairs with alkali metal cations by tetra-substituted lower rim calix[4]arene tryptophan derivative. Supramol. Chem. 2016, 28, 608–615. [Google Scholar] [CrossRef]
- Escobar, L.; Ballester, P. Molecular Recognition in Water Using Macrocyclic Synthetic Receptors. Chem. Rev. 2021, 121, 2445–2514. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Sedgwick, A.C.; Hirao, T.; Sessler, J.L. Supramolecular fluorescent sensors: An historical overview and update. Coord. Chem. Rev. 2020, 427, 213560. [Google Scholar] [CrossRef]
- Wu, Y.; Bertran, M.T.; Rowley, J.; Calder, E.D.D.; Joshi, D.; Walport, L.J. Fluorescent Amino Acid Initiated de novo Cyclic Peptides for the Label-Free Assessment of Cell Permeability. ChemMedChem 2021, 16, 3185–3188. [Google Scholar] [CrossRef]
- Goshisht, M.K.; Tripathi, N. Fluorescence-based sensors as an emerging tool for anion detection: Mechanism, sensory materials and applications. J. Mater. Chem. C 2021, 9, 9820–9850. [Google Scholar] [CrossRef]
- Mendive-Tapia, L.; Wang, J.; Vendrell, M. Fluorescent cyclic peptides for cell imaging. Pept. Sci. 2020, 113, e24181. [Google Scholar] [CrossRef]
- McNaughton, D.A.; Fares, M.; Picci, G.; Gale, P.A.; Caltagirone, C. Advances in fluorescent and colorimetric sensors for anionic species. Coord. Chem. Rev. 2020, 427, 213573. [Google Scholar] [CrossRef]
- Vidović, N.; Horvat, G.; Riva, D.; Rinkovec, T.; Cindro, N.; Tomišić, V.; Speranza, G. Chloride-Assisted Peptide Macrocyclization. Org. Lett. 2020, 22, 2129–2134. [Google Scholar] [CrossRef] [PubMed]
- Gans, P.; Sabatini, A.; Vacca, A. Investigation of equilibria in solution. Determination of equilibrium constants with the HYPERQUAD suite of programs. Talanta 1996, 43, 1739–1753. [Google Scholar] [CrossRef]
- Bregović, N.; Cindro, N.; Frkanec, L.; Užarević, K.; Tomišić, V. Thermodynamic Study of Dihydrogen Phosphate Dimerisation and Complexation with Novel Urea- and Thiourea-Based Receptors. Chem. Eur. J. 2014, 20, 15863–15871. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.J.C.; Van Der Spoel, D.; Van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Lindahl, E.; Hess, B.; Van Der Spoel, D. GROMACS 3.0: A package for molecular simulation and trajectory analysis. J. Mol. Model. 2001, 7, 306–317. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [Green Version]
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; Van Der Spoel, D.; et al. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Páll, S.; Abraham, M.J.; Kutzner, C.; Hess, B.; Lindahl, E. Tackling Exascale Software Challenges in Molecular Dynamics Simulations with GROMACS. Lect. Notes Comput. Sci. 2015, 8759, 3–27. [Google Scholar]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Tirado-Rives, J. Potential energy functions for atomic-level simulations of water and organic and biomolecular systems. Proc. Natl. Acad. Sci. USA 2005, 102, 6665–6670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodda, L.; De Vaca, I.C.; Tirado-Rives, J.; Jorgensen, W.L. LigParGen web server: An automatic OPLS-AA parameter generator for organic ligands. Nucleic Acids Res. 2017, 45, W331–W336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodda, L.S.; Vilseck, J.Z.; Tirado-Rives, J.; Jorgensen, W.L. 1.14*CM1A-LBCC: Localized Bond-Charge Corrected CM1A Charges for Condensed-Phase Simulations. J. Phys. Chem. B 2017, 121, 3864–3870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swope, W.C.; Andersen, H.C.; Berens, P.H.; Wilson, K.R. A computer simulation method for the calculation of equilibrium constants for the formation of physical clusters of molecules: Application to small water clusters. J. Chem. Phys. 1982, 76, 637–649. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An Nlog(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Nosé, S. A molecular dynamics method for simulations in the canonical ensemble. Mol. Phys. 1984, 52, 255–268. [Google Scholar] [CrossRef]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef] [Green Version]
- Martyna, G.J.; Tuckerman, M.E.; Tobias, D.J.; Klein, M.L. Explicit reversible integrators for extended systems dynamics. Mol. Phys. 1996, 87, 1117–1157. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Pazos, I.M.; Roesch, R.M.; Gai, F. Quenching of p-cyanophenylalanine fluorescence by various anions. Chem. Phys. Lett. 2013, 563, 93–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bulheller, B.M.; Hirst, J.D. DichroCalc--circular and linear dichroism online. Bioinformatics 2009, 25, 539–540. [Google Scholar] [CrossRef] [PubMed]
- Besley, N.A.; Hirst, J.D. Theoretical Studies toward Quantitative Protein Circular Dichroism Calculations. J. Am. Chem. Soc. 1999, 121, 9636–9644. [Google Scholar] [CrossRef]
- Marcus, Y. Ion Properties; CRC Press: Boca Raton, FL, USA, 1997. [Google Scholar]
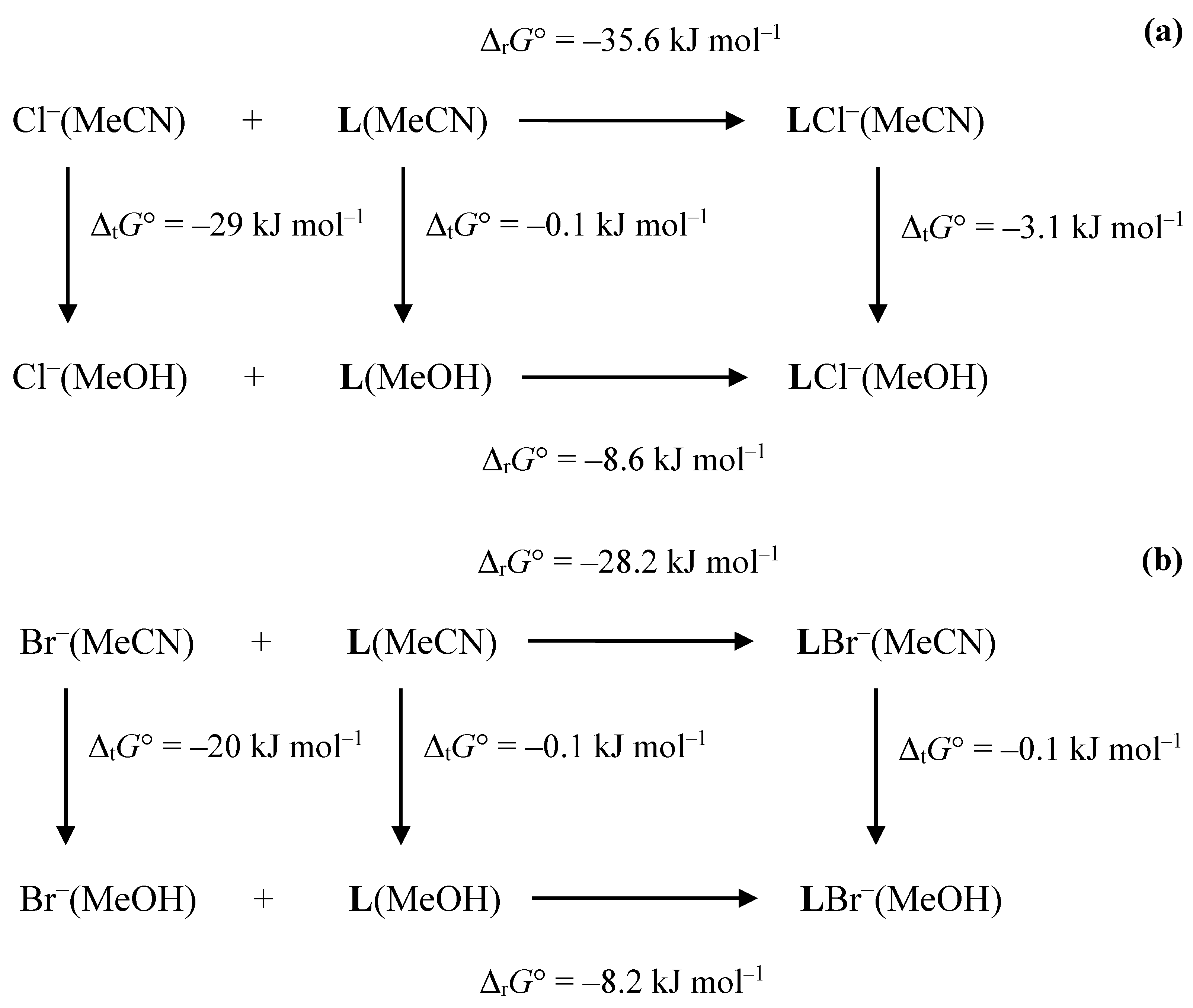

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Anion | MeCN | MeOH |
|---|---|---|
| Cl− | 6.06 ± 0.04 d | 1.50 ± 0.01 a |
| 1.57 b | ||
| 1.64 c | ||
| Br− | 4.97 ± 0.10 a | 1.42 ± 0.04 a |
| 1.75 b | ||
| 1.60 c | ||
| I− | 3.39 c | 1.72 ± 0.01 a |
| 1.28 b | ||
| 1.24 c | ||
| H2PO4− | 4.18 ± 0.08 a | 3.19 ± 0.04 a |
| 2.95 b | ||
| 3.32 c | ||
| HSO4− | 2.88 ± 0.12 a | |
| 4.31 ± 0.07 a | 2.43 b | |
| 2.46 c | ||
| NO3− | 3.27 ± 0.03 a 3.43 c | 1.58 ± 0.04 a |
| 1.51 b | ||
| 1.11 c | ||
| SCN− | 2.91 ± 0.07 a 2.92 c | 1.30 ± 0.01 a |
| 0.76 b | ||
| 0.69 c |
| Anion | Cl− | Br− | I− | H2PO4− | HSO4− | NO3− | SCN− |
|---|---|---|---|---|---|---|---|
| N(-H) | 4.98 | 4.97 | 4.85 | 4.93 | 4.33 | 4.90 | 4.93 |
| Anion | Cl− | Br− | I− | H2PO4− | HSO4− | NO3− | SCN− |
|---|---|---|---|---|---|---|---|
| N(-H) | 4.94 | 4.91 | 4.80 | 4.95 | 4.25 | 4.76 | 4.98 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petters, I.; Modrušan, M.; Vidović, N.; Crnolatac, I.; Cindro, N.; Piantanida, I.; Speranza, G.; Horvat, G.; Tomišić, V. Anion-Sensing Properties of Cyclopentaphenylalanine. Molecules 2022, 27, 3918. https://doi.org/10.3390/molecules27123918
Petters I, Modrušan M, Vidović N, Crnolatac I, Cindro N, Piantanida I, Speranza G, Horvat G, Tomišić V. Anion-Sensing Properties of Cyclopentaphenylalanine. Molecules. 2022; 27(12):3918. https://doi.org/10.3390/molecules27123918
Chicago/Turabian StylePetters, Ivan, Matija Modrušan, Nikolina Vidović, Ivo Crnolatac, Nikola Cindro, Ivo Piantanida, Giovanna Speranza, Gordan Horvat, and Vladislav Tomišić. 2022. "Anion-Sensing Properties of Cyclopentaphenylalanine" Molecules 27, no. 12: 3918. https://doi.org/10.3390/molecules27123918
APA StylePetters, I., Modrušan, M., Vidović, N., Crnolatac, I., Cindro, N., Piantanida, I., Speranza, G., Horvat, G., & Tomišić, V. (2022). Anion-Sensing Properties of Cyclopentaphenylalanine. Molecules, 27(12), 3918. https://doi.org/10.3390/molecules27123918