
Azides in the Synthesis of Various Heterocycles

 ,
,  , , , ,
, , , ,  and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Some Synthetic Procedures of Organic Azides
2.1. From Diazonium Salts
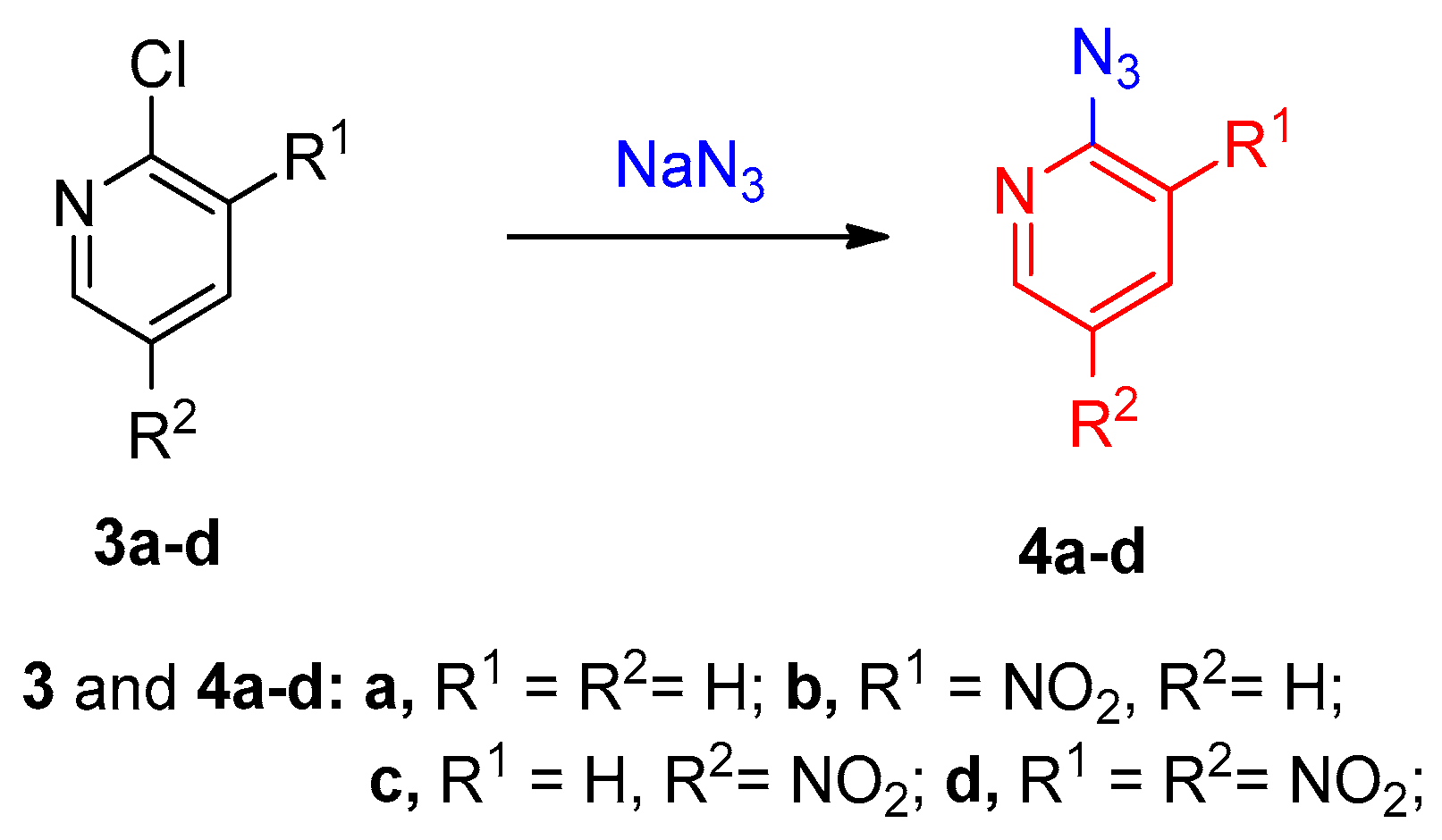
2.2. Via SNAr Reactions (Nucleophilic Aromatic Substitution Reactions)
2.3. From Lithium-Reagent
2.4. From Aryl Hydrazines
3. Chemistry of Azides
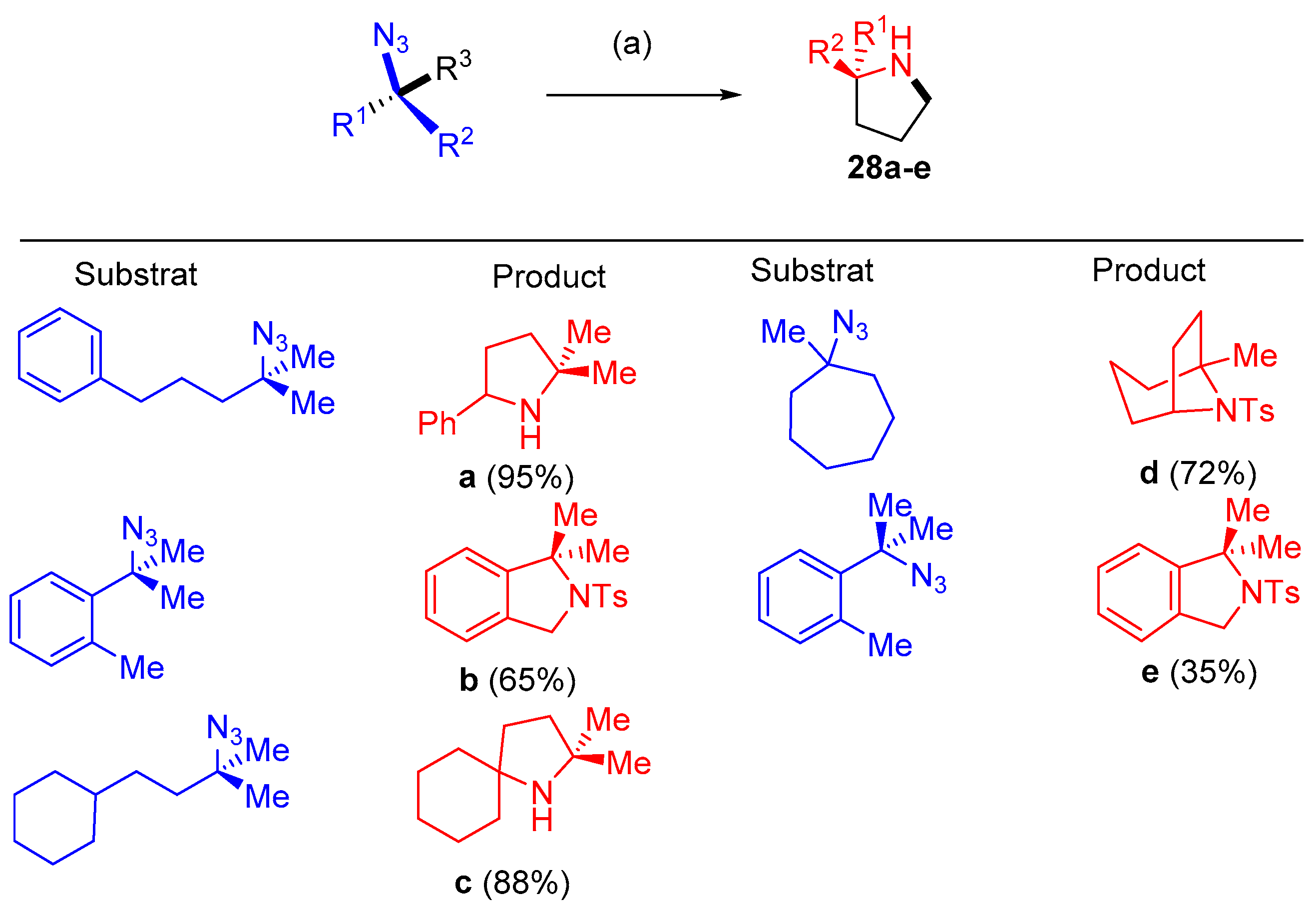
3.1. Azide as Aminating Group
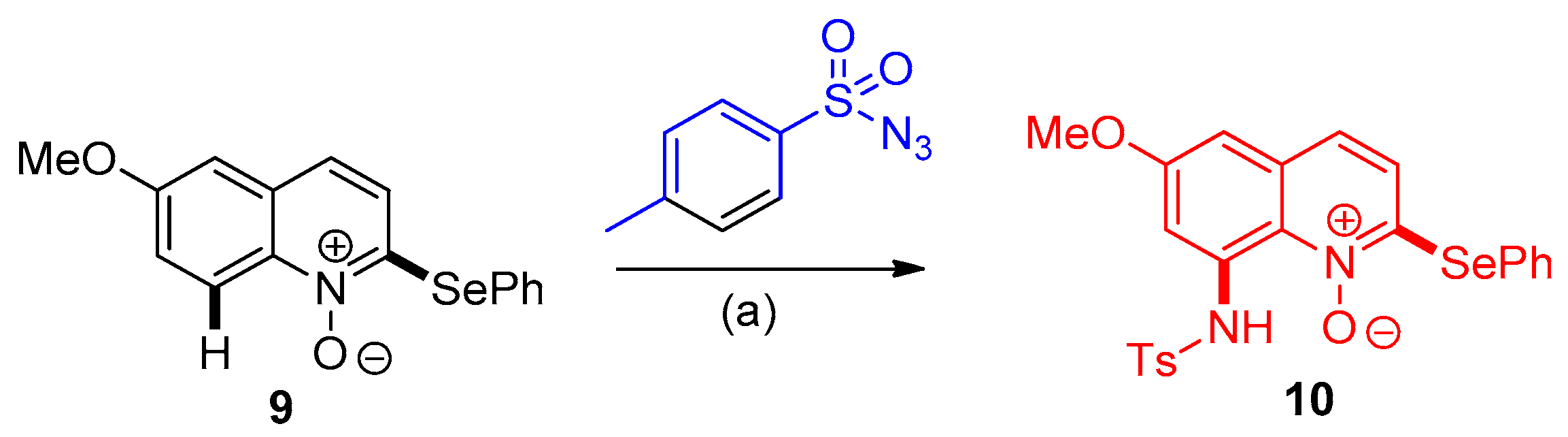
3.1.1. Synthesis of 8-Aminoquinoline
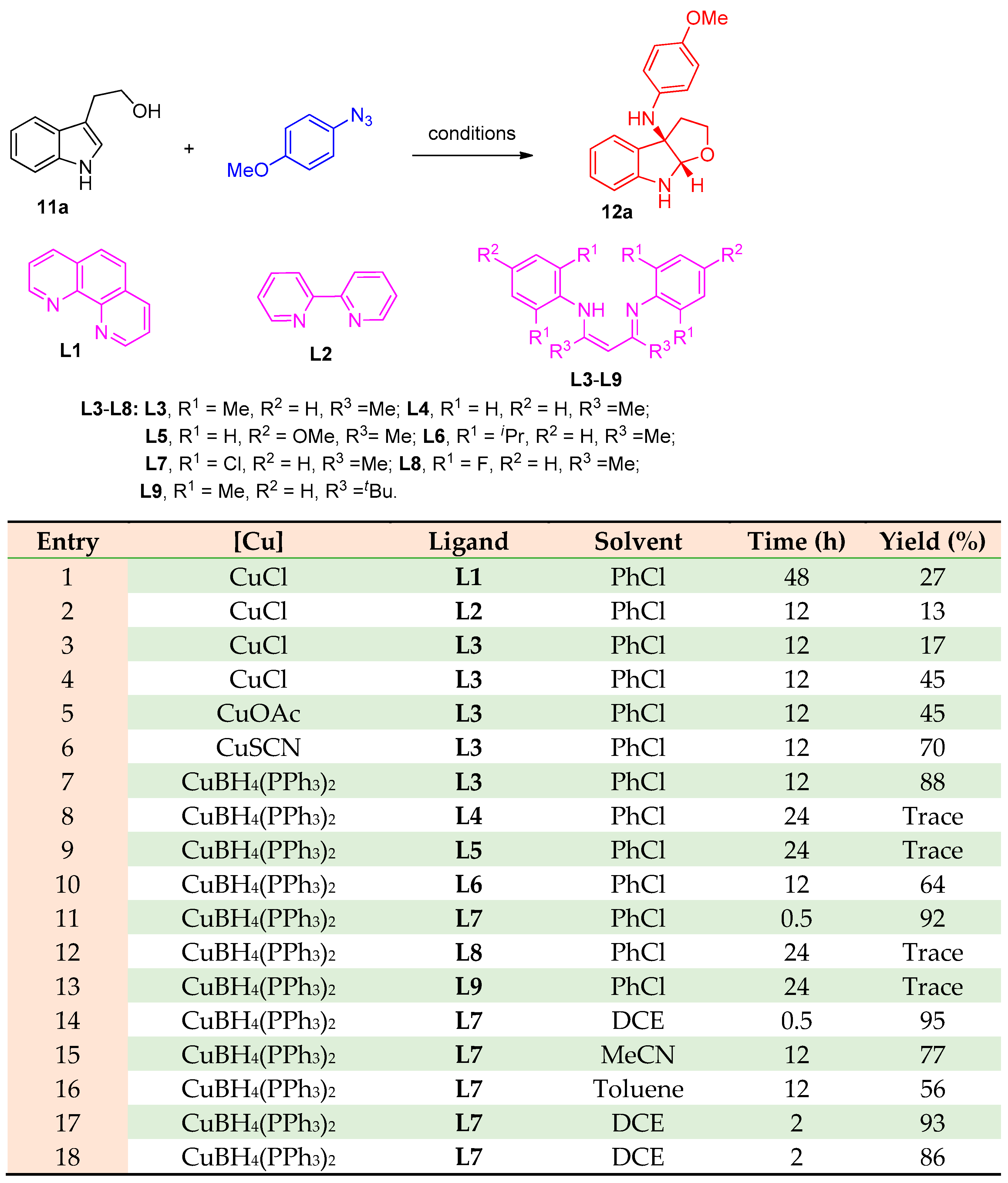
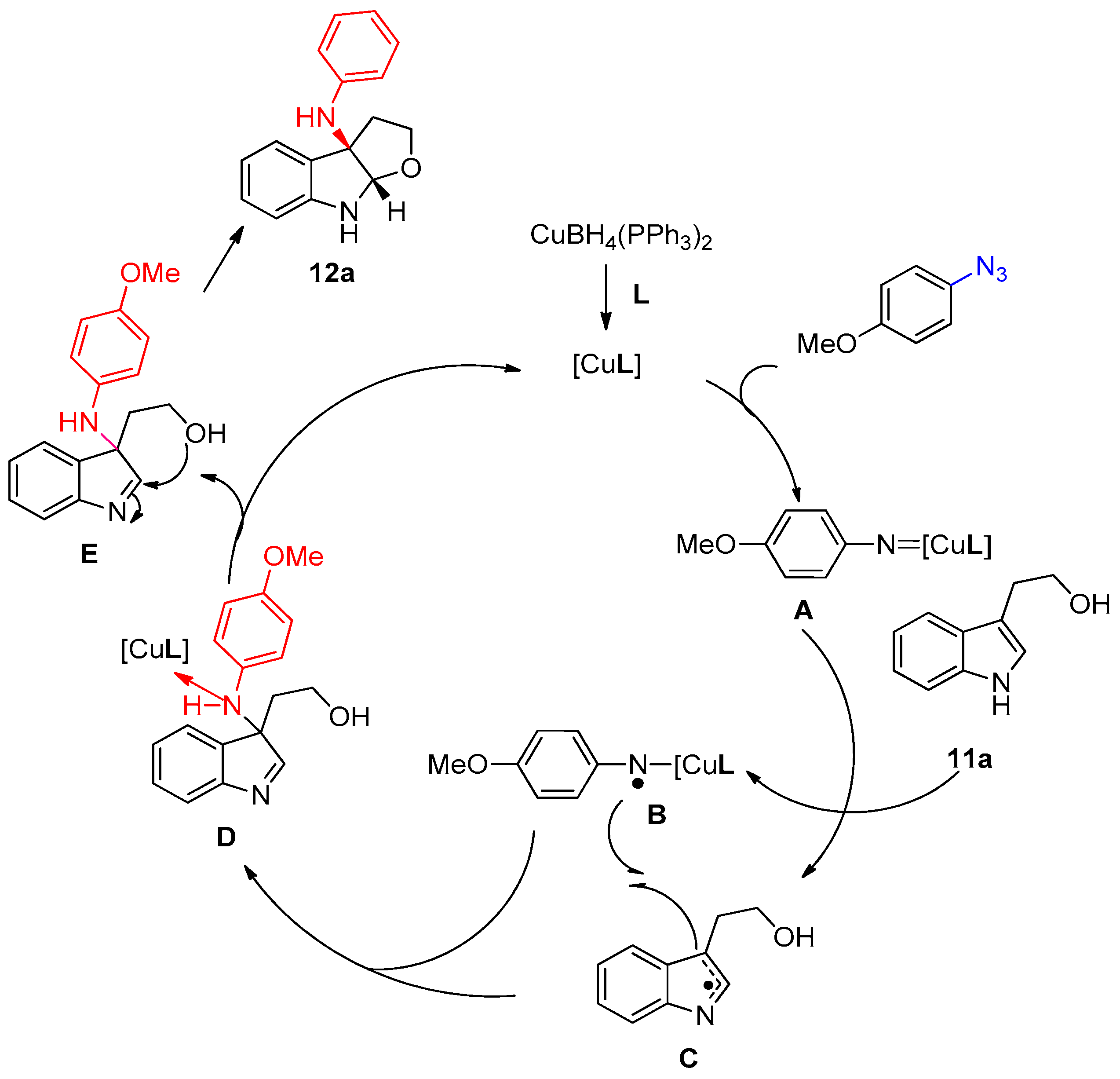
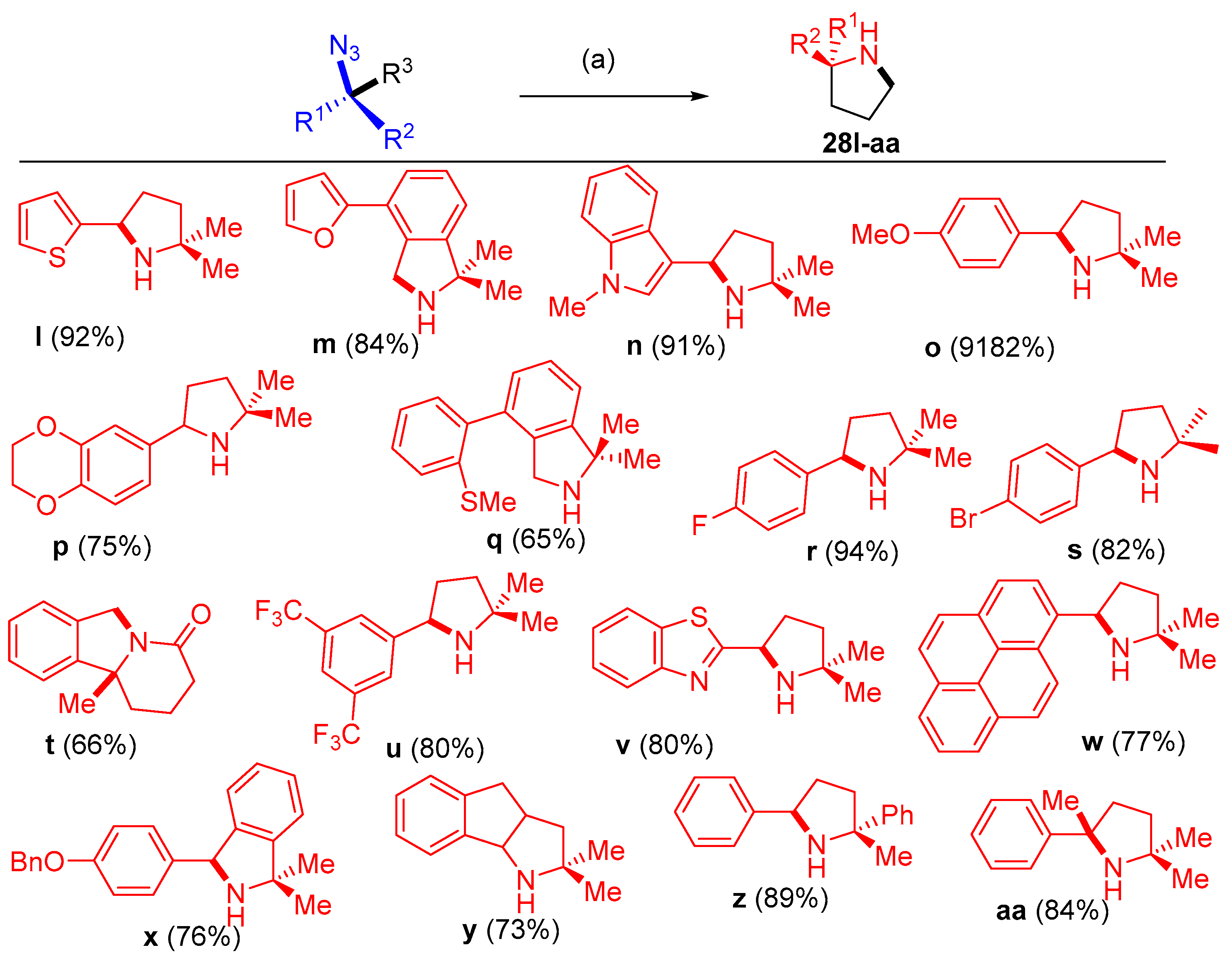
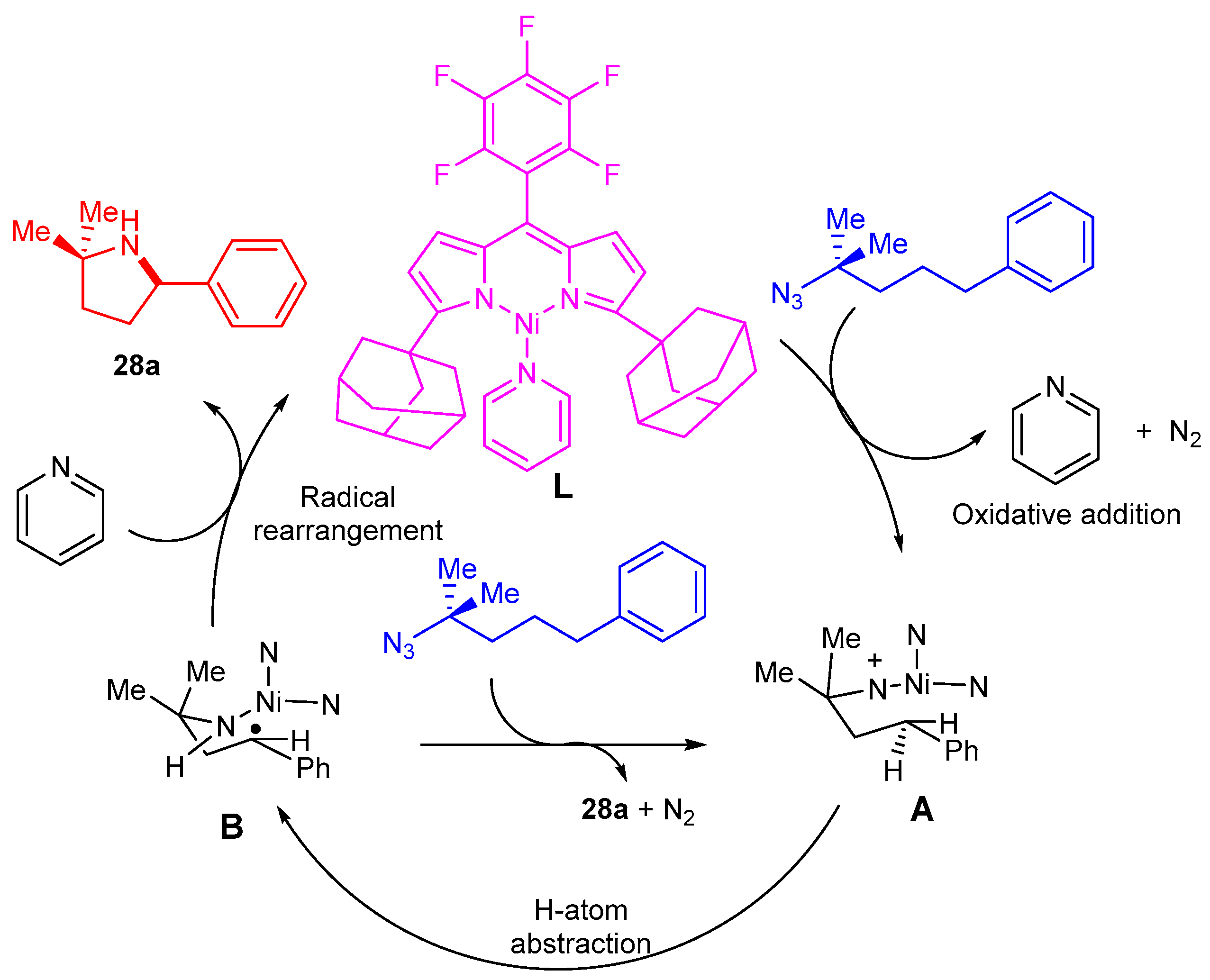
3.1.2. Synthesis of Amino Furo/Pyrroloindole Derivatives
3.2. Azidation
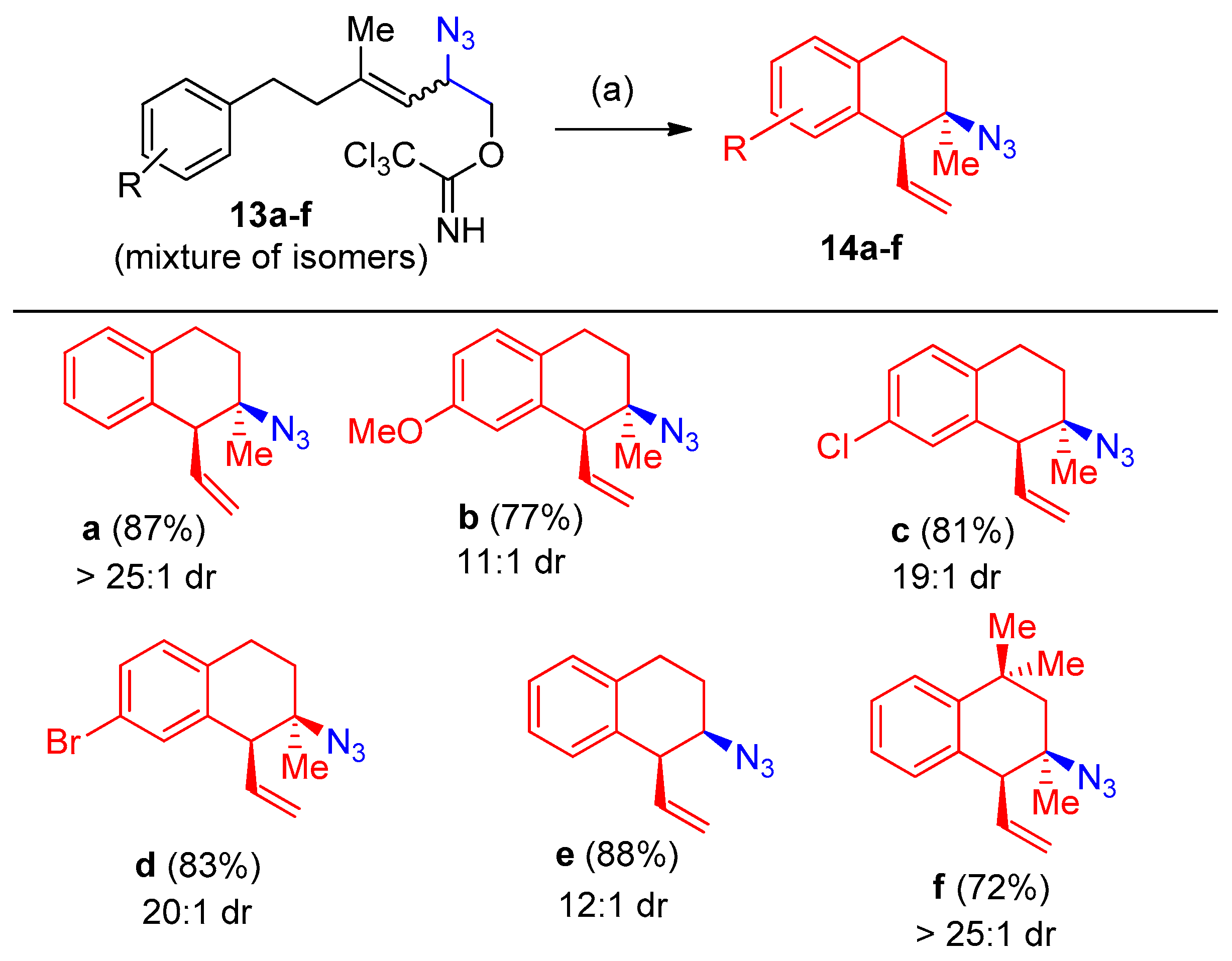
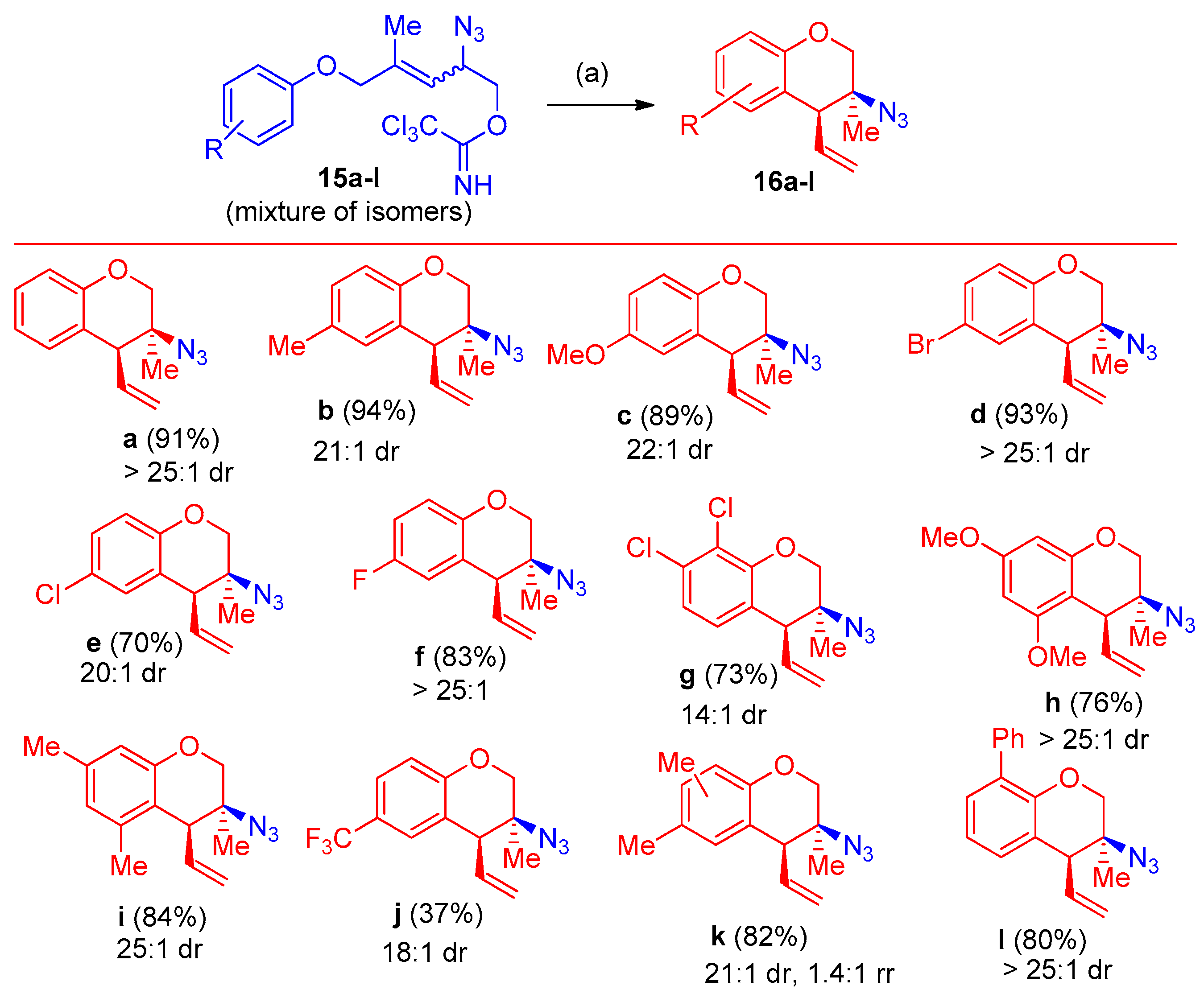
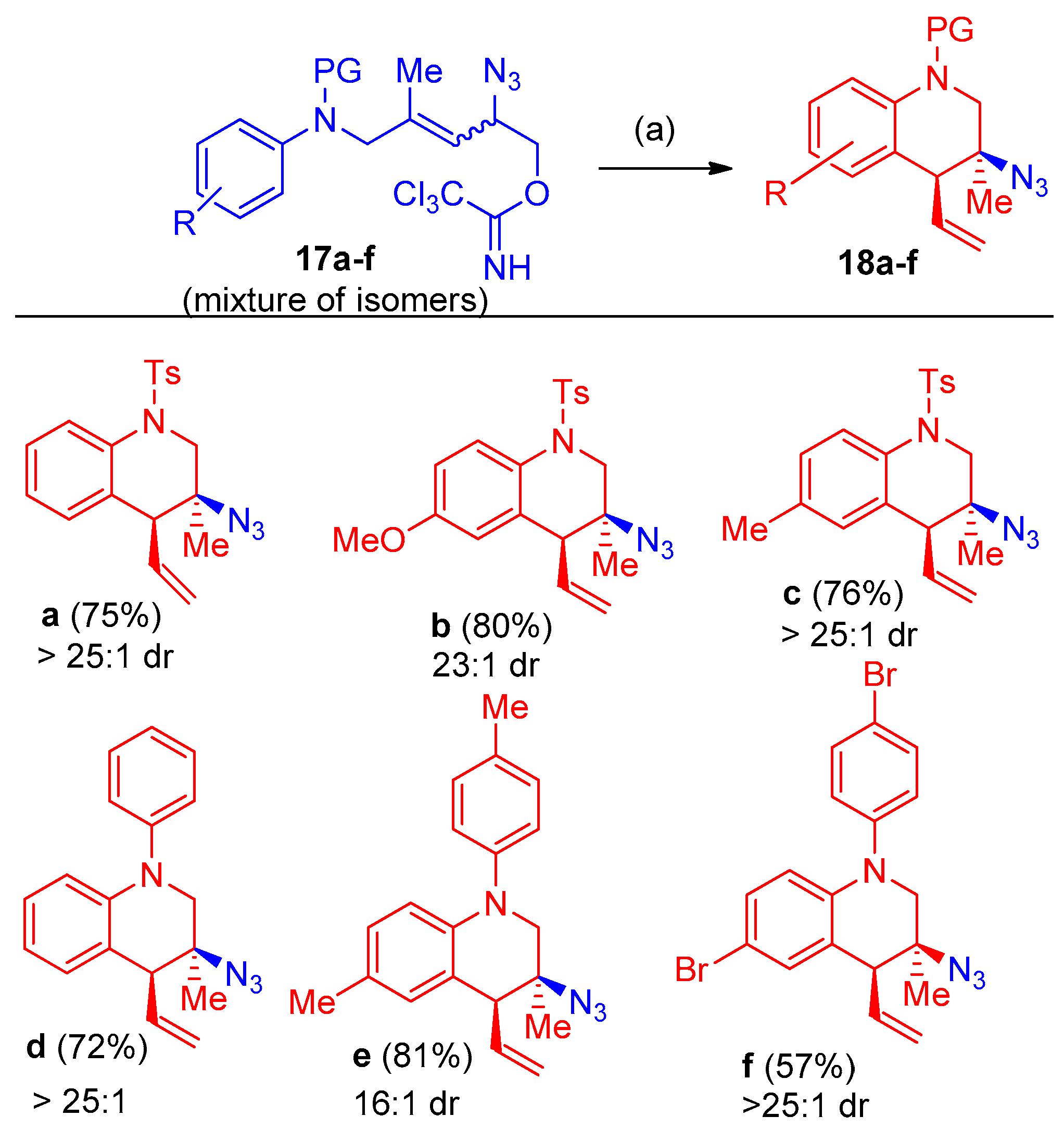
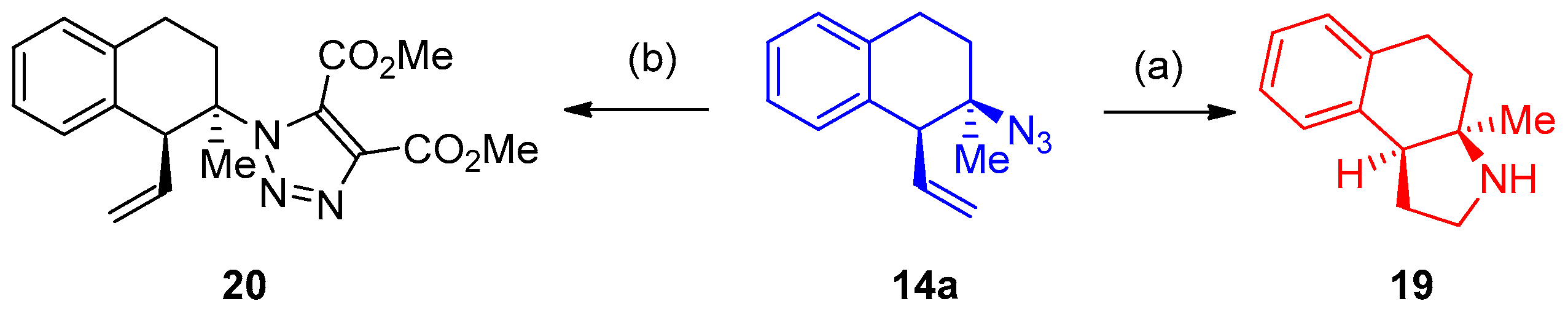
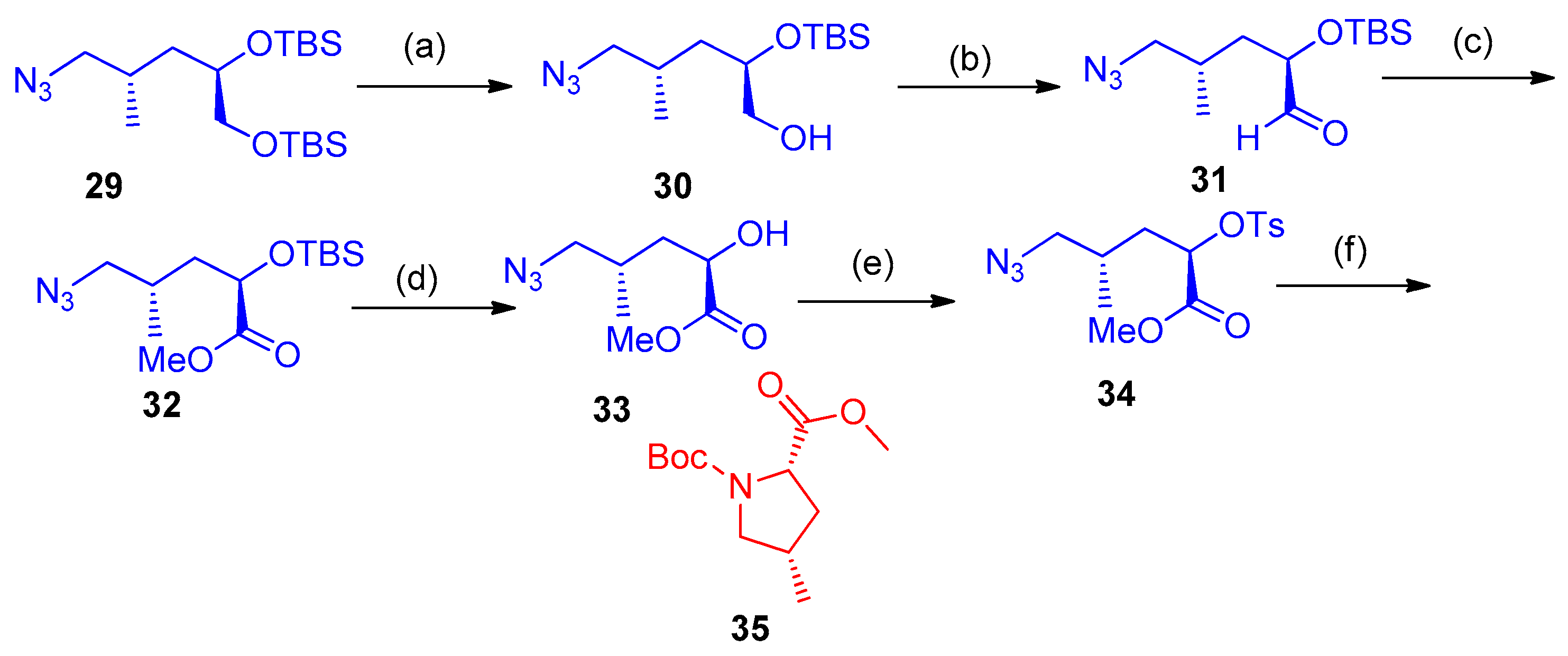
Synthesis of 3-Azido-Tetralins, Chromanes, and -Tetrahydroquinolines
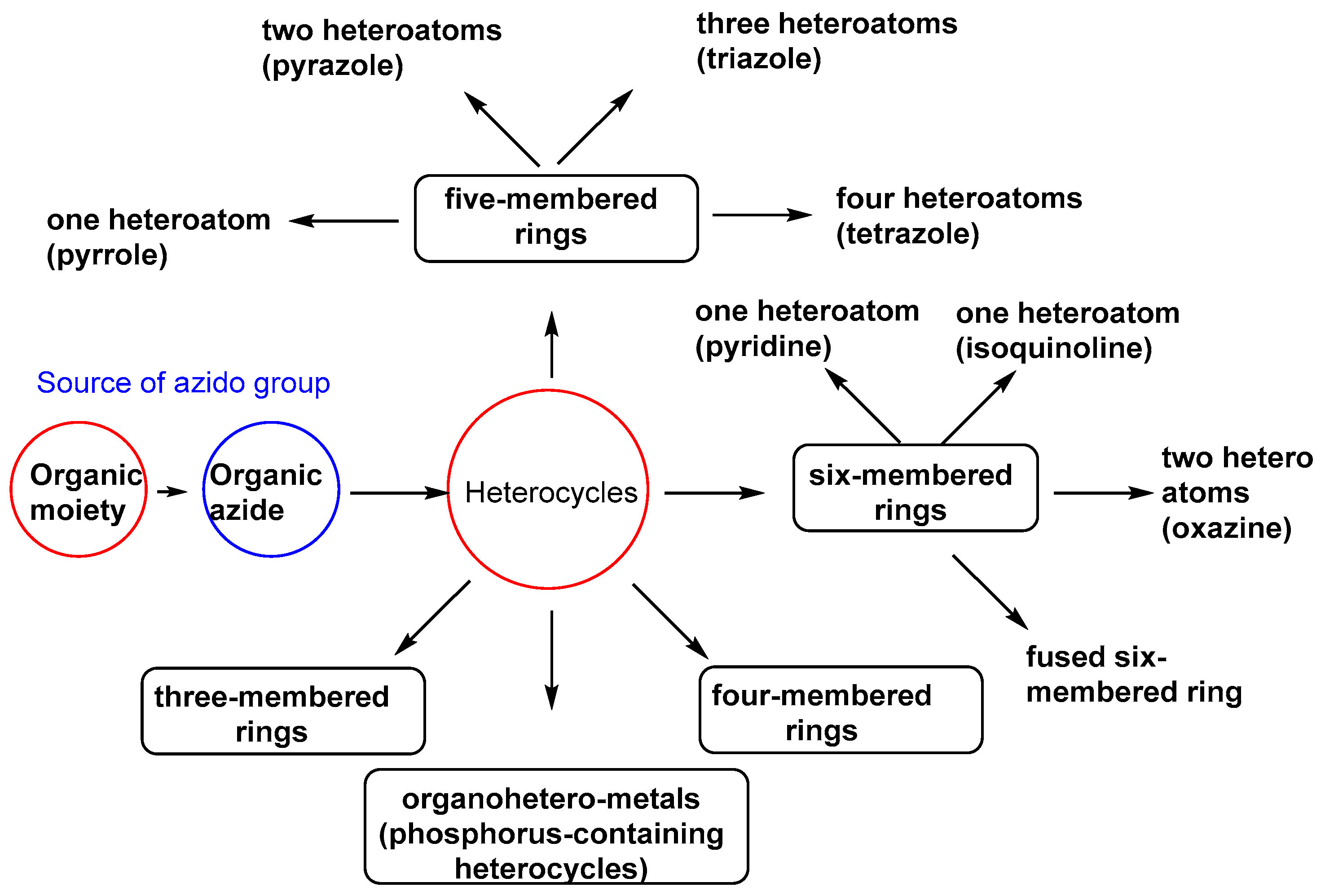
4. Organic Azides in the Synthesis of Heterocycles
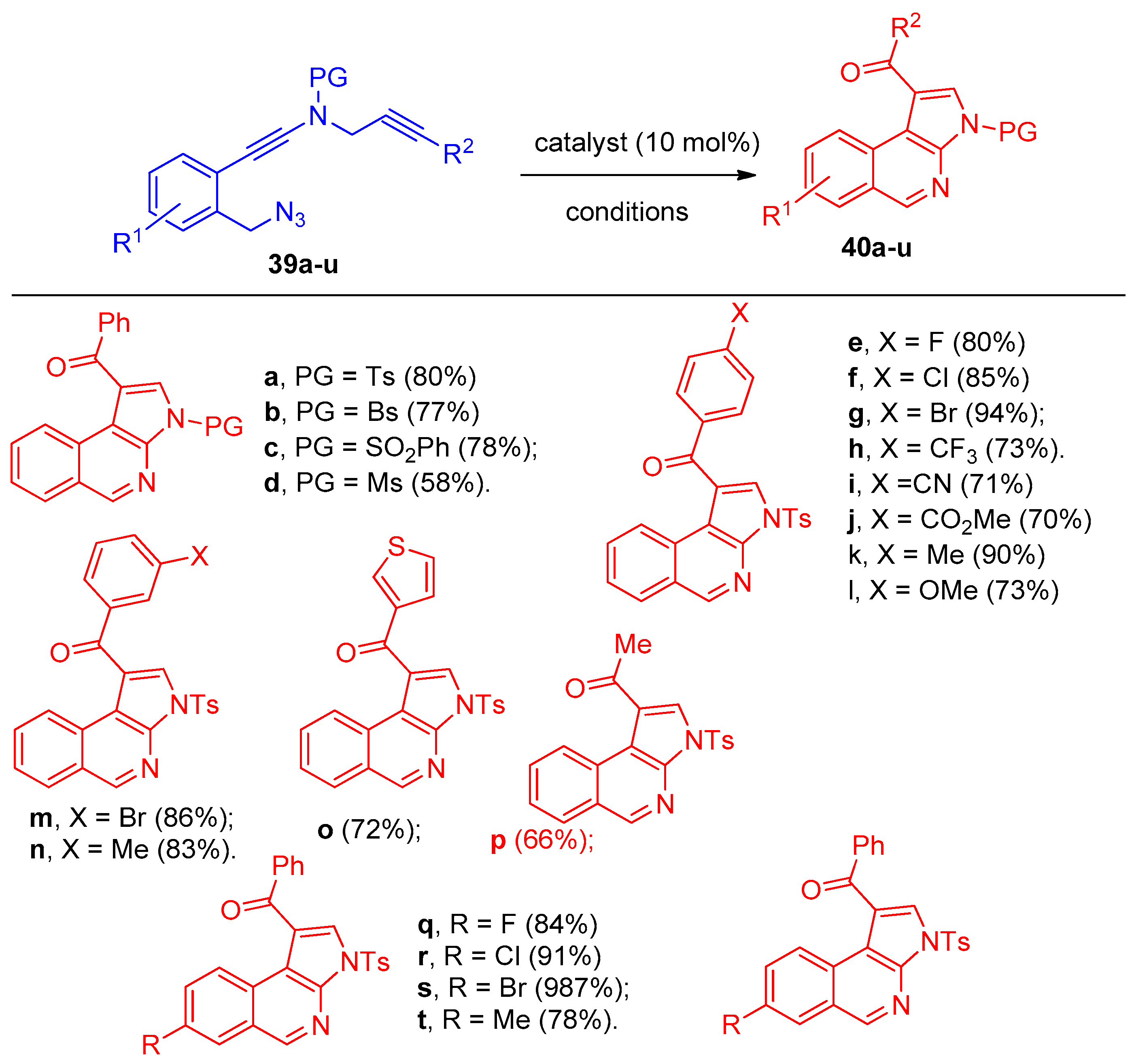
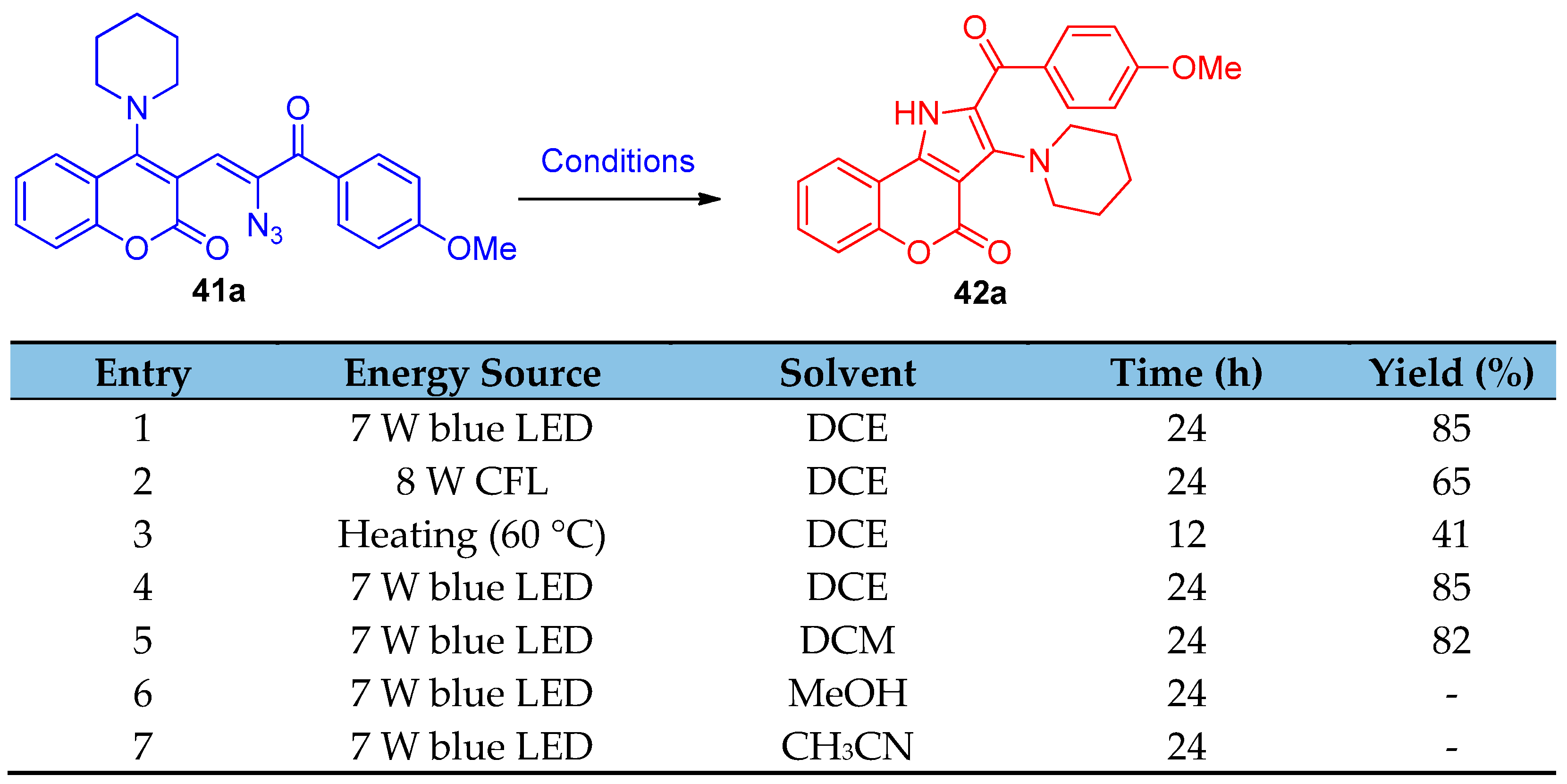
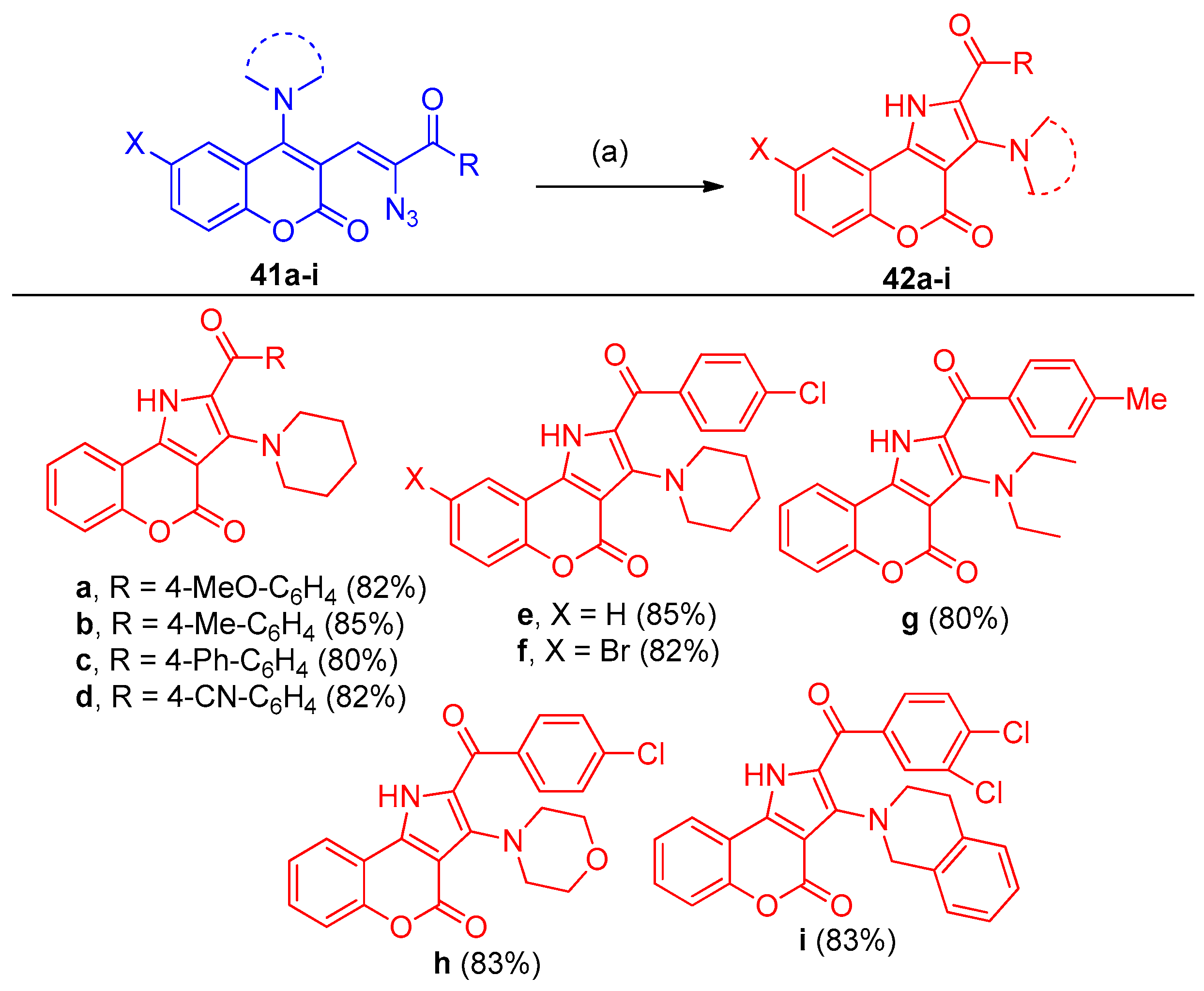
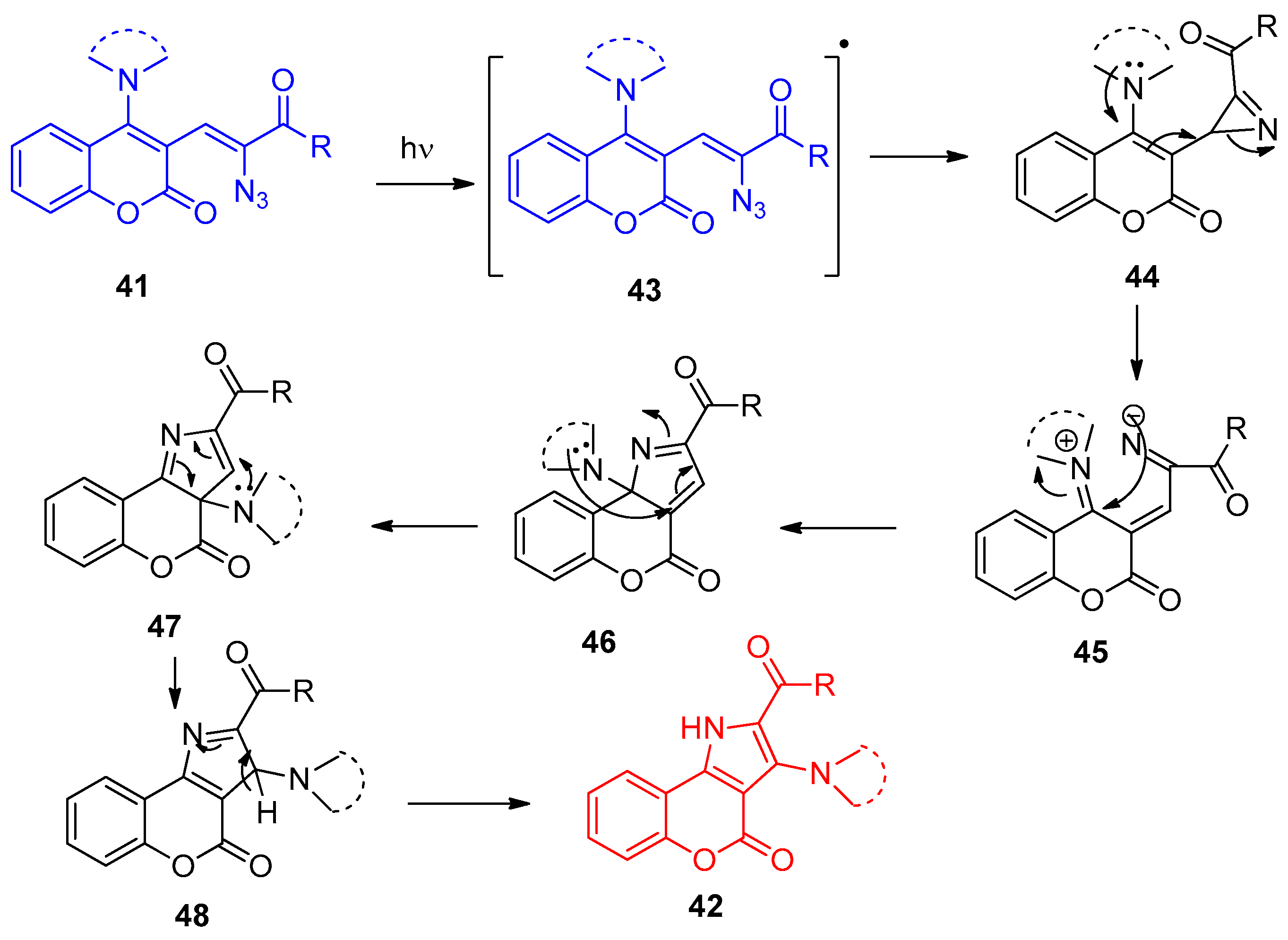
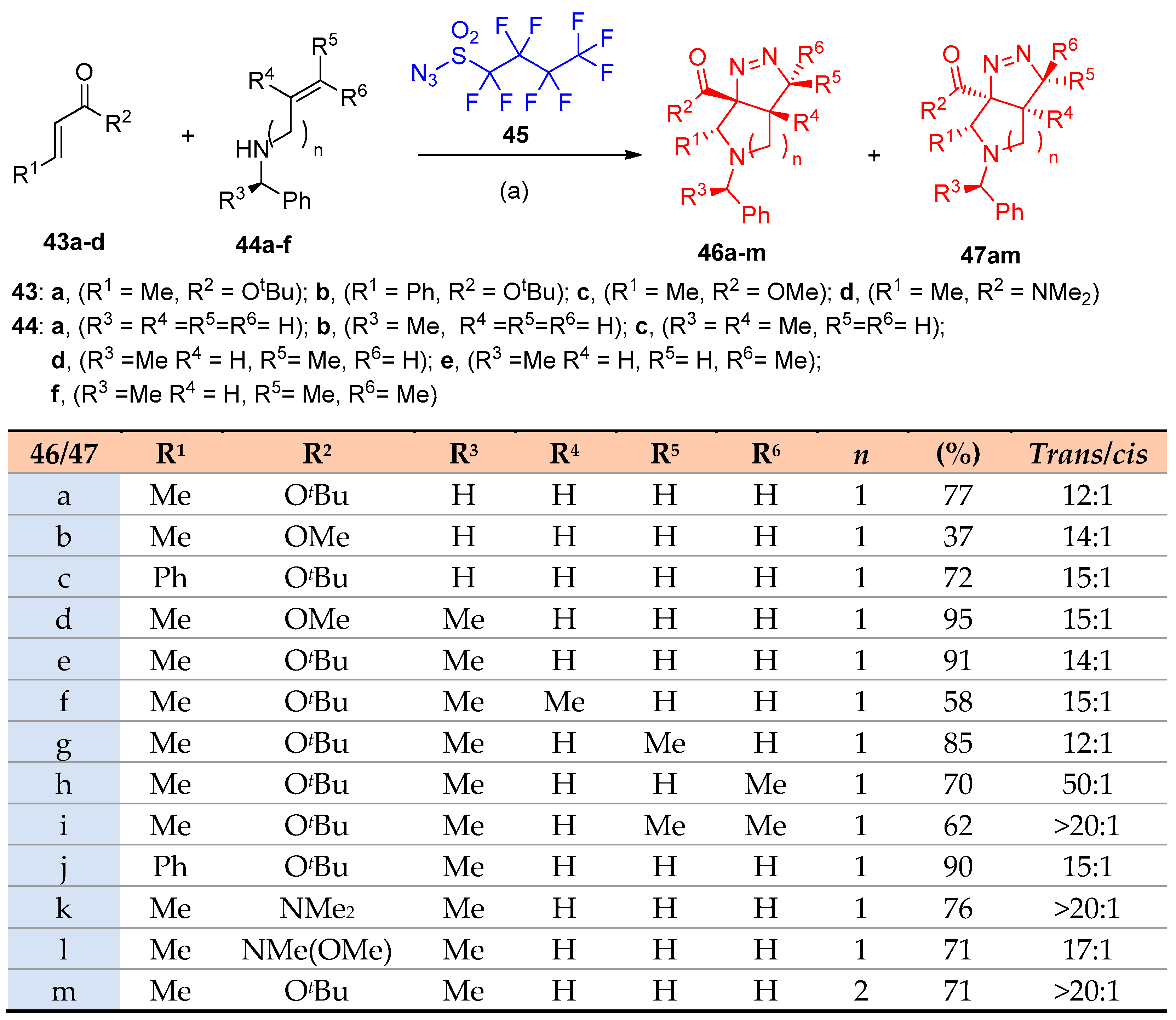
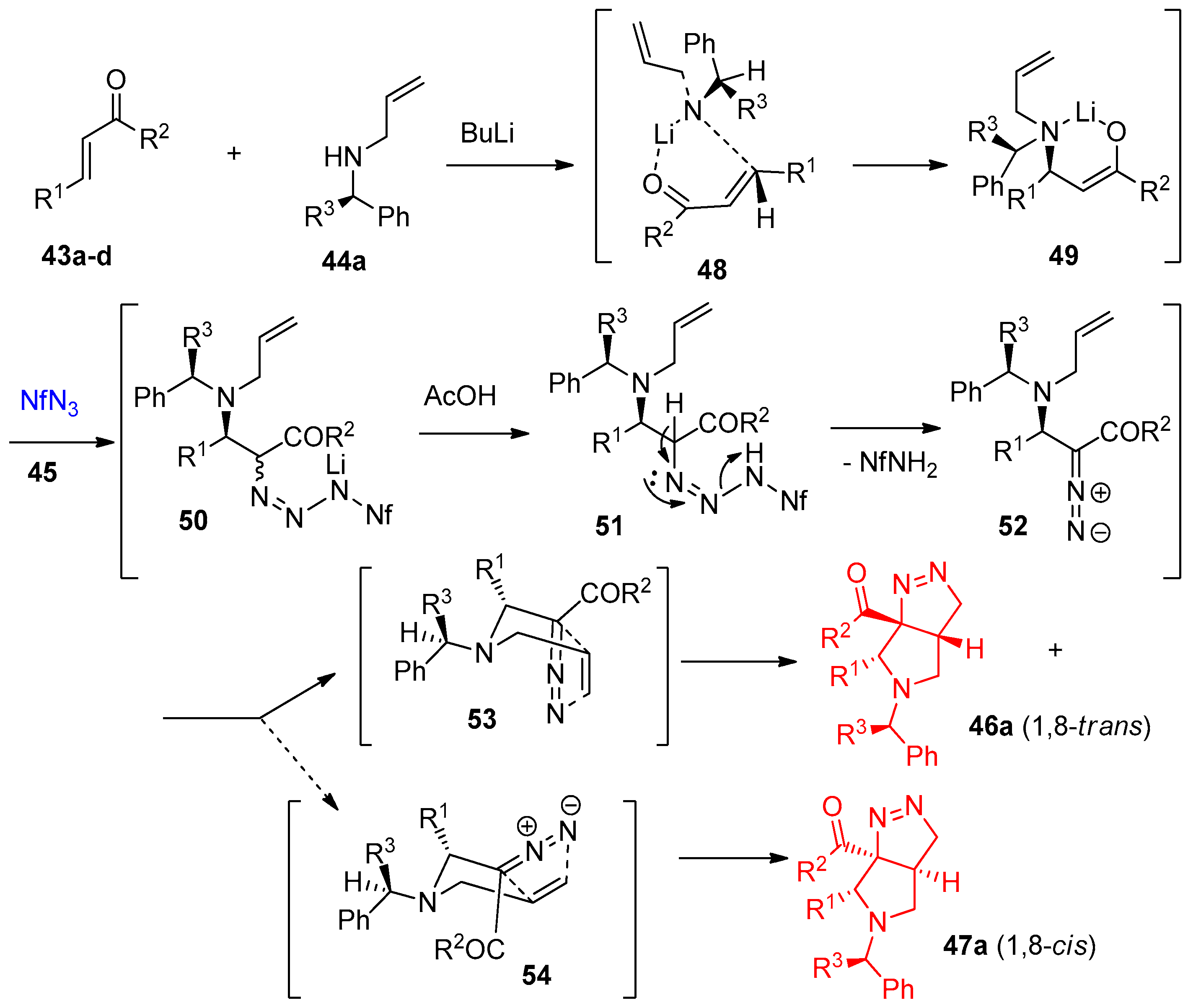
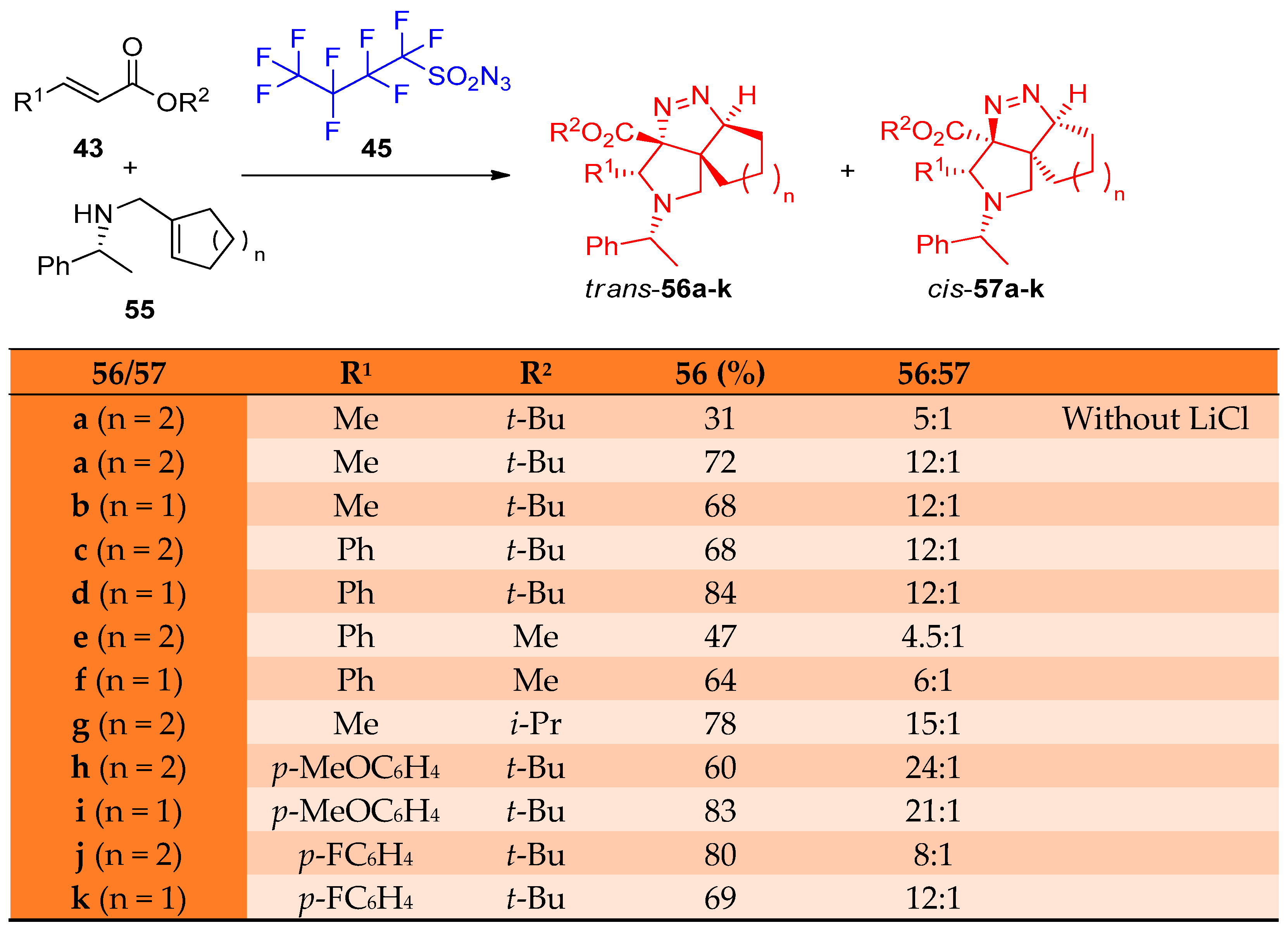
4.1. Synthesis of the Pyrrole Ring
4.2. Synthesis of the Pyrazole Ring
4.3. Synthesis of Heterocycles Containing Two Heteroatoms
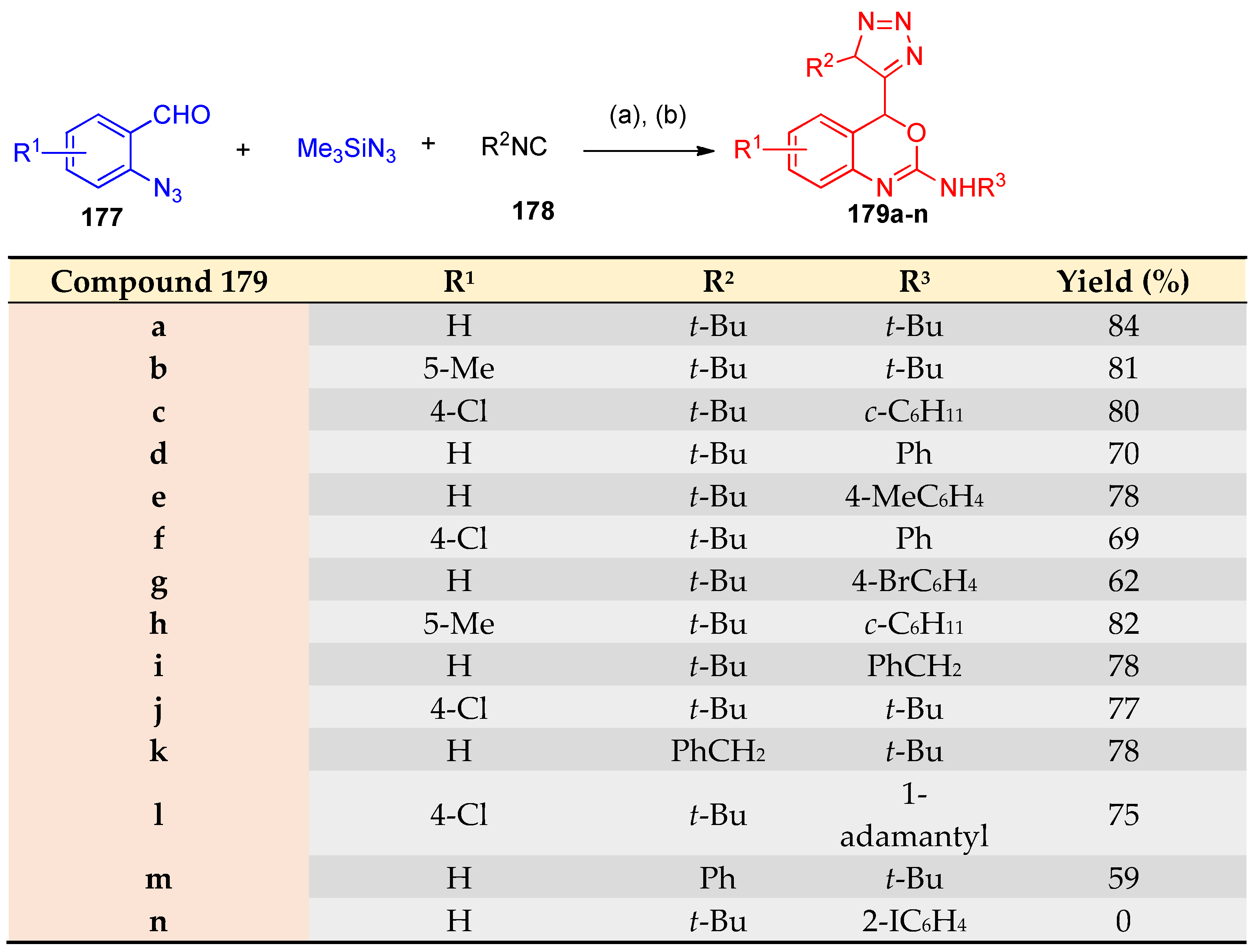
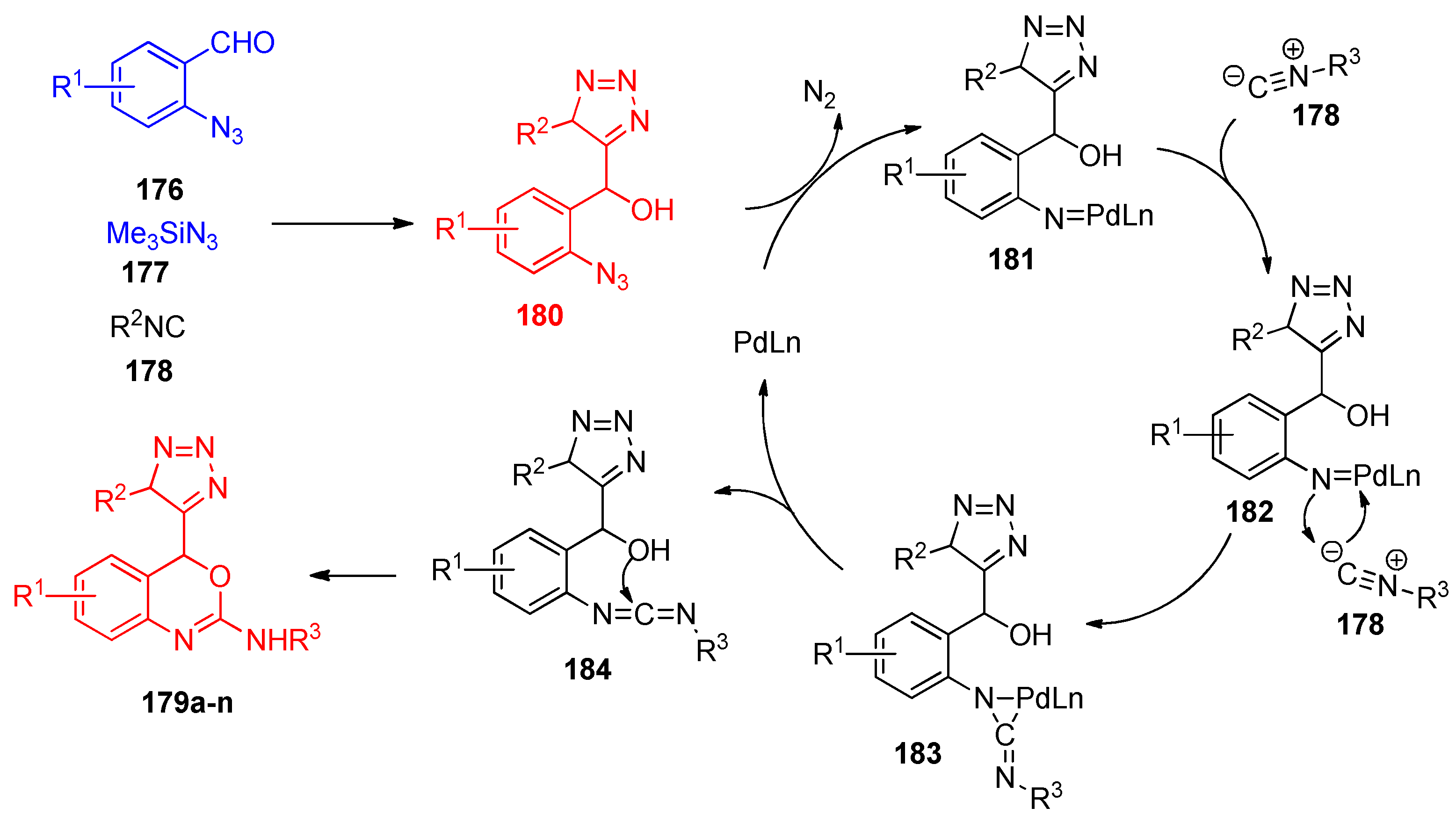
4.4. Synthesis of Oxazole, Thiazole, and Oxazine Derivatives
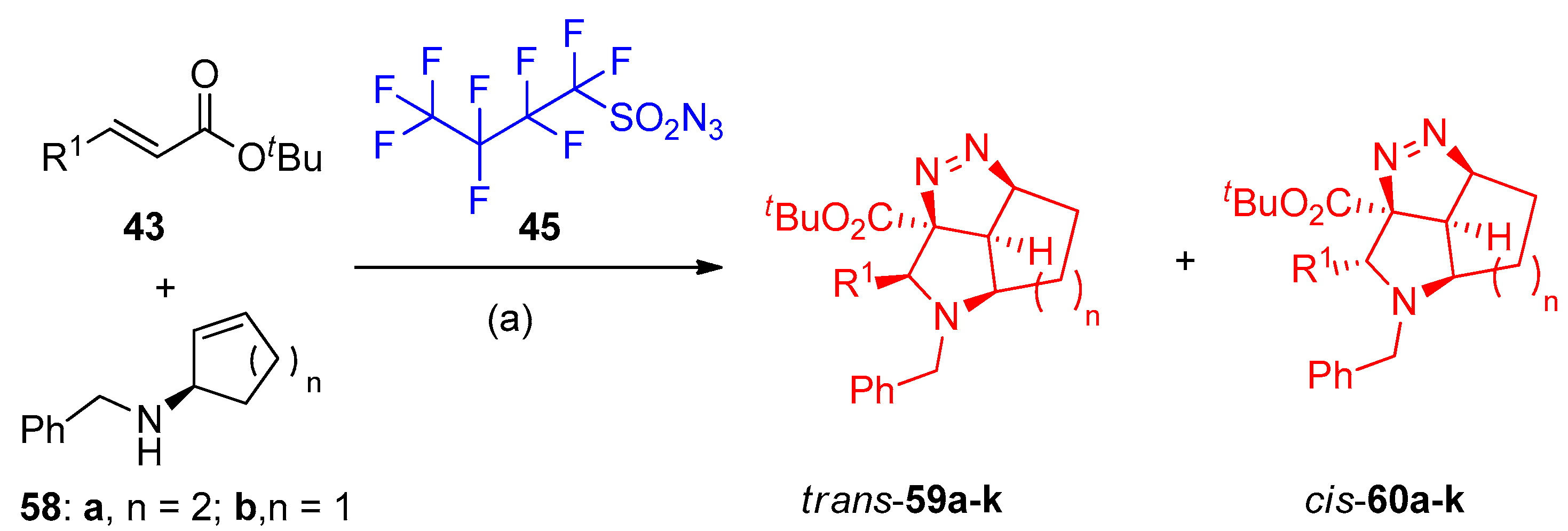
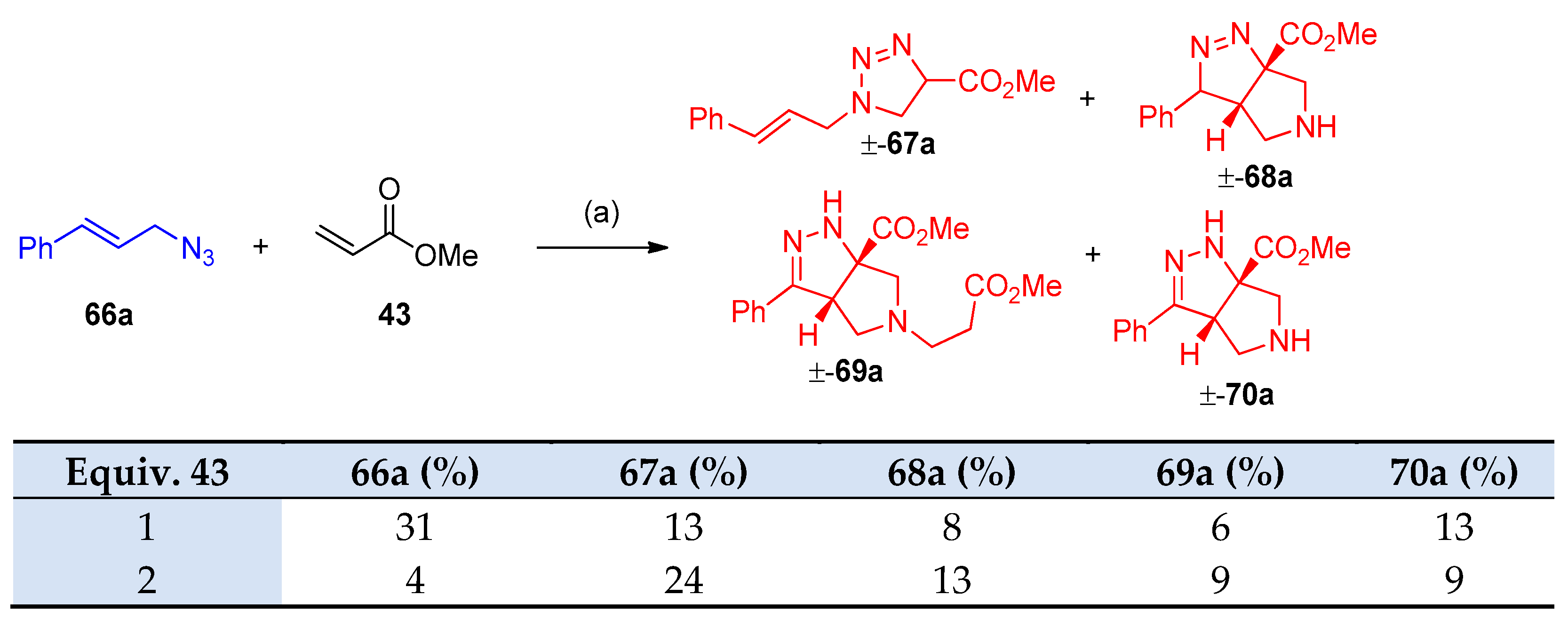
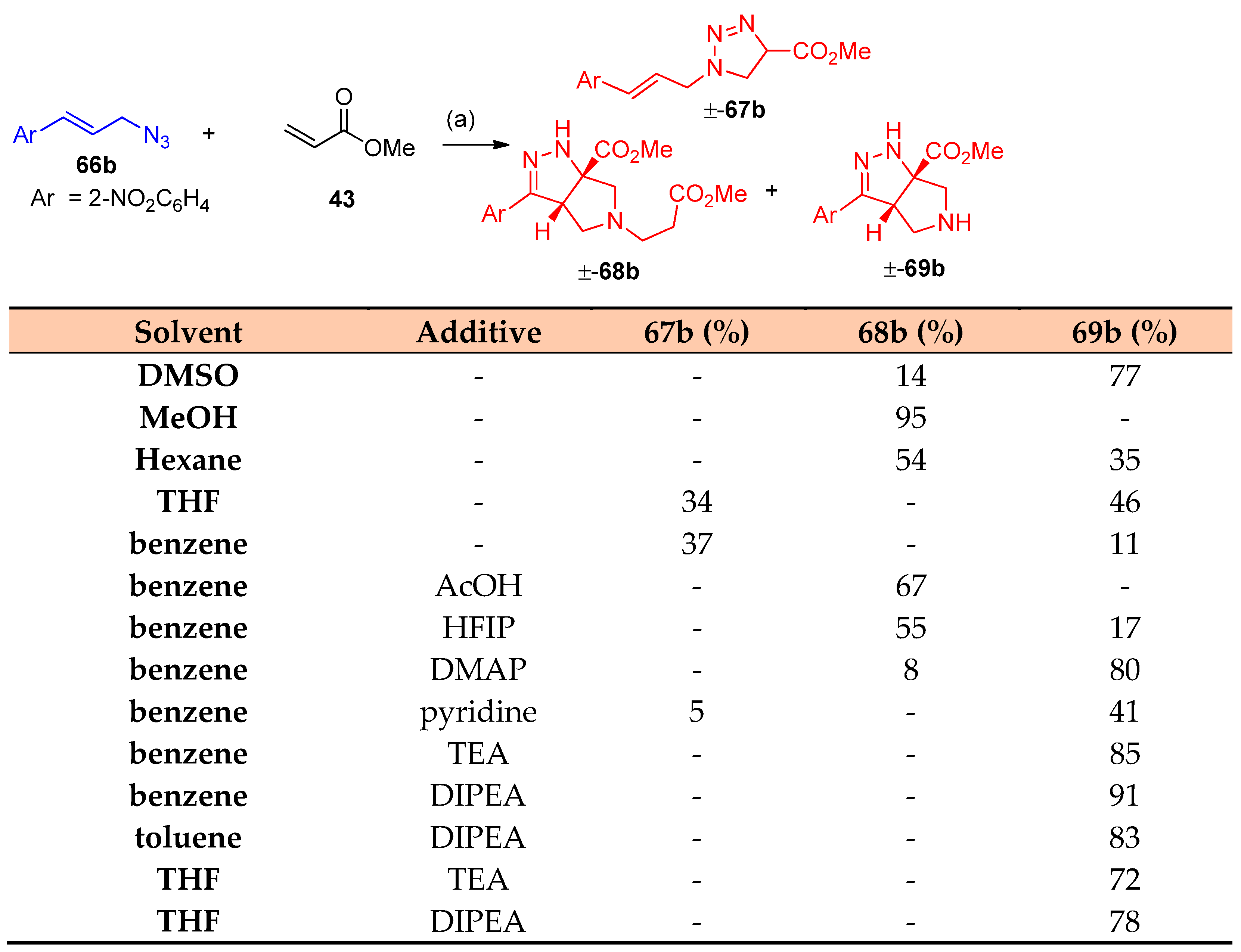
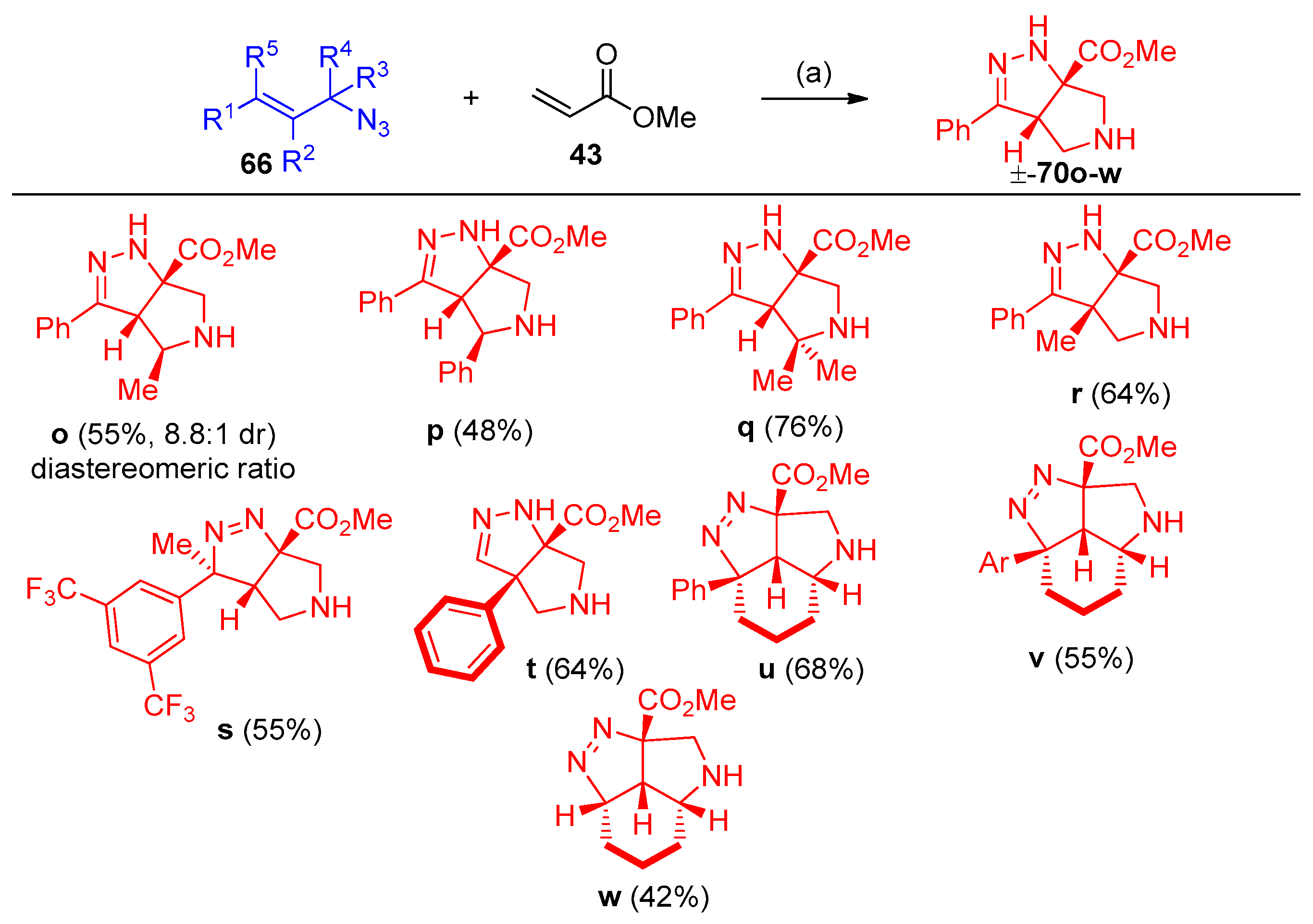
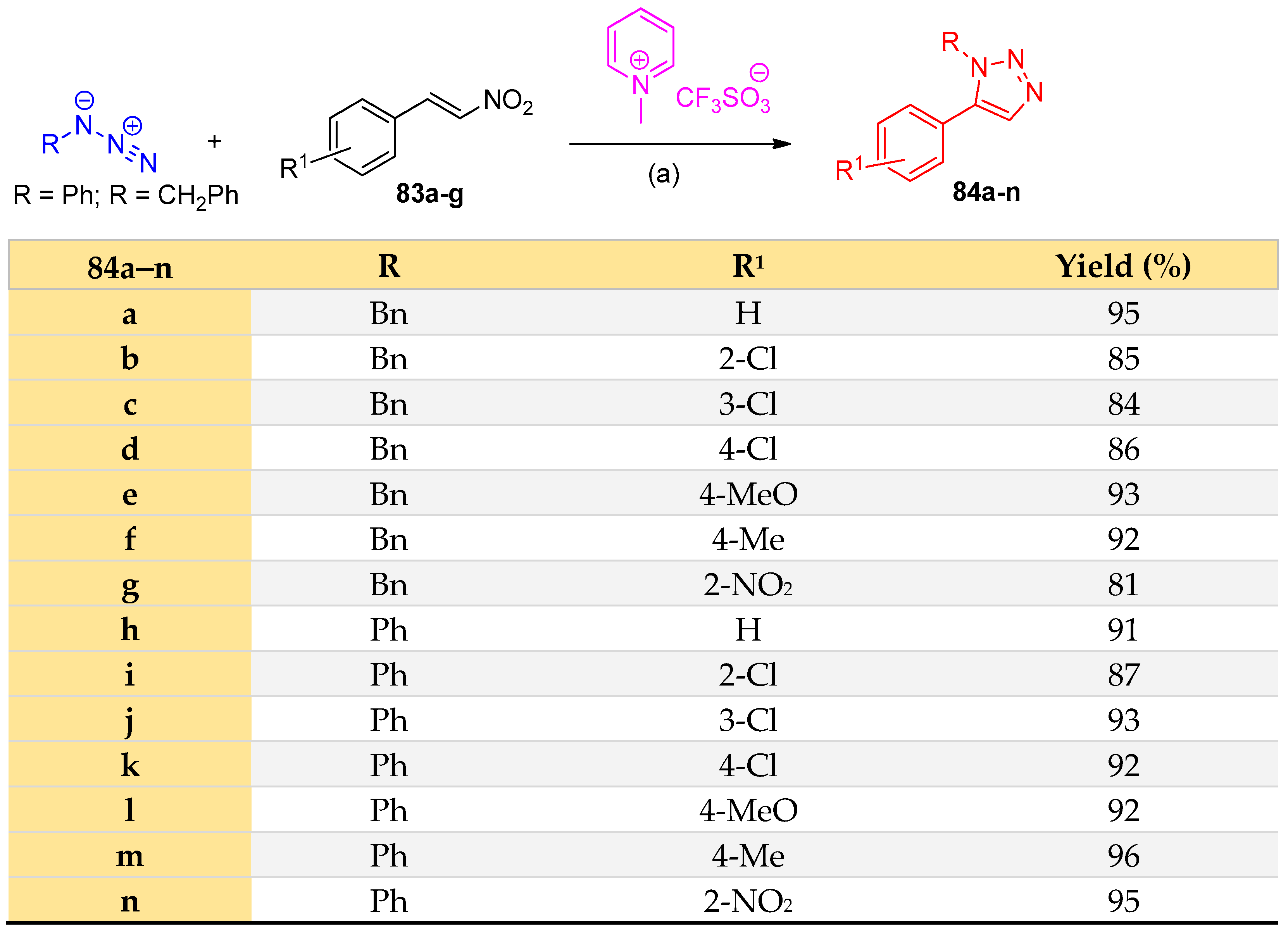
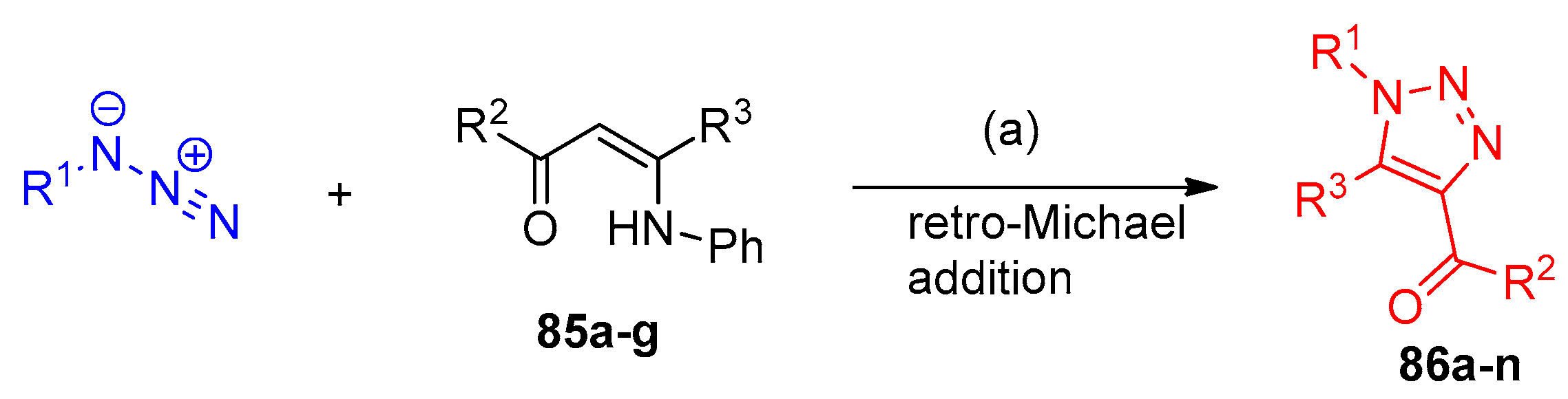
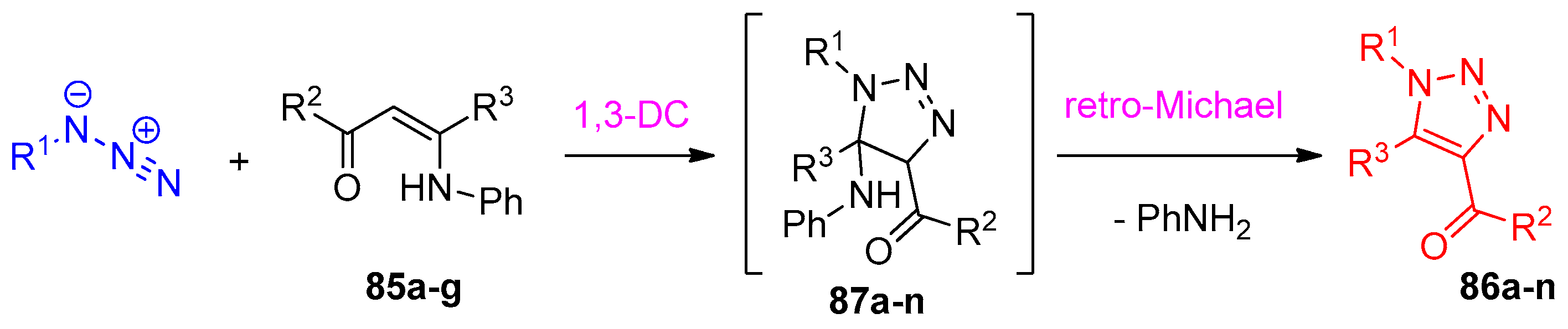
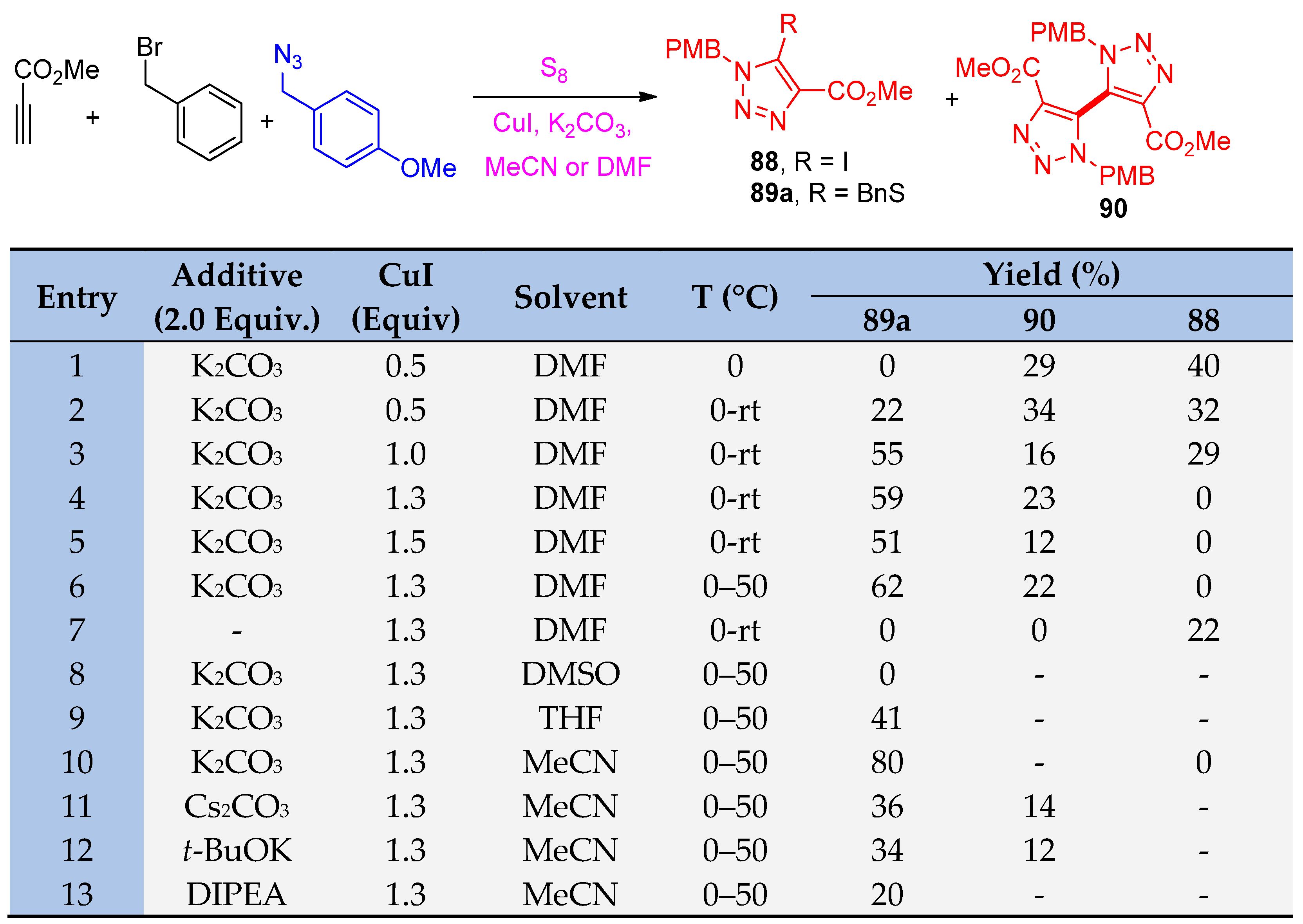
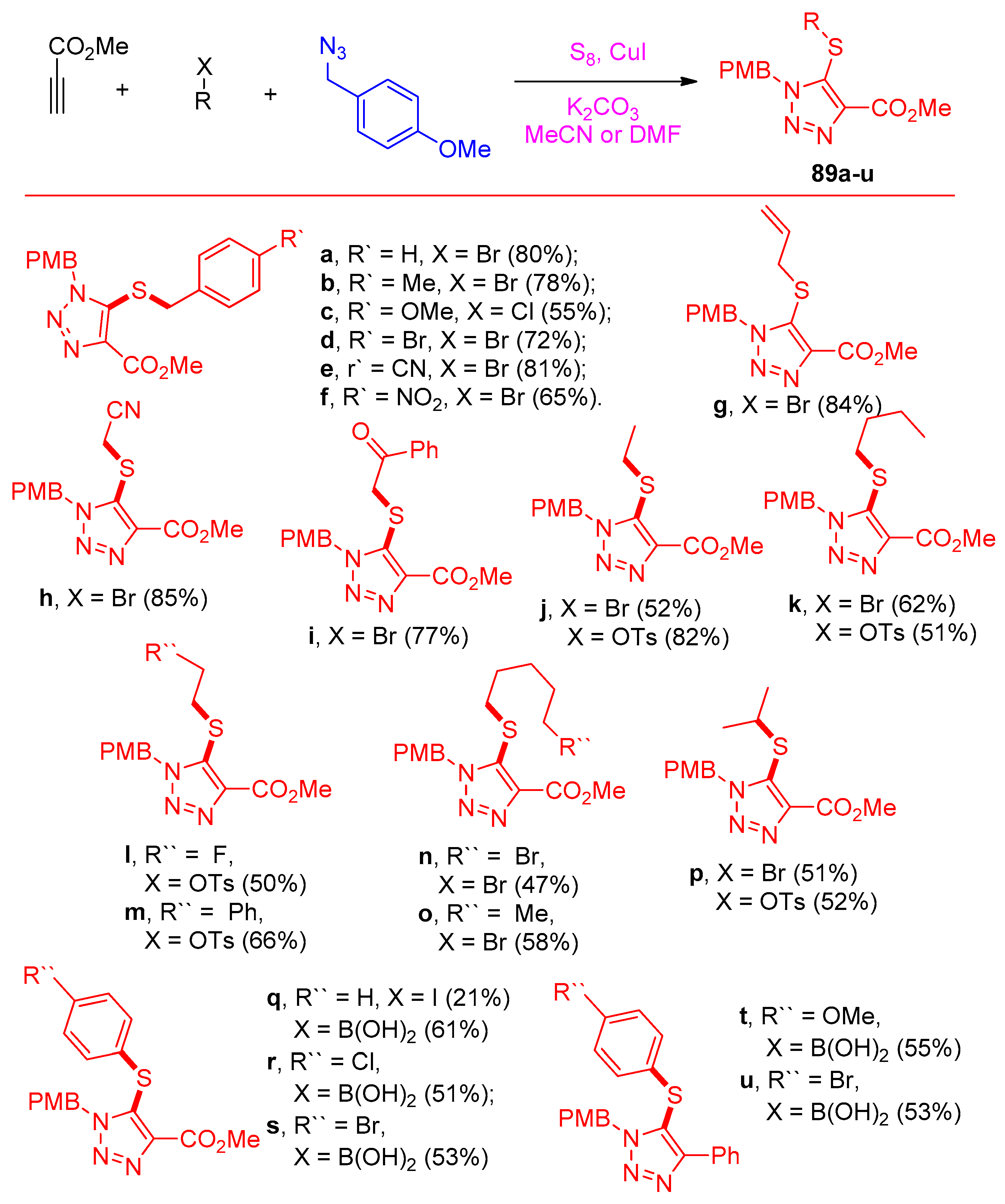
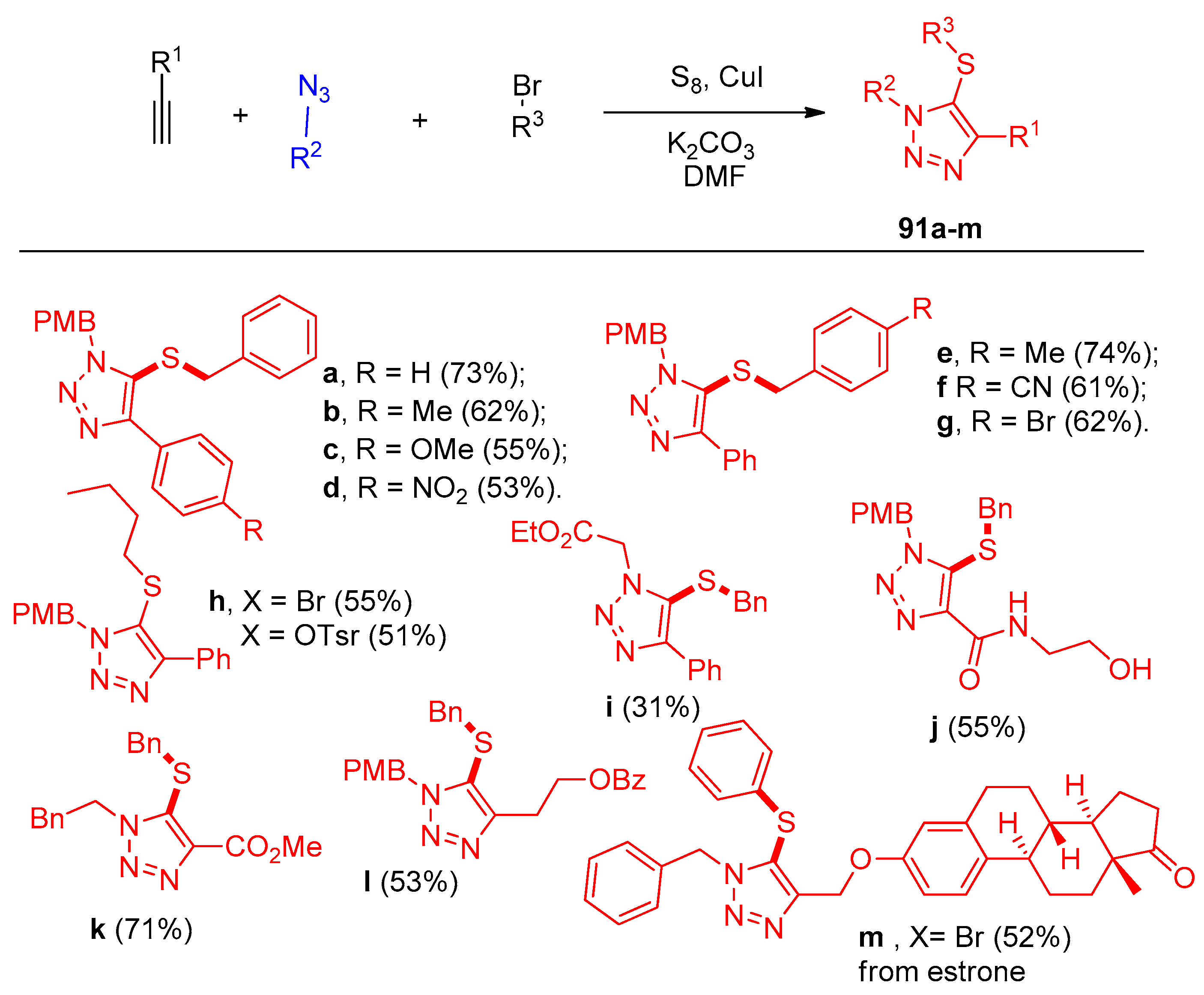
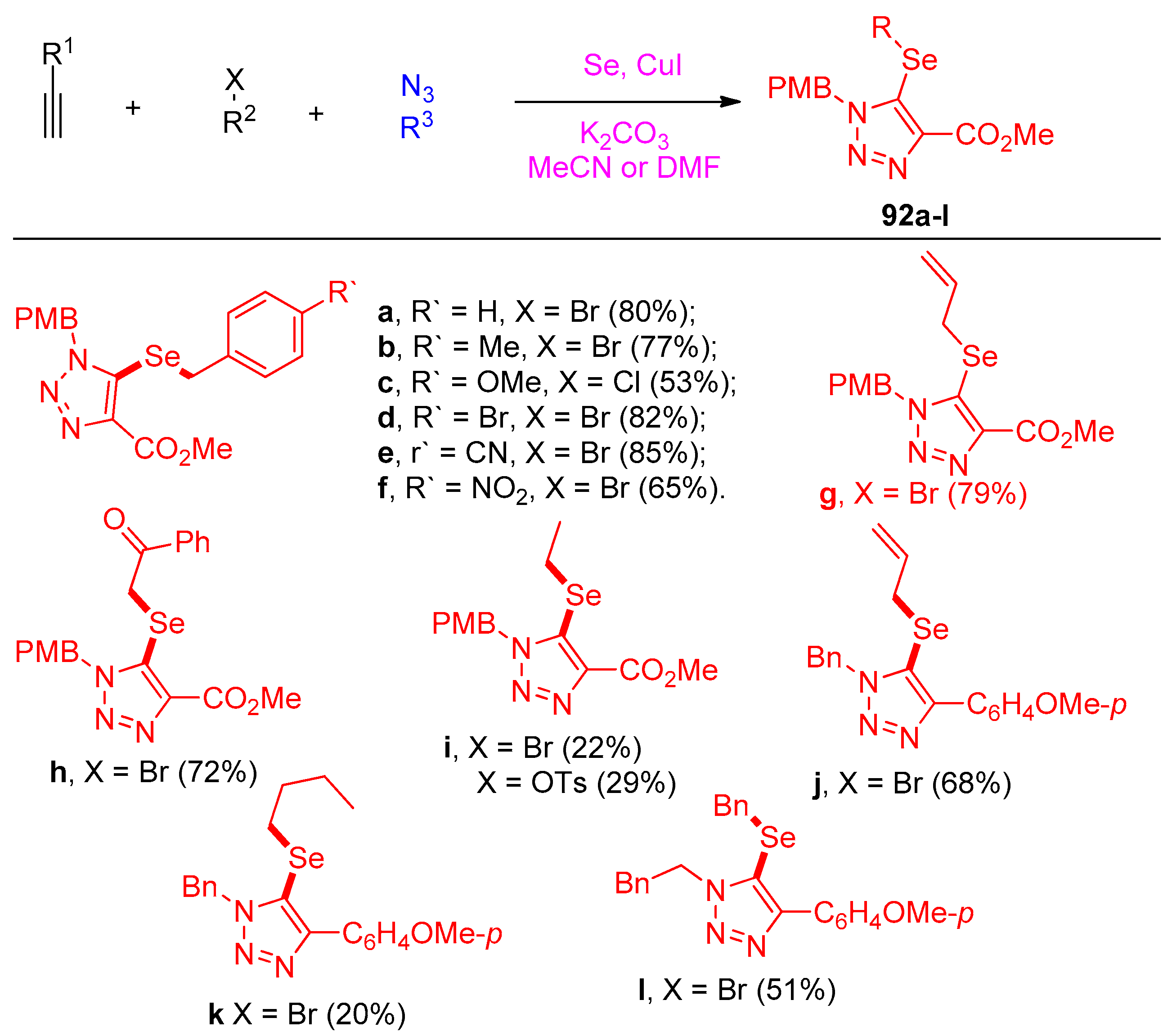
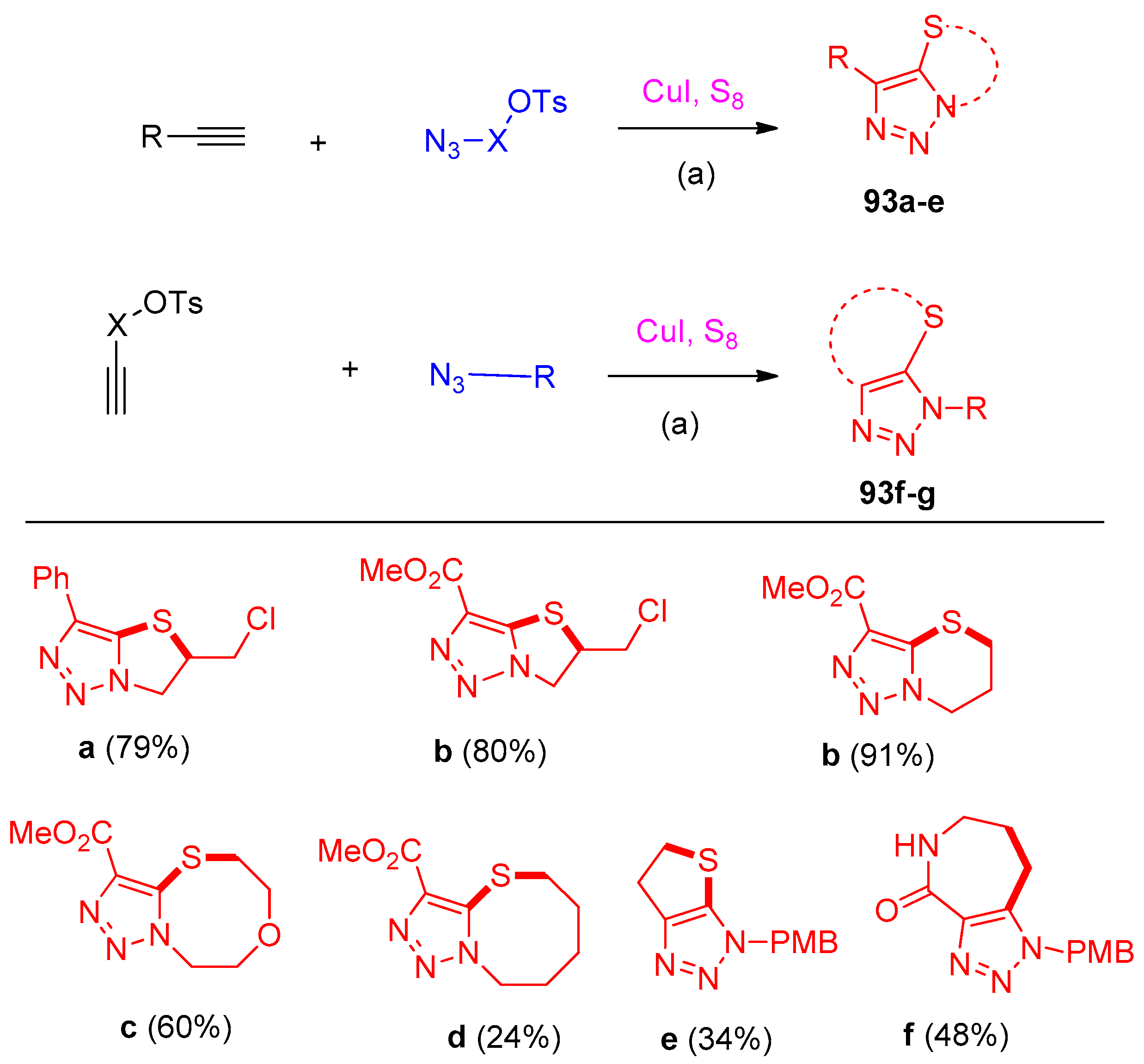
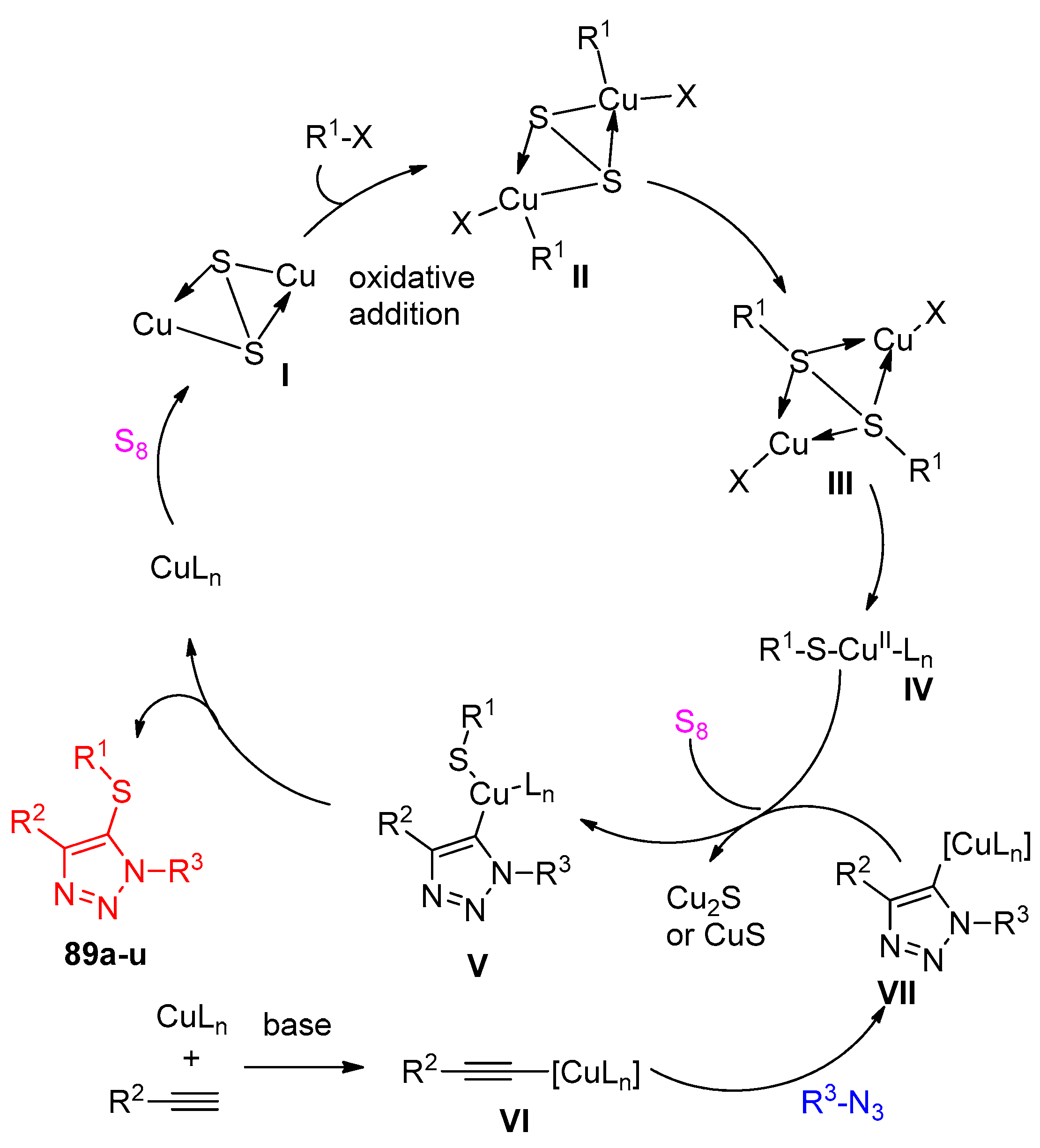
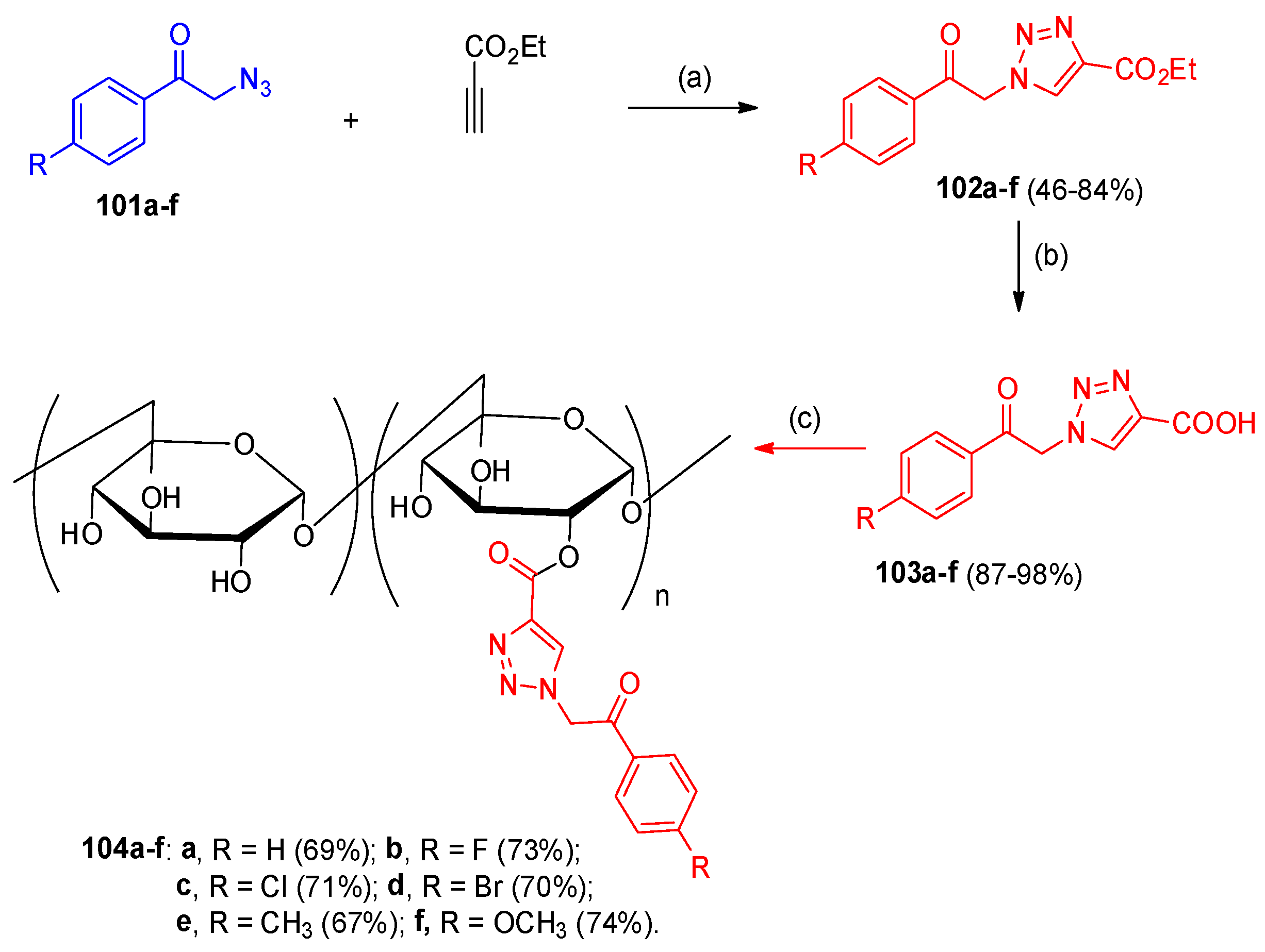
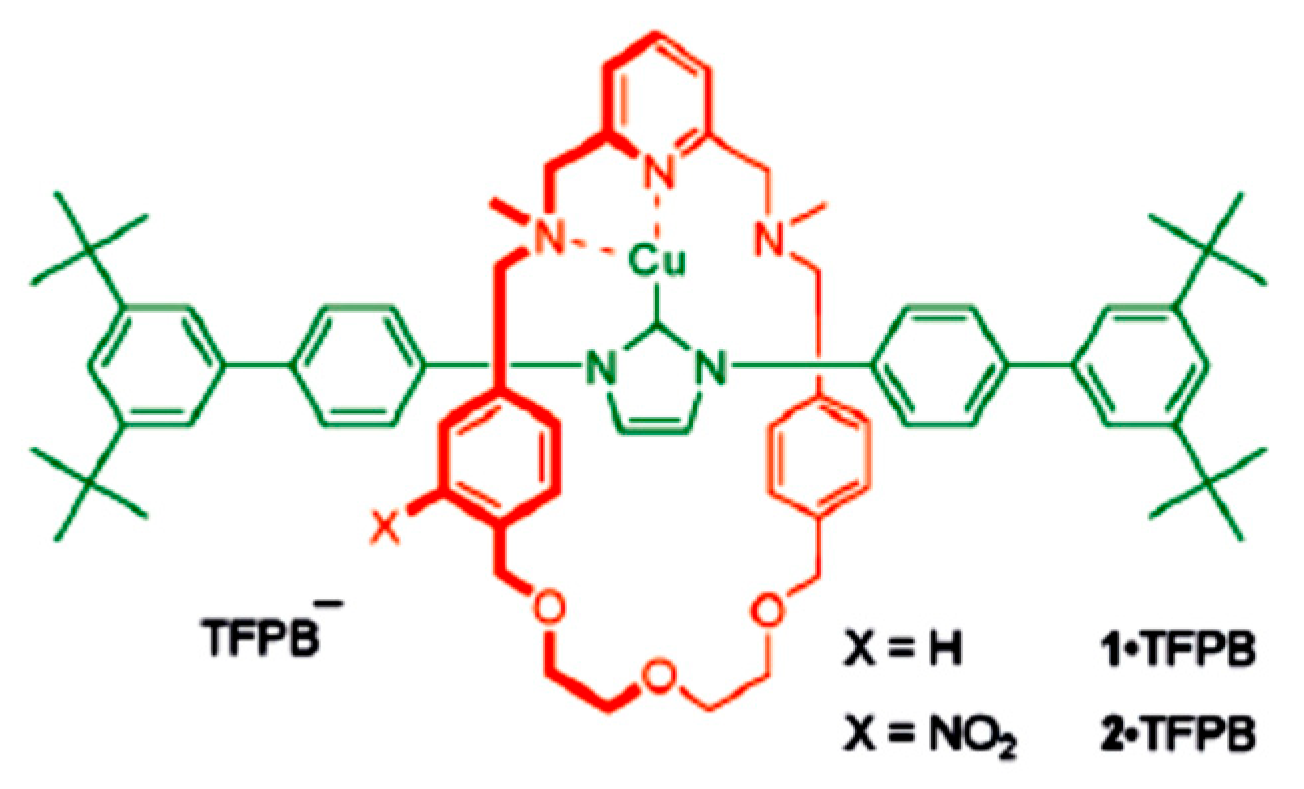
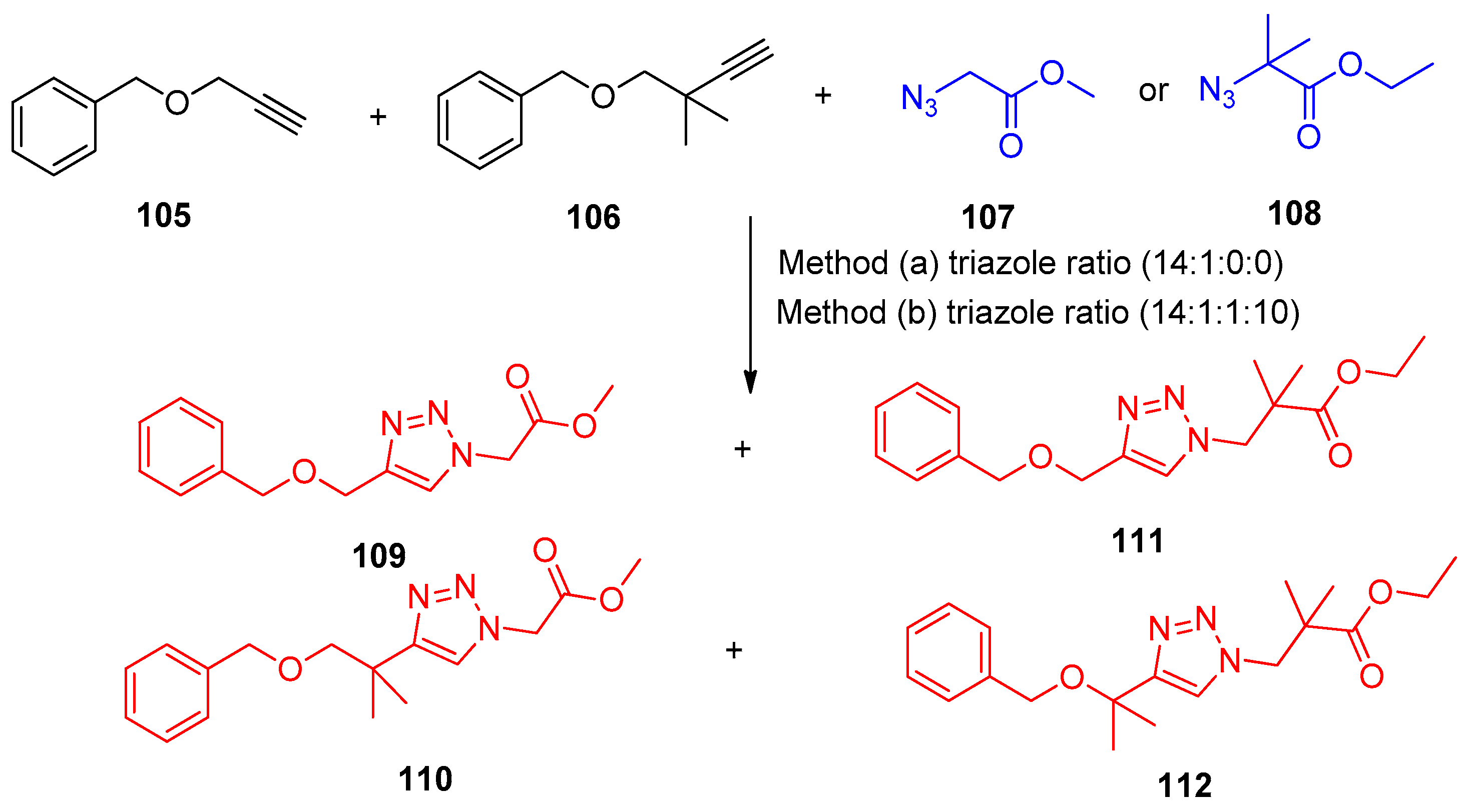
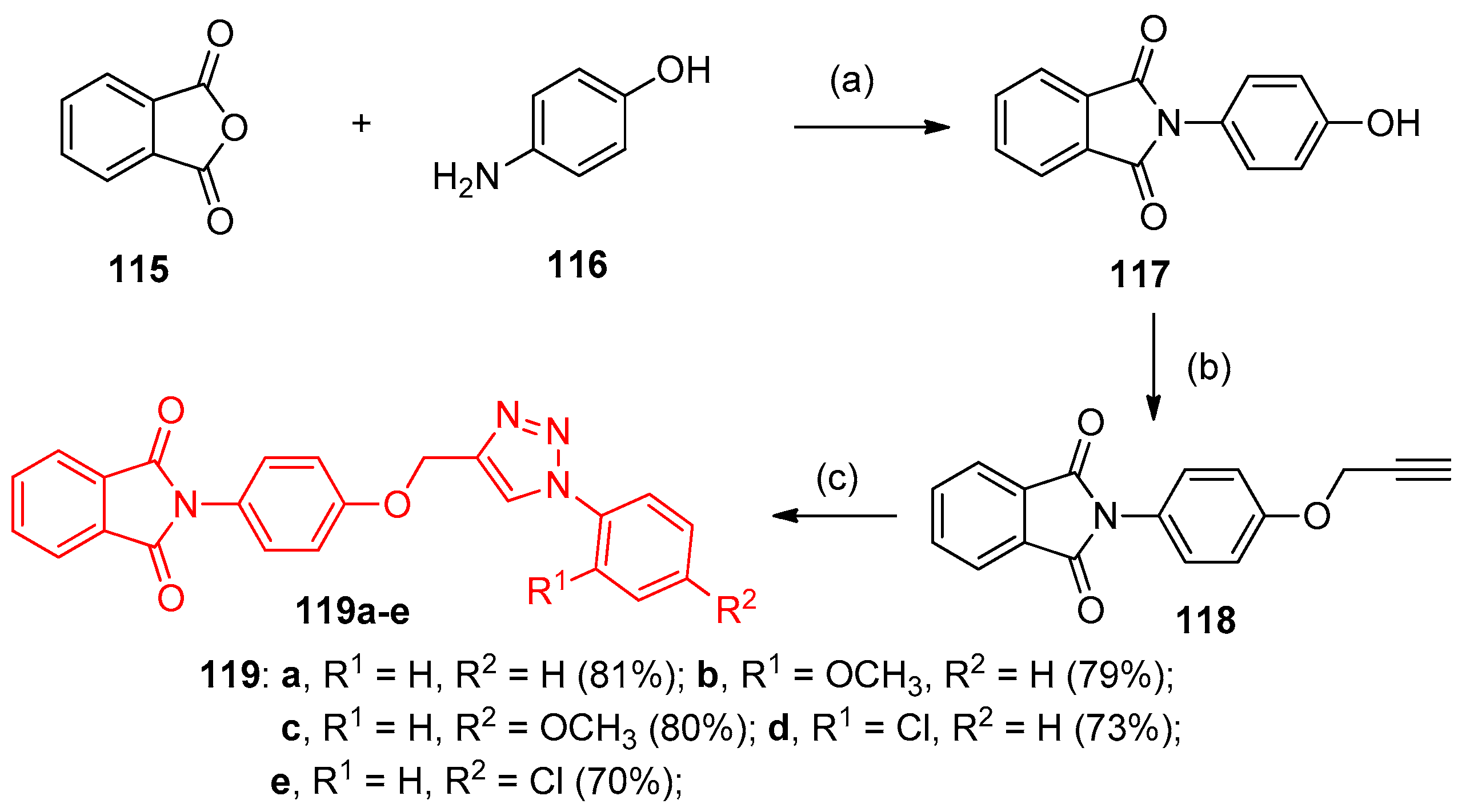
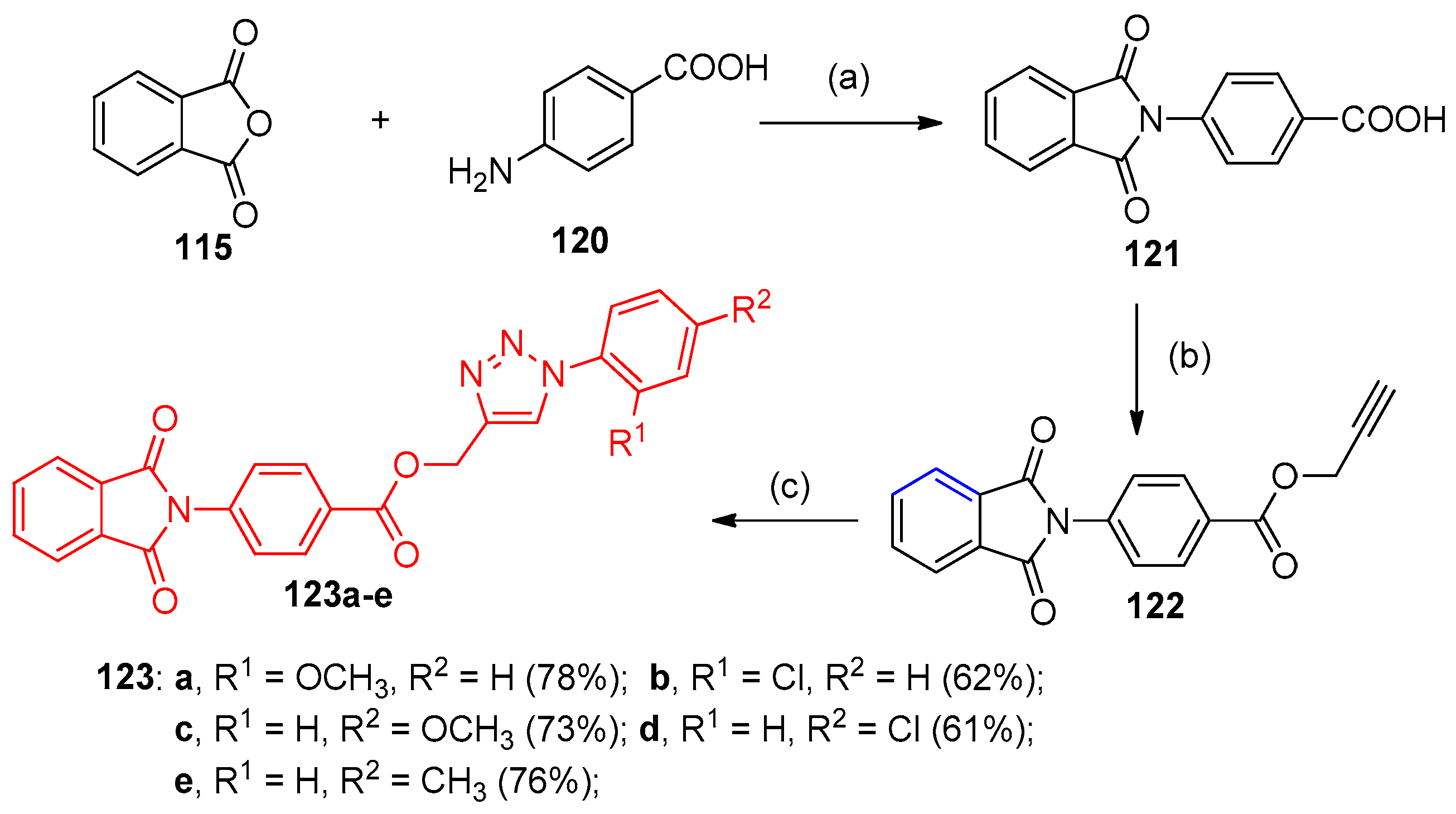
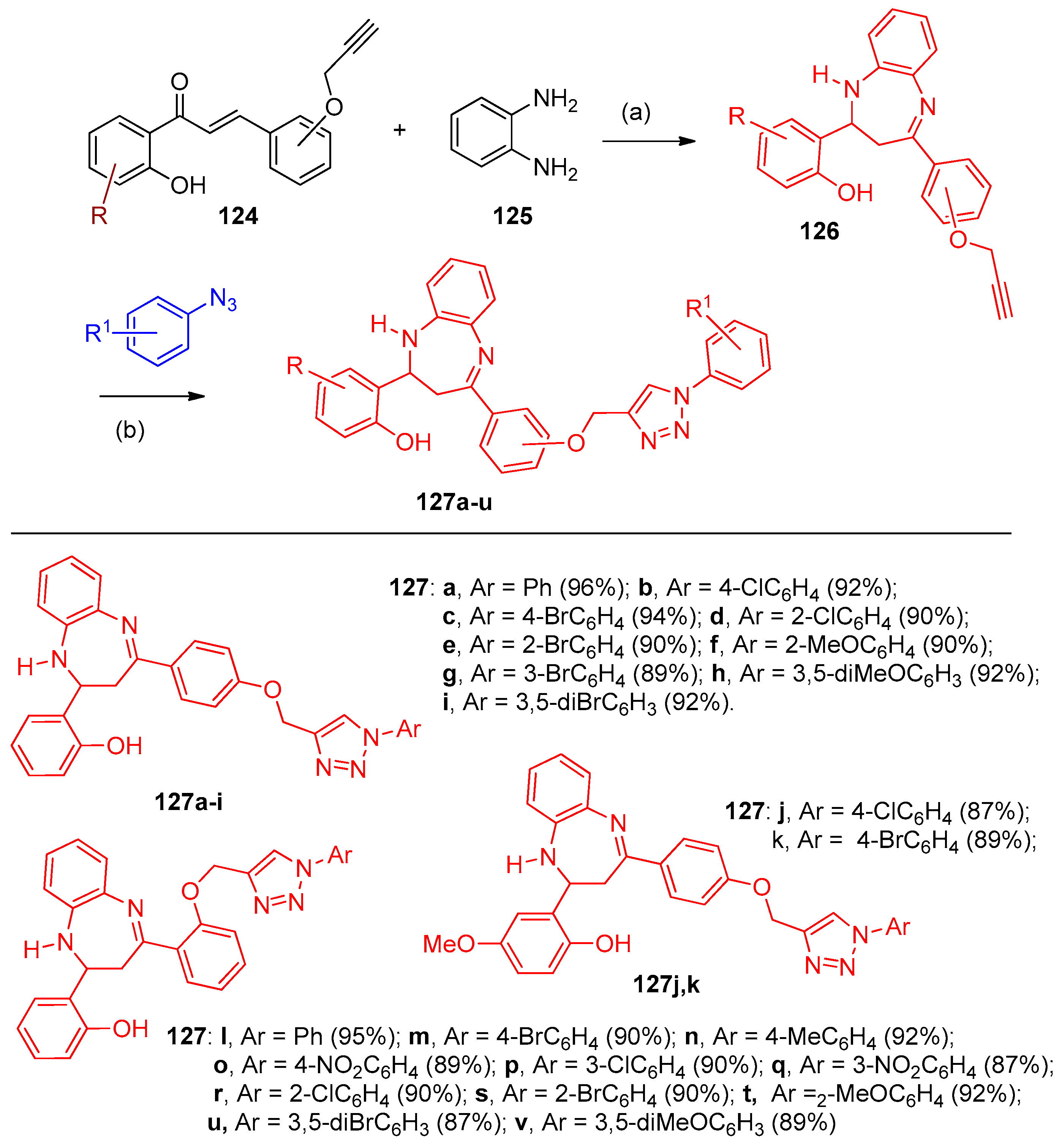
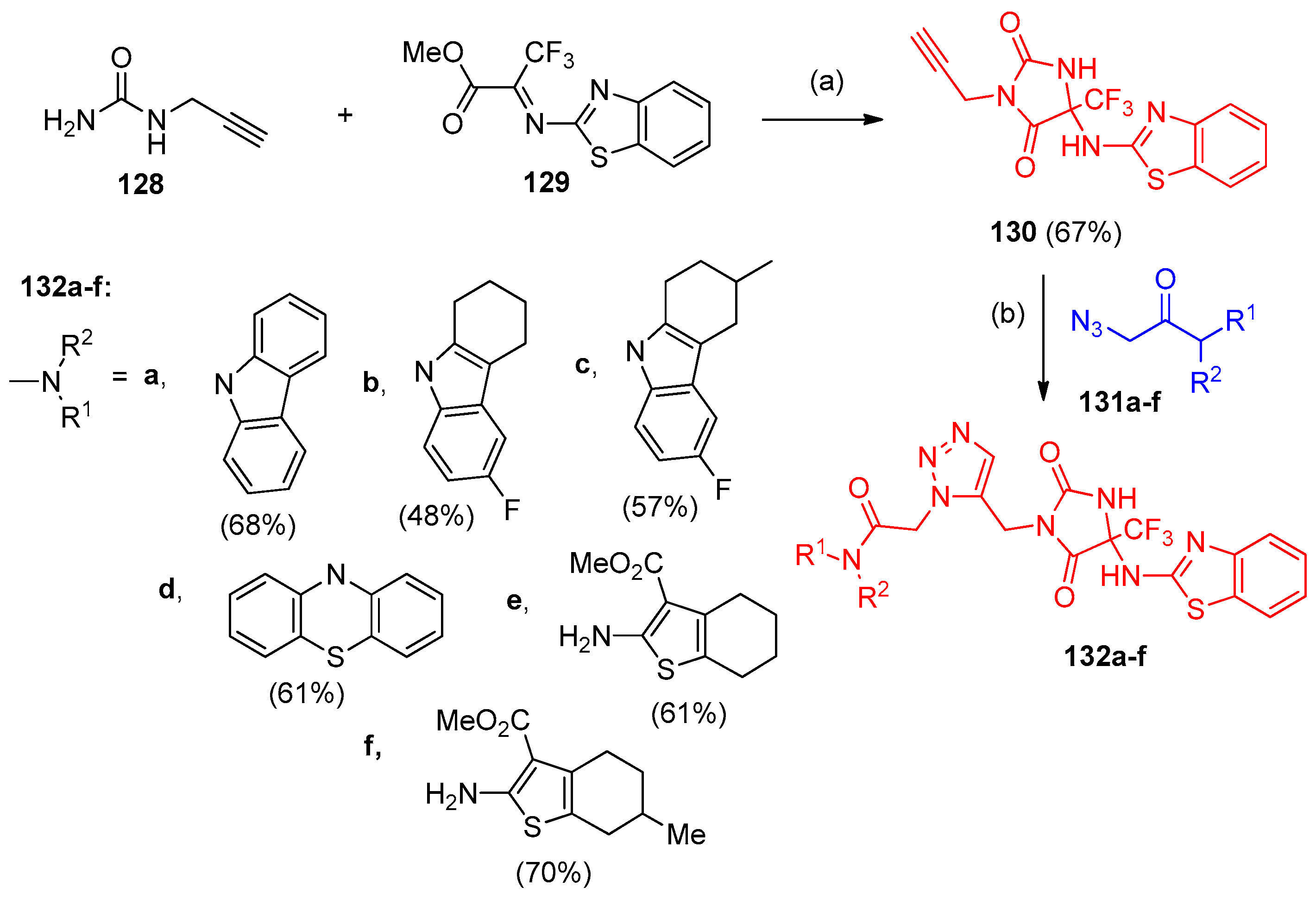
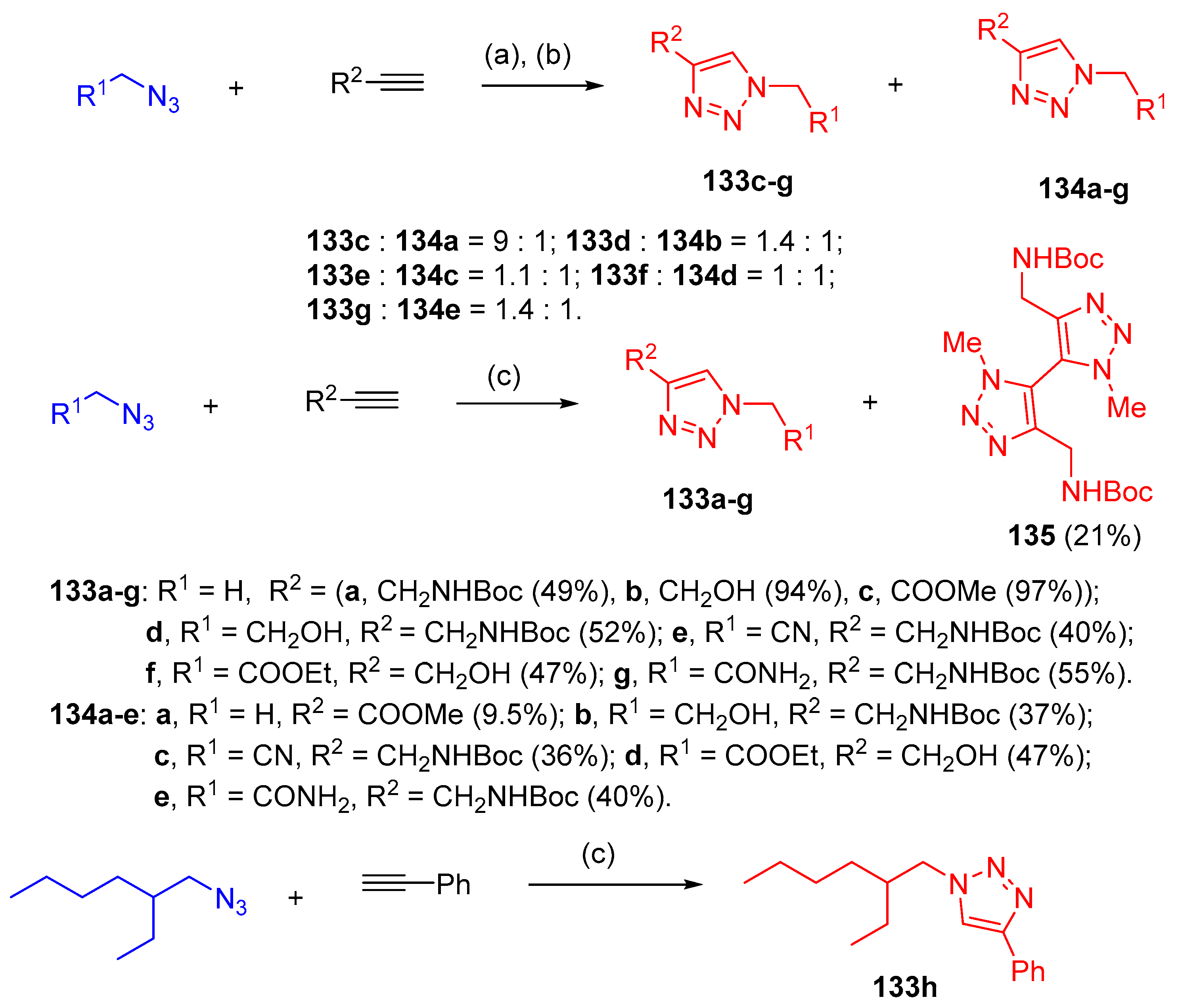
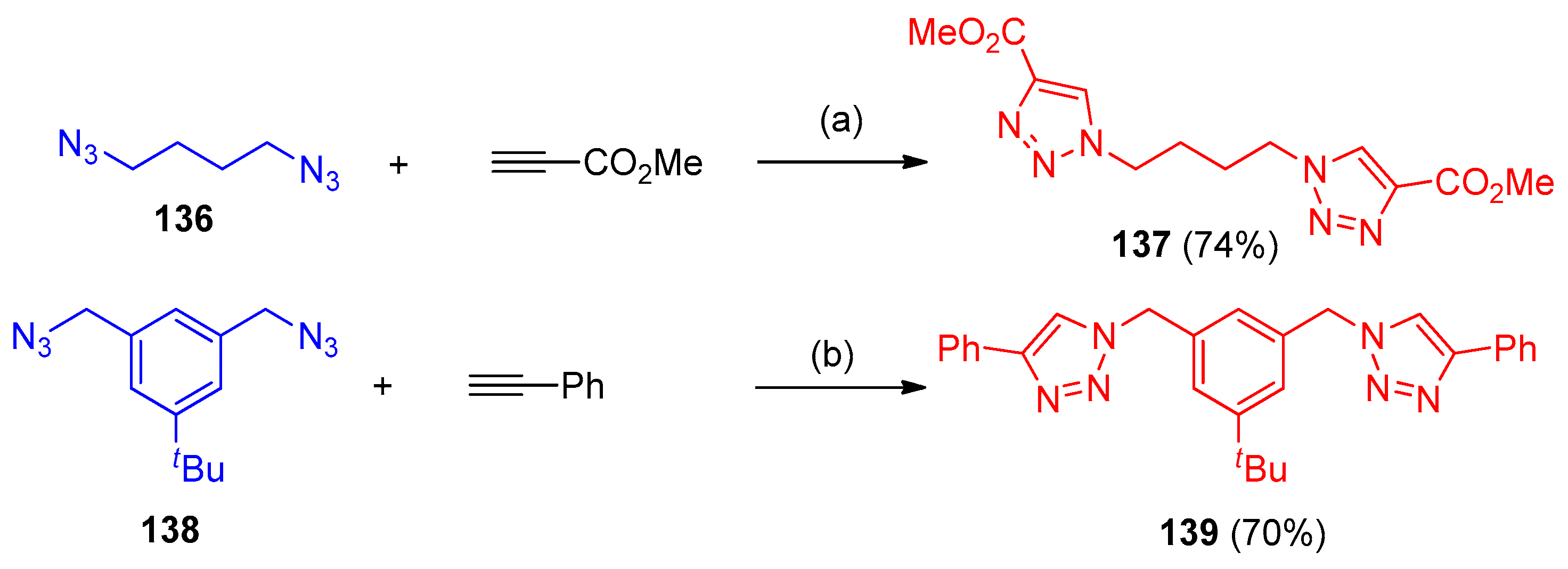
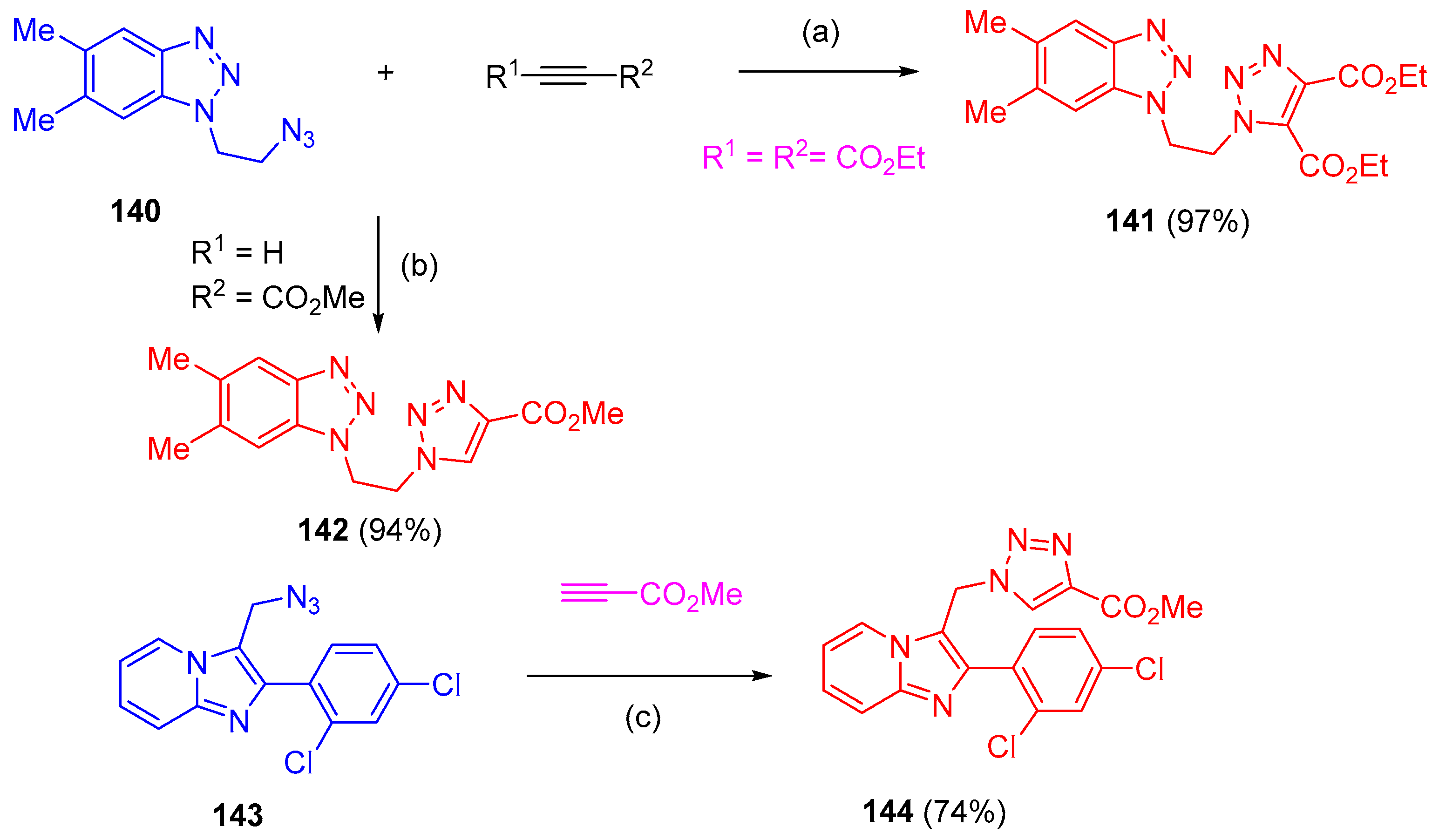
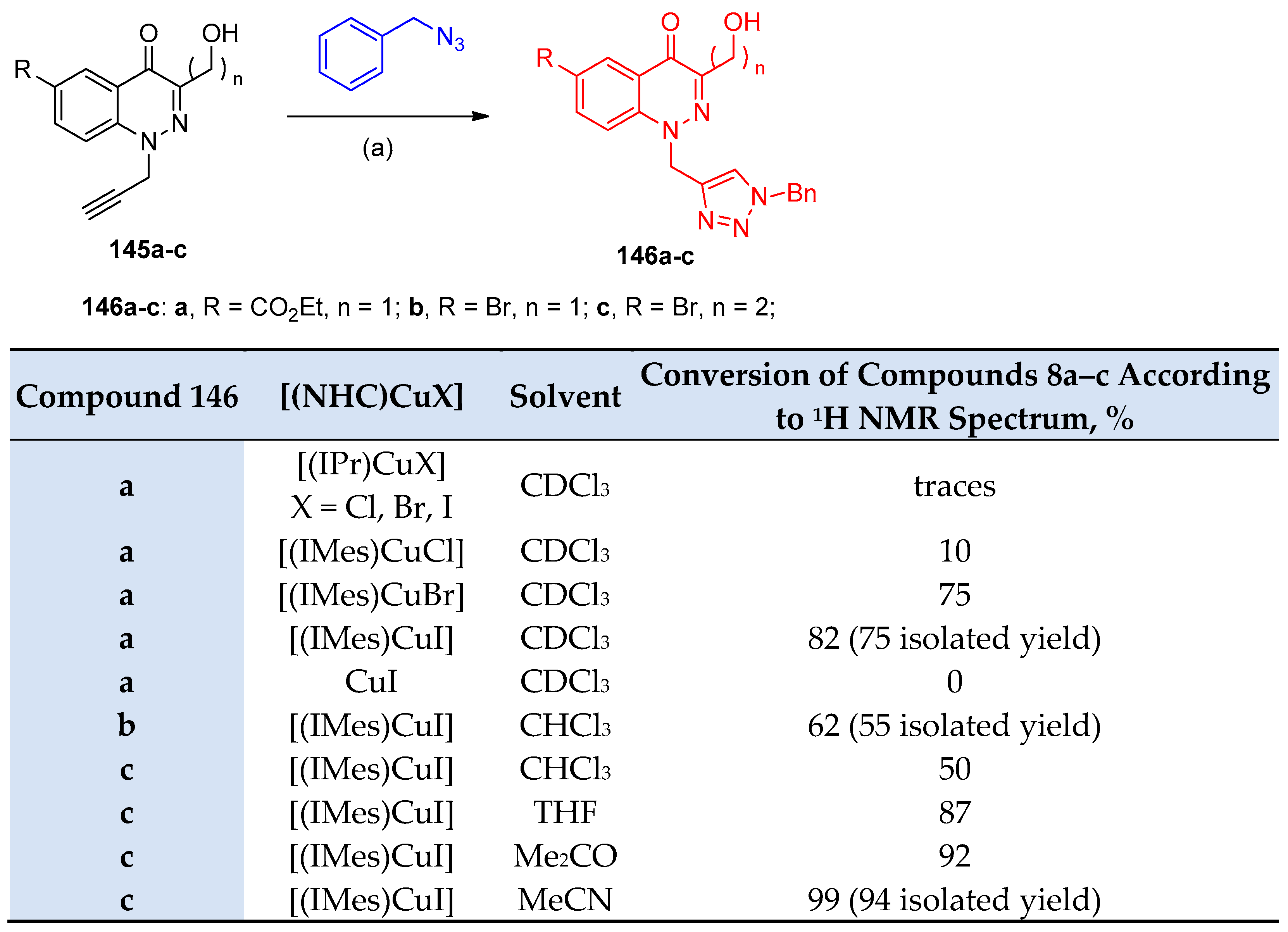
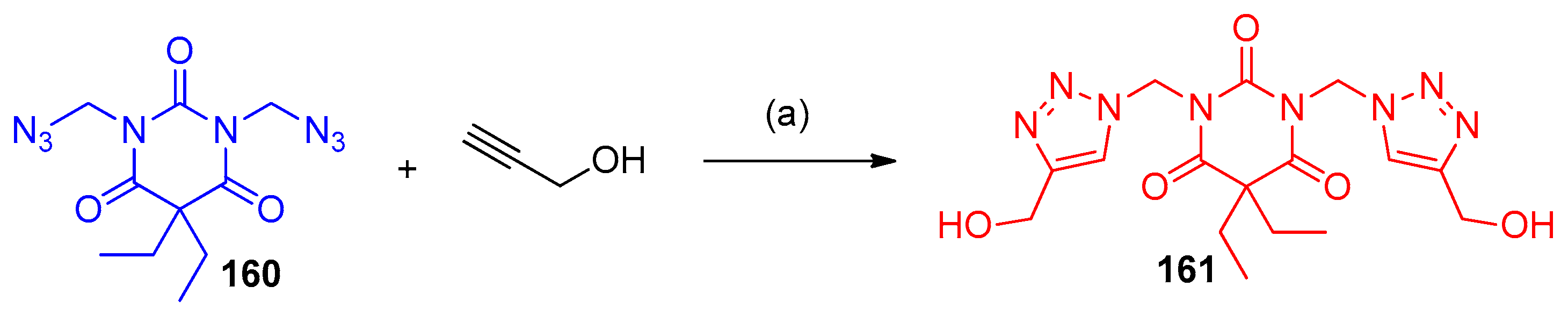
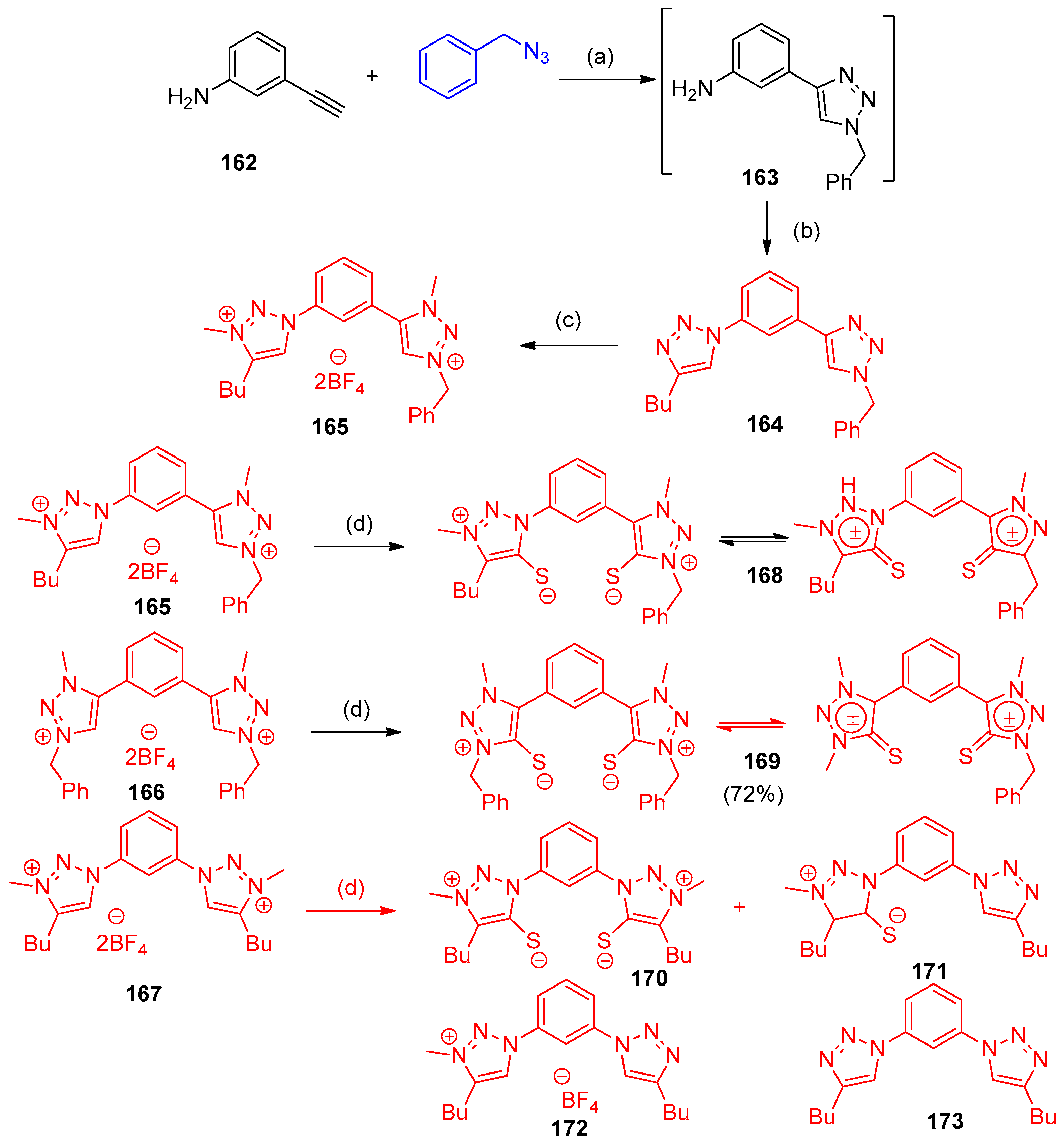
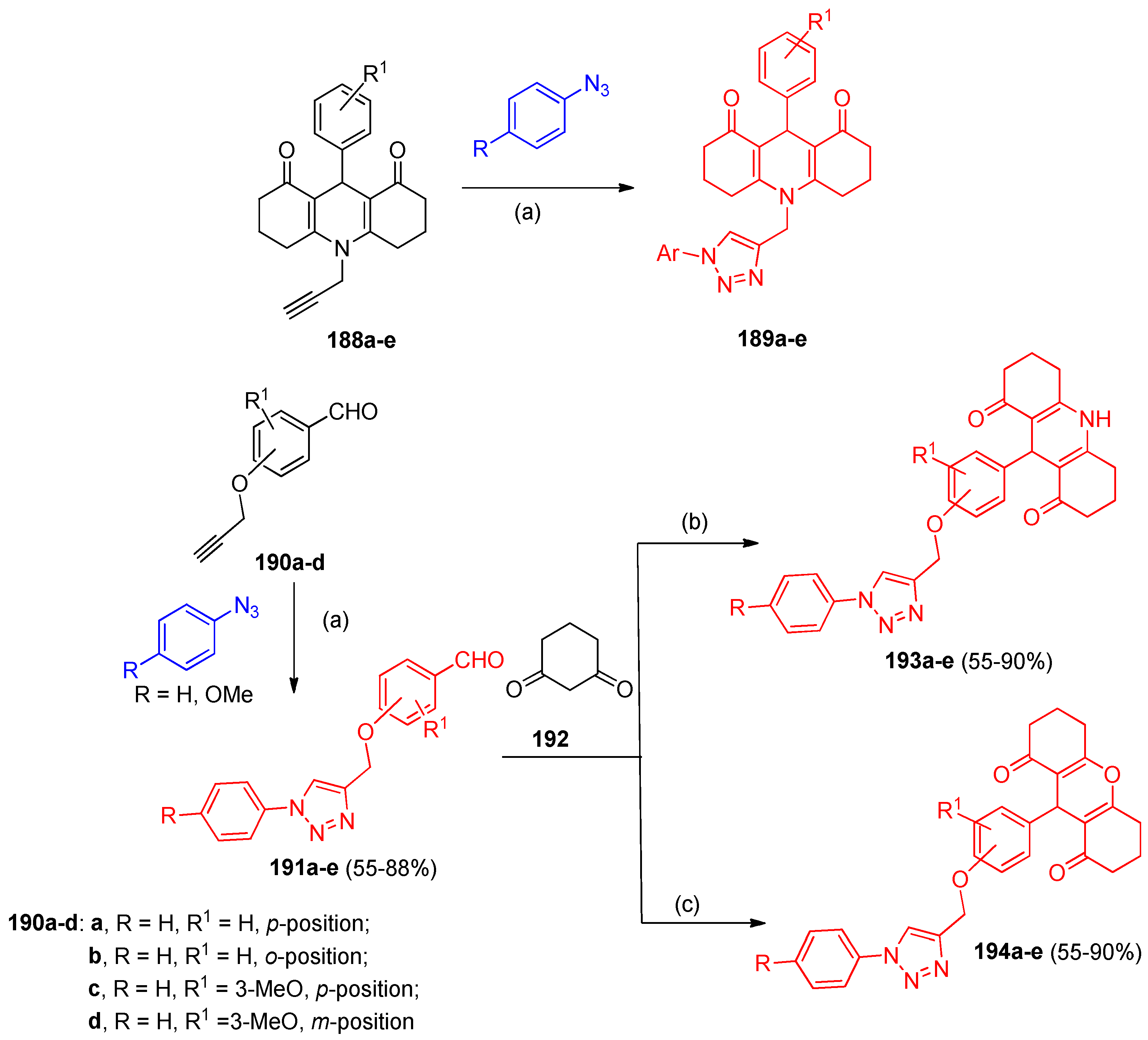
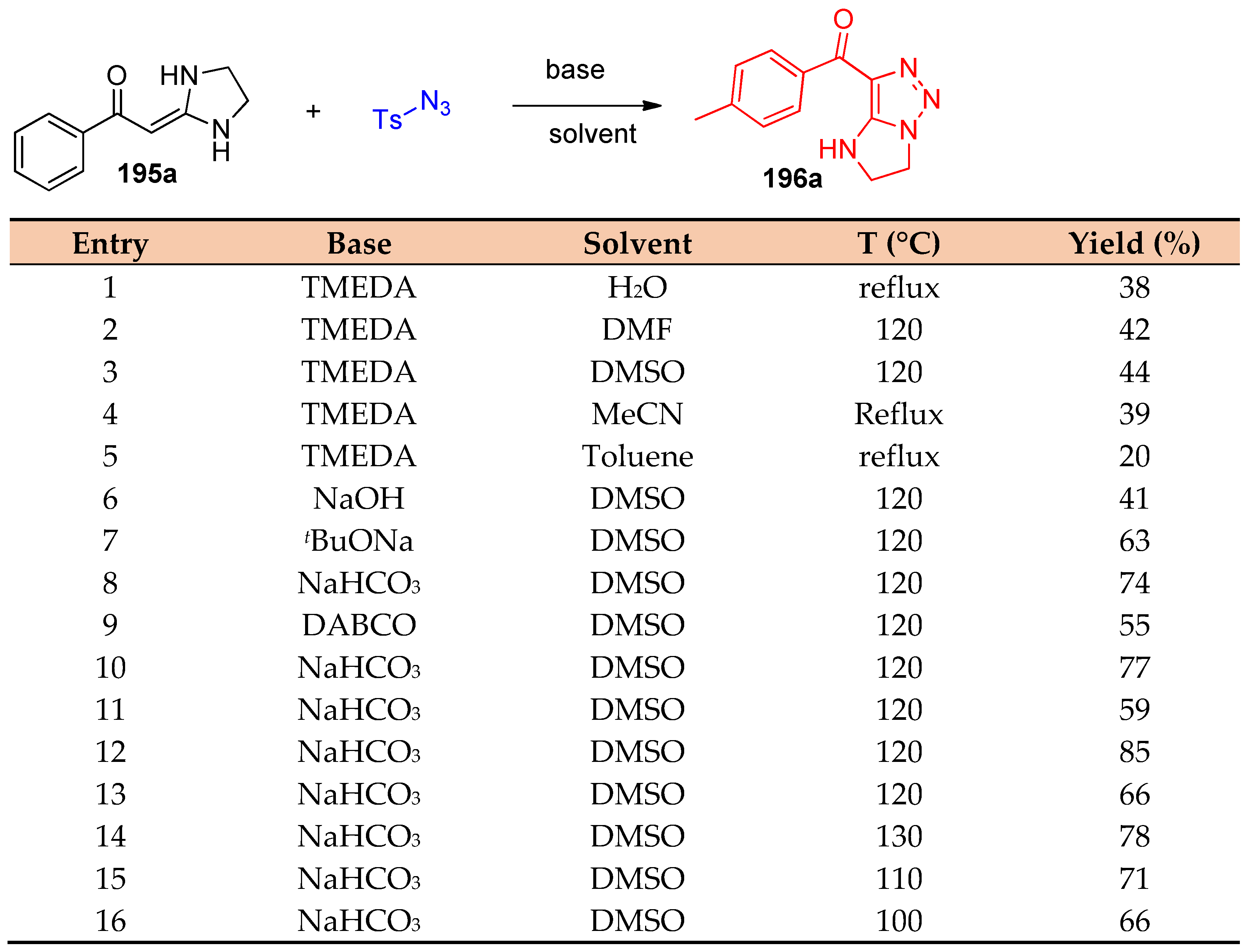
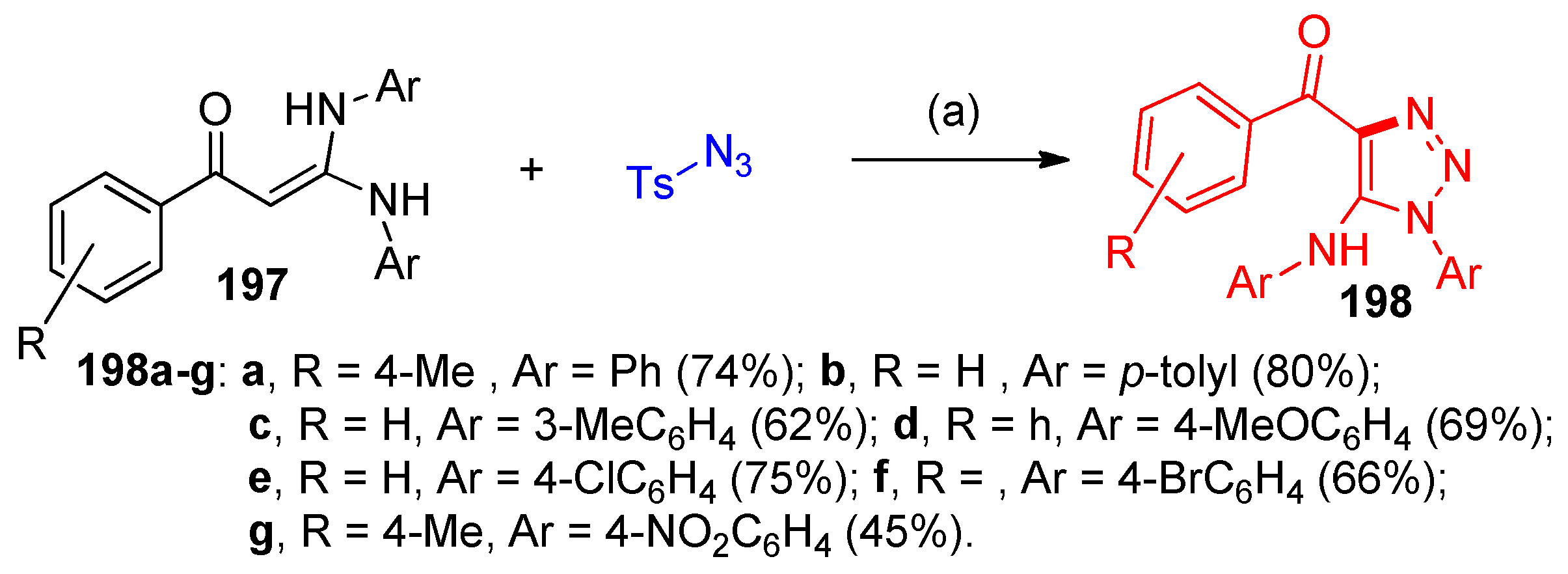
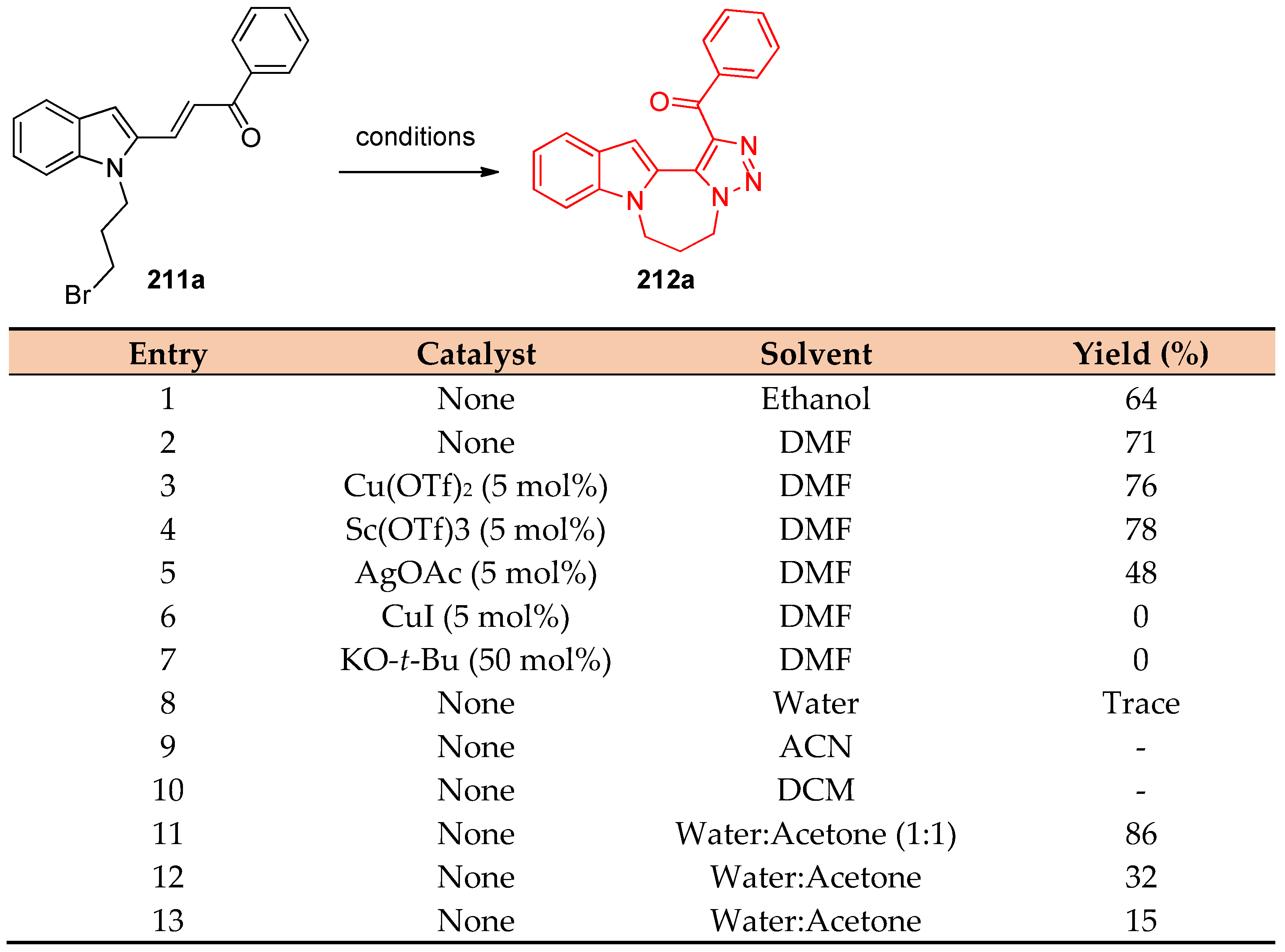
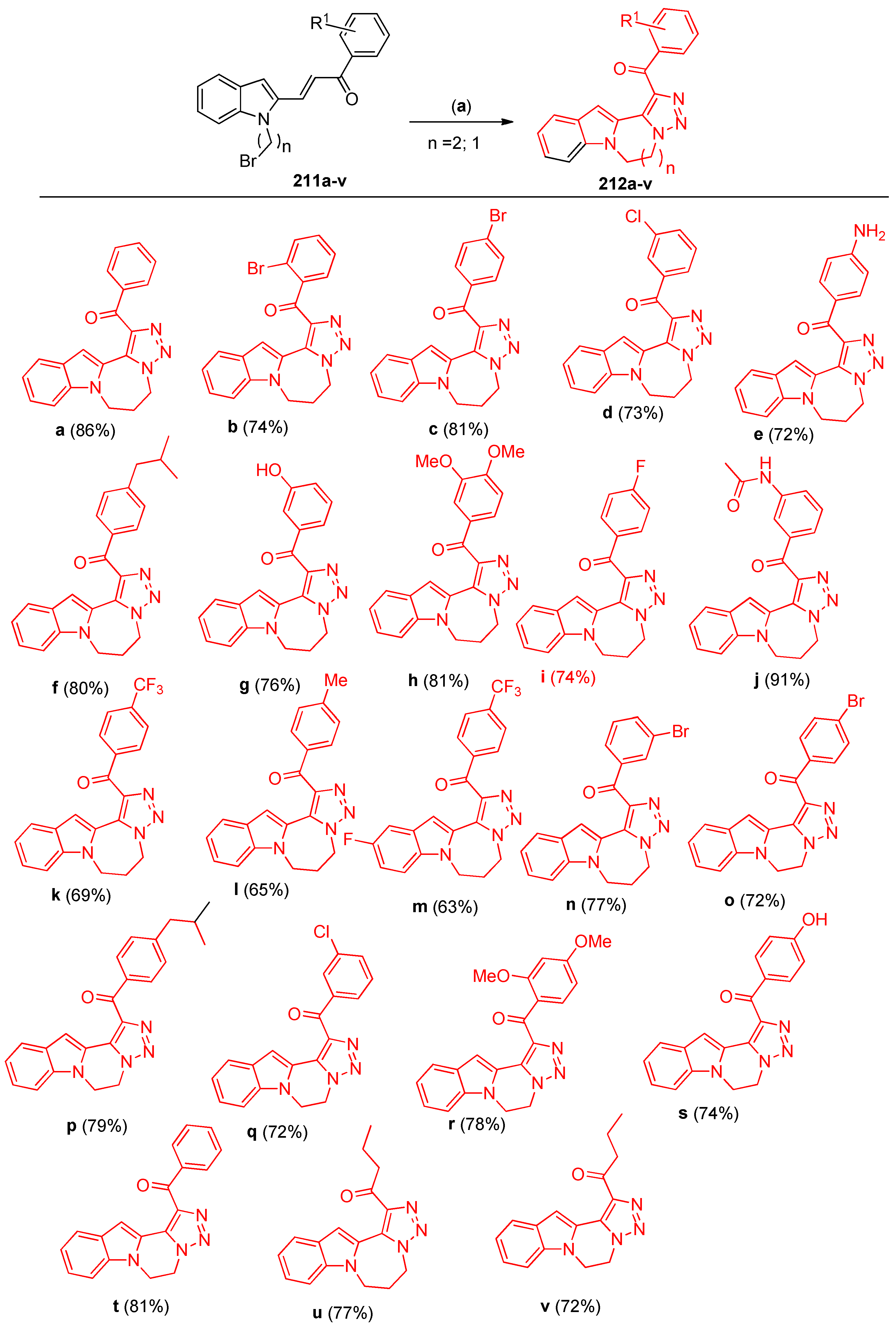
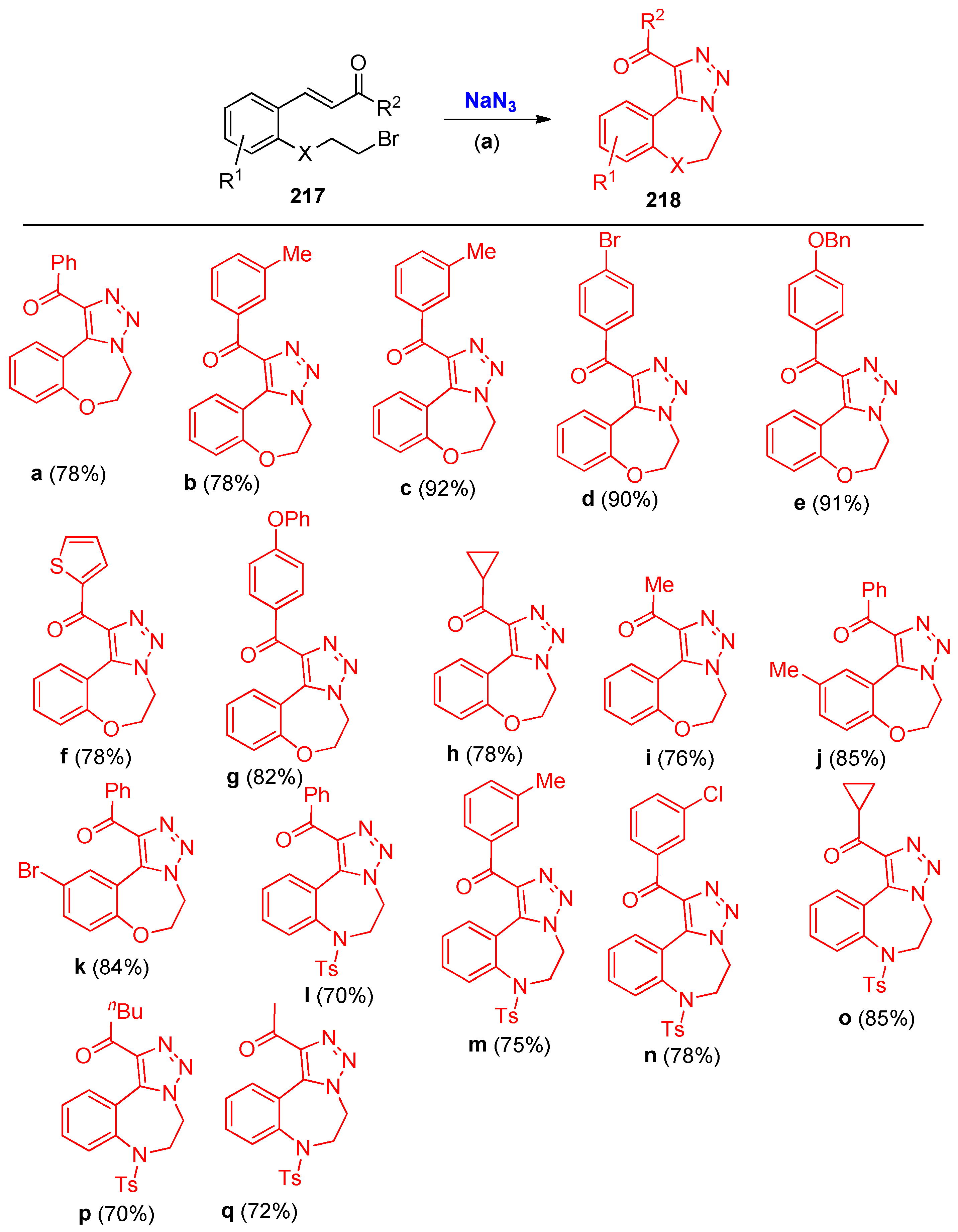
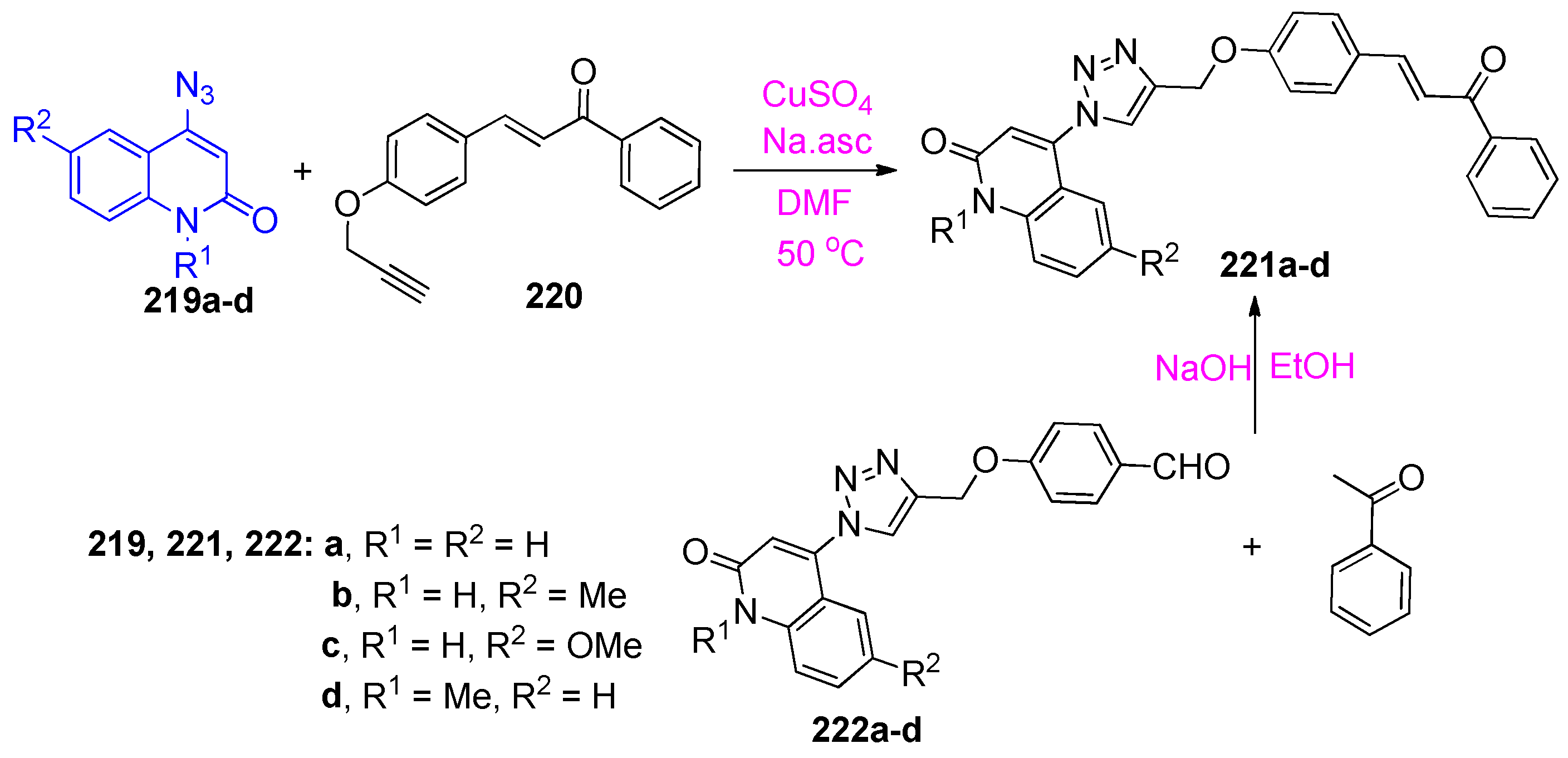
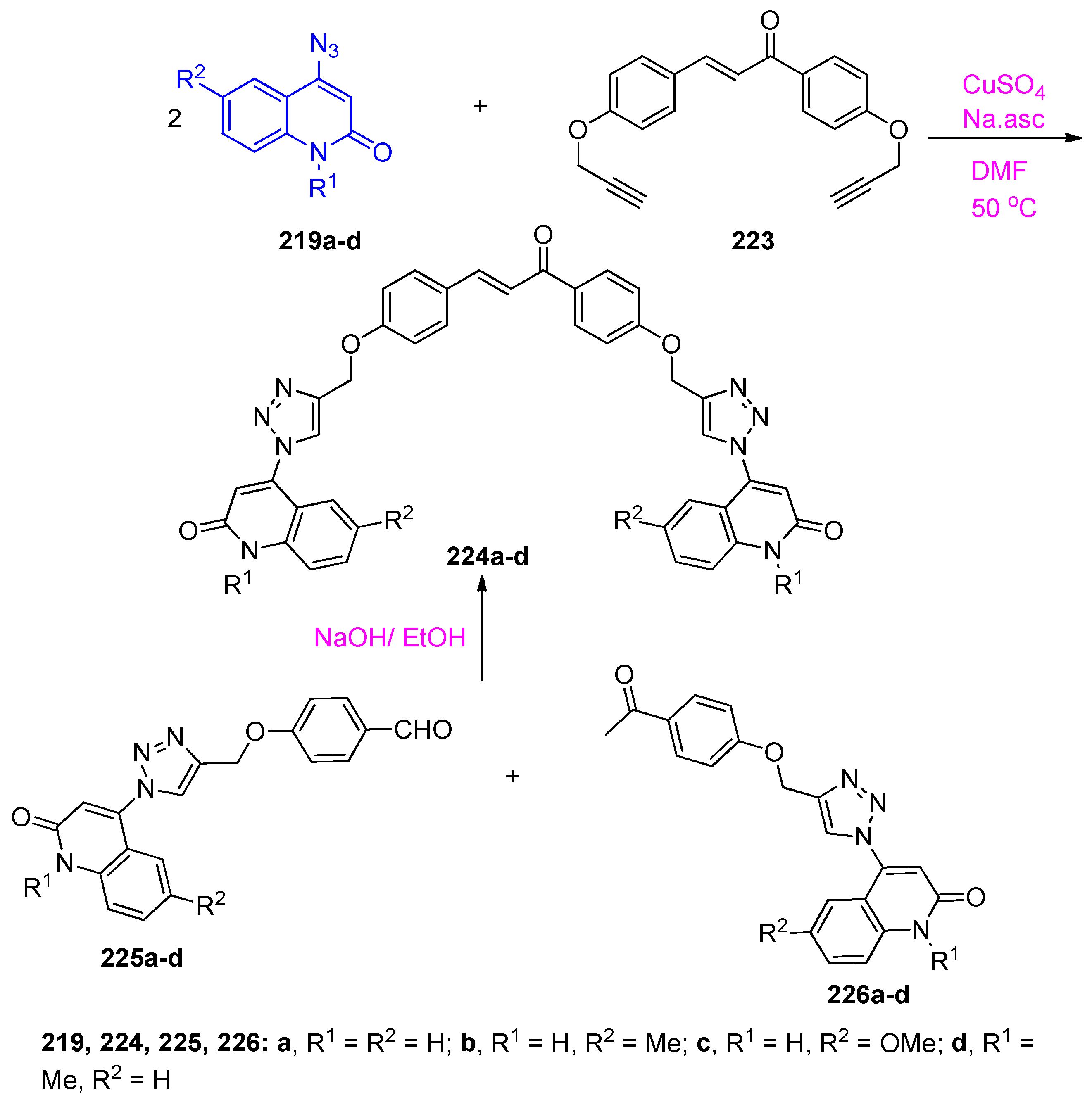
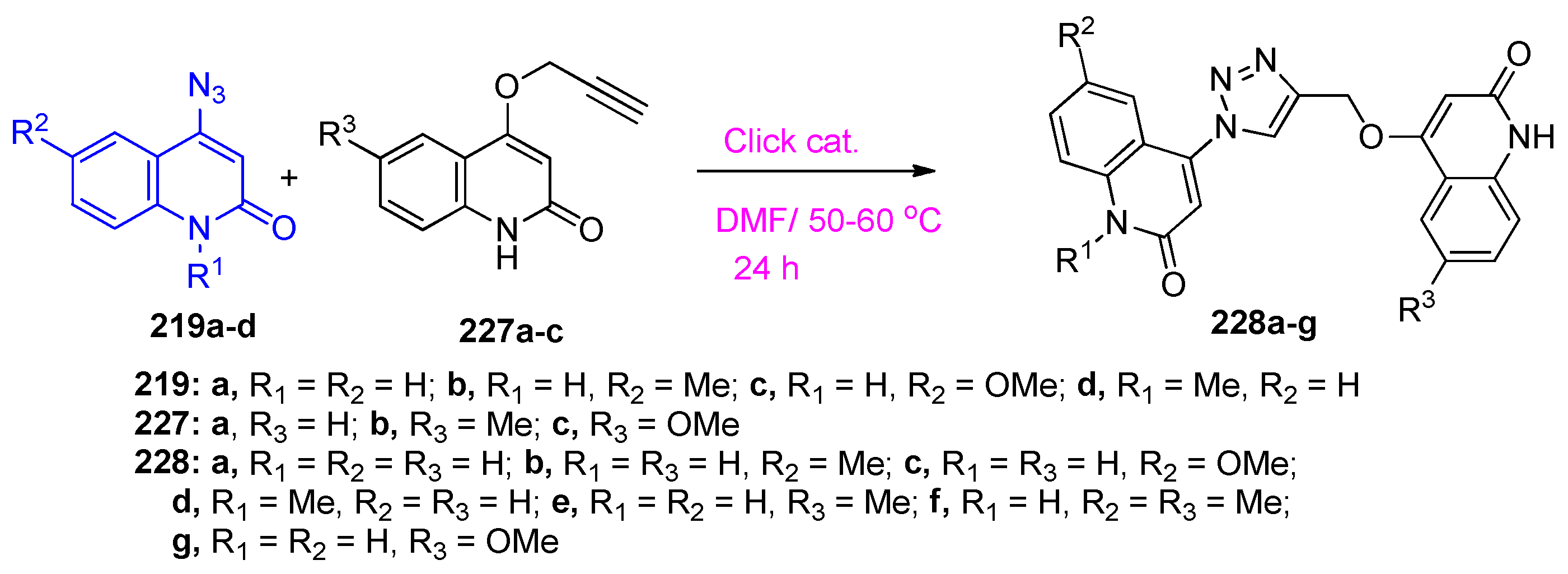
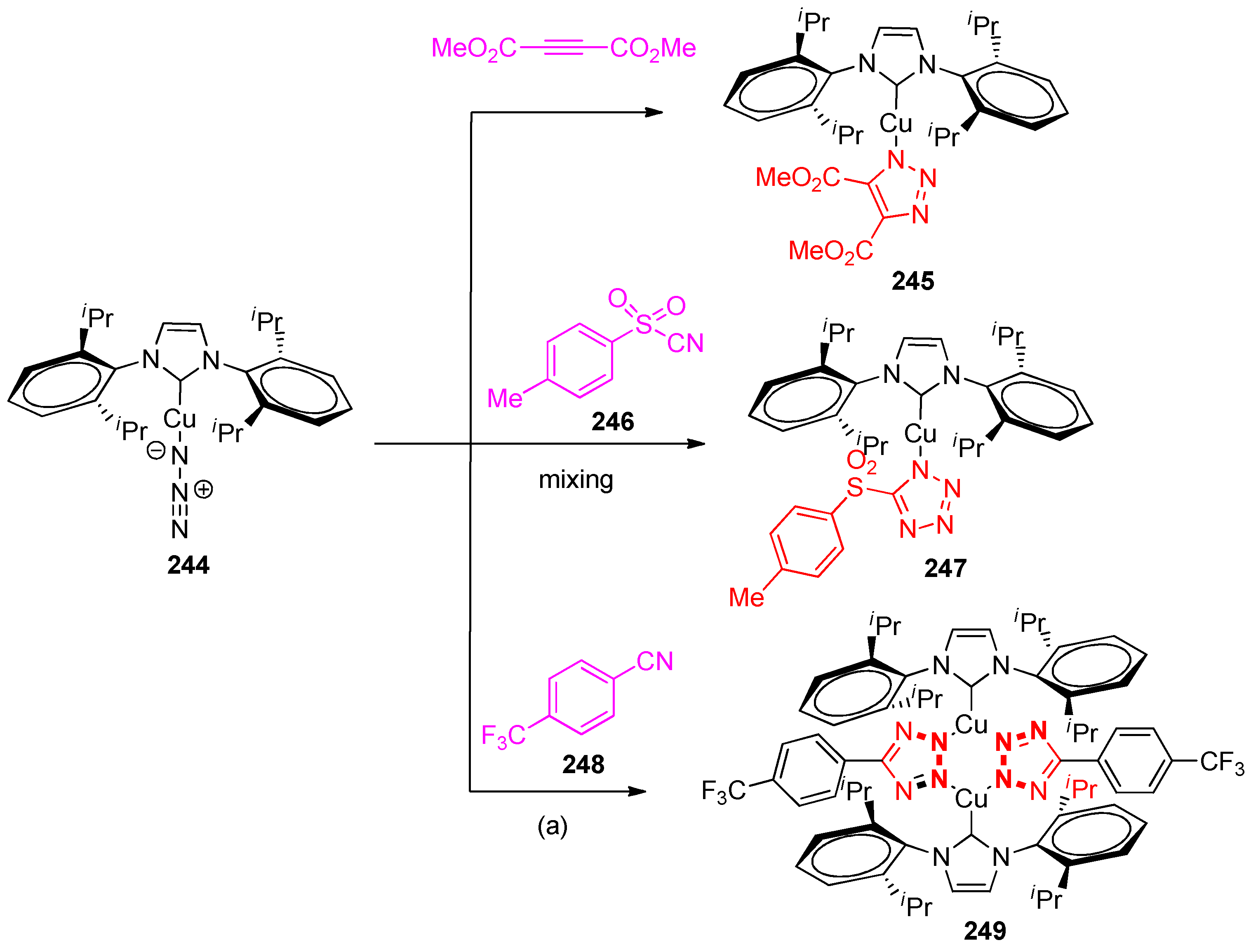
4.5. Synthesis of the Triazole Ring
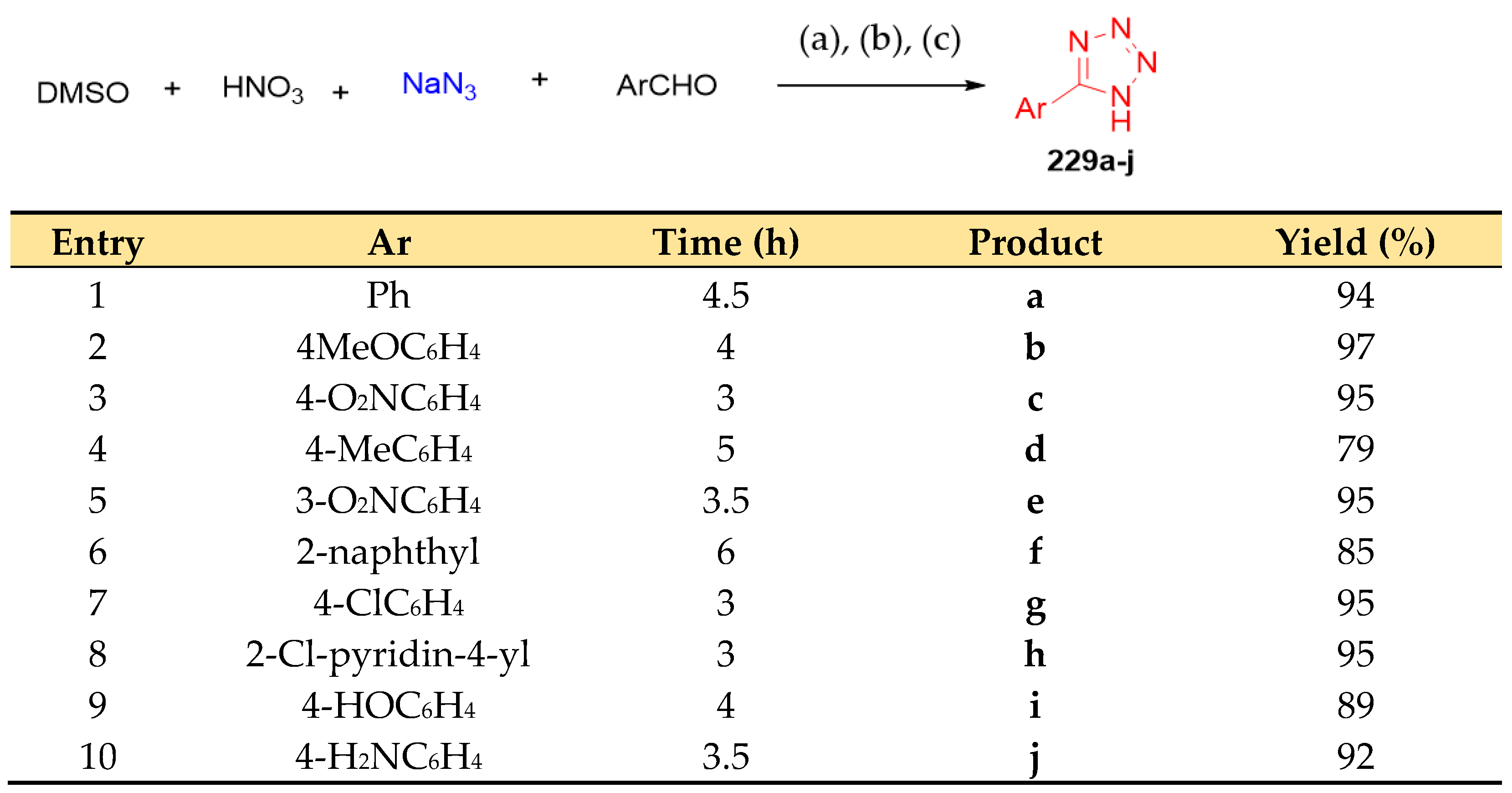
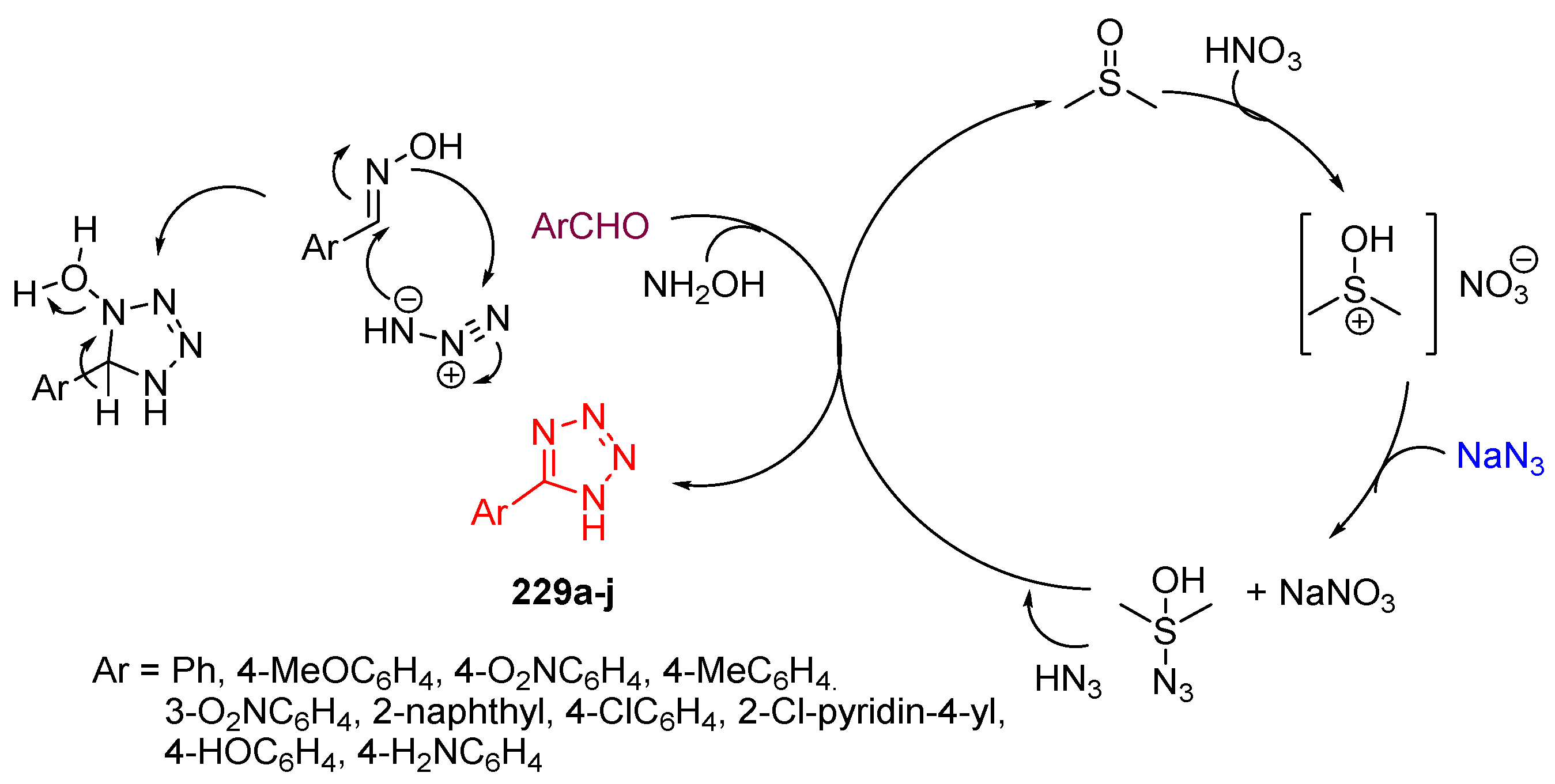
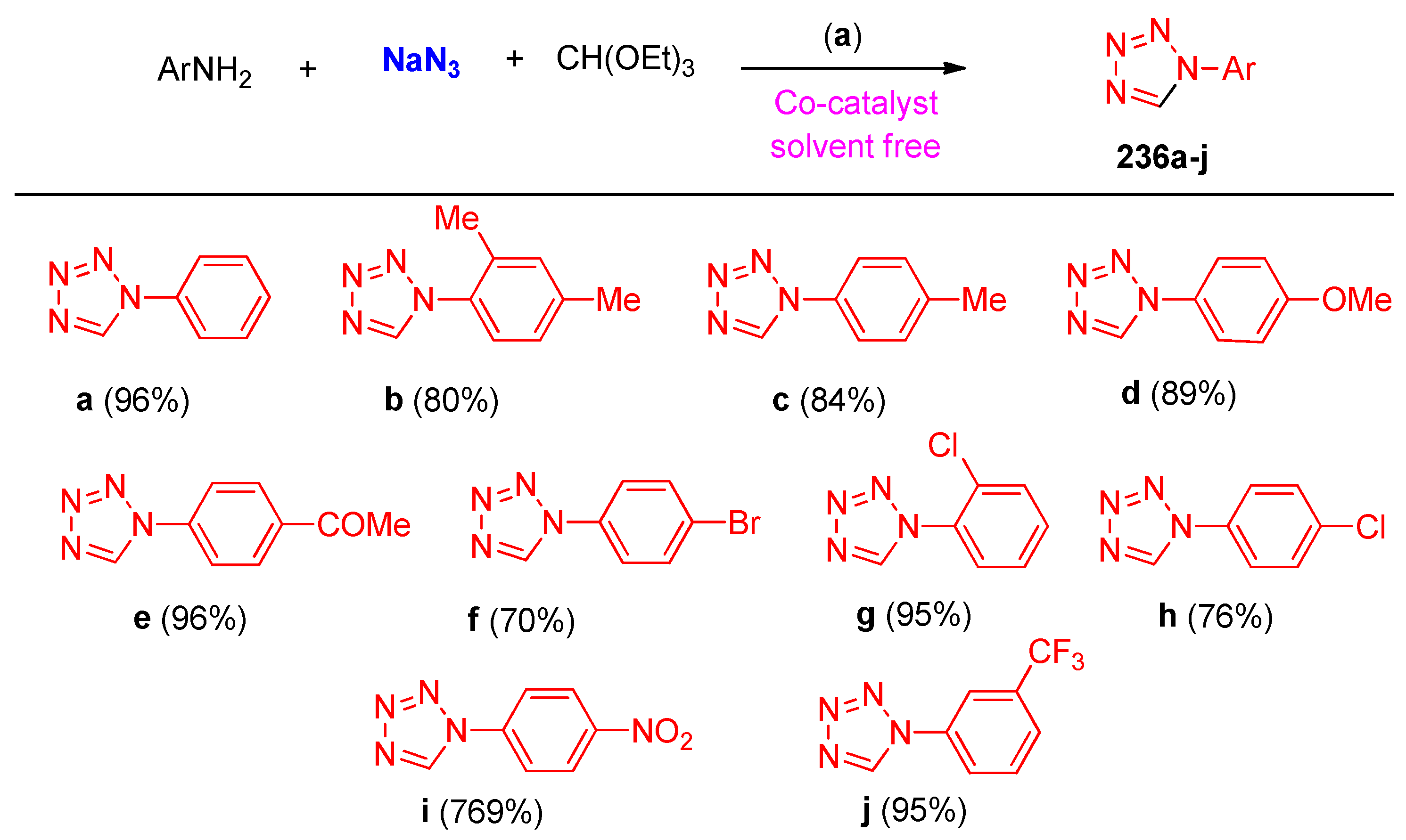
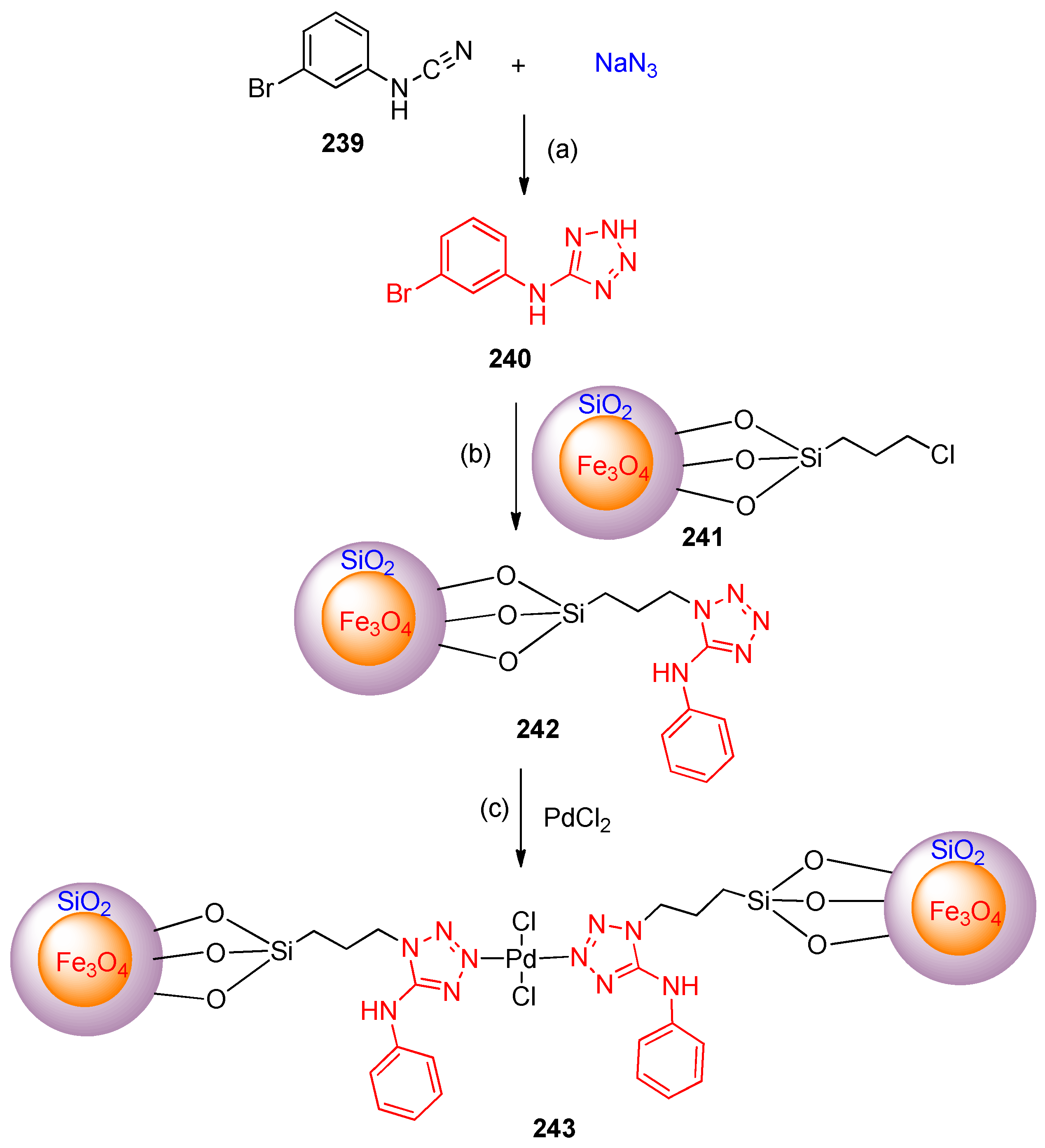
4.6. Synthesis of Tetrazole Ring
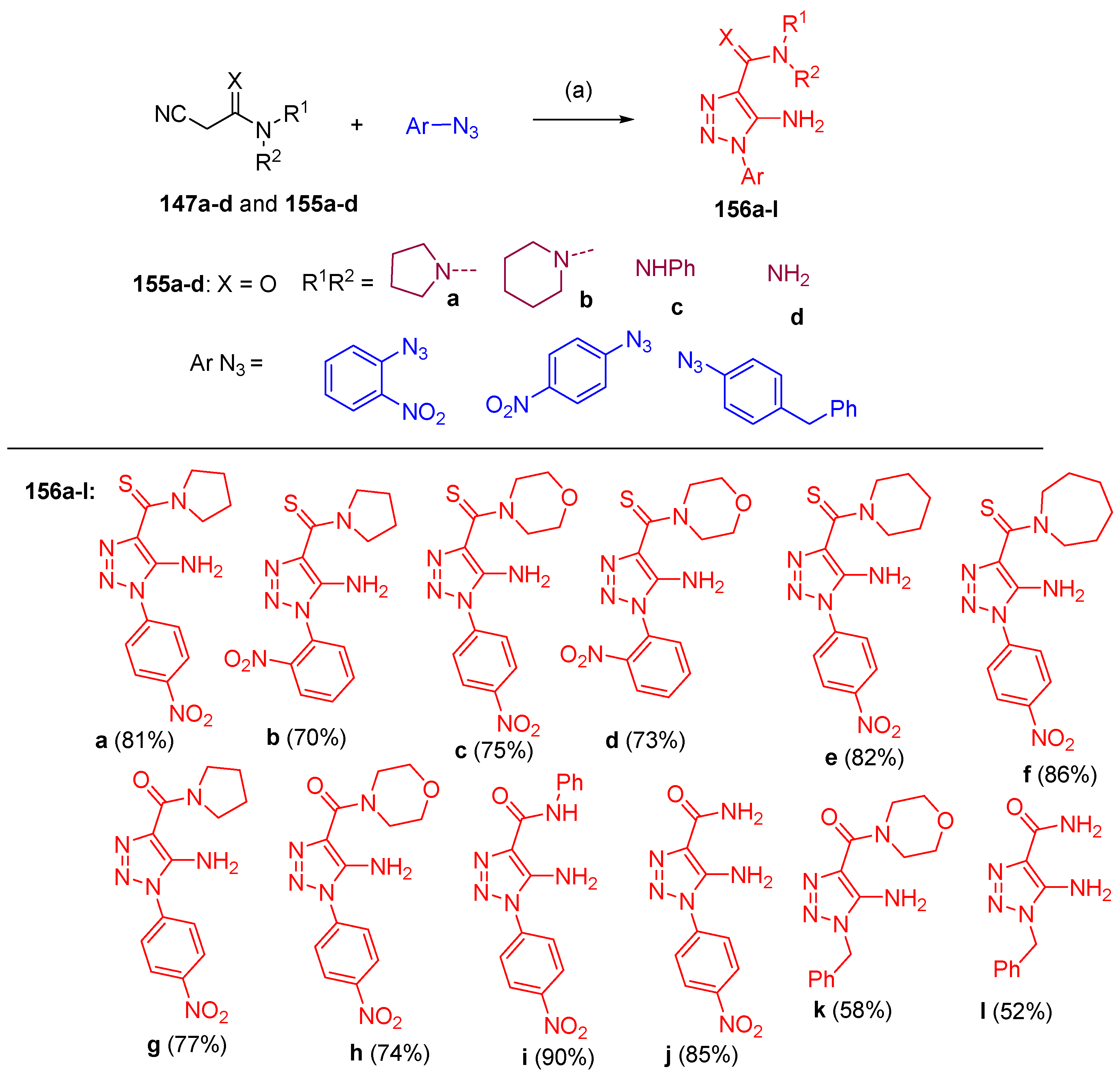
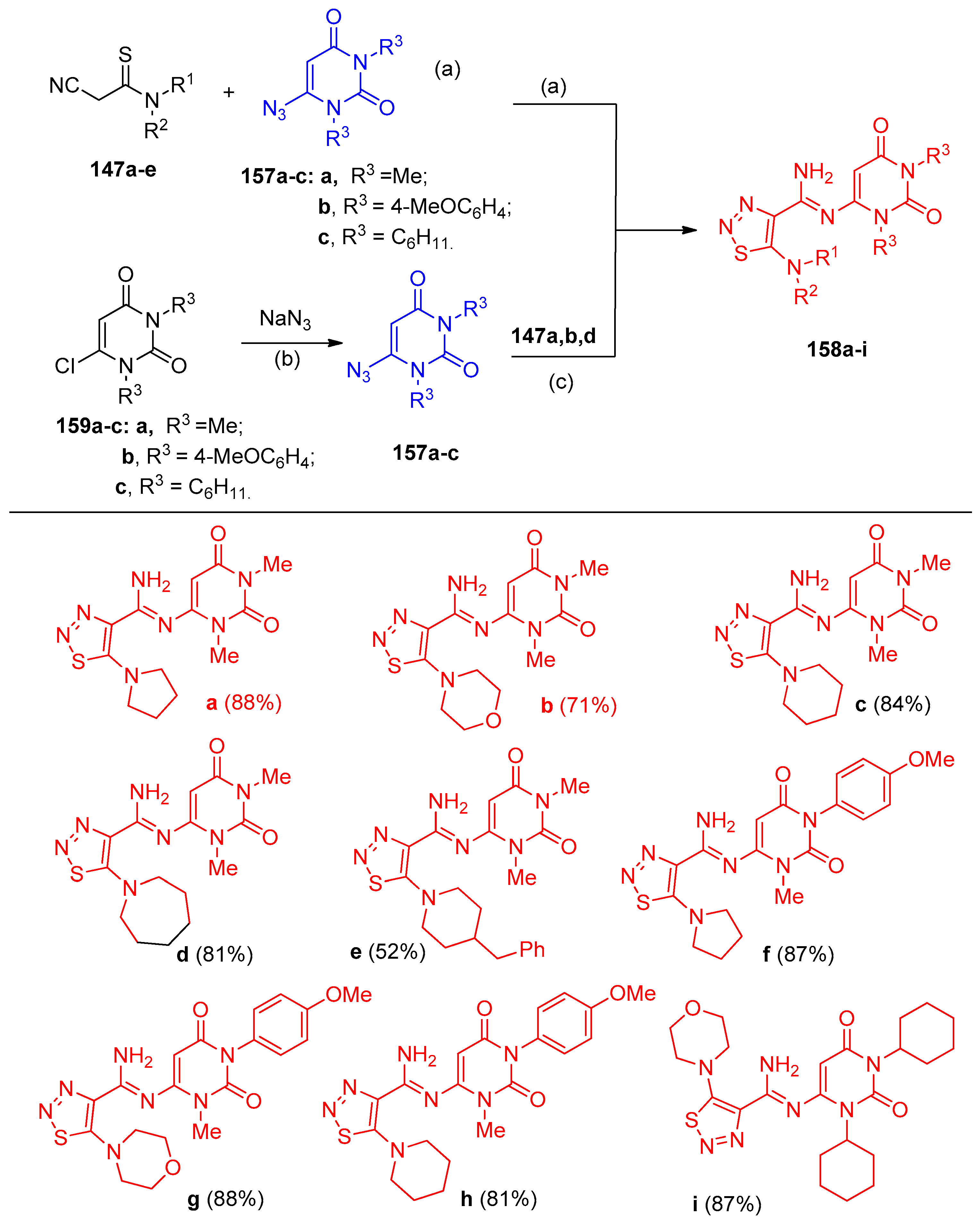
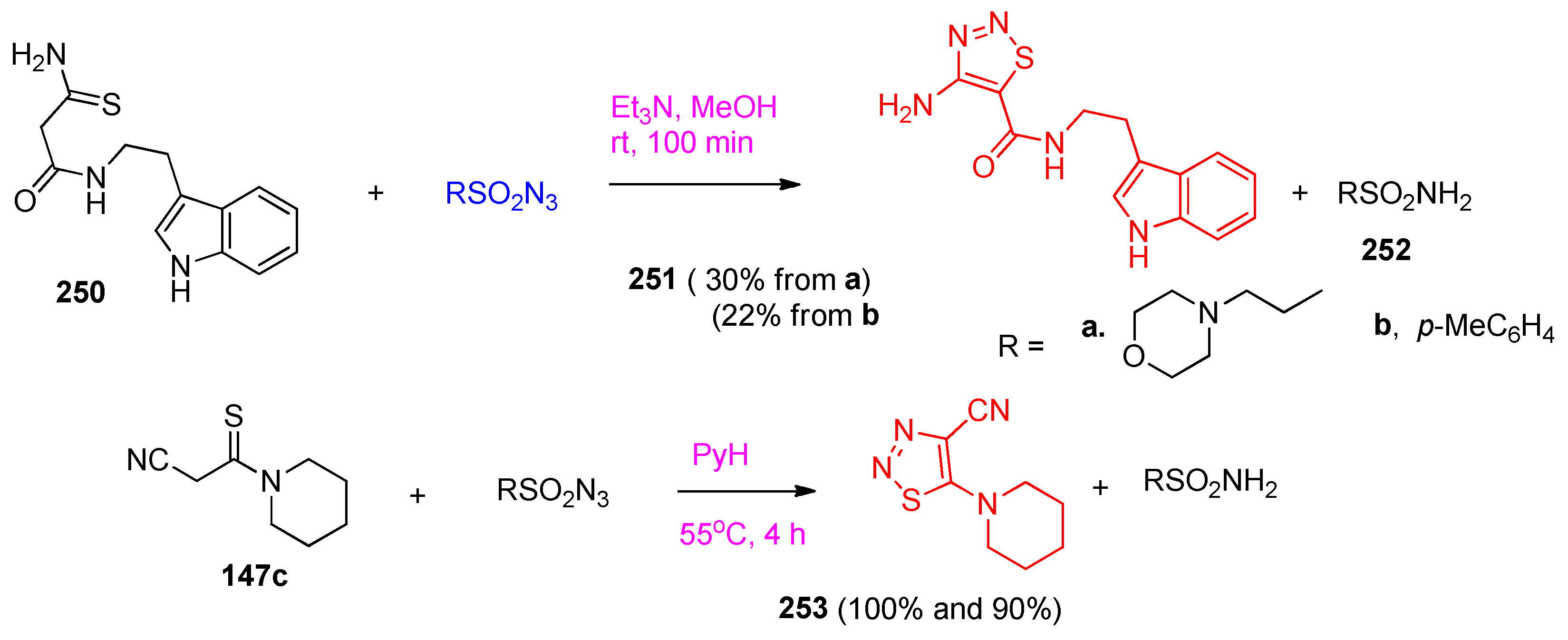
4.7. Synthesis of Thiadiazole
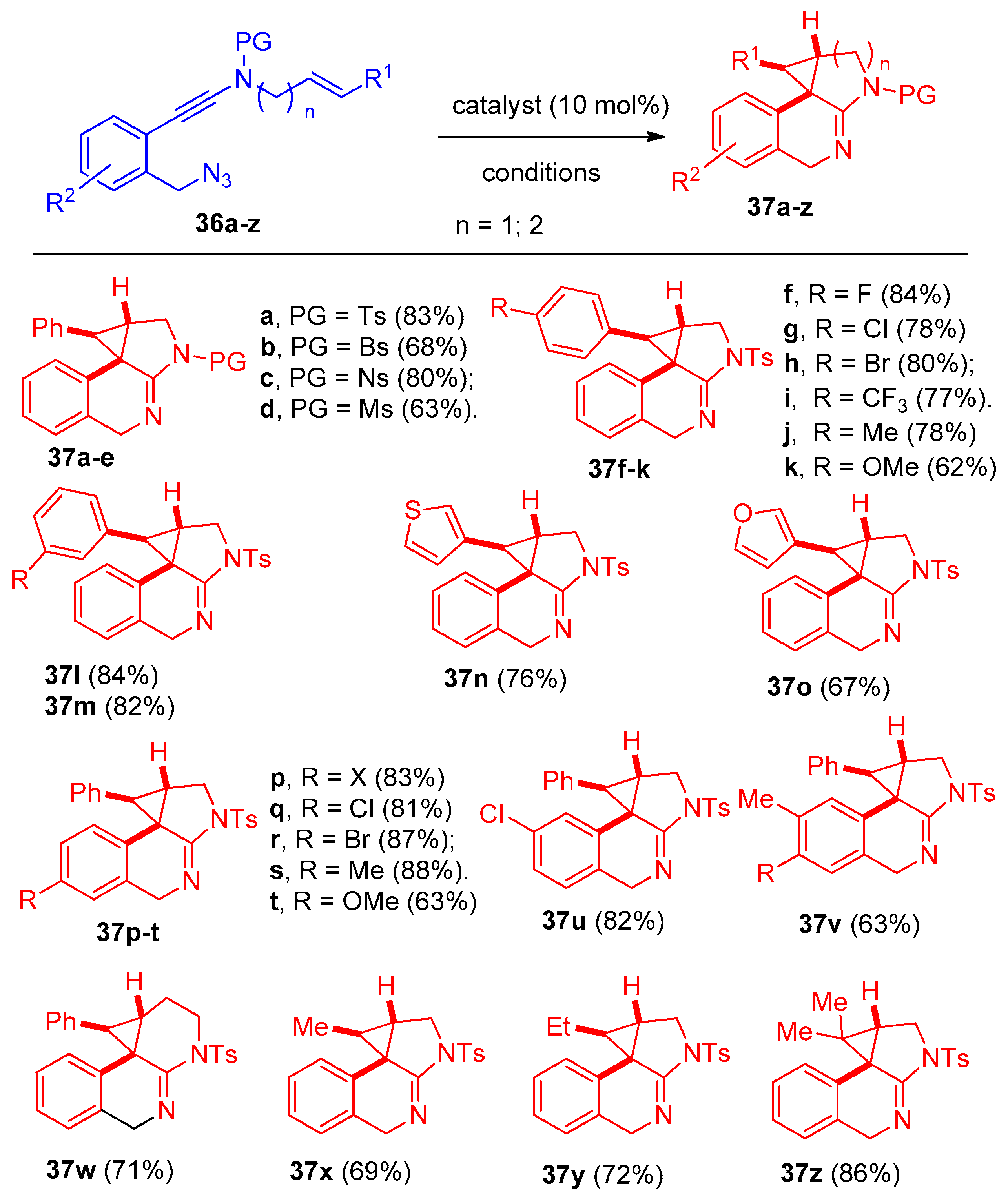
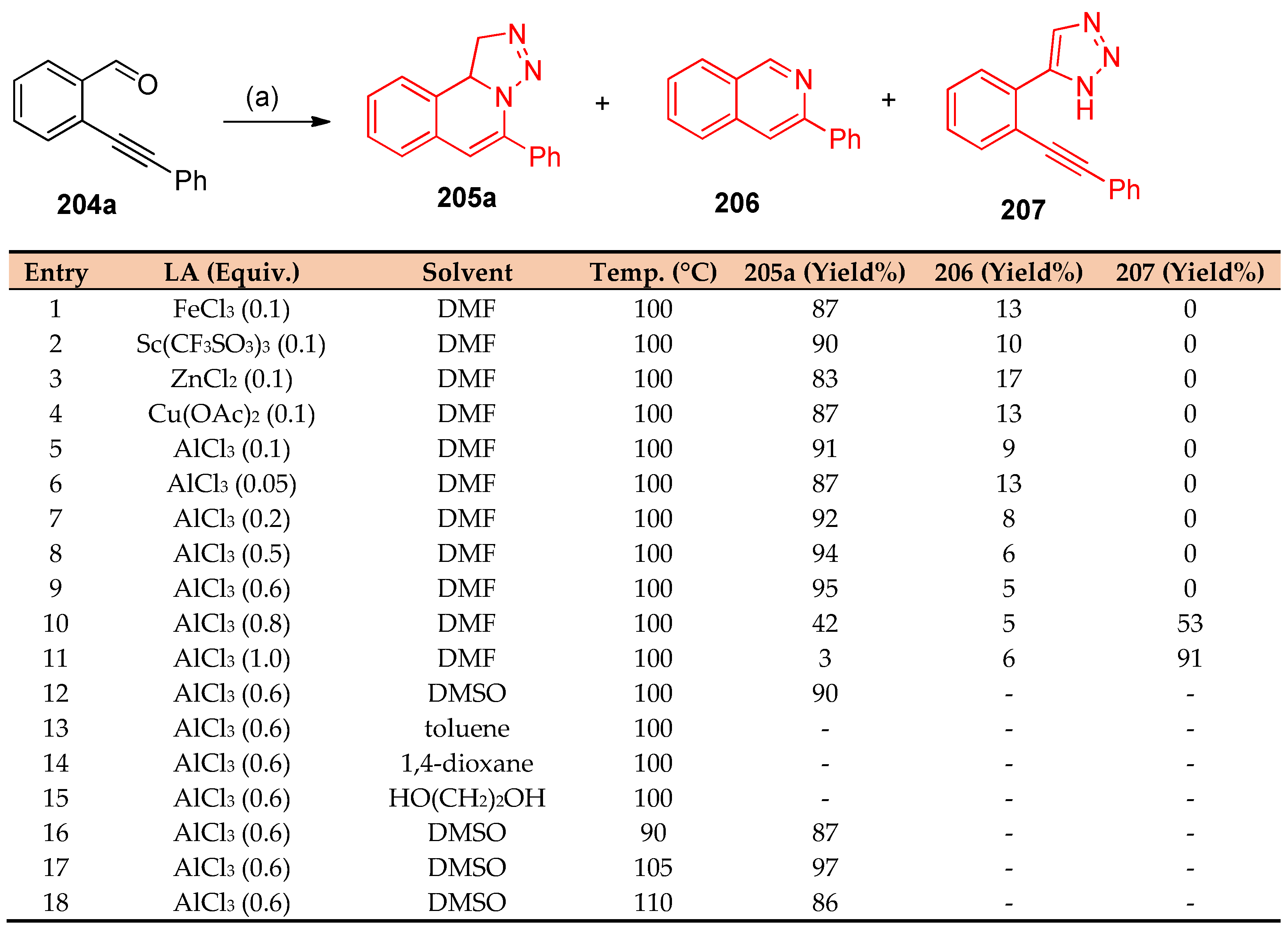
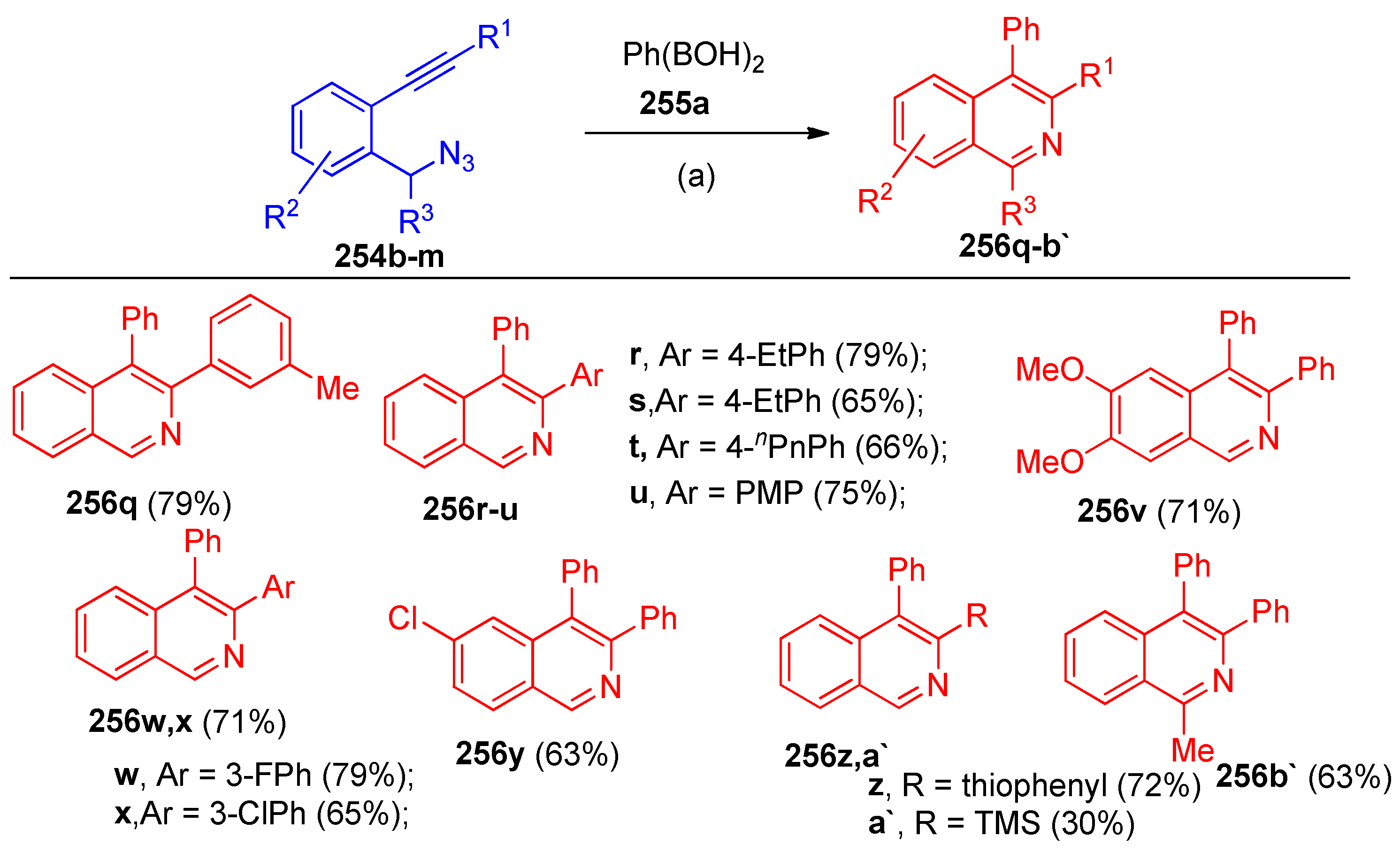
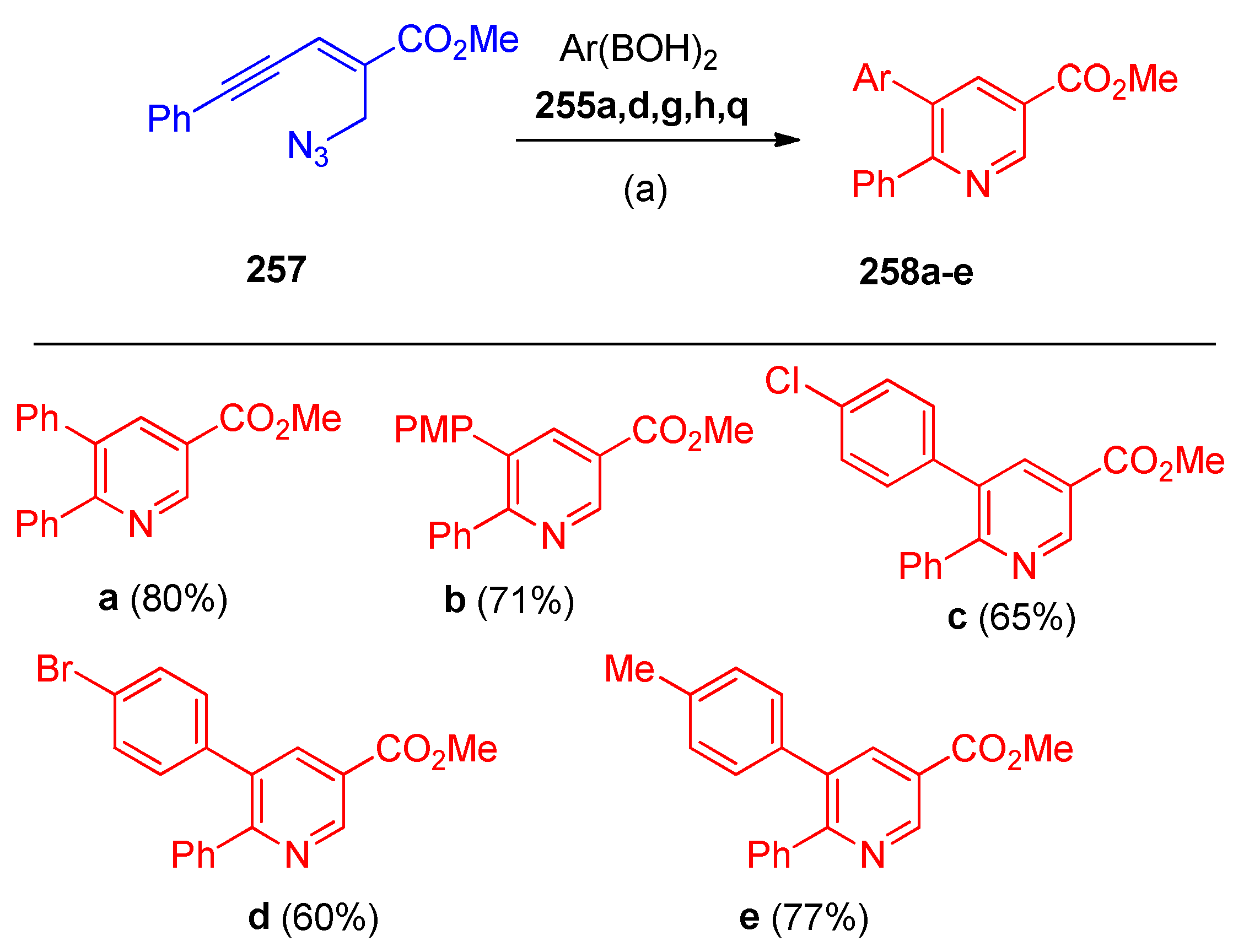
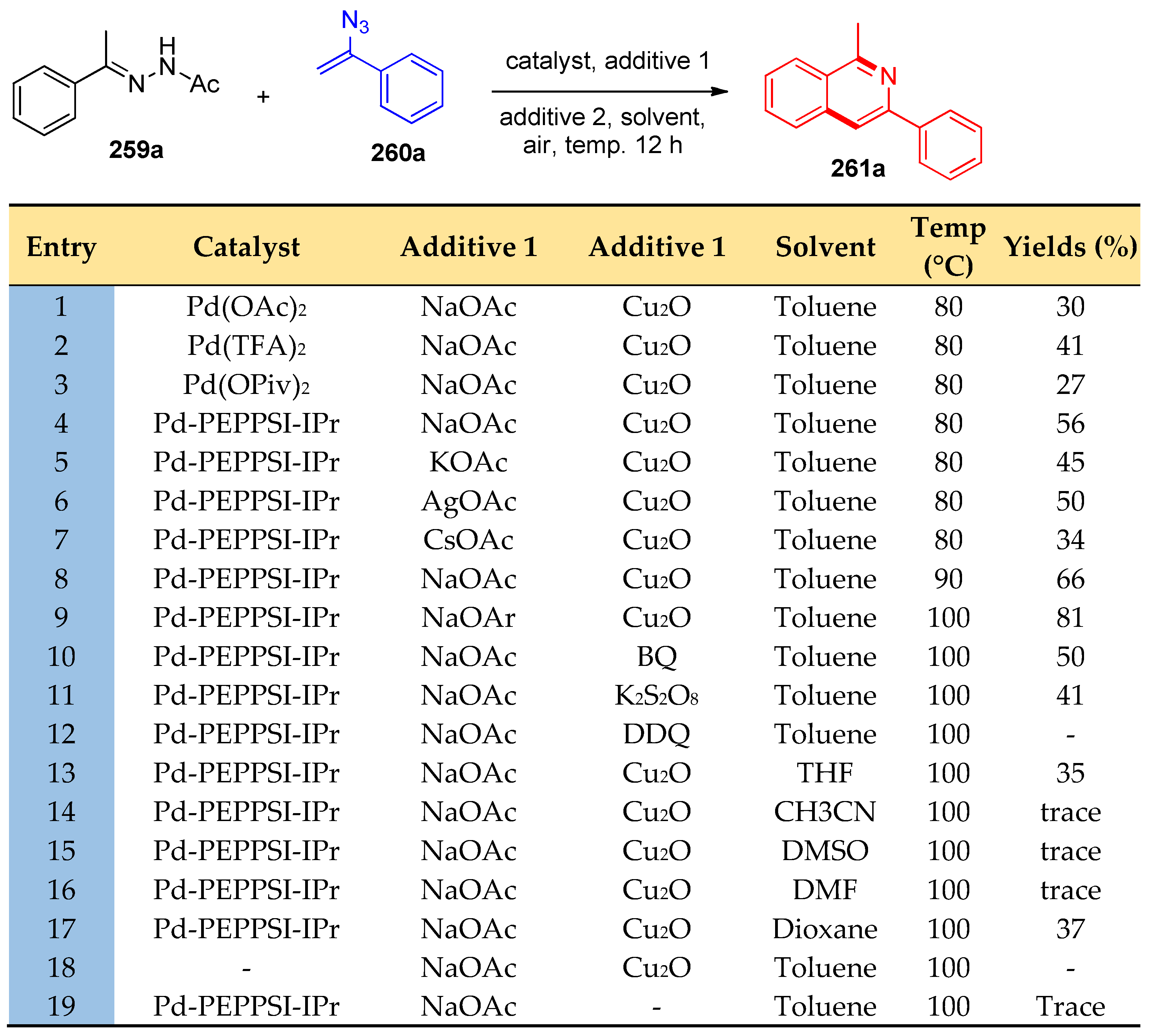
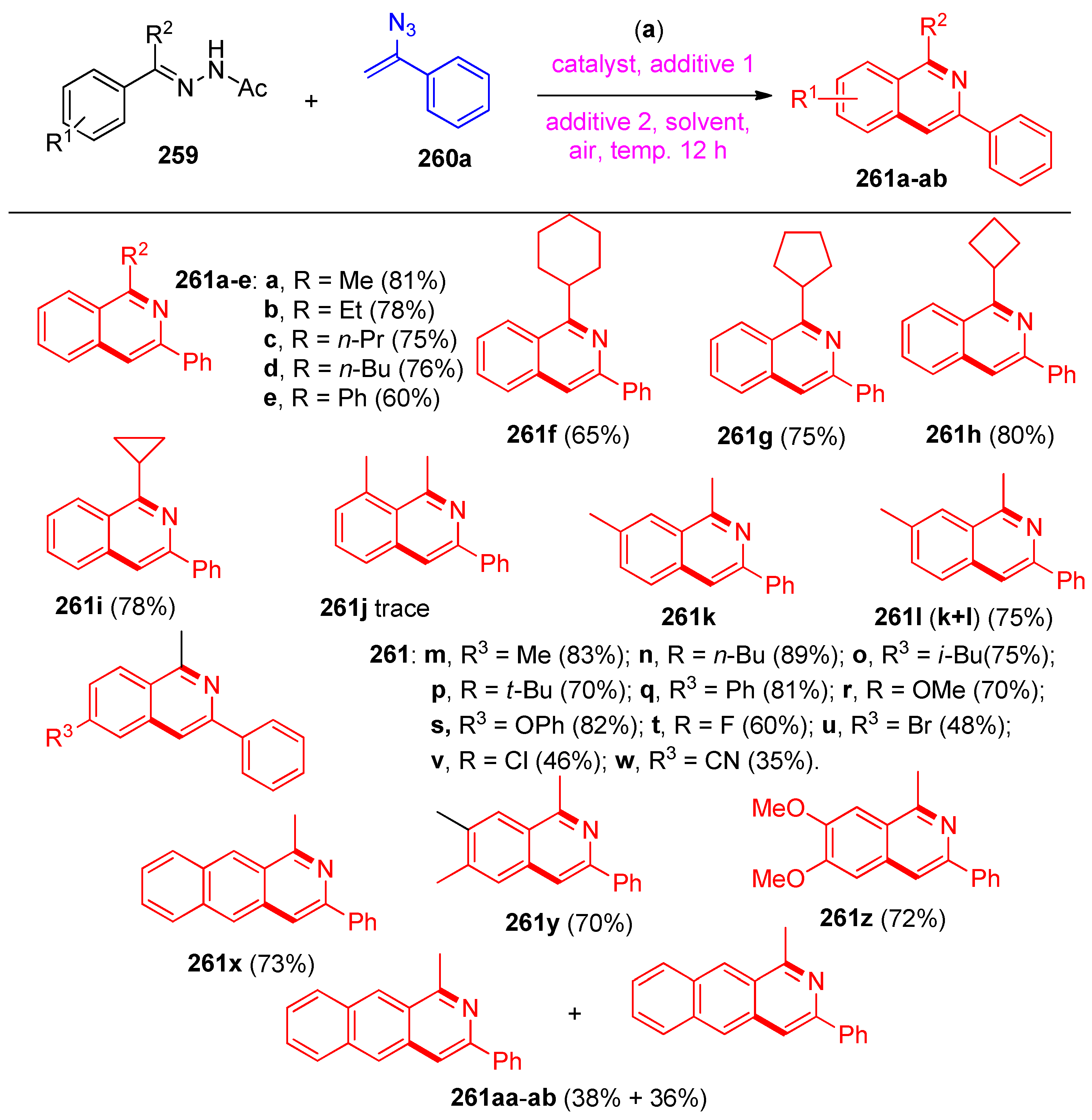
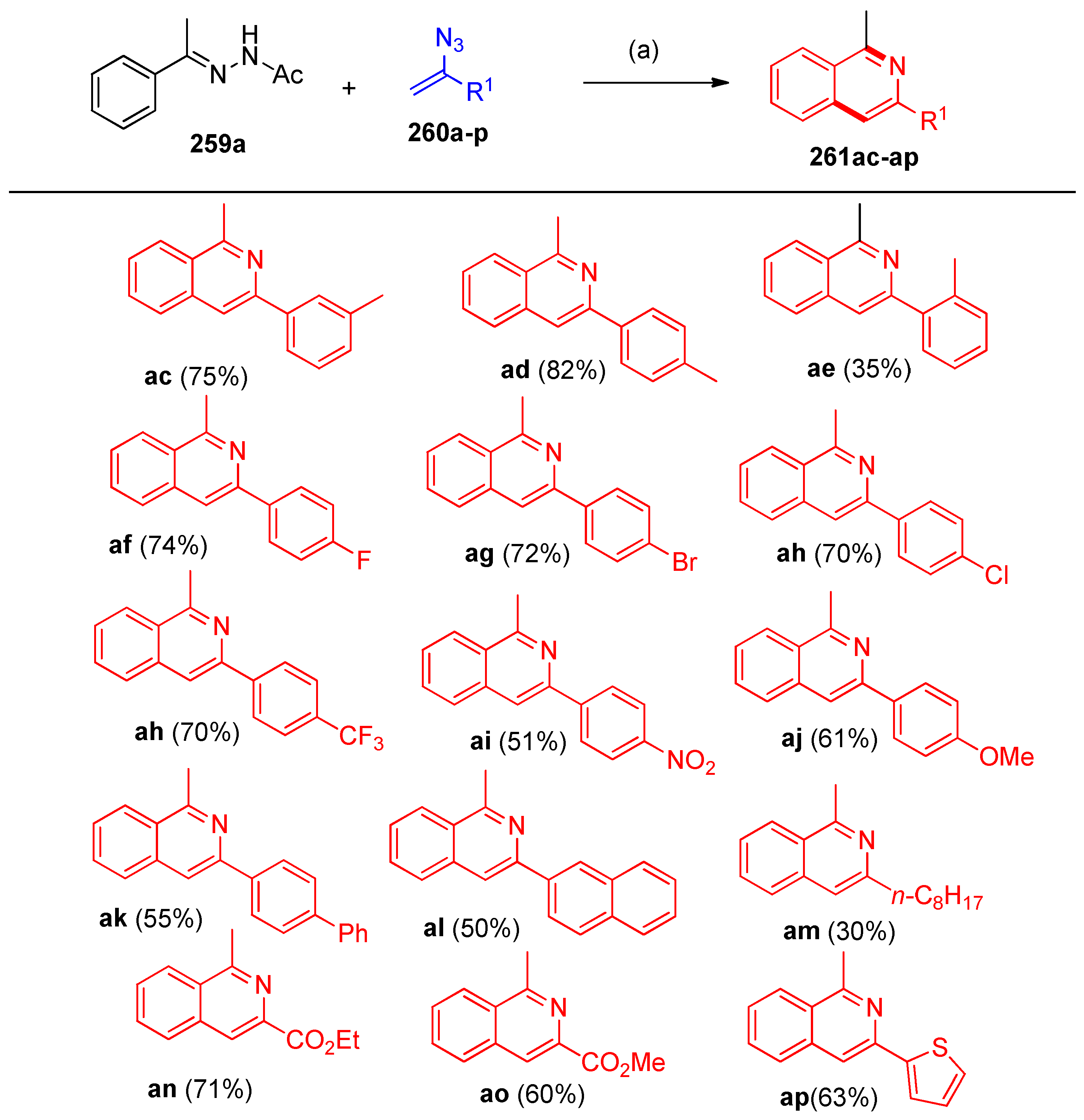
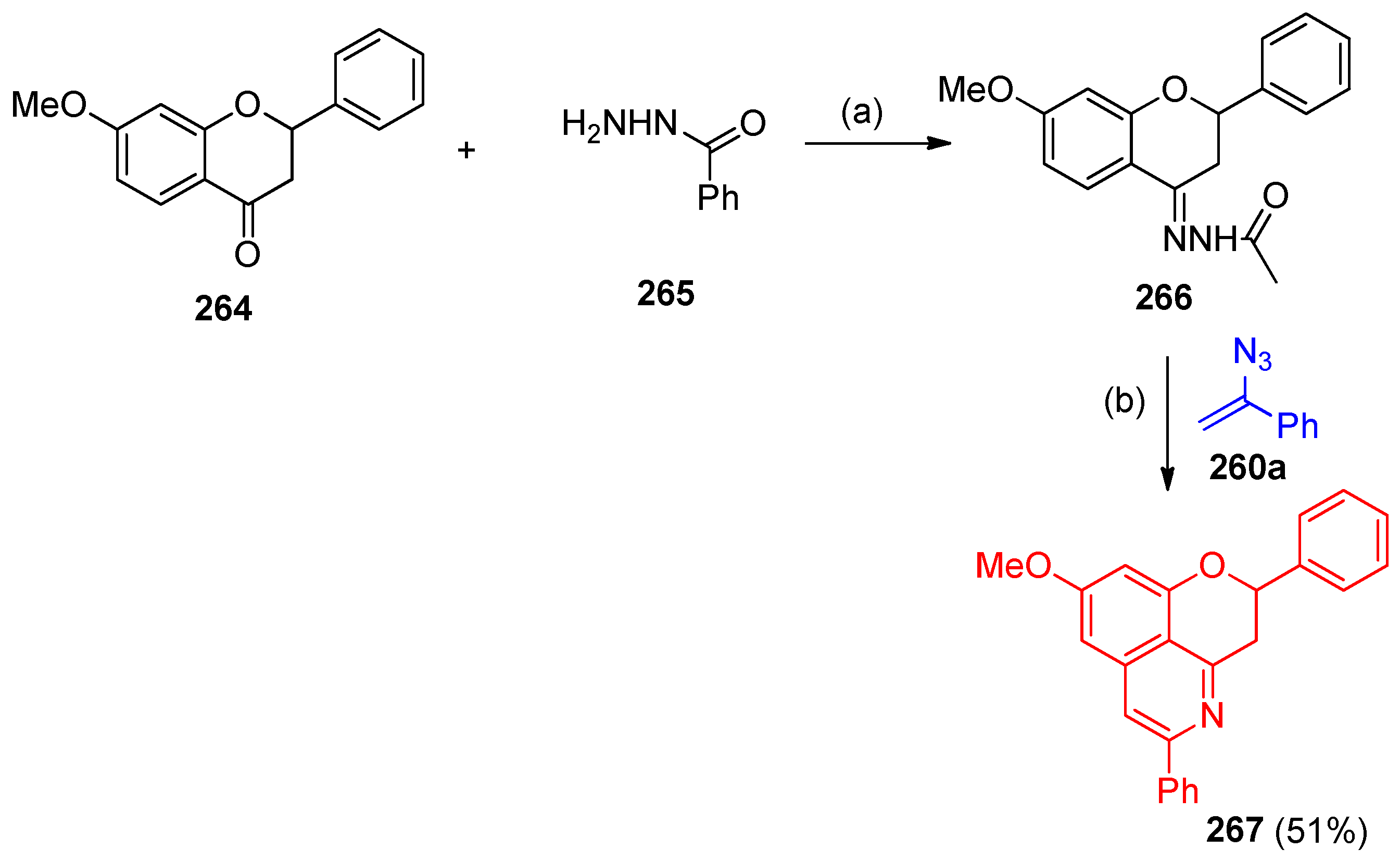
4.8. Synthesis of Pyridine and Isoquinoline Derivatives
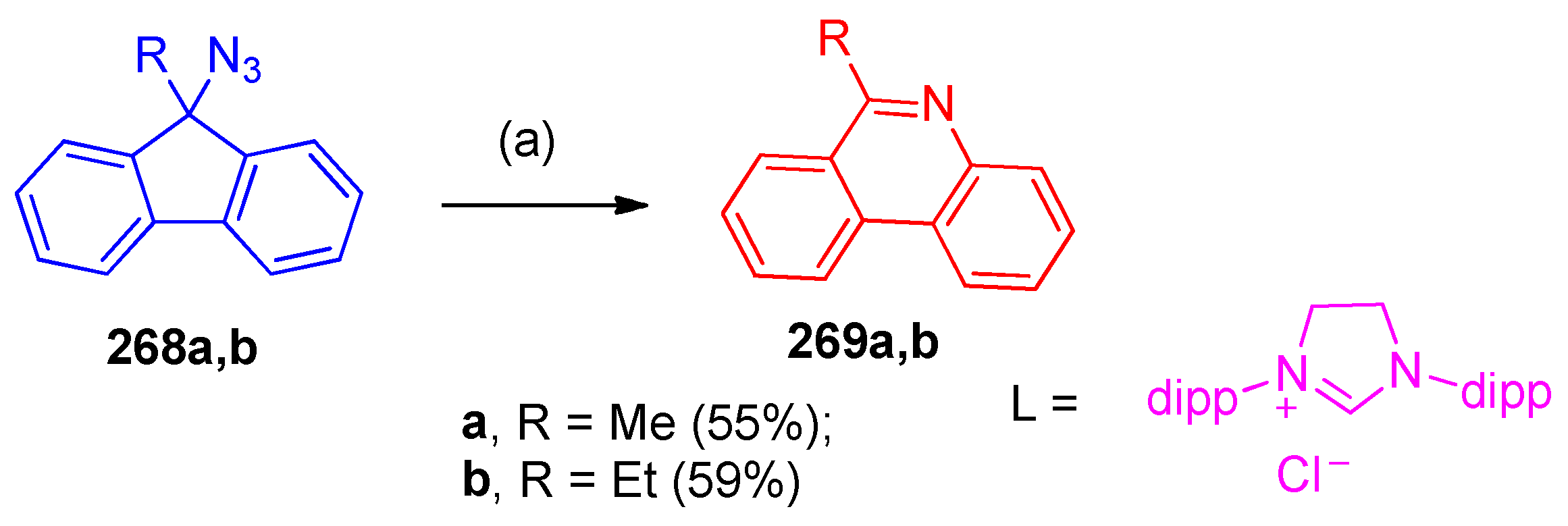
4.9. Synthesis of Phenanthridine
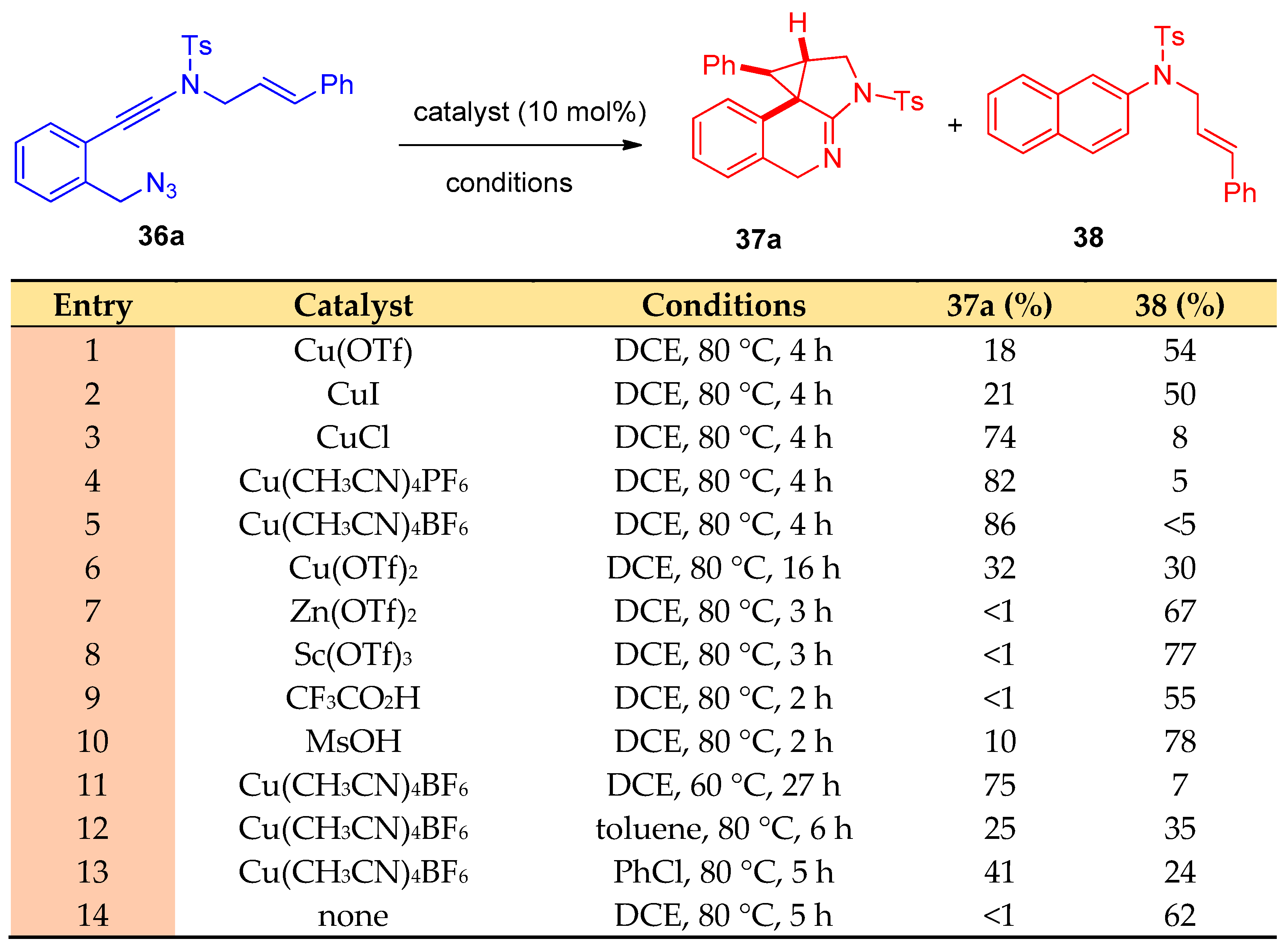
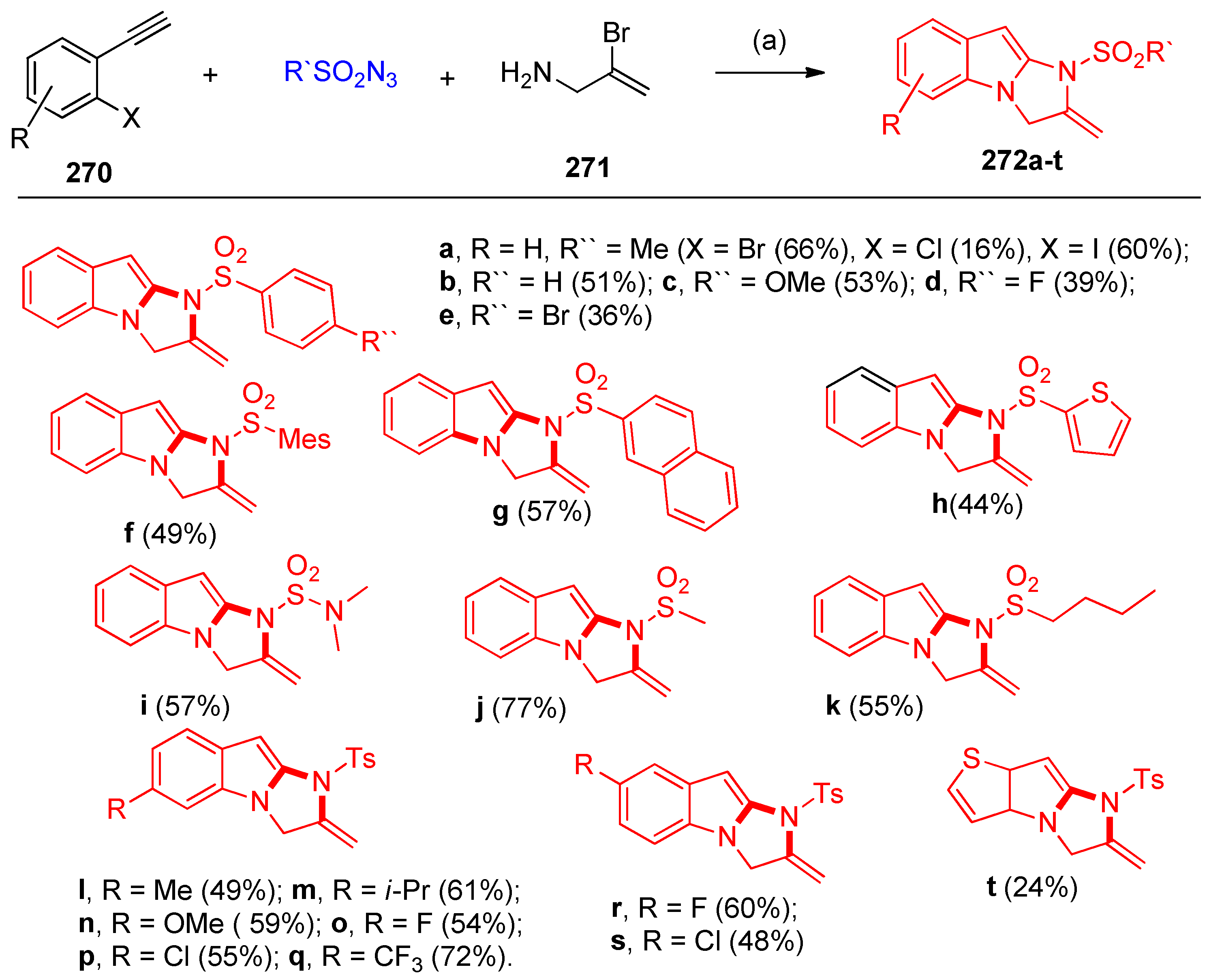
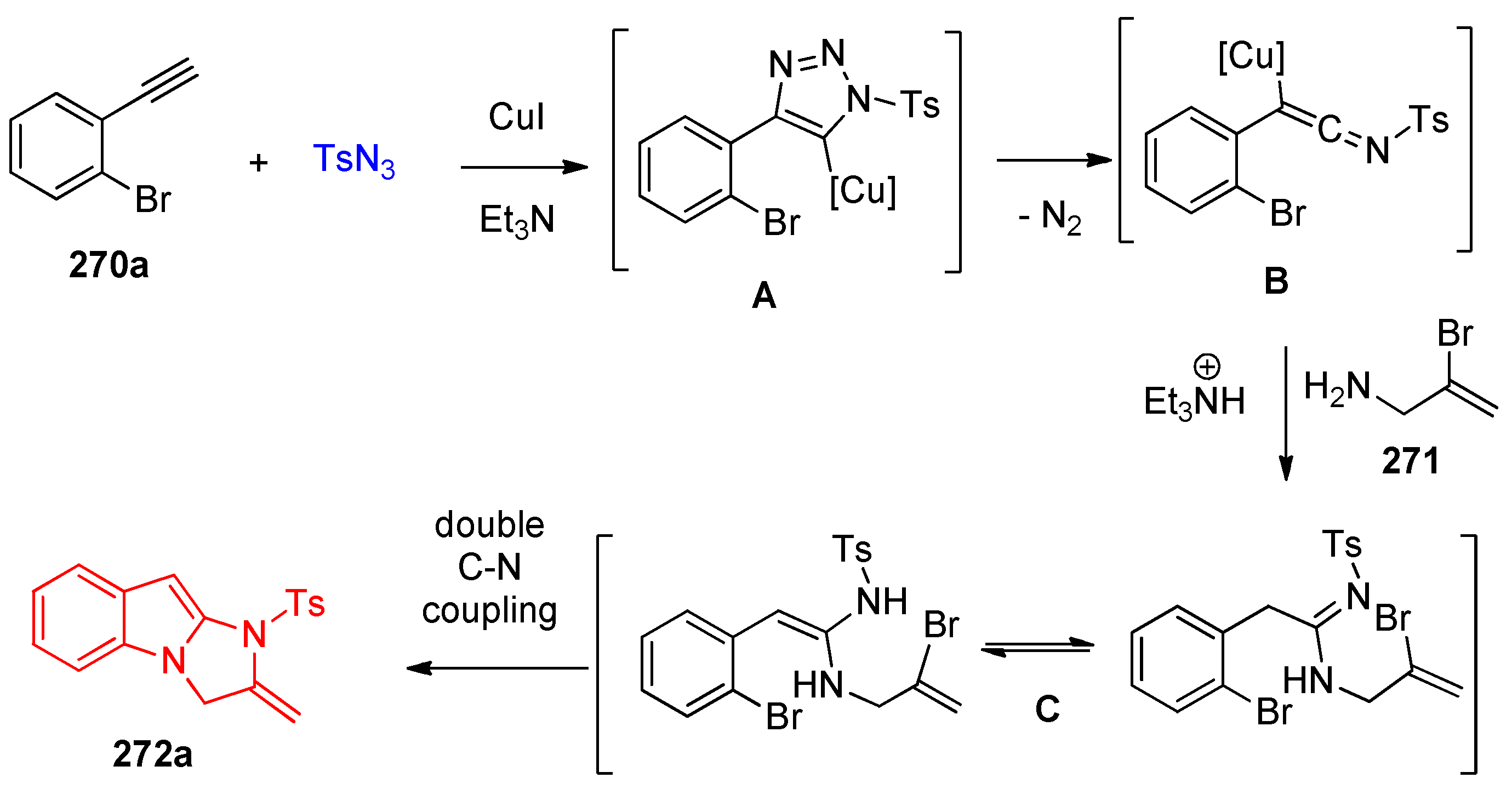
4.10. Synthesis of Imidazoindoles
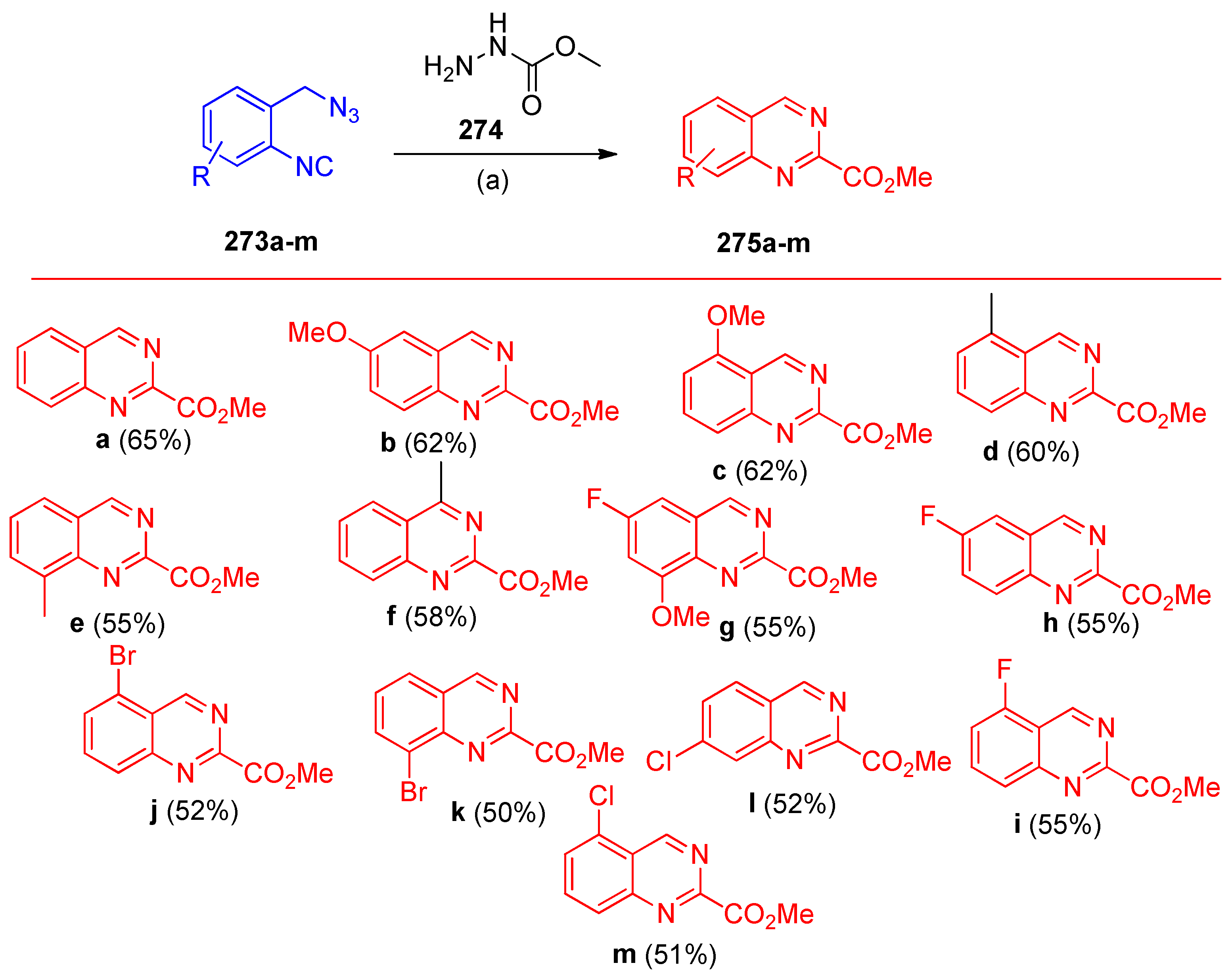
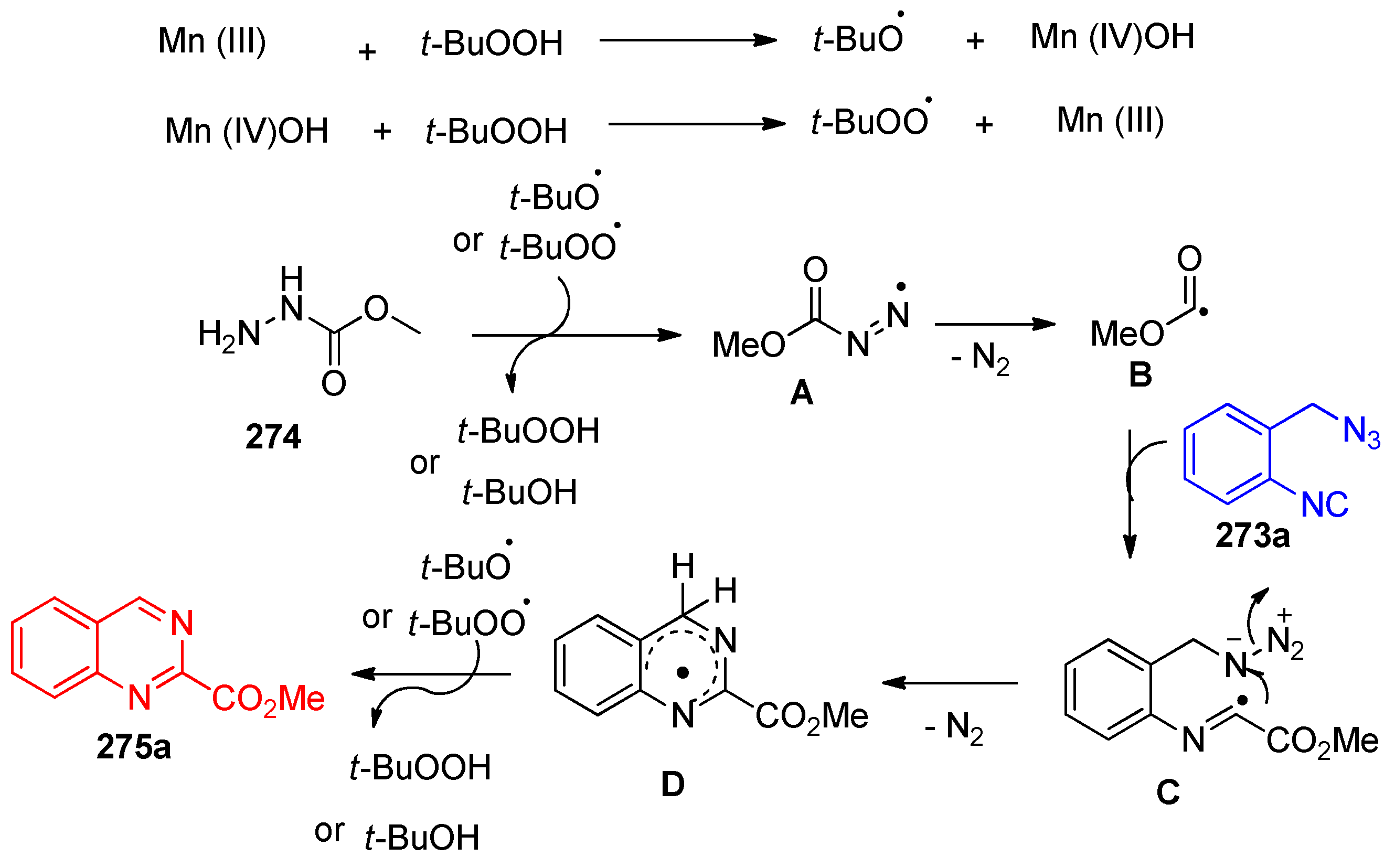
4.11. Synthesis of Quinazoline Derivatives
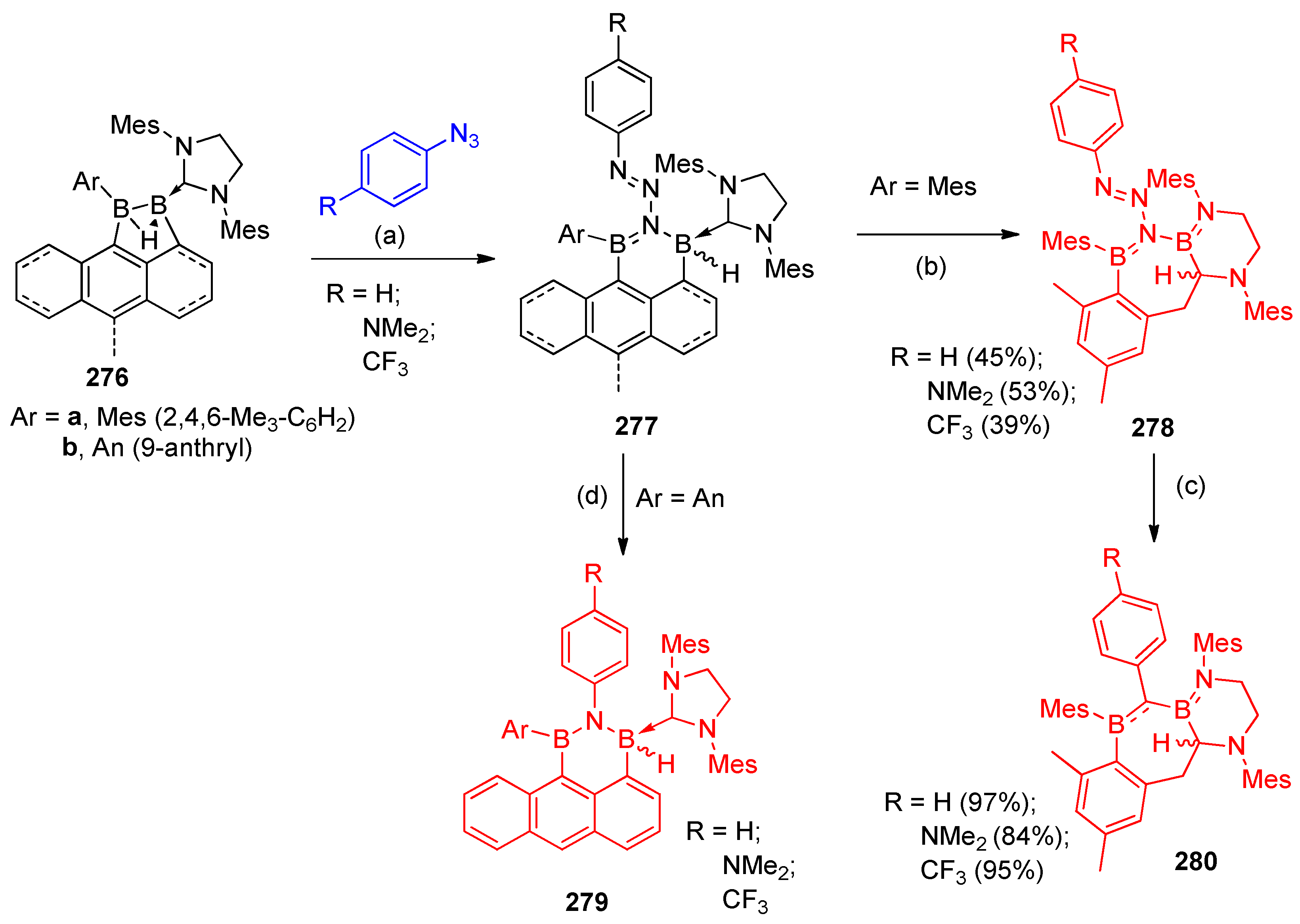
4.12. Synthesis of Borane Containing Heterocycles
4.12.1. Synthesis of Diborylaniline and Diboryl-Fused Pyrimidine
4.12.2. Synthesis of Triazaphosphaborolidine
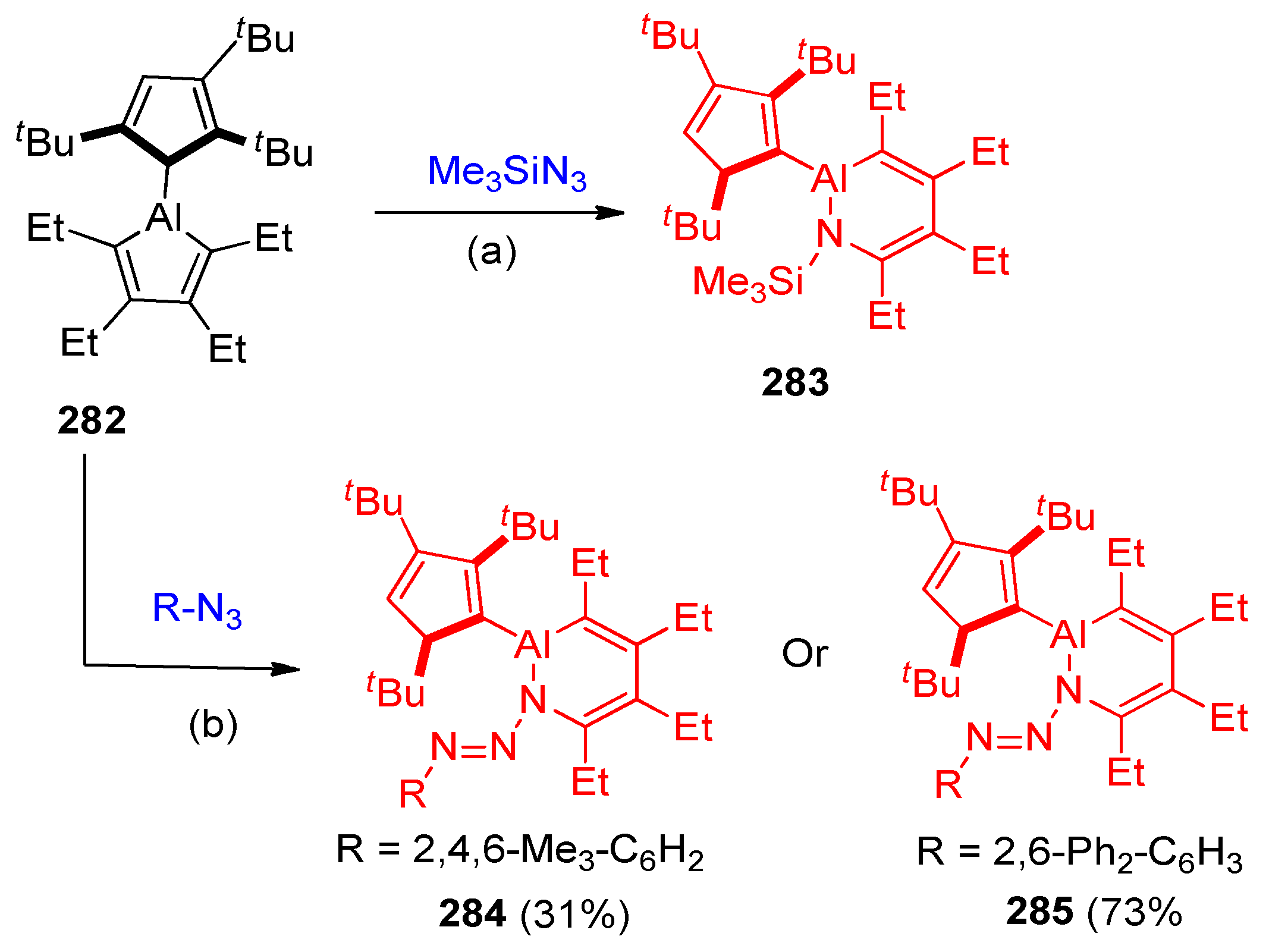
4.13. Synthesis of Aluminum-Containing Heterocyclic
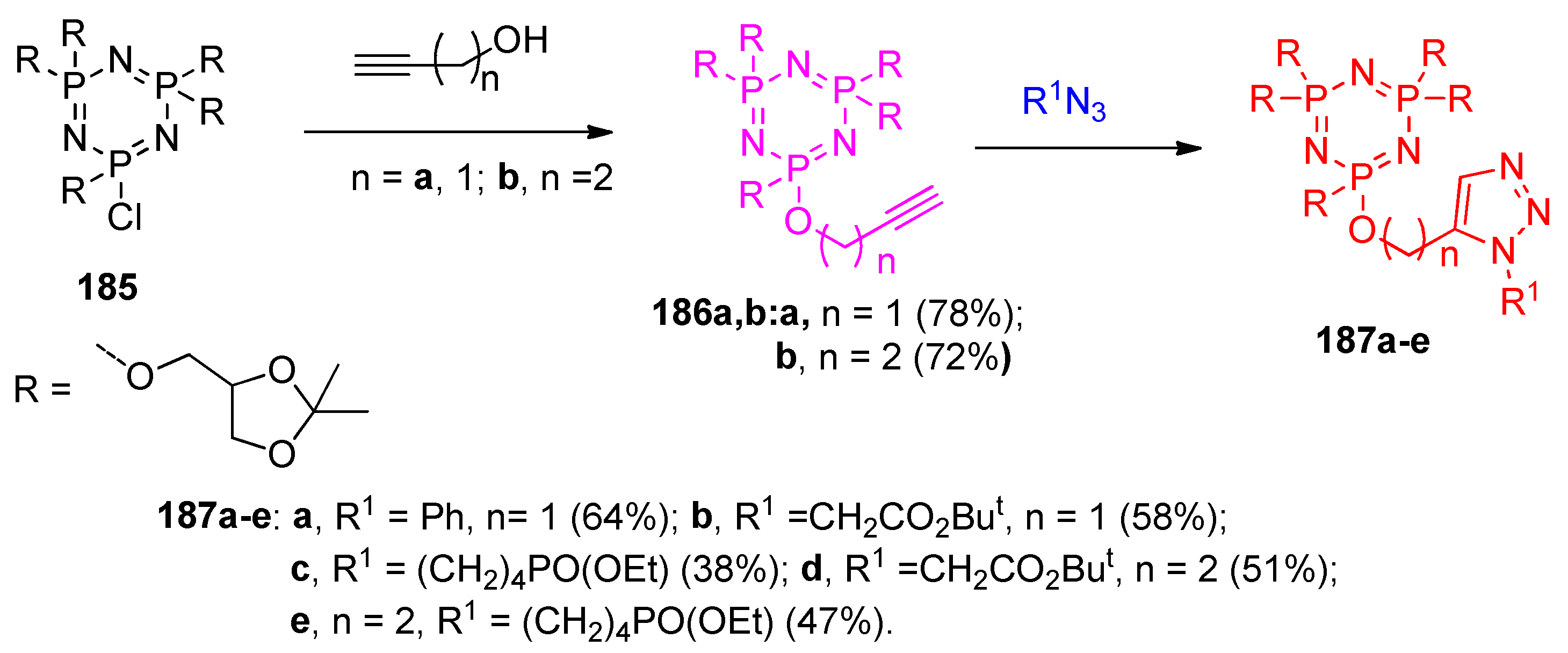
4.14. Synthesis of Phosphorus-Containing Heterocycles
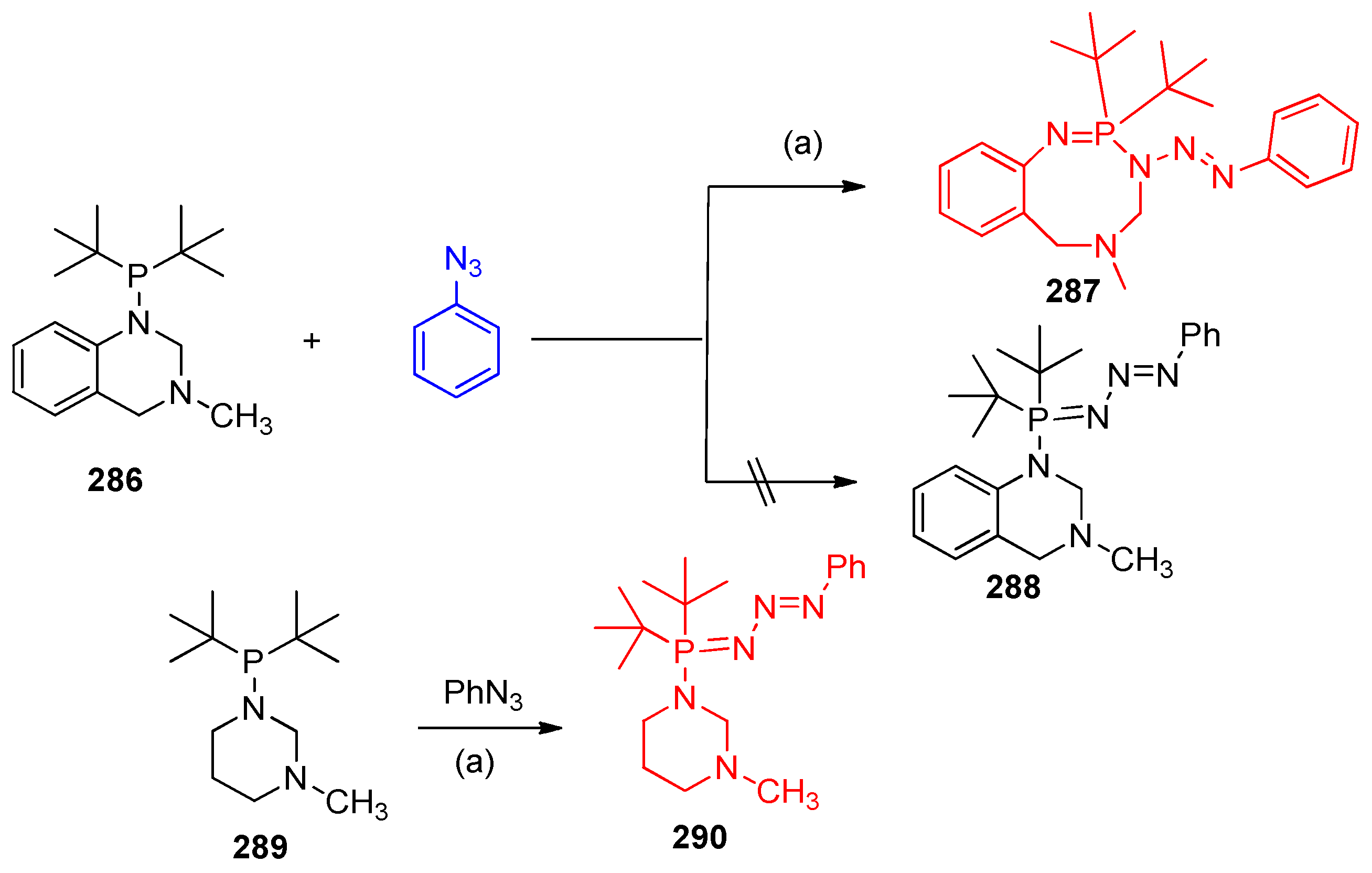
4.14.1. Synthesis of Triazphosphocine
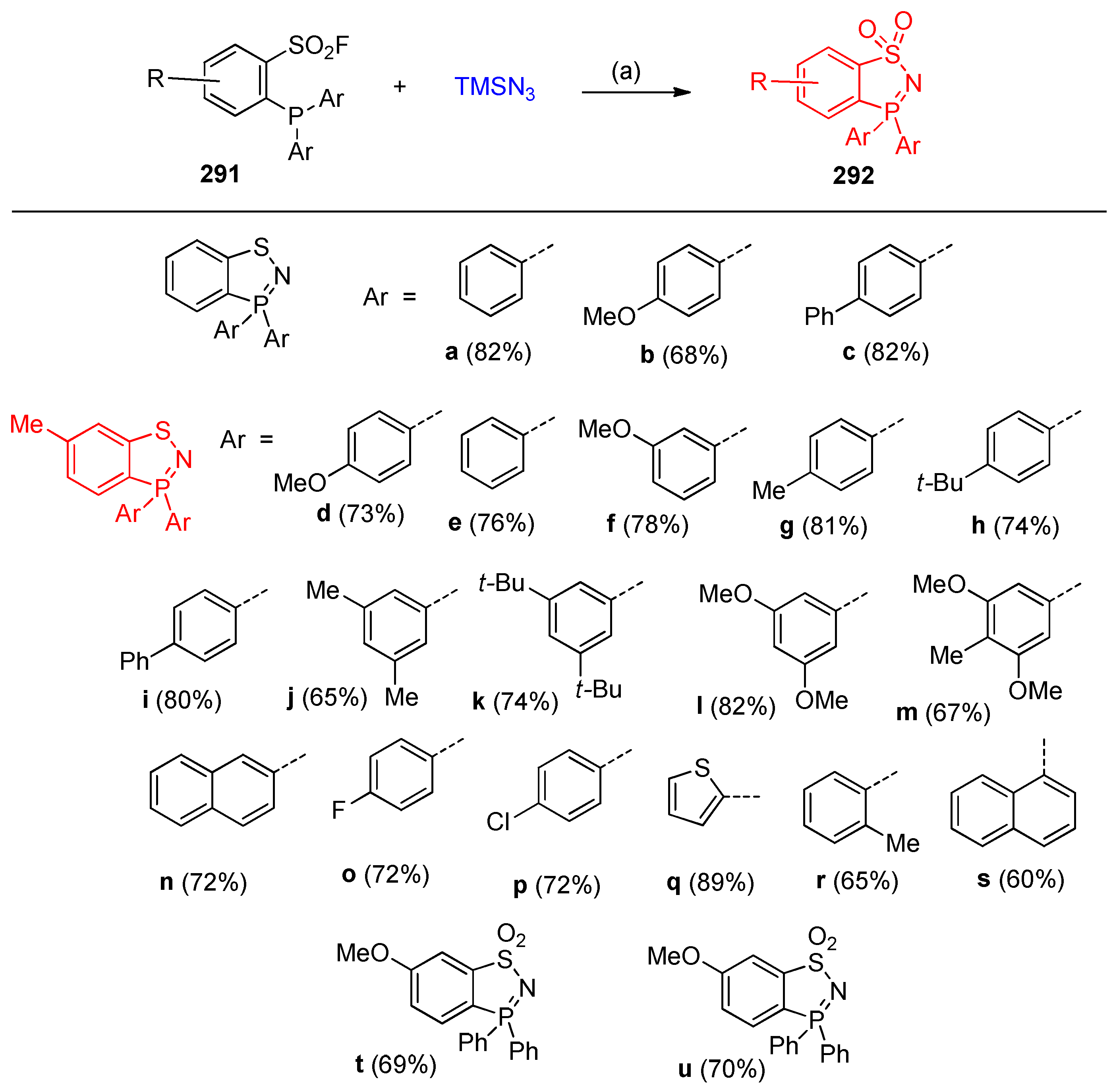
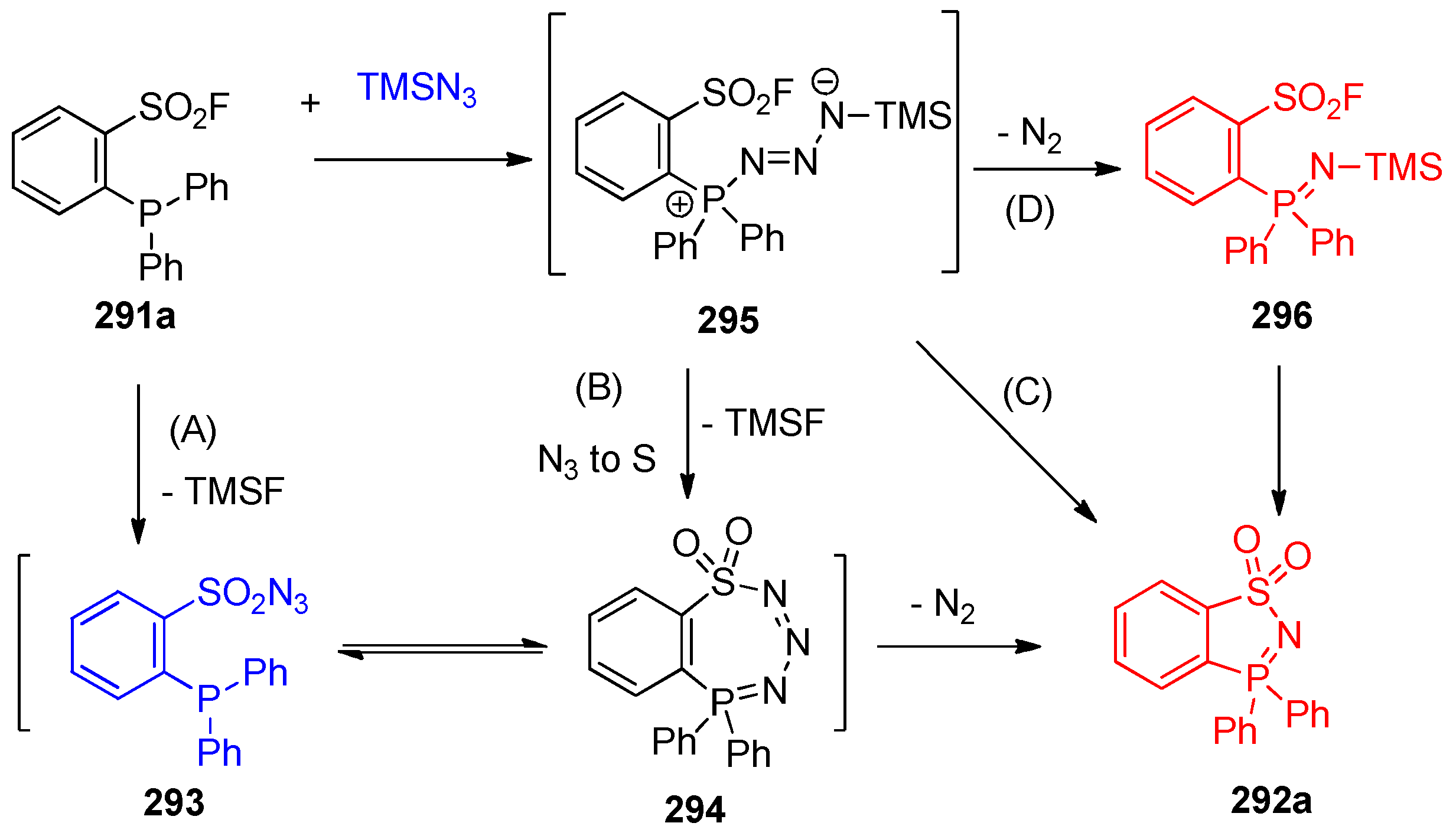
4.14.2. Synthesis of Benzothiazaphosphole
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
Hazardous Information
References
- Nguyen, M.T.; Sengupta, D.; Ha, T.-K. Another Look at the Decomposition of Methyl Azide and Methanimine: How Is HCN Formed? J. Phys. Chem. 1996, 100, 6499–6503. [Google Scholar] [CrossRef]
- Breuning, A.; Vicik, R.; Schirmeister, T. An improved synthesis of aziridine-2,3-dicarboxylates via azido alcohols—epimerization studies. Tetrahedron Asymmetry 2003, 14, 3301–3312. [Google Scholar] [CrossRef]
- Bräse, S.; Banert, K. Organic azides: Syntheses and applications. In Chichester; John Wiley: West Sussex, UK, 2010; p. 507. ISBN 978-0-470-68252-4. [Google Scholar]
- Kölmel, D.K.; Jung, N.; Bräse, S. Azides-Diazonium ions-Triazenes: Versatile nitrogen-rich functional groups. Austr. J. Chem. 2014, 67, 328–336. [Google Scholar] [CrossRef]
- Monguchi, Y.; Sajiki, H. Catalytic Reduction in Organic. Synthesis 2018, 217, 353. [Google Scholar] [CrossRef]
- Gololobov, Y.G.; Kasukhin, L.F. Recent advances in the Staudinger reaction. Tetrahedron 1992, 48, 1353–1406. [Google Scholar] [CrossRef]
- Fresneda, P.M.; Molina, P. Application of Iminophosphorane-Based Methodologies for the Synthesis of Natural Products. Synlett 2004, 35, 1–17. [Google Scholar] [CrossRef]
- Palacios, F.; Alonso, C.; Aparicio, D.; Rubiales, G.; de Los, S.; Jesús, M. The aza-Wittig reaction: An efficient tool for the construction of carbon–nitrogen double bonds. Tetrahedron 2007, 63, 523–575. [Google Scholar] [CrossRef]
- Álvares, Y.S.P.; Alves, M.J.; Azoia, N.G.; Bickley, J.F.; Gilchrist, T.L. Diastereoselective synthesis of aziridines from (1R)-10-(N,N-dialkyl-sulfamoyl)isobornyl 2H-azirine-3-carboxylates. J. Chem. Soc. Perkin Trans. 2022, 1, 1911–1919. [Google Scholar] [CrossRef]
- Capitosti, S.M.; Hansen, T.P.; Brown, M.L. Facile Synthesis of an Azido-Labeled Thalidomide Analogue. Org. Lett. 2003, 5, 2865–2867. [Google Scholar] [CrossRef]
- Lowe-Ma, C.K.; Nissan, R.A.; Wilson, W.S. Tetrazolo [1,5-a]pyridines and Furazano [4,5-b]pyridine 1-Oxides. J. Org. Chem. 1990, 55, 3755–3761. [Google Scholar] [CrossRef]
- Stadlbauer, W.; Fiala, W.; Fischer, M.; Hojas, G. Thermal cyclization of 4-azido-3-nitropyridines to furoxanes. J. Heterocycl. Chem. 2000, 37, 1253–1256. [Google Scholar] [CrossRef]
- Gavenonis, J.; Tilley, T.D. Tantalum Alkyl and Silyl Complexes of the Bulky (Terphenyl)imido Ligand [2,6-(2,4,6-Me3C6H2)2C6H3N]2- ([Ar*N]2-). Generation and Reactivity of [(Ar*N)(Ar*NH)Ta(H)(OSO2CF3)], Which Reversibly Transfers Hydride to an Aromatic Ring of the Arylamide Ligand. Organometallics 2002, 21, 5549–5563. [Google Scholar] [CrossRef]
- Kim, Y.H.; Kim, K.; Shim, S.B. Reaction of arylhydrazines with nitric oxide in the presence of oxygen. Tetrahedron Lett. 1986, 27, 4749–4752. [Google Scholar] [CrossRef]
- Lee, S.H.; Kwon, N.Y.; Lee, J.Y.; An, W.; Jung, Y.H.; Mishra, N.K.; Ghosh, P.; Park, J.S.; Kim, I.S. Transition-Metal-Free and Site-Selective Selenylation of Heterocyclic N-Oxides in Anisole as a Green Solvent. Eur. J. Org. Chem. 2020, 2020, 4886–4892. [Google Scholar] [CrossRef]
- Zhang, G.-Y.; Peng, Y.; Xue, J.; Fan, Y.-H.; Deng, Q.-H. Copper-catalyzed nitrene transfer/cyclization cascade to synthesize 3a-nitrogenous furoindolines and pyrroloindolines. Org. Chem. Front. 2019, 6, 3934–3938. [Google Scholar] [CrossRef]
- Porter, M.R.; Shaker, R.M.; Calcanas, C.; Topczewski, J.J. Stereoselective Dynamic Cyclization of Allylic Azides: Synthesis of Tetralins, Chromanes, and Tetrahydroquinolines. J. Am. Chem. Soc. 2018, 140, 1211–1214. [Google Scholar] [CrossRef]
- Dong, Y.; Clarke, R.M.; Porter, G.J.; Betley, T.A. Efficient C−H Amination Catalysis Using Nickel-Dipyrrin Complexes. J. Am. Chem. Soc. 2020, 142, 10996–11005. [Google Scholar] [CrossRef]
- Sun, K.; Tao, C.; Long, B.; Zeng, X.; Wu, Z.; Zhang, R. Highly stereoselective gram scale synthesis of all the four diastereoisomers of Boc-protected 4-methylproline carboxylates. RSC Adv. 2019, 9, 32017–32020. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Wang, Z.-S.; Zhai, T.-Y.; Luo, C.; Zhang, Y.-P.; Chen, Y.-B.; Deng, C.; Liu, R.S.; Ye, L.-W. Copper-Catalyzed Azide–Ynamide Cyclization to Generate α-Imino Copper Carbenes: Divergent and Enantioselective Access to Polycyclic N-Heterocycles. Angew. Chem. Int. Ed. 2020, 59, 2–9. [Google Scholar] [CrossRef]
- Fu, J.G.; Zanoni, G.; Anderson, E.A.; Bi, X. α-Substituted vinyl azides: An emerging functionalized alkene. Chem. Soc. Rev. 2017, 46, 7208–7228. [Google Scholar] [CrossRef]
- Borra, S.; Borkotoky, L.; Newar, U.D.; Das, B.; Maurya, R.A. Photocatalyst-Free Visible-Light Enabled Synthesis of Substituted Pyrroles from α-Keto Vinyl Azides. Adv. Synth. Catal. 2020, 362, 3364–3368. [Google Scholar] [CrossRef]
- Quiclet-Sire, B.; Zard, S.Z. Observations on the Reaction of Hydrazones with Iodine: Interception of the Diazo Intermediates. Chem. Commun. 2006, 1831–1832. [Google Scholar] [CrossRef]
- Kapras, V.; Pohl, R.; Císařová, I.; Jahn, U. Asymmetric Domino Aza-Michael Addition/[3+2] Cycloaddition Reactions as a Versatile Approach to α,β,γ-Triamino Acid Derivatives. Org. Lett. 2014, 16, 1088–1091. [Google Scholar] [CrossRef] [PubMed]
- Just, D.; Hernandez-Guerra, D.; Kritsch, S.; Pohl, R.; Císařová, I.; Jones, P.G.; Mackman, R.; Bahador, G.; Jahn, U. Lithium Chloride Catalyzed Asymmetric Domino Aza-Michael Addition/[3+2] Cycloaddition Reactions for the Synthesis of Spiro- and Bicyclic α,β,γ-Triamino Acid Derivatives. Eur. J. Org. Chem. 2018, 2018, 5213–5221. [Google Scholar] [CrossRef]
- Yang, C.; Shen, H. One Pot Multiple-Steps Reactions of Allyl Azide and Alkenes Carrying Electron-Withdrawing Groups. Tetrahedron Lett. 1993, 34, 4051–4054. [Google Scholar] [CrossRef]
- Yang, C.-H.; Sherf, H.-J.; Wang, R.-H.; Wang, J.-C. 2,3,7-Triazabicyclo [3.3.0]Octenes Prepared by Tandem Cascade Reaction of Allyl Azides and Olefinic Dipolarophiles. J. Chin. Chem. Soc. 2002, 49, 95–102. [Google Scholar] [CrossRef]
- Carlson, A.S.; Liu, E.-C.; Topczewski, J.J. A Cascade Reaction of Cinnamyl Azides with Acrylates Directly Generates Tetrahydro-Pyrrolo-Pyrazole Heterocycles. J. Org. Chem. 2020, 85, 6044–6059. [Google Scholar] [CrossRef] [PubMed]
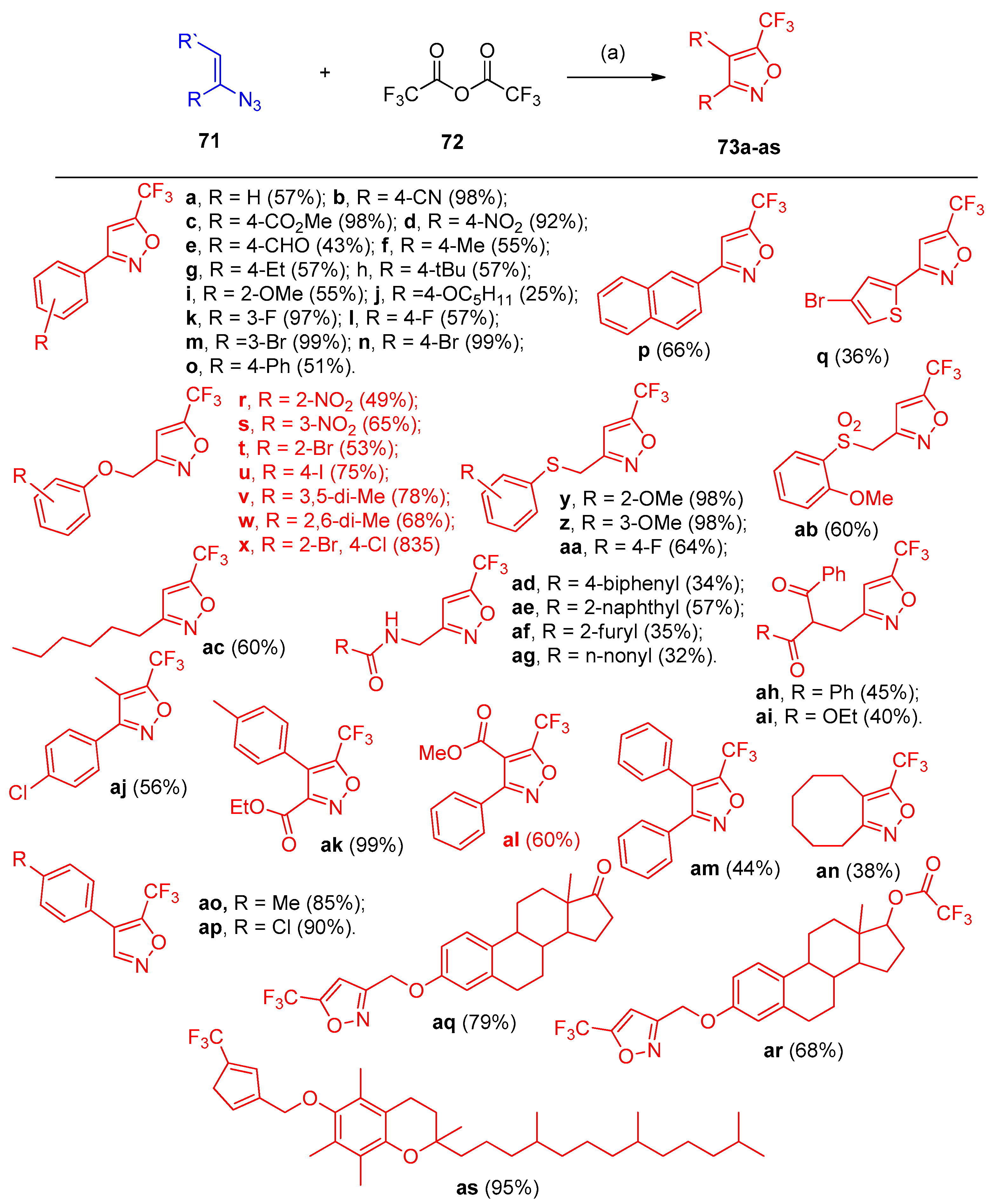
- Wu, W.; Chen, Q.; Tian, Y.; Xu, Y.; Huang, Y.; You, Y.; Weng, Z. Synthesis of polysubstituted 5-trifluoromethyl isoxazoles via denitrogenative cyclization of vinyl azides with trifluoroacetic anhydride. Org. Chem. Front. 2020, 7, 1878–1883. [Google Scholar] [CrossRef]
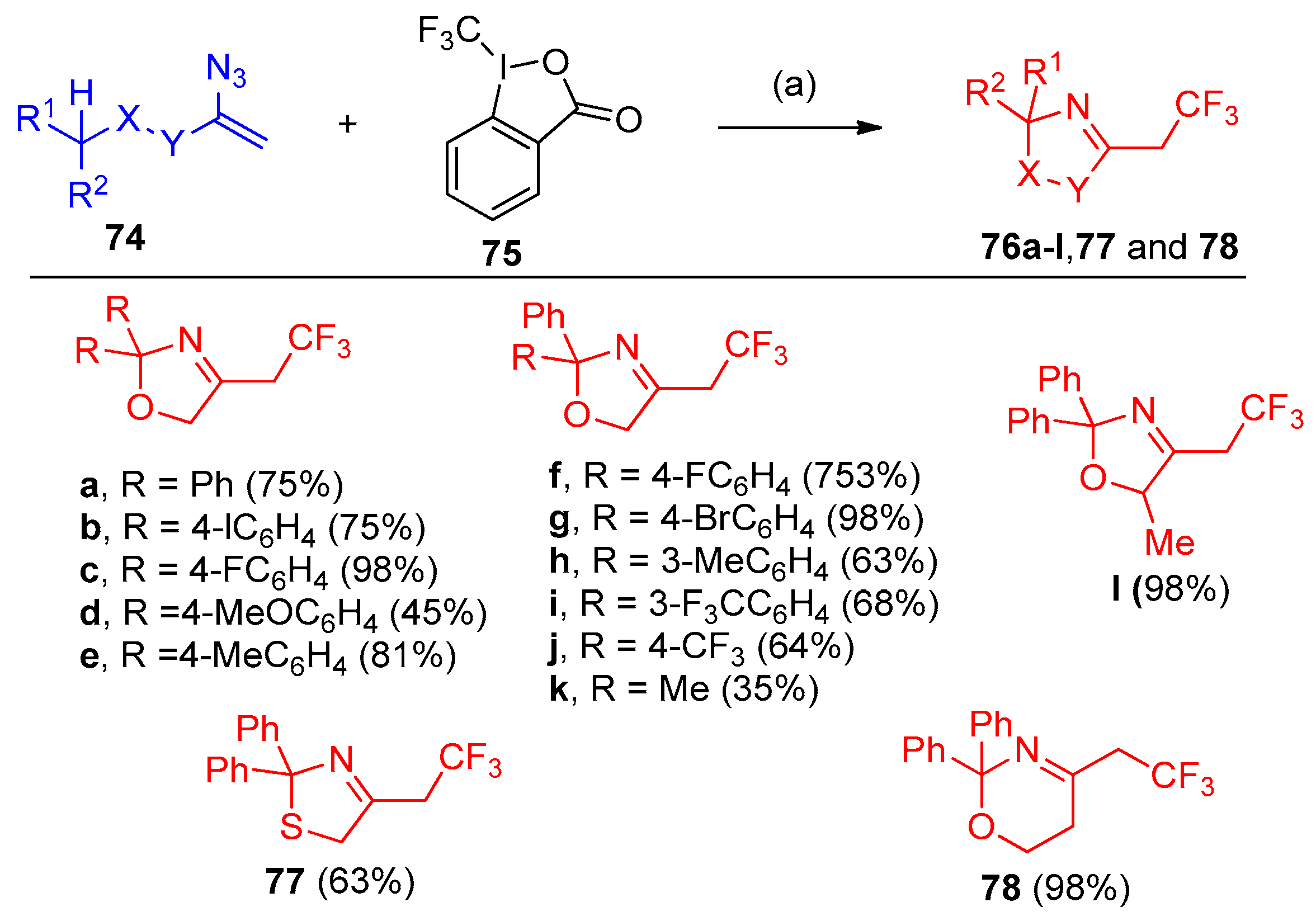
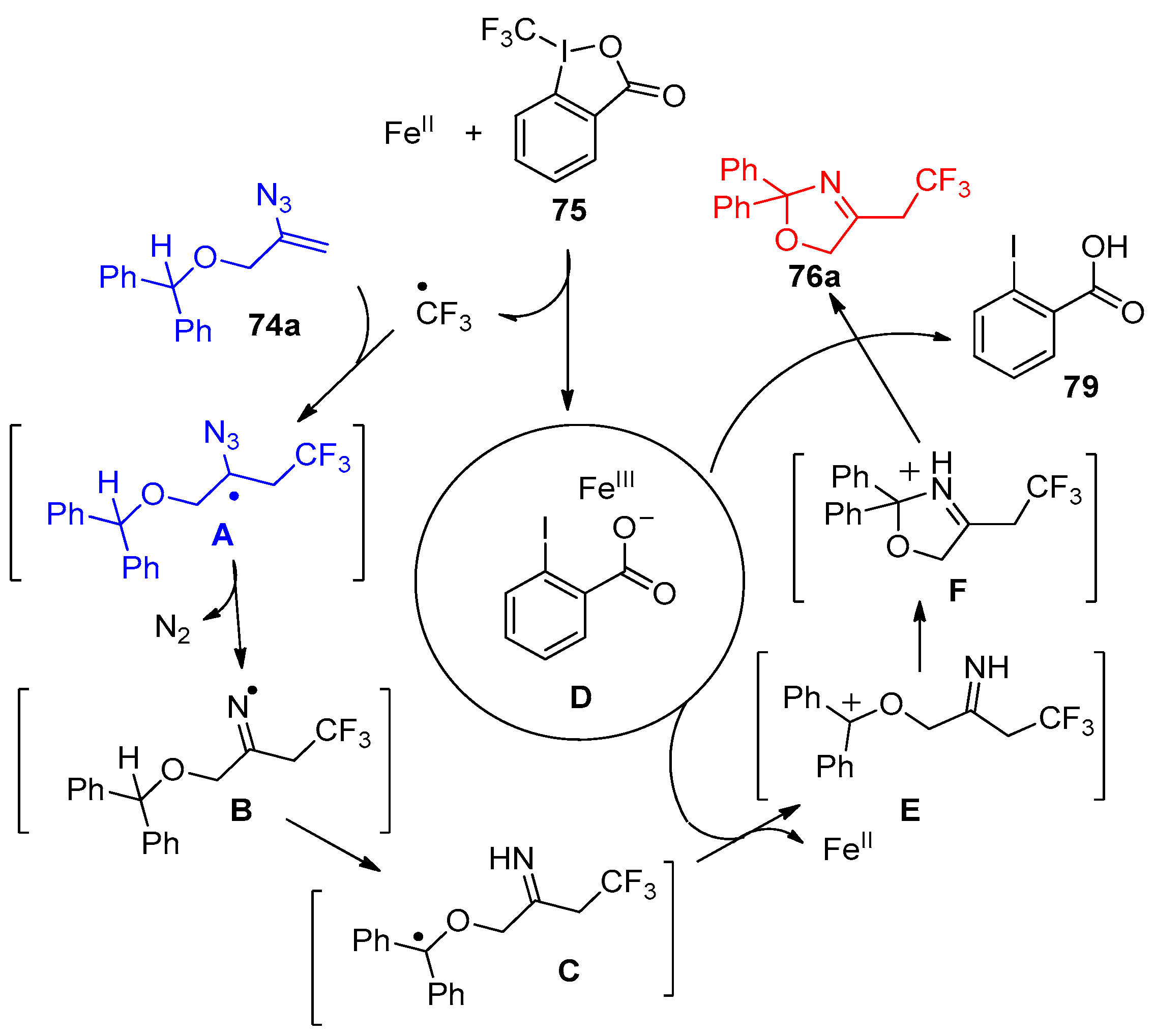
- Terhorst, S.; Tiwari, D.P.; Meister, D.; BPetran, B.; Rissanen, K.; Bolm, C. Syntheses of Trifluoroethylated N-Heterocycles from Vinyl Azides and Togni’s Reagent Involving 1,n-Hydrogen-Atom Transfer Reactions. Org. Lett. 2020, 22, 4766–4770. [Google Scholar] [CrossRef]
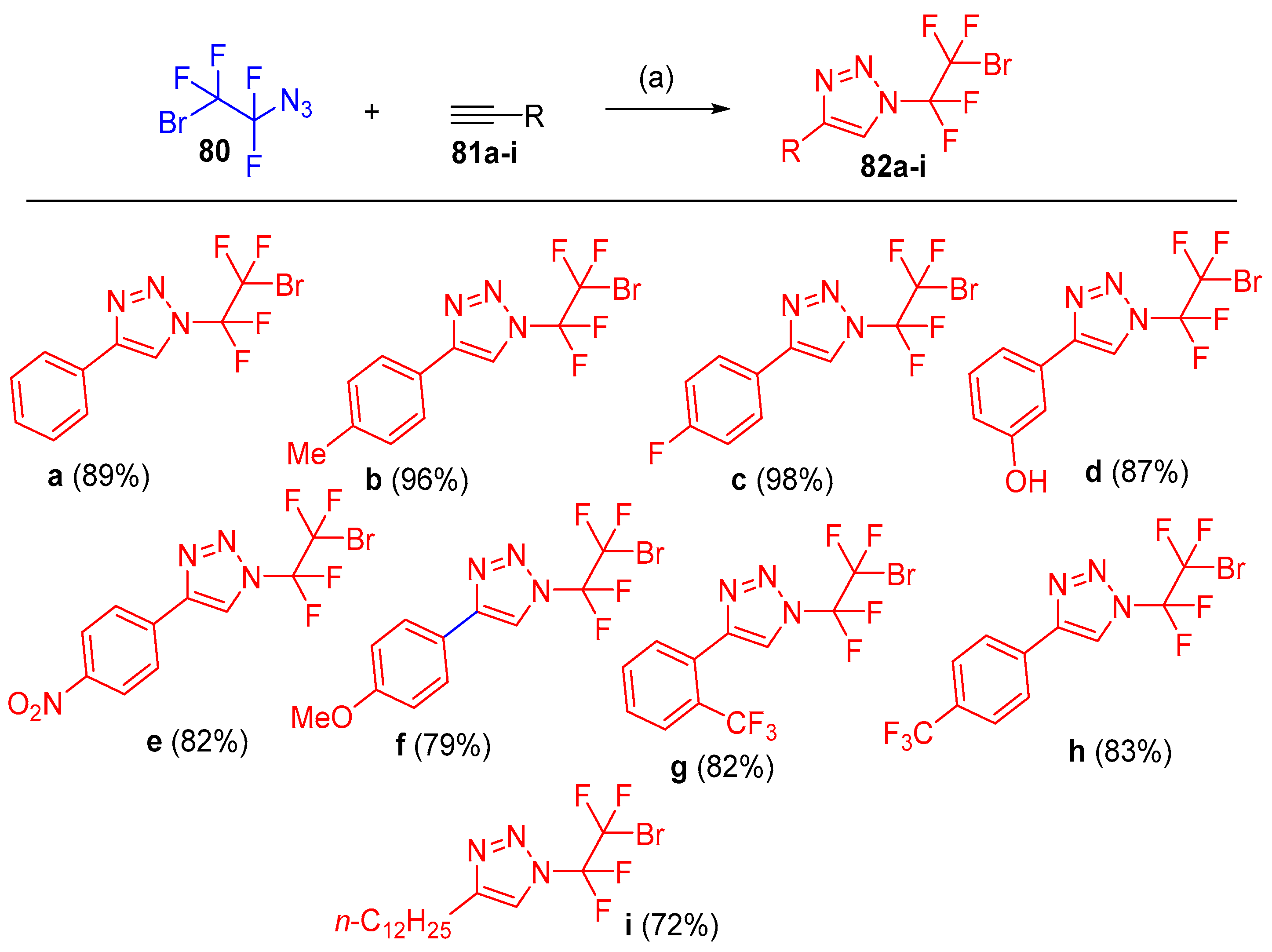
- Tichý, D.; Košťál, V.; Motornov, V.; Klimánková, I.; Beier, P. Preparation of 1-Azido-2-Bromo-1,1,2,2-Tetrafluoroethane and Its Use in the Synthesis of N-Fluoroalkylated Nitrogen Heterocycles. J. Org. Chem. 2020, 85, 11482–11489. [Google Scholar] [CrossRef]
- De Nino, A.; Merino, P.; Algieri, V.; Nardi, M.; Di Gioia, M.L.; Russo, B.; Tallarida, M.A.; Maiuolo, L. Synthesis of 1,5-Functionalized 1,2,3-Triazoles Using Ionic Liquid/Iron(III) Chloride as an Efficient and Reusable Homogeneous Catalyst. Catalysts 2018, 8, 364. [Google Scholar] [CrossRef]
- De Nino, A.; Algieri, V.; Tallarida, M.A.; Costanzo, P.; Pedrón, M.; Tejero, T.; Merino, P.; Maiuolo, L. Regioselective Synthesis of 1,4,5-Trisubstituted-1,2,3-Triazoles from Aryl Azides and Enaminones. Eur. J. Org. Chem. 2019, 2019, 5725–5731. [Google Scholar] [CrossRef]
- Zhang, L.-L.; Li, Y.-T.; Gao, T.; Guo, S.S.; Yang, B.; Meng, Z.-H.; Dai, Q.P.; Xu, Z.-B.; Wu, Q.P. Efficient Synthesis of Diverse 5-Thio- or 5-Selenotriazoles: One-Pot Multicomponent Reaction from Elemental Sulfur or Selenium. Synthesis 2019, 51, 4170–4182. [Google Scholar] [CrossRef] [Green Version]
- Taia, A.; Essaber, M.; Oubella, A.; Aatif, A.; Bodiguel, J.; Grégoire, B.J.; Itto, M.Y.A.; Morjani, H. Synthesis, characterization, and biological evaluation of new heterocyclic systems 1,2,3-triazole-isoxazoline from eugenol by the mixed condensation reactions. Synth. Commu. 2020, 50, 2052–2065. [Google Scholar] [CrossRef]
- Shafran, Y.M.; Silaichev, P.S.; Bakulev, V.A. β-(Cycloalkylamino)ethanesulfonyl azides as novel water-soluble reagents for the synthesis of diazo compounds and heterocycles. Chem. Heterocycl. Compd. 2019, 55, 1251–1261. [Google Scholar] [CrossRef]
- Stanciu, M.C.; Belei, D.; Bicu, E.; Tuchilus, C.G.; Nichifor, M. Novel amphiphilic dextran esters with antimicrobial activity. Internat. J. Biol. Macromol. 2020, 150, 746–755. [Google Scholar] [CrossRef]
- Denis, M.; Goldup, S.M. The active template approach to interlocked molecules. Nat. Rev. Chem. 2017, 1, 61. [Google Scholar] [CrossRef]
- Liu, Y.; Zhao, W.; Chen, C.-H.; Flood, A.H. Chloride capture using a C-H hydrogen-bonding cage. Science 2019, 365, 159–161. [Google Scholar]
- Hsueh, F.-C.; Tsai, C.-Y.; Lai, C.-C.; Liu, Y.-H.; Peng, S.-M.; Chiu, S.-H. N Heterocyclic Carbene Copper(I) Rotaxanes Mediate Sequential Click Ligations with All Reagents Premixed. Angew. Chem. 2020, 132, 11374–11378. [Google Scholar] [CrossRef]
- Boddu, L.; Potlapati, V.; Subhashini, N.J.P. Synthesis of novel isoindolone-based medium-sized macromolecules and triazole containing heterocyclic compounds. J. Heterocycl. Chem. 2019, 56, 3197–3205. [Google Scholar] [CrossRef]
- Gupta, A.; Sarkar, F.K.; Sarkar, R.; Jamatia, R.; Lee, C.Y.; Gupta, G.; Pal, A.K. Development of a new catalytic and sustainable methodology for the synthesis of benzodiazepine triazole scaffold using magnetically separable CuFe2O4@MIL-101(Cr) nano-catalyst in aqueous medium. Appl. Organomet. Chem. 2020, 34, e5782. [Google Scholar] [CrossRef]
- Sokolov, V.B.; Aksinenko, A.Y.; Goreva, T.V.; Epishina, T.A.; Bachurin, S.O. Modification of the drug “riluzole” with an alkyne-azide “click”-reaction with pharmacologically active fragments. Russ. Chem. Bull. Inter. 2019, 68, 2241–2244. [Google Scholar] [CrossRef]
- Pokhodylo, N.T.; Tupychak, M.A.; Shyyka, O.Y.; Obushak, M.D. Some Aspects of the Azide–Alkyne 1,3-Dipolar Cycloaddition Reaction. Russ. J. Org. Chem. 2019, 55, 1310–1321. [Google Scholar] [CrossRef]
- Mikhaylov, V.N.; Pavlov, A.O.; Ogorodnov, Y.V.; Spiridonova, D.V.; Sorokoumov, V.N.; Balova, I.A. N-Propargylation and copper(I)-catalyzed azide-alkyne cycloaddition as a convenient strategy for directed post-synthetic modification of 4-oxo-1,4-dihydrocinnoline derivatives. Chem. Heterocycl. Comp. 2020, 56, 915–922. [Google Scholar] [CrossRef]
- Filimonov, V.O.; Dianova, L.N.; Beryozkina, T.V.; Mazur, D.; Beliaev, N.A.; Volkova, N.N.; Ilkin, V.G.; Dehaen, W.; Lebedev, A.T.; Bakulev, V.A. Water/Alkali-Catalyzed Reactions of Azides with 2-Cyanothioacetamides. Eco-Friendly Synthesis of Monocyclic and Bicyclic 1,2,3-Thiadiazole-4-carbimidamides and 5-Amino-1,2,3-triazole-4-carbothioamides. J. Org. Chem. 2019, 84, 13430–13446. [Google Scholar] [CrossRef]
- Hussain, B.J.; Salim, A.T. Synthesis of mannich and triazole derivatives for barbiturate derivatives with some analytical applications. J. Phys. Conf. Series 2019, 1294, 052058–052073. [Google Scholar] [CrossRef] [Green Version]
- Kilpin, K.J.; Paul, U.S.D.; Lee, A.-L.; Crowley, J.D. Gold(I) ‘‘click’’ 1,2,3-triazolylidenes: Synthesis, self-assembly and catalysis. Chem. Commun. 2011, 47, 328–330. [Google Scholar] [CrossRef]
- Crowley, J.D.; Bandeen, P.H.; Hanton, L.R. A one pot multi-component CuAAC “click” approach to bidentate and tridentate pyridyl-1,2,3-triazole ligands: Synthesis, X-ray structures and copper(II) and silver(I) complexes. Polyhedron 2010, 29, 70. [Google Scholar] [CrossRef]
- Yan, X.; Wang, H.; Guo, S. Employing Aryl-Linked Bis-mesoionic Carbenes as a Pincer-Type Platform to A cess Ambient-Stable Palladium(IV) Complexes. Angew. Chem. Int. Ed. 2019, 58, 16907. [Google Scholar] [CrossRef]
- Yan, X.; Zhang, B.; Zhang, X.; Wang, H.; Duan, Y.-A.; Guo, S. Symmetrical and Non-symmetrical Pd (II) Pincer Complexes Bearing Mesoionic N-heterocyclic Thiones: Synthesis, Characterizations and Catalytic Properties. Appl. Organomet. Chem. 2020, 34, e5885. [Google Scholar] [CrossRef]
- Yoshida, S.; Shiraishi, A.; Kanno, K.; Matsushita, T.; Johmoto, K.; Uekusa, H.; Hosoya, T. Enhanced clickability of doubly sterically-hindered aryl azides. Sci. Rep. 2011, 1, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, S.; Goto, S.; Nishiyama, Y.; Hazama, Y.; Kondo, M.; Matsushita, T.; Hosoya, T. Effect of Resonance on the Clickability of Alkenyl Azides in the Strain-promoted Cycloaddition with Dibenzo-fused Cyclooctynes. Chem. Lett. 2019, 48, 1038–1041. [Google Scholar] [CrossRef] [Green Version]
- Ugi, I.; Meyr, R.; Isonitrile, V. Erweiterter Anwendungsbereich der Passerini-Reaktion. Chem. Ber. 1961, 94, 2229. [Google Scholar] [CrossRef]
- Neochoritis, C.G.; Zhao, T.; Dömling, A. Tetrazoles via Multicomponent Reactions. Chem. Rev. 2019, 119, 1970–2042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Q.-X.; Mu, Z.-Y.; Yao, G.; Zhang, J.-A.; He, H.-T.; Pang, Y.-L.; Xiong, J. Efficient Synthesis of 4H-3, 1-Benzoxazine Derivatives via One-Pot Sequential Passerini-Azide/Palladium-Catalyzed Azide–Isocyanide Coupling/Cyclization Reaction. Synlett 2020, 31, 1003–1006. [Google Scholar] [CrossRef]
- Morgalyuk, V.P.; Strelkova, T.S.; Kononevich, Y.N.; Brel, V.K. Synthesis of 1,3,3,5,5-Penta [1-(2,2-dimethyl-1,3-dioxolan-4-yl)methoxy]-1-[(1H-1,2,3-triazol-4-yl)alkoxy]cyclotriphosphazenes. Rus. J. Gen. Chem. 2019, 89, 1620–1624. [Google Scholar] [CrossRef]
- Naouri, A.; Djemoui, A.; Ouahran, M.R.; Lahrech, M.B.; Lemouari, N.; Rocha, D.H.A.; Albuquerque, H.; Mendes, R.F.; Paz, F.A.A.; Helguero, L.A.; et al. Multicomponent and 1,3-dipolar cycloaddition synthesis of triazoleand isoxazole-acridinedione/xanthenedione heterocyclic hybrids: Cytotoxic effects on human cancer cells. J. Mol. Struct. 2020, 1217, 128325–128333. [Google Scholar] [CrossRef]
- Deng, L.; Liu, Y.; Zhu, Y.; Wan, I.P. Transition-Metal-Free Annulation of Enamines and Tosyl Azide toward N-Heterocycle Fused and 5-Amino-1,2,3-Triazoles. Eur. J. Org. Chem. 2020, 2020, 5606–5609. [Google Scholar] [CrossRef]
- Gulevskaya, A.V.; Tyaglivy, A.S.; Pozharskii, A.F.; Nelina-Nemtseva, J.I.; Steglenko, D.V. Heterocyclization of Enediynes Promoted by Sodium Azide: A Case of Ambiguity of X-ray Data and Structure Revision. Org. Lett. 2014, 16, 1582–1585. [Google Scholar] [CrossRef]
- Ning, Y.; Wu, N.; Yu, H.; Liao, P.; Li, X.; Bi, X. Silver-Catalyzed Tandem Hydroazidation/Alkyne–Azide Cycloaddition of Diynes with TMS-N3: An Easy Access to 1,5-Fused 1,2,3-Triazole Frameworks. Org. Lett. 2015, 17, 2198–2201. [Google Scholar] [CrossRef]
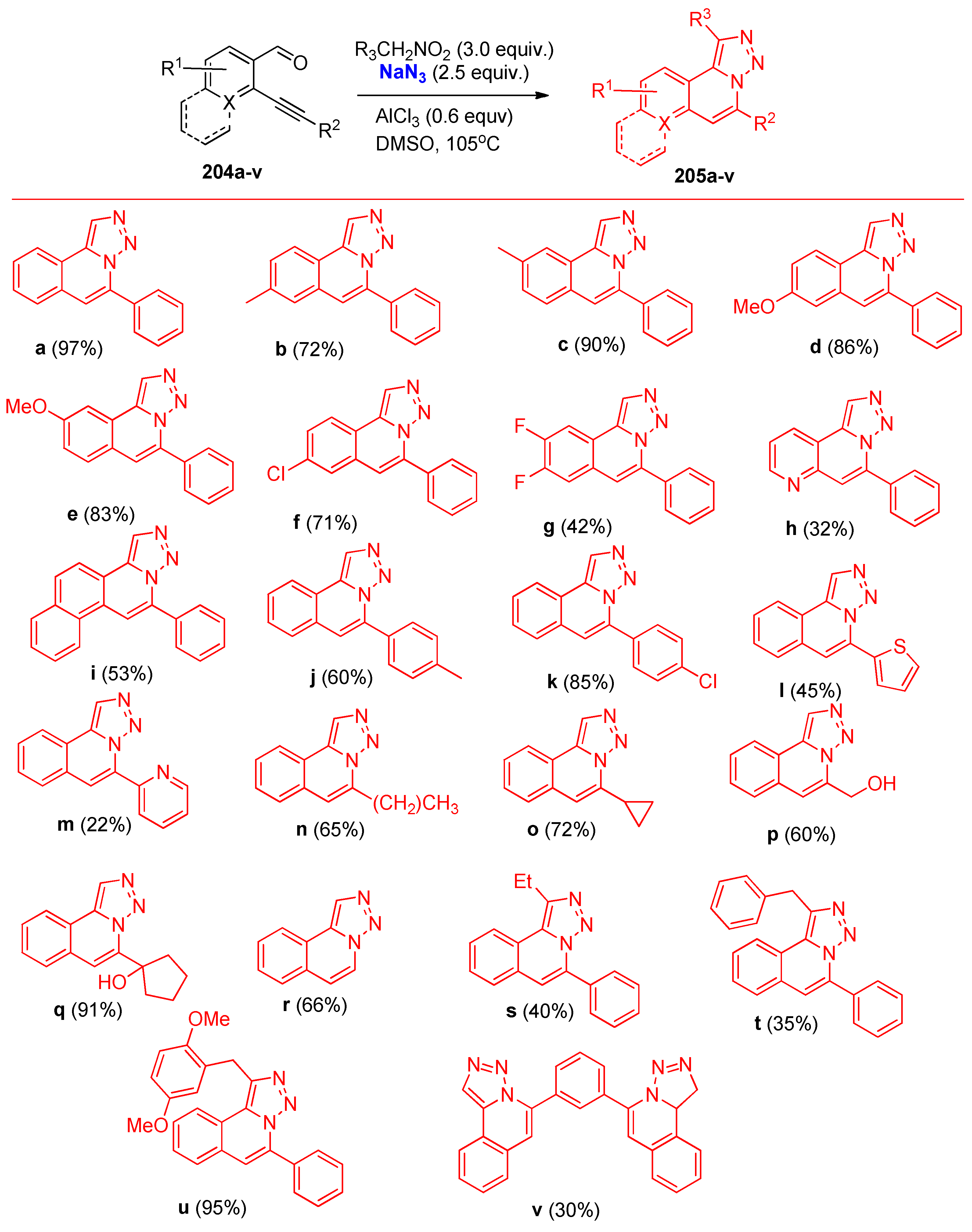
- Tao, S.; Hu, Q.; Li, H.; Ma, S.; Chen, Y. Synthesis of [1,2,3]Triazolo [5,1-a]isoquinoline Derivatives via a Selective Cascade Cyclization Sequence of 1,2-bis(Phenylethynyl)benzene Derivatives. Synth. Commun. 2015, 45, 1354–1361. [Google Scholar] [CrossRef]
- Zhao, S.; Yu, R.; Chen, W.; Liu, M.; Wu, H. Efficient Approach to Mesoionic Triazolo [5,1-a]isoquinolium through Rhodium-Catalyzed Annulation of Triazoles and Internal Alkynes. Org. Lett. 2015, 17, 2828–2831. [Google Scholar] [CrossRef] [PubMed]
- Fan, M.; Liu, Y.; Hu, Q.; Jia, L.; Chen, Y. Facile Synthesis of [1,2,3]Triazolo [5,1-a]isoquinolines through a Copper-Catalyzed Tandem Sonogashira Coupling/Cyclization Reaction. Eur. J. Org. Chem. 2016, 2016, 5470–5473. [Google Scholar] [CrossRef]
- Chen, Y.; Zhou, S.; Ma, S.; Liu, W.; Pan, Z.; Shi, X. A facile synthesis of 5- amino-[1,2,3]triazolo[5,1-a]isoquinoline derivatives through copper-catalyzed cascade reactions. Org. Biomol. Chem. 2013, 11, 8171–8174. [Google Scholar] [CrossRef]
- Hu, Y.Y.; Hu, J.; Wang, X.C.; Guo, L.N.; Shu, X.Z.; Niu, Y.N.; Liang, Y.M. Copper-catalyzed tandem synthesis of [1,2,3]triazolo [5,1-a]isoquinolines and their transformation to 1,3-disubstituted isoquinolines. Tetrahedron 2010, 66, 80–86. [Google Scholar] [CrossRef]
- Maurya, R.A.; Adiyala, P.R.; Chandrasekhar, D.; Reddy, C.N.; Kapure, J.S.; Kamal, A. Rapid Access to Novel 1,2,3-Triazolo-Heterocyclic Scaffolds via Tandem Knoevenagel Condensation/Azide–Alkyne 1,3-Dipolar Cycloaddition Reaction in One Pot. ACS Comb. Sci. 2014, 16, 466–477. [Google Scholar] [CrossRef]
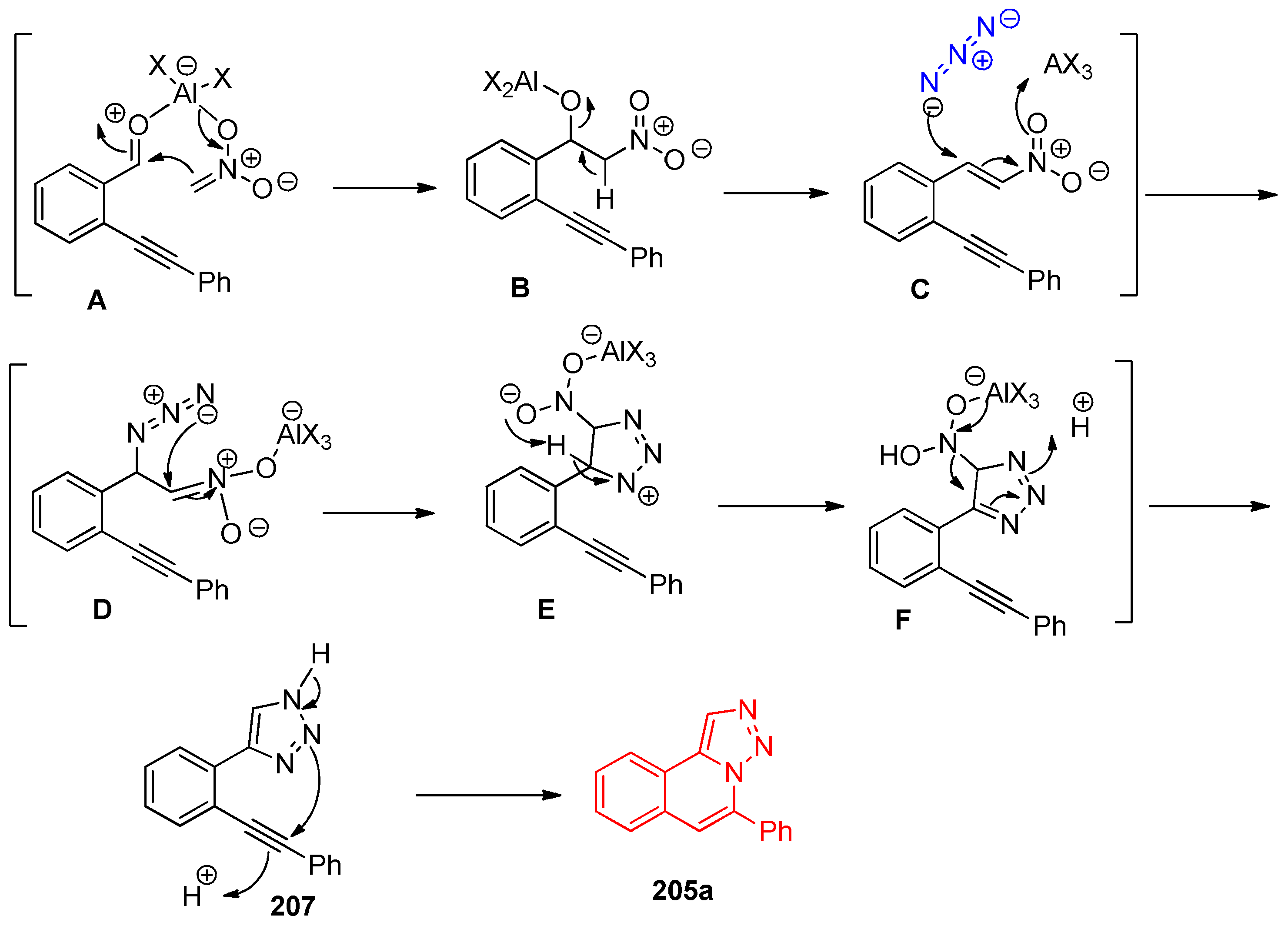
- Wu, Y.; Meng, Y.; Liu, Z.; Zhang, M.; Song, C. AlCl3-promoted three-component cascade reaction for rapid access to [1,2,3]triazolo [5,1-a]isoquinolines. Tetrahedron Lett. 2019, 60, 151287. [Google Scholar] [CrossRef]
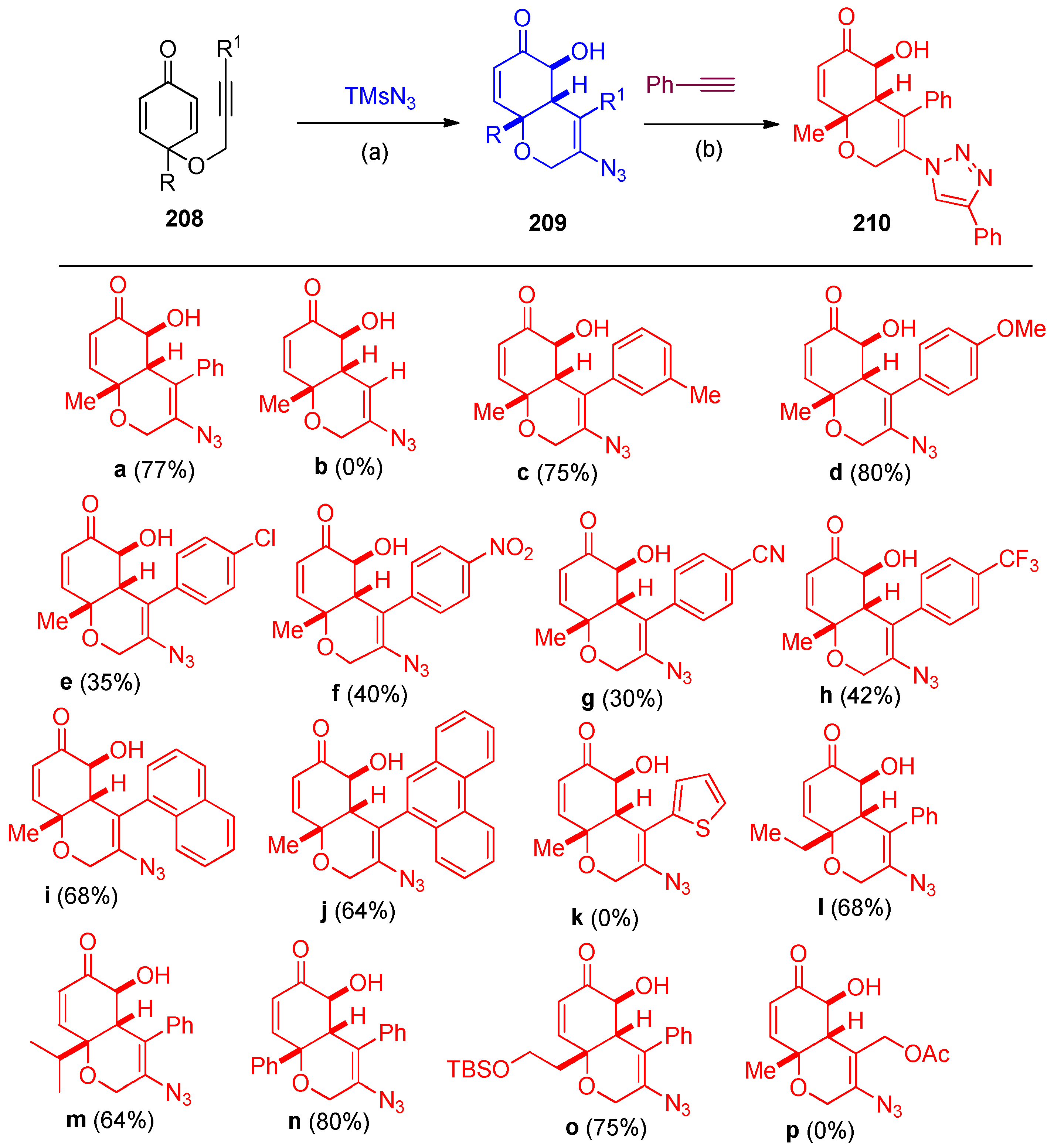
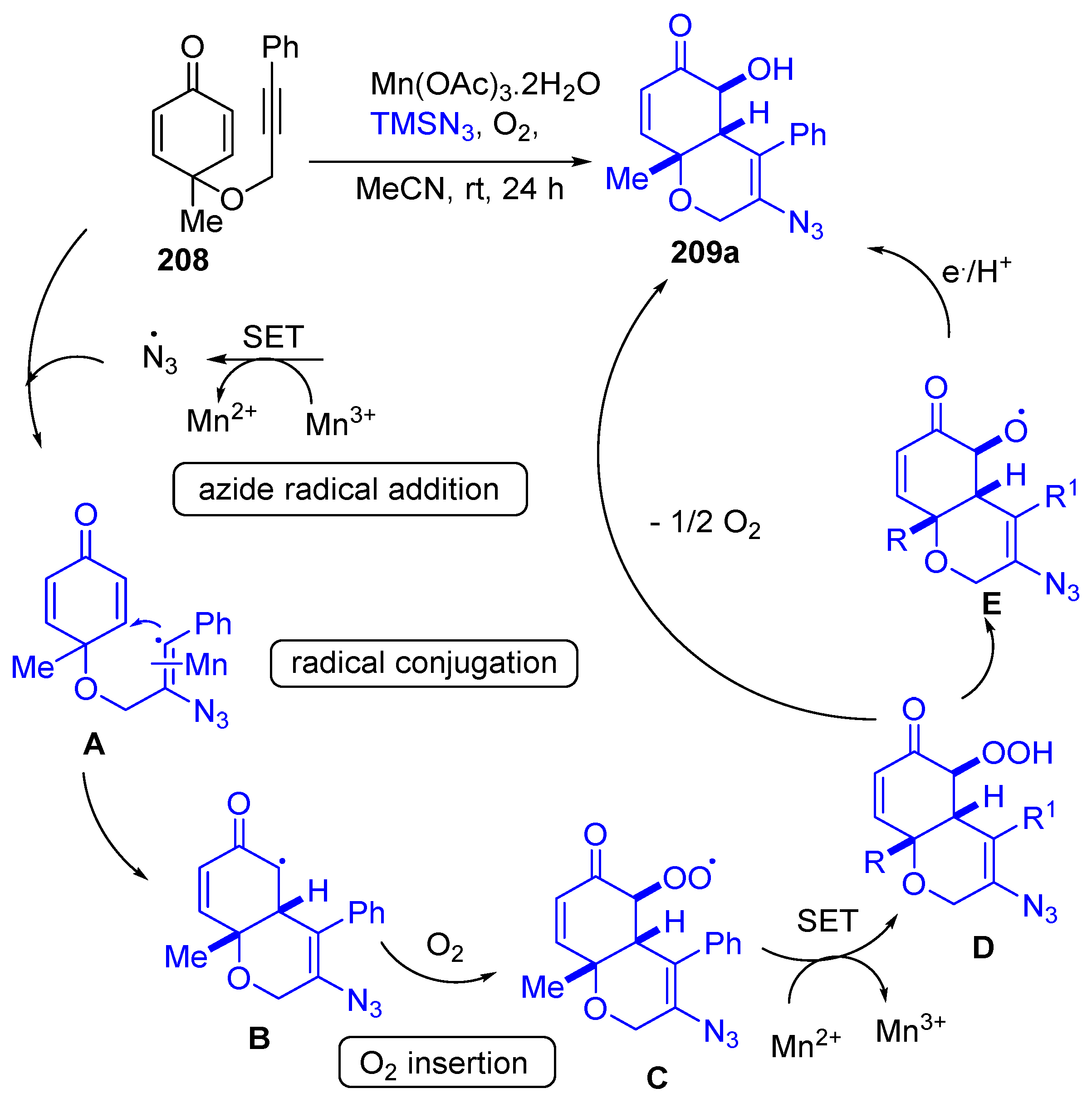
- Pal, P.; Mainkar, P.S.; Nayani, K.; Chandrasekhar, S. Mn-catalyzed radical initiated domino transformation of alkynylated cyclohexadienones with TMSN3 and O2 to bicyclic azido alcohols. Chem. Commun. 2020, 56, 3453–3456. [Google Scholar] [CrossRef]
- Gour, J.; Gatadi, S.; Akunuri, R.; Yaddanapudi, M.V.; Nengroo, M.A.; Datta, D.; Chopra, S.; Nanduri, S. Catalyst-free facile synthesis of polycyclic indole/pyrrole substituted-1,2,3-triazoles. Org. Biomol. Chem. 2019, 17, 8153–8165. [Google Scholar] [CrossRef]
- Yan, Y.-M.; Li, H.-Y.; Zhang, M.; Wang, R.-X.; Zhou, C.-G.; Ren, Z.-X.; Ding, M.-W. One-Pot Synthesis of [1,2,3]Triazolo [1,5-a]quinoxalin-4(5H)-ones by a Metal-Free Sequential Ugi-4CR/Alkyne–Azide Cycloaddition Reaction. Synlett 2020, 31, 73–76. [Google Scholar] [CrossRef]
- Gangaprasad, D.; Raj, J.P.; Karthikeyan, K.; Rengasamy, R.; Kesavan, M.; Vajjiravel, M.; Elangovan, J. A new route to 1,2,3-triazole fused benzooxazepine and benzodiazepine analogues through metal-free intramolecular azide-olefin oxidative cycloaddition. Tetrahedron Lett. 2019, 60, 151164–151166. [Google Scholar] [CrossRef]
- Alshammari, M.B.; Aly, A.A.; Brown, A.B.; Bakht, M.A.; Shawky, A.M.; Abdelhakem, A.M.; El-Sheref, E.M. An Efficient Click Synthesis of Chalcones Derivatized with Two 1-(2-quinolon-4-yl)-1,2,3-triazoles. Z. Für Nat. 2021, 76, 395–403. [Google Scholar] [CrossRef]
- El-Sheref, E.M.; Aly, A.A.; Alshammari, M.B.; Brown, A.B.; Abdel-Hafez, S.M.N. Design, Synthesis, Molecular Docking, Antiapoptotic and Caspase-3 Inhibition of New 1,2,3-Triazole/Bis-2(1H)-Quinolinone Hybrids. Molecules 2020, 25, 5057. [Google Scholar] [CrossRef] [PubMed]
- Voitekhovich, S.V.; Grigoriev, Y.V.; Grigoriev, Y.V.; Lyakhov, A.S.; Matulis, V.E.; Ivashkevich, L.S.; Bogomyakov, A.S.; Lavrenova, L.G.; Ivashkevich, O.A. 1-(1,2,4-Triazol-3-yl)-1H-tetrazoles and their complexation with copper(II) chloride. Polyhedron 2020, 176, 114299. [Google Scholar] [CrossRef]
- Abdullahi, J.A.; Rabeaa, M.A. Grinding-Assisted Synthesis of Some Heterocyclic Compounds. Asian J. Chem. 2020, 32, 1713–1718. [Google Scholar] [CrossRef]
- Sarrafiouna, F.; Jamehbozorgib, S.; Ramezani, M. Synthesis of Tetrazoles Catalyzed by Novel Cobalt Magnetic Nanoparticles. Russ. J. Org. Chem. 2019, 55, 1777–1784. [Google Scholar] [CrossRef]
- Saber, A.F.; Kamal El-Dean, A.M.; Redwan, S.M.; Zaki, R.M. Synthesis, spectroscopic characterization, and in vitro antimicrobial activity of fused pyrazolo [40,30:4,5]thieno [3,2-d]pyrimidine. J. Chin. Chem. Soc. 2020, 67, 1239–1246. [Google Scholar] [CrossRef]
- Sajjadi, M.; Nasrollahzadeh, M.; Sajadi, S.M. Green synthesis of Ag/Fe3O4 nanocomposite using Euphorbia peplus Linn leaf extract and evaluation of its catalytic activity. J. Colloid Interface Sci. 2017, 497, 1–13. [Google Scholar] [CrossRef]
- Nasrollahzadeh, M.; Sajjadi, M.; Varma, R.S. Magnetically recoverable nanocatalyst based on N-heterocyclic ligands: Efficient treatment of environmental pollutants in aqueous media. Clean Technol. Environ. Policy 2020, 22, 423–440. [Google Scholar] [CrossRef]
- Trose, M.; Nahra, F.; Cordes, D.B.; Slawin, A.M.Z.; Cazin, C.S.I. Cu–NHC azide complex: Synthesis and reactivity. Chem. Commun. 2019, 55, 12068–12071. [Google Scholar] [CrossRef]
- Singam, M.K.R.; Nagireddy, A.; Rajesh, M.; Ganesha, V.; Reddy, M.S. Ni-Catalyzed electrophile driven regioselective arylative cyclization of ortho-functional diaryl acetylenes for the synthesis of pyridine and indene derivatives. Org. Chem. Front. 2020, 7, 30–34. [Google Scholar] [CrossRef]
- Nie, B.; Wu, W.; Zeng, W.; Ren, Q.; Zhang, J.; Zhang, Y.; Jiang, H. Synthesis of Isoquinoline Derivatives via Palladium-Catalyzed C-H/C-N Bond Activation of N-Acyl Hydrazones with α-Substituted Vinyl Azides. Adv. Synth. Catal. 2020, 362, 1362–1369. [Google Scholar] [CrossRef]
- Wei, K.; Yang, T.; Chen, Q.; Liang, S.; Yu, W. Iron-catalysed 1,2-aryl migration of tertiary azides. Chem. Commun. 2020, 56, 11685–11688. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Liu, D.; Zhou, B.; Liu, Y. One-Pot Copper-Catalyzed Three-Component Reaction of Sulfonyl Azides, Alkynes, and Allylamines To Access 2,3-Dihydro-1H-imidazo [1,2-a]indoles. Synthesis 2020, 52, 1417–1424. [Google Scholar] [CrossRef]
- Kumar, G.R.Y.; Begum, N.S.; Imran, K.M. Mn-mediated oxidative radical cyclization of 2-(azidomethyl)phenyl isocyanides with carbazate: Access to quinazoline-2-carboxylates. New J. Chem. 2020, 44, 7001–7006. [Google Scholar] [CrossRef]
- Prieschl, D.; Arrowsmith, M.; Dietz, M.; Rempel, A.; Müller, M.; Braunschweig, H. Synthesis of polyheterocyclic 1,1-diboryltriazenes by γ-nitrogen insertion of azides into activated B–B single bonds. Chem. Commun. 2020, 56, 5681–5684. [Google Scholar] [CrossRef]
- LaFortune, J.H.W.; Trofimova, A.; Cummings, H.; Westcott, S.A.; Stephan, D.W. Phosphinoboration of Diazobenzene: Intramolecular FLP Synthon for PN2B-Derived Heterocycles. Chem. Eur. J. 2019, 25, 12521–12525. [Google Scholar] [CrossRef]
- Drescher, R.; Lin, S.; Hofmann, A.; Lenczyk, C.; Kachel, S.; Krummenacher, L.Z.; Braunschweig, H. Ring expansion of alumoles with organic azides: Selective formation of six-membered aluminum–nitrogen heterocycles. Chem. Sci. 2020, 11, 5559–5564. [Google Scholar] [CrossRef]
- Marchenko, A.; Koidan, G.; Hurieva, A.N.; Shishkina, S.; Rusanov, E.; Kostyuk, A. “Ring-enlargement of N-phosphanyl-1,2,3,4-tetrahydroquinazolines. J. Org. Chem. 2020, 85, 14467–14472. [Google Scholar] [CrossRef]
- Luo, W.; Wang, Z.; Cao, X.; Liang, D.; Wei, M.; Yin, K.; Li, L. Construction of Benzo-1,2,3-thiazaphosphole Heterocycles by Annulations of ortho-Phosphinoarenesulfonyl Fluorides with Trimethylsilyl Azide. J. Org. Chem. 2020, 85, 14785–14794. [Google Scholar] [CrossRef]
- Schock, M.; Bräse, S. Reactive & Efficient: Organic Azides as Cross-Linkers in Material Sciences. Molecules 2020, 25, 1009. [Google Scholar]
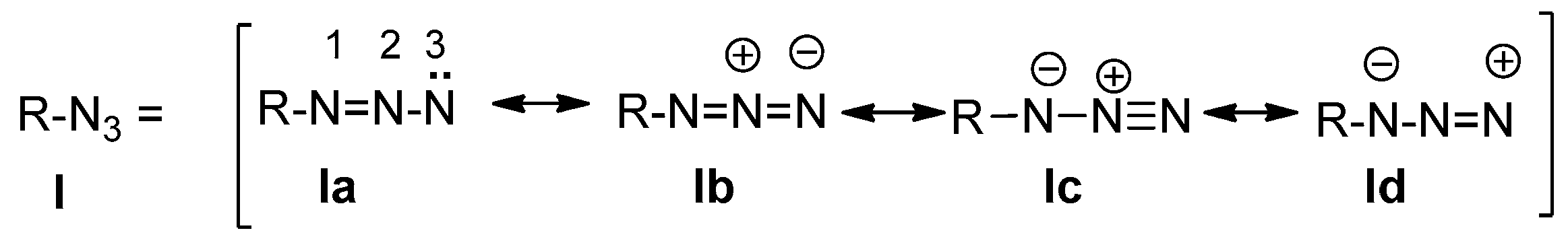


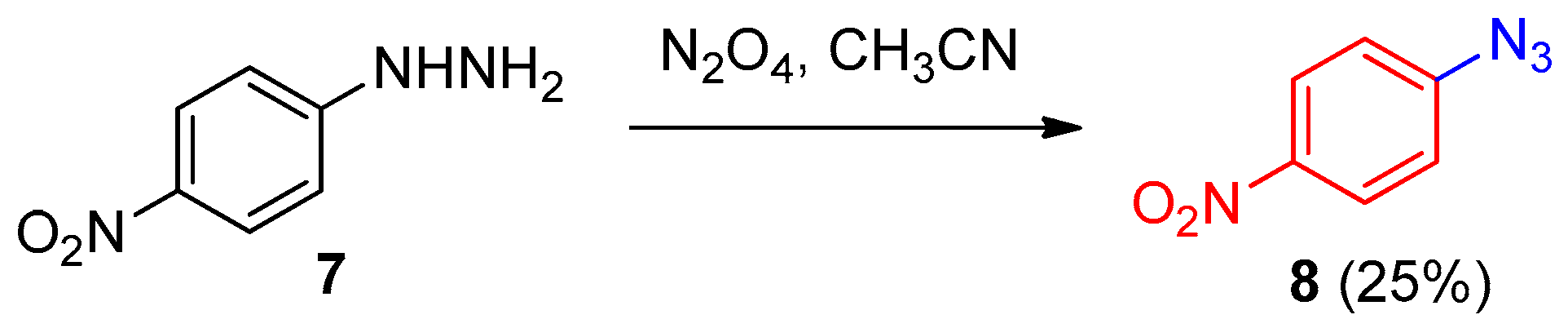






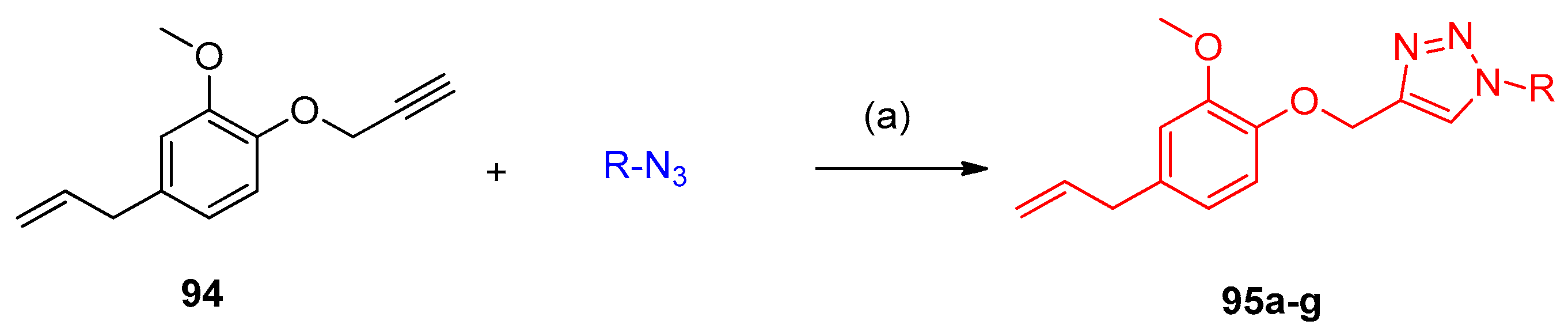















Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nayl, A.A.; Aly, A.A.; Arafa, W.A.A.; Ahmed, I.M.; Abd-Elhamid, A.I.; El-Fakharany, E.M.; Abdelgawad, M.A.; Tawfeek, H.N.; Bräse, S. Azides in the Synthesis of Various Heterocycles. Molecules 2022, 27, 3716. https://doi.org/10.3390/molecules27123716
Nayl AA, Aly AA, Arafa WAA, Ahmed IM, Abd-Elhamid AI, El-Fakharany EM, Abdelgawad MA, Tawfeek HN, Bräse S. Azides in the Synthesis of Various Heterocycles. Molecules. 2022; 27(12):3716. https://doi.org/10.3390/molecules27123716
Chicago/Turabian StyleNayl, AbdElAziz A., Ashraf A. Aly, Wael A. A. Arafa, Ismail M. Ahmed, Ahmed I. Abd-Elhamid, Esmail M. El-Fakharany, Mohamed A. Abdelgawad, Hendawy N. Tawfeek, and Stefan Bräse. 2022. "Azides in the Synthesis of Various Heterocycles" Molecules 27, no. 12: 3716. https://doi.org/10.3390/molecules27123716
APA StyleNayl, A. A., Aly, A. A., Arafa, W. A. A., Ahmed, I. M., Abd-Elhamid, A. I., El-Fakharany, E. M., Abdelgawad, M. A., Tawfeek, H. N., & Bräse, S. (2022). Azides in the Synthesis of Various Heterocycles. Molecules, 27(12), 3716. https://doi.org/10.3390/molecules27123716