
Irreversible Antagonists for the Adenosine A2B Receptor †

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Results
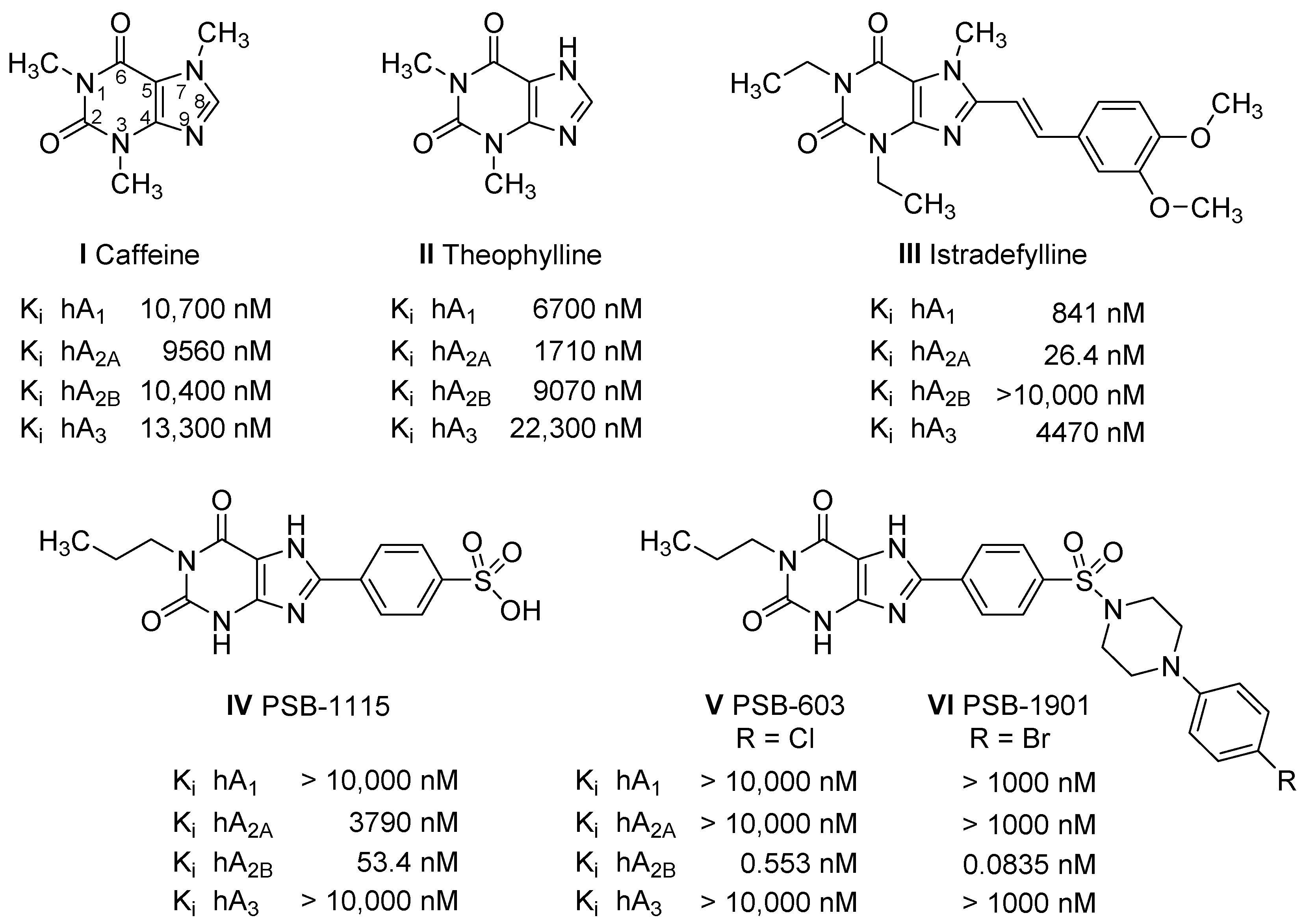
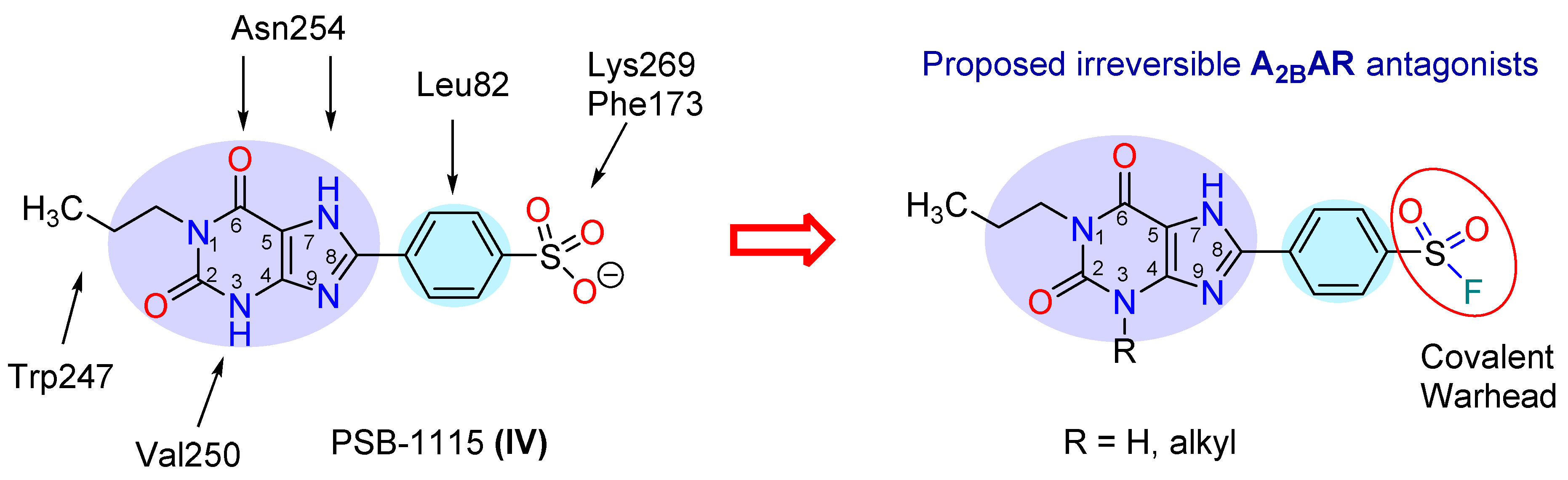
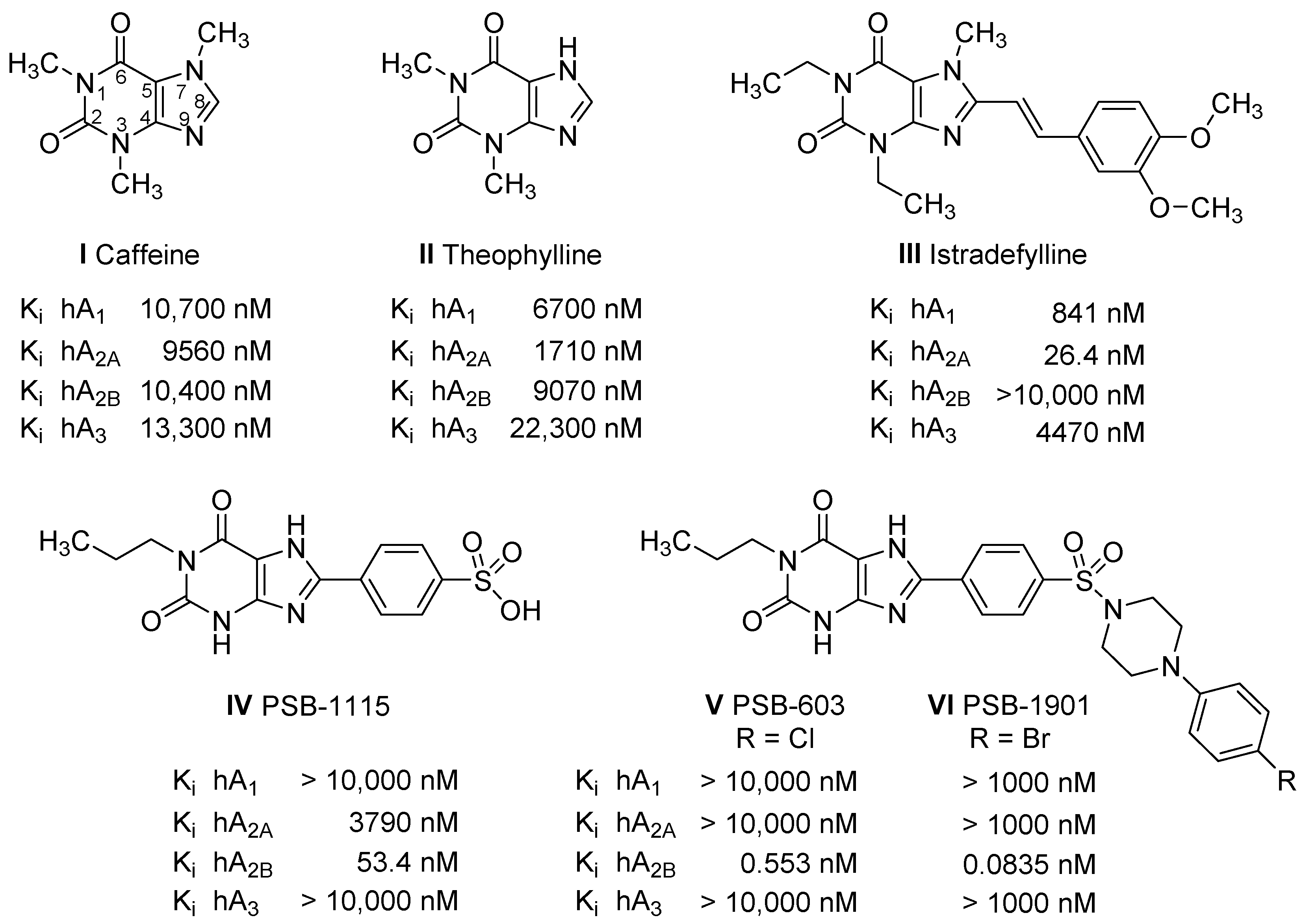
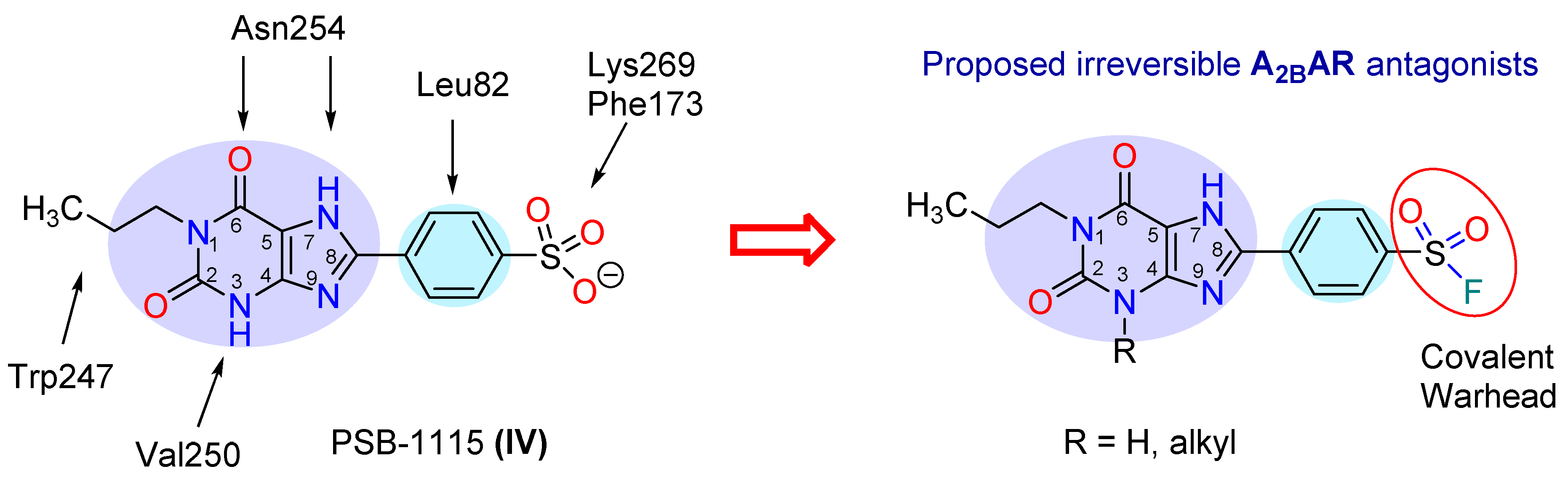
2.1. Compound Design
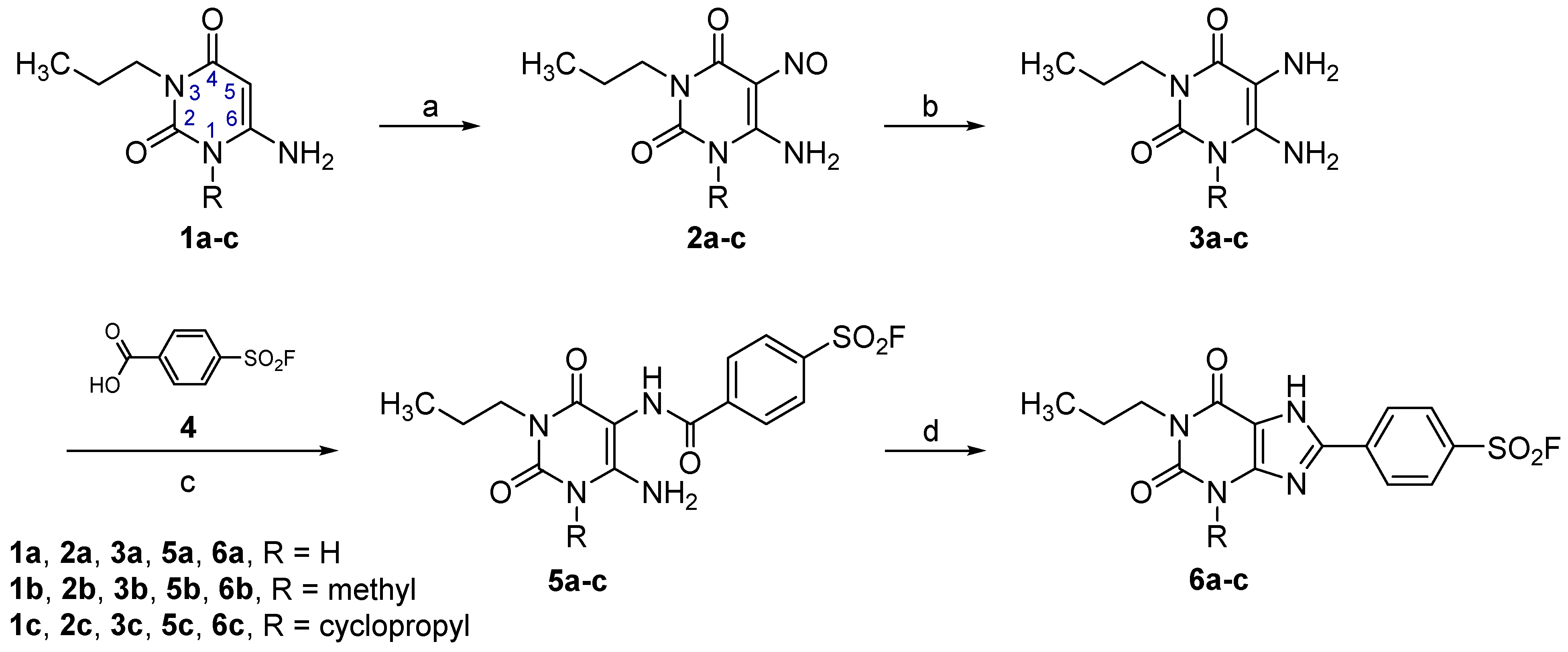
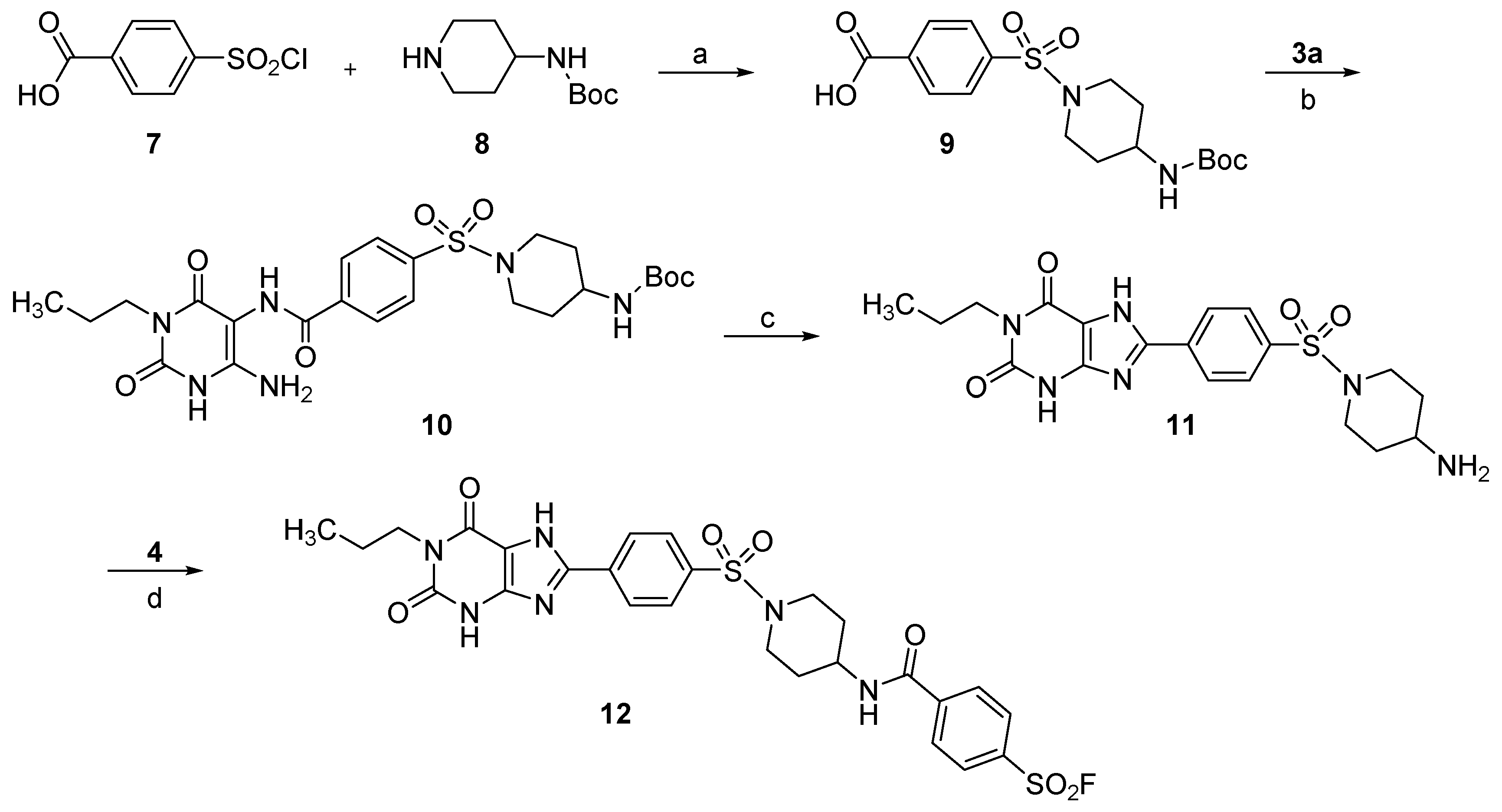
2.2. Syntheses
2.3. Pharmacological Characterization
2.3.1. Radioligand Binding
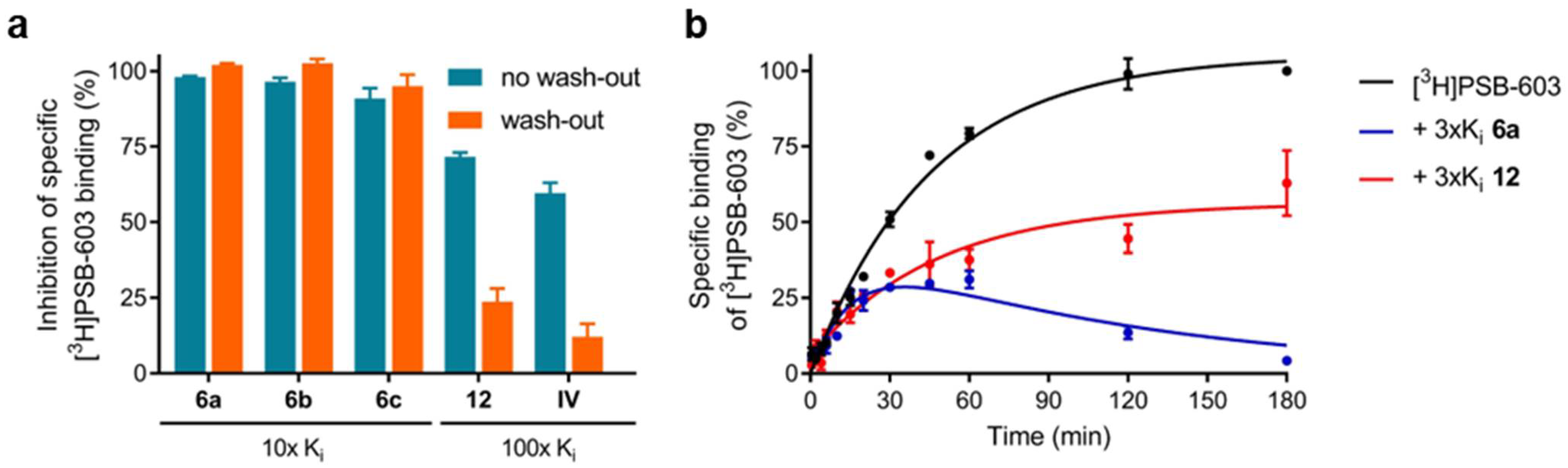
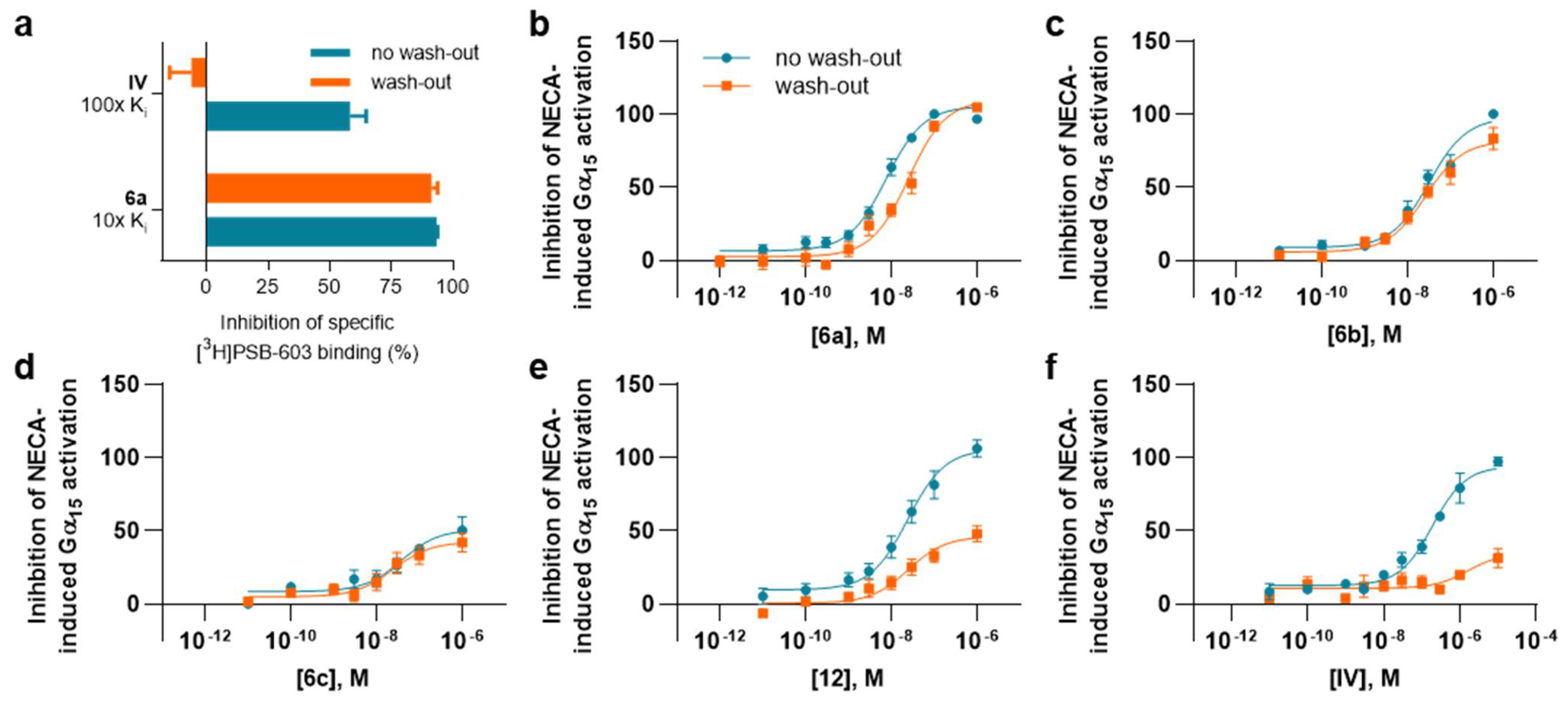
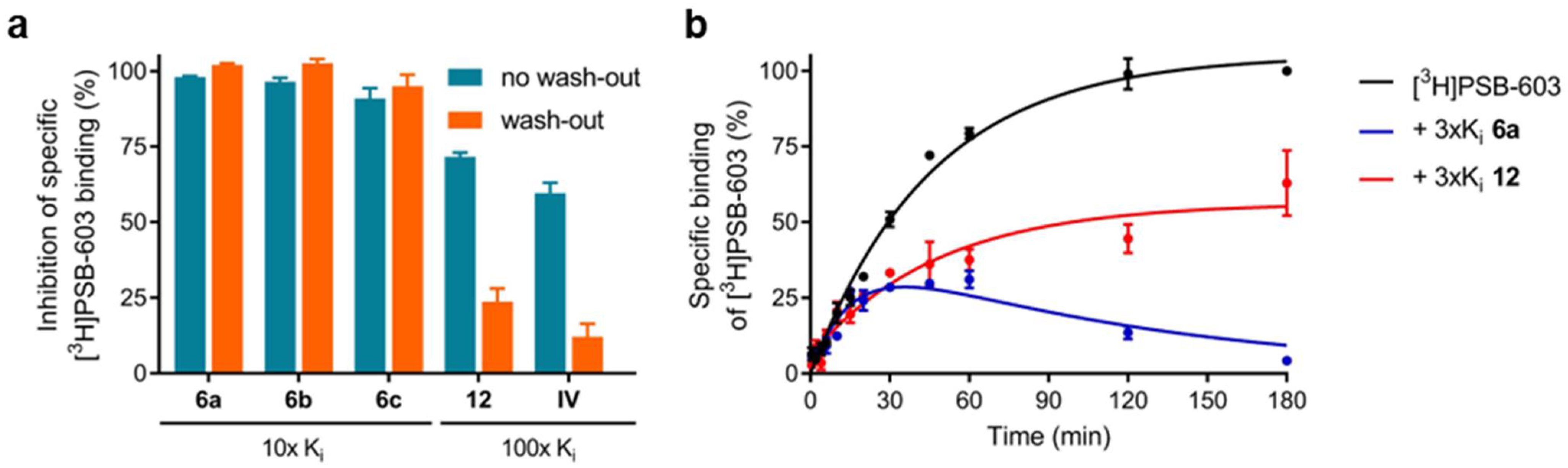
2.3.2. Assessment of Reversible Versus Covalent Binding
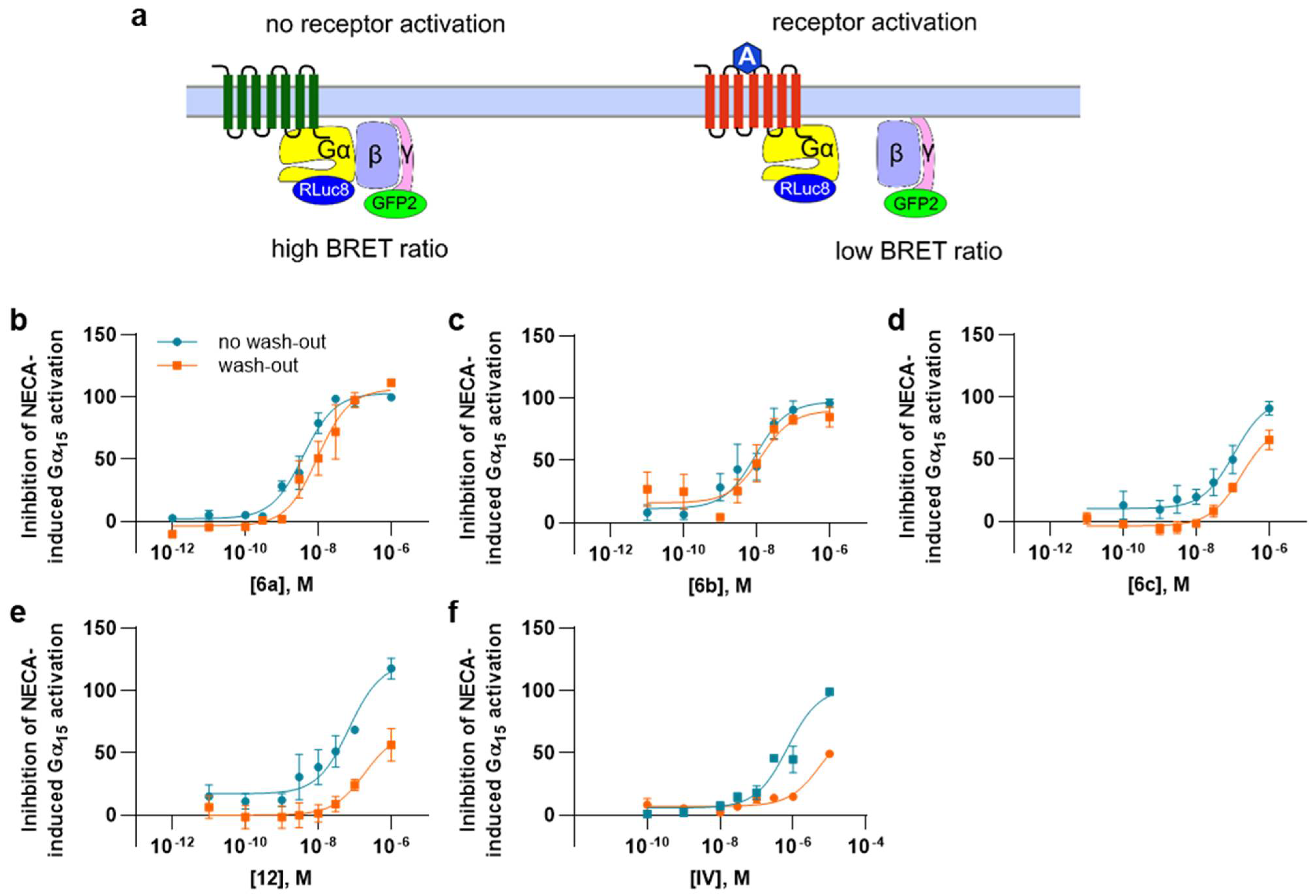
2.3.3. Functional Assays
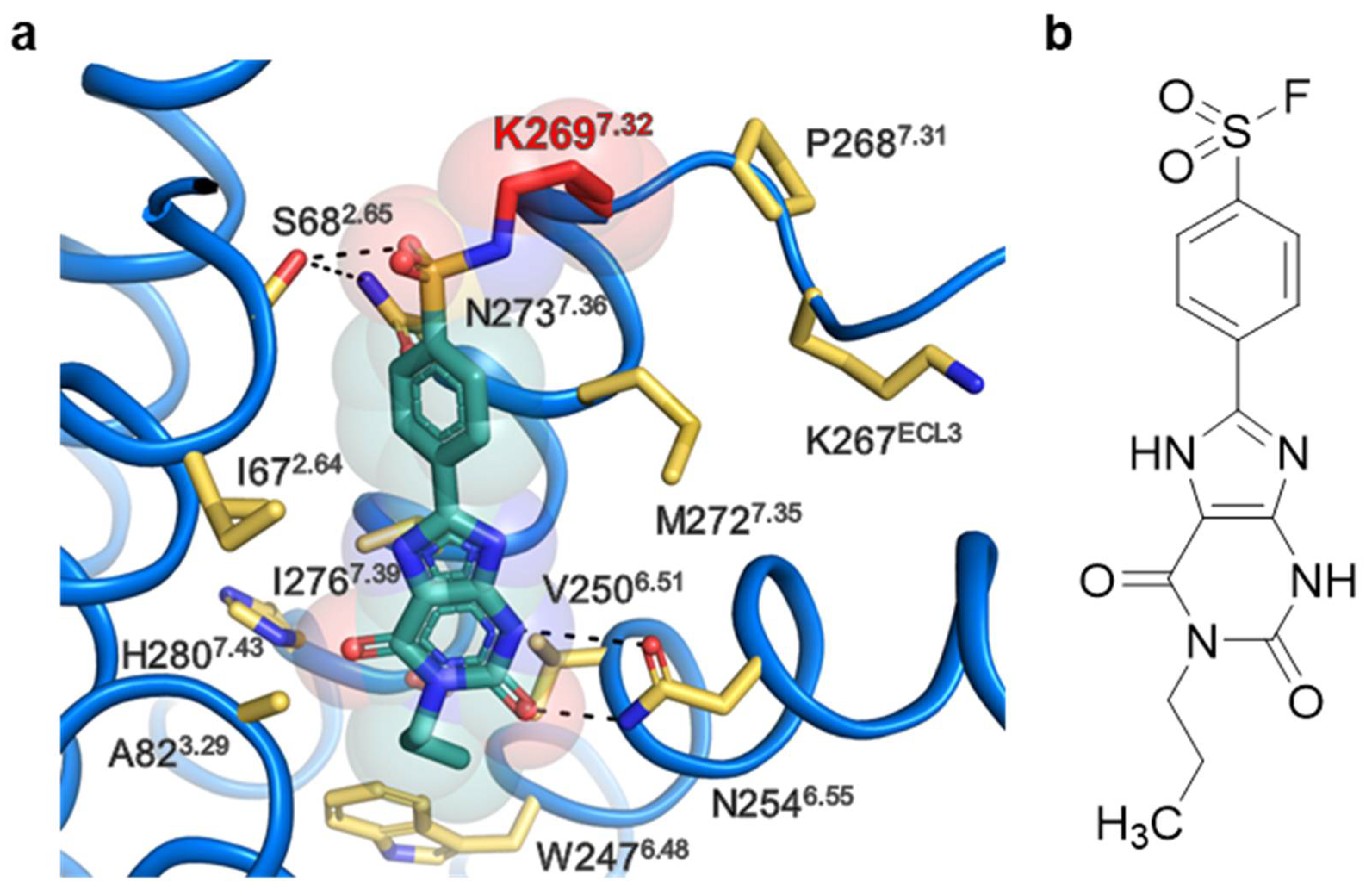
2.4. Extracellular Lysine K2697.32 as Potential Interaction Partner for Irreversible Binding of 6a (PSB-21500)
2.4.1. Docking Studies of Compound 6a
2.4.2. A2BAR Mutant K2697.32L
3. Discussion
4. Materials and Methods
4.1. Chemistry
4.1.1. General
4.1.2. General Procedure A
4.1.3. General Procedure B
4.1.4. General Procedure C
4.1.5. General Procedure D
4.1.6. Chemical Analysis
4.2. Receptor Expression and Membrane Preparation
4.3. Radioligand Binding Experiments
4.4. Wash-Out Experiments
4.5. Functional G Protein Activation Experiments Using BRET² Assays
4.6. Docking Experiments
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- IJzerman, A.P.; Jacobson, K.A.; Müller, C.E.; Cronstein, B.N.; Cunha, R.A. International Union of Basic and Clinical Pharmacology. CXII: Adenosine receptors: A further update. Pharmacol. Rev. 2022, 74, 340–372. [Google Scholar] [CrossRef] [PubMed]
- Pereira-Figueiredo, D.; Nascimento, A.A.; Cunha-Rodrigues, M.C.; Brito, R.; Calaza, K.C. Caffeine and its neuroprotective role in ischemic events: A mechanism dependent on adenosine receptors. Cell. Mol. Neurobiol. 2021. [Google Scholar] [CrossRef]
- Diener, H.C.; Gaul, C.; Lehmacher, W.; Weiser, T. Aspirin, paracetamol (acetaminophen) and caffeine for the treatment of acute migraine attacks: A systemic review and meta-analysis of randomized placebo-controlled trials. Eur. J. Neurol. 2022, 29, 350–357. [Google Scholar] [CrossRef]
- Mahemuti, G.; Zhang, H.; Li, J.; Tieliwaerdi, N.; Ren, L. Efficacy and side effects of intravenous theophylline in acute asthma: A systematic review and meta-analysis. Drug Des. Devel. Ther. 2018, 12, 99–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borea, P.A. The Adenosine Receptors; Springer International Publishing AG: Cham, Swizterland, 2018; ISBN 9783319908083. [Google Scholar]
- Abo-Salem, O.M.; Hayallah, A.M.; Bilkei-Gorzo, A.; Filipek, B.; Zimmer, A.; Müller, C.E. Antinociceptive effects of novel A2B adenosine receptor antagonists. J. Pharmacol. Exp. Ther. 2004, 308, 358–366. [Google Scholar] [CrossRef] [Green Version]
- Mustafa, S.J.; Nadeem, A.; Fan, M.; Zhong, H.; Belardinelli, L.; Zeng, D. Effect of a specific and selective A2B adenosine receptor antagonist on adenosine agonist AMP and allergen-induced airway responsiveness and cellular influx in a mouse model of asthma. J. Pharmacol. Exp. Ther. 2007, 320, 1246–1251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenner, P.; Mori, A.; Aradi, S.D.; Hauser, R.A. Istradefylline-A first generation adenosine A2A antagonist for the treatment of Parkinson’s disease. Expert. Rev. Neurother. 2021, 21, 317–333. [Google Scholar] [CrossRef] [PubMed]
- Franco, R.; Rivas-Santisteban, R.; Navarro, G.; Reyes-Resina, I. Adenosine receptor antagonists to combat cancer and to boost anti-cancer chemotherapy and immunotherapy. Cells 2021, 10, 2831. [Google Scholar] [CrossRef]
- Gao, Z.-G.; Jacobson, K.A. A2B adenosine receptor and cancer. Int. J. Mol. Sci. 2019, 20, 5139. [Google Scholar] [CrossRef] [Green Version]
- Willingham, S.B.; Hotson, A.N.; Miller, R.A. Targeting the A2AR in cancer; early lessons from the clinic. Curr. Opin. Pharmacol. 2020, 53, 126–133. [Google Scholar] [CrossRef]
- Sun, C.; Wang, B.; Hao, S. Adenosine-A2A receptor pathway in cancer immunotherapy. Front. Immunol. 2022, 13, 837230. [Google Scholar] [CrossRef] [PubMed]
- Leão Batista Simões, J.; Fornari Basso, H.; Cristine Kosvoski, G.; Gavioli, J.; Marafon, F.; Elias Assmann, C.; Barbosa Carvalho, F.; Dulce Bagatini, M. Targeting purinergic receptors to suppress the cytokine storm induced by SARS-CoV-2 infection in pulmonary tissue. Int. Immunopharmacol. 2021, 100, 108150. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Claff, T.; Klapschinski, T.A.; Tiruttani Subhramanyam, U.K.; Vaaßen, V.J.; Schlegel, J.G.; Vielmuth, C.; Voß, J.H.; Labahn, J.; Müller, C.E. Single stabilizing point mutation enables high-resolution co-crystal structures of the adenosine A2A receptor with preladenant conjugates. Angew. Chem. Int. Ed. Engl. 2022, 61, e202115545. [Google Scholar] [CrossRef]
- Rosenbaum, D.M.; Zhang, C.; Lyons, J.A.; Holl, R.; Aragao, D.; Arlow, D.H.; Rasmussen, S.G.F.; Choi, H.-J.; Devree, B.T.; Sunahara, R.K.; et al. Structure and function of an irreversible agonist-β2 adrenoceptor complex. Nature 2011, 469, 236–240. [Google Scholar] [CrossRef] [Green Version]
- Glukhova, A.; Thal, D.M.; Nguyen, A.T.; Vecchio, E.A.; Jörg, M.; Scammells, P.J.; May, L.T.; Sexton, P.M.; Christopoulos, A. Structure of the adenosine A1 receptor reveals the basis for subtype selectivity. Cell 2017, 168, 867–877.e13. [Google Scholar] [CrossRef] [Green Version]
- Jiang, J.; Seel, C.J.; Temirak, A.; Namasivayam, V.; Arridu, A.; Schabikowski, J.; Baqi, Y.; Hinz, S.; Hockemeyer, J.; Müller, C.E. A2B adenosine receptor antagonists with picomolar potency. J. Med. Chem. 2019, 62, 4032–4055. [Google Scholar] [CrossRef]
- Hayallah, A.M.; Sandoval-Ramírez, J.; Reith, U.; Schobert, U.; Preiss, B.; Schumacher, B.; Daly, J.W.; Müller, C.E. 1,8-disubstituted xanthine derivatives: Synthesis of potent A2B-selective adenosine receptor antagonists. J. Med. Chem. 2002, 45, 1500–1510. [Google Scholar] [CrossRef]
- Yan, L.; Müller, C.E. Preparation, properties, reactions, and adenosine receptor affinities of sulfophenylxanthine nitrophenyl esters: Toward the development of sulfonic acid prodrugs with peroral bioavailability. J. Med. Chem. 2004, 47, 1031–1043. [Google Scholar] [CrossRef]
- Rüsing, D.; Müller, C.E.; Verspohl, E.J. The impact of adenosine and A2B receptors on glucose homoeostasis. J. Pharm. Pharmacol. 2006, 58, 1639–1645. [Google Scholar] [CrossRef]
- Borrmann, T.; Hinz, S.; Bertarelli, D.C.G.; Li, W.; Florin, N.C.; Scheiff, A.B.; Müller, C.E. 1-alkyl-8-(piperazine-1-sulfonyl)phenylxanthines: Development and characterization of adenosine A2B receptor antagonists and a new radioligand with subnanomolar affinity and subtype specificity. J. Med. Chem. 2009, 52, 3994–4006. [Google Scholar] [CrossRef] [PubMed]
- Müller, C.E.; Baqi, Y.; Namasivayam, V. Agonists and antagonists for purinergic receptors. Methods Mol. Biol. 2020, 2041, 45–64. [Google Scholar] [CrossRef] [PubMed]
- Sherbiny, F.F.; Schiedel, A.C.; Maass, A.; Müller, C.E. Homology modelling of the human adenosine A2B receptor based on X-ray structures of bovine rhodopsin, the beta2-adrenergic receptor and the human adenosine A2A receptor. J. Comput. Aided Mol. Des. 2009, 23, 807–828. [Google Scholar] [CrossRef] [PubMed]
- Ballesteros, J.A.; Weinstein, H. [19] Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. In Receptor Molecular Biology; Sealfon, S.C., Ed.; Academic Press: San Diego, LA, USA, 1995; pp. 366–428. ISBN 9780121852955. [Google Scholar]
- Yang, X.; Dong, G.; Michiels, T.J.M.; Lenselink, E.B.; Heitman, L.; Louvel, J.; IJzerman, A.P. A covalent antagonist for the human adenosine A2A receptor. Purinergic Signal. 2017, 13, 191–201. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; van Veldhoven, J.P.D.; Offringa, J.; Kuiper, B.J.; Lenselink, E.B.; Heitman, L.H.; van der Es, D.; IJzerman, A.P. Development of covalent ligands for G protein-coupled receptors: A case for the human adenosine A3 receptor. J. Med. Chem. 2019, 62, 3539–3552. [Google Scholar] [CrossRef] [Green Version]
- Müller, C.E.; Shi, D.; Manning, M.; Daly, J.W. Synthesis of paraxanthine analogs (1,7-disubstituted xanthines) and other xanthines unsubstituted at the 3-position: Structure-activity relationships at adenosine receptors. J. Med. Chem. 1993, 36, 3341–3349. [Google Scholar] [CrossRef]
- Müller, C.E. Synthesis of 3-substituted 6-aminouracils. Tetrahedron Lett. 1991, 32, 6539–6540. [Google Scholar] [CrossRef]
- Müller, C.E. General synthesis and properties of 1-monosubstituted xanthines. Synthesis 1993, 1993, 125–128. [Google Scholar] [CrossRef]
- Priego, E.-M.; Camarasa, M.-J.; Pérez-Pérez, M.-J. Efficient synthesis of N-3-substituted 6-aminouracil derivatives via N6-[(dimethylamino)methylene] protection. Synthesis 2001, 2001, 478–482. [Google Scholar] [CrossRef]
- Gehringer, M.; Laufer, S.A. Emerging and re-emerging warheads for targeted covalent inhibitors: Applications in medicinal chemistry and chemical biology. J. Med. Chem. 2019, 62, 5673–5724. [Google Scholar] [CrossRef]
- Prime, M.E.; Brookfield, F.A.; Courtney, S.M.; Gaines, S.; Marston, R.W.; Ichihara, O.; Li, M.; Vaidya, D.; Williams, H.; Pedret-Dunn, A.; et al. Irreversible 4-aminopiperidine transglutaminase 2 inhibitors for Huntington’s disease. ACS Med. Chem. Lett. 2012, 3, 731–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.C.; Ji, X.; Melman, N.; Linden, J.; Jacobson, K.A. Anilide derivatives of an 8-phenylxanthine carboxylic congener are highly potent and selective antagonists at human A2B adenosine receptors. J. Med. Chem. 2000, 43, 1165–1172. [Google Scholar] [CrossRef] [PubMed]
- Seibt, B.F.; Schiedel, A.C.; Thimm, D.; Hinz, S.; Sherbiny, F.F.; Müller, C.E. The second extracellular loop of GPCRs determines subtype-selectivity and controls efficacy as evidenced by loop exchange study at A2 adenosine receptors. Biochem. Pharmacol. 2013, 85, 1317–1329. [Google Scholar] [CrossRef] [PubMed]
- Katritch, V.; Kufareva, I.; Abagyan, R. Structure based prediction of subtype-selectivity for adenosine receptor antagonists. Neuropharmacology 2011, 60, 108–115. [Google Scholar] [CrossRef] [Green Version]
- Alnouri, M.W.; Jepards, S.; Casari, A.; Schiedel, A.C.; Hinz, S.; Müller, C.E. Selectivity is species-dependent: Characterization of standard agonists and antagonists at human, rat, and mouse adenosine receptors. Purinergic Signal. 2015, 11, 389–407. [Google Scholar] [CrossRef] [Green Version]
- Voss, J.H.; Mahardhika, A.B.; Inoue, A.; Müller, C.E. Agonist-dependent coupling of the promiscuous adenosine A2B receptor to Gα protein subunits. ACS Pharmacol. Transl. Sci. 2022, 5, 373–386. [Google Scholar] [CrossRef]
- Olsen, R.H.J.; DiBerto, J.F.; English, J.G.; Glaudin, A.M.; Krumm, B.E.; Slocum, S.T.; Che, T.; Gavin, A.C.; McCorvy, J.D.; Roth, B.L.; et al. TRUPATH, an open-source biosensor platform for interrogating the GPCR transducerome. Nat. Chem. Biol. 2020, 16, 841–849. [Google Scholar] [CrossRef]
- Michino, M.; Abola, E.; Brooks, C.L.; Dixon, J.S.; Moult, J.; Stevens, R.C. Community-wide assessment of GPCR structure modelling and ligand docking: GPCR Dock 2008. Nat. Rev. Drug Discov. 2009, 8, 455–463. [Google Scholar] [CrossRef] [Green Version]
- Katritch, V.; Rueda, M.; Lam, P.C.-H.; Yeager, M.; Abagyan, R. GPCR 3D homology models for ligand screening: Lessons learned from blind predictions of adenosine A2a receptor complex. Proteins 2010, 78, 197–211. [Google Scholar] [CrossRef] [Green Version]
- Edupuganti, R.; Wang, Q.; Tavares, C.D.J.; Chitjian, C.A.; Bachman, J.L.; Ren, P.; Anslyn, E.V.; Dalby, K.N. Synthesis and biological evaluation of pyrido2,3-dpyrimidine-2,4-dione derivatives as eEF-2K inhibitors. Bioorg. Med. Chem. 2014, 22, 4910–4916. [Google Scholar] [CrossRef]
- Elzein, E.; Kalla, R.V.; Li, X.; Perry, T.; Gimbel, A.; Zeng, D.; Lustig, D.; Leung, K.; Zablocki, J. Discovery of a novel A2B adenosine receptor antagonist as a clinical candidate for chronic inflammatory airway diseases. J. Med. Chem. 2008, 51, 2267–2278. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Chu, W.; Qi, Q.; Xun, L. New insights into the QuikChange™ process guide the use of Phusion DNA polymerase for site-directed mutagenesis. Nucleic Acids Res. 2015, 43, e12. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
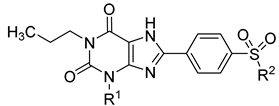 | A1AR vs. Radioligand [3H]CCPA | A2AAR vs. Radioligand [3H]MSX-2 | A2BAR vs. Radioligand [3H]PSB-603 | A3AR vs. Radioligand [3H]PSB-11 | ||
|---|---|---|---|---|---|---|
| Name | R1 | R2 | Ki ± SEM (nM), (or % inhibition at 1 µM) | |||
| PSB-1115 (IV) | H | OH | >10,000 2 | 3790 ± 520 3 | 152 ± 12 [53.4] 4 | >10,000 2 |
| 6a PSB-21500 | H | F | 406 ± 194 | ~1000 (53%) | 10.6 ± 0.9 | >1000 (27%) |
| 6b PSB-21501 | methyl | F | 293 ± 92 | 372 ± 48 | 15.9 ± 1.0 | >1000 (13%) |
| 6c PSB-21502 | cyclo- propyl | F | 41.5 ± 2.8 | 72.9 ± 8.1 | 26.0 ± 2.8 | >1000 (33%) |
| 12 PSB-21503 | H | 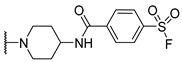 | >1000 (22%) | >1000 (32%) | 7.37 ± 0.83 | >1000 (15%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Temirak, A.; Schlegel, J.G.; Voss, J.H.; Vaaßen, V.J.; Vielmuth, C.; Claff, T.; Müller, C.E. Irreversible Antagonists for the Adenosine A2B Receptor. Molecules 2022, 27, 3792. https://doi.org/10.3390/molecules27123792
Temirak A, Schlegel JG, Voss JH, Vaaßen VJ, Vielmuth C, Claff T, Müller CE. Irreversible Antagonists for the Adenosine A2B Receptor. Molecules. 2022; 27(12):3792. https://doi.org/10.3390/molecules27123792
Chicago/Turabian StyleTemirak, Ahmed, Jonathan G. Schlegel, Jan H. Voss, Victoria J. Vaaßen, Christin Vielmuth, Tobias Claff, and Christa E. Müller. 2022. "Irreversible Antagonists for the Adenosine A2B Receptor" Molecules 27, no. 12: 3792. https://doi.org/10.3390/molecules27123792
APA StyleTemirak, A., Schlegel, J. G., Voss, J. H., Vaaßen, V. J., Vielmuth, C., Claff, T., & Müller, C. E. (2022). Irreversible Antagonists for the Adenosine A2B Receptor. Molecules, 27(12), 3792. https://doi.org/10.3390/molecules27123792