
Effects of Smoking on Inflammatory-Related Cytokine Levels in Human Serum

Abstract
1. Introduction
2. Materials and Methods
2.1. Study Design and Participants
2.2. Serum Isolation
2.3. Assessment of Inflammatory Markers
2.4. Statistical Analysis
3. Results
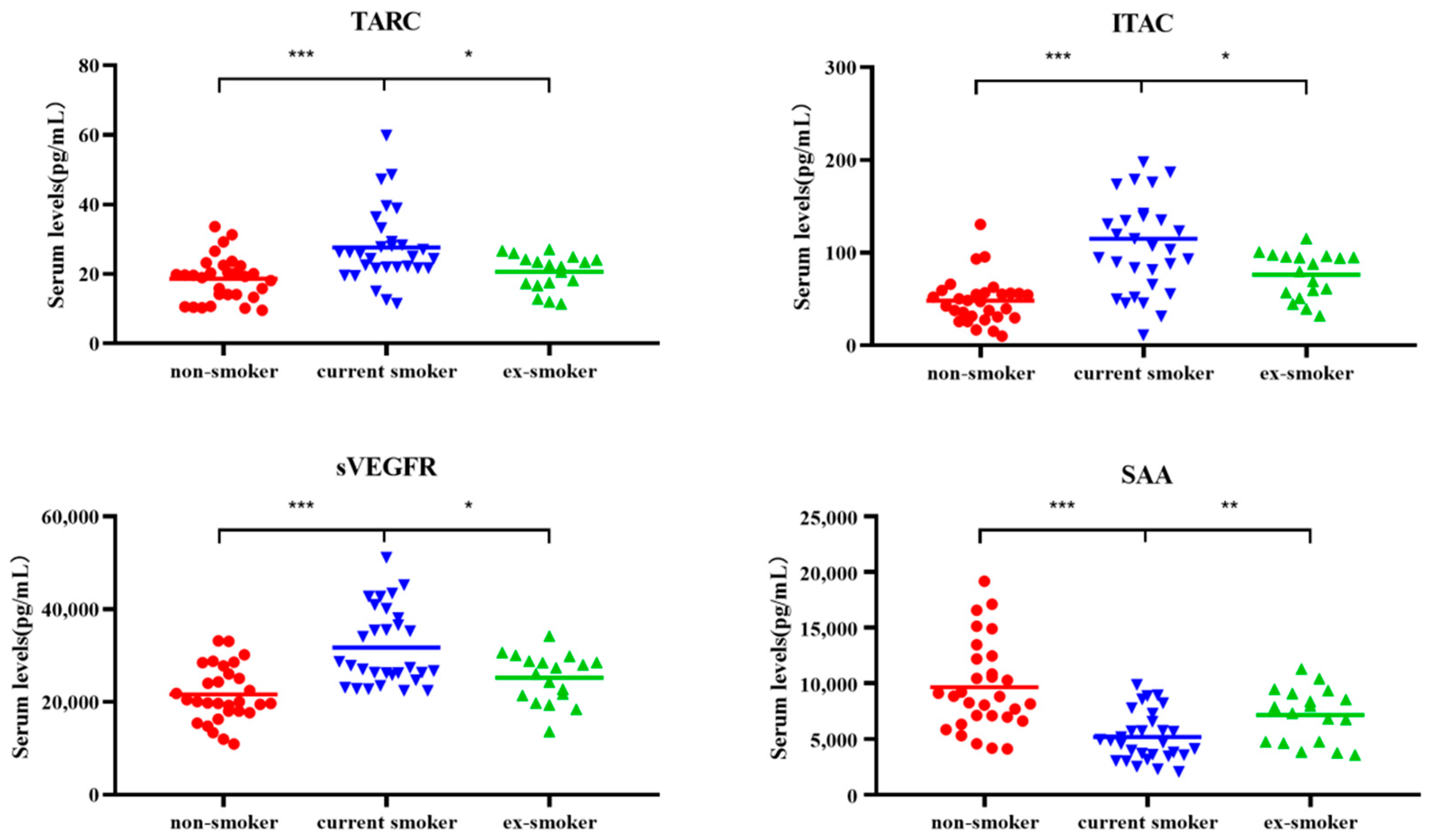
3.1. Cytokine Levels Significantly Increased in Non-Smokers, Current Smokers, and Ex-Smokers
3.2. Changes in Four Types of Cytokines
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Wong, M.M.; Fish, E.N. Chemokines: Attractive mediators of the immune response. Semin. Immunol. 2003, 15, 5–14. [Google Scholar] [CrossRef]
- Furman, D.; Campisi, J.; Verdin, E.; Carrera-Bastos, P.; Targ, S.; Franceschi, C.; Ferrucci, L.; Gilroy, D.W.; Fasano, A.; Miller, G.W.; et al. Chronic inflammation in the etiology of disease across the life span. Nat. Med. 2019, 25, 1822–1832. [Google Scholar] [CrossRef] [PubMed]
- Cooper, G.S.; Bynum, M.L.; Somers, E.C. Recent insights in the epidemiology of autoimmune diseases: Improved prevalence estimates and understanding of clustering of diseases. J. Autoimmun. 2009, 33, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Hartel, C.; Adam, N.; Strunk, T.; Temming, P.; Muller-Steinhardt, M.; Schultz, C. Cytokine responses correlate differentially with age in infancy and early childhood. Clin. Exp. Immunol. 2005, 142, 446–453. [Google Scholar] [CrossRef]
- Benahmed, M.; Meresse, B.; Arnulf, B.; Barbe, U.; Mention, J.J.; Verkarre, V.; Allez, M.; Cellier, C.; Hermine, O.; Cerf-Bensussan, N. Inhibition of TGF-beta signaling by IL-15: A new role for IL-15 in the loss of immune homeostasis in celiac disease. Gastroenterology 2007, 132, 994–1008. [Google Scholar] [CrossRef]
- Lee, J.; Taneja, V.; Vassallo, R. Cigarette smoking and inflammation: Cellular and molecular mechanisms. J. Dent. Res. 2012, 91, 142–149. [Google Scholar] [CrossRef]
- Oberg, M.; Jaakkola, M.S.; Woodward, A.; Peruga, A.; Pruss-Ustun, A. Worldwide burden of disease from exposure to second-hand smoke: A retrospective analysis of data from 192 countries. Lancet 2011, 377, 139–146. [Google Scholar] [CrossRef]
- Mathers, C.D.; Loncar, D. Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med. 2006, 3, e442. [Google Scholar] [CrossRef]
- Thun, M.J.; DeLancey, J.O.; Center, M.M.; Jemal, A.; Ward, E.M. The global burden of cancer: Priorities for prevention. Carcinogenesis 2010, 31, 100–110. [Google Scholar] [CrossRef]
- Ezzati, M.; Lopez, A.D. Estimates of global mortality attributable to smoking in 2000. Lancet 2003, 362, 847–852. [Google Scholar] [CrossRef]
- Sokolowska, M.; Quesniaux, V.F.J.; Akdis, C.A.; Chung, K.F.; Ryffel, B.; Togbe, D. Acute Respiratory Barrier Disruption by Ozone Exposure in Mice. Front. Immunol. 2019, 10, 2169. [Google Scholar] [CrossRef] [PubMed]
- Durazzo, T.C.; Mattsson, N.; Weiner, M.W.; Alzheimer’s Disease Neuroimaging Initiative. Smoking and increased Alzheimer’s disease risk: A review of potential mechanisms. Alzheimers Dement. 2014, 10 (Suppl. 3), S122–S245. [Google Scholar] [CrossRef] [PubMed]
- Shein, M.; Jeschke, G. Comparison of Free Radical Levels in the Aerosol from Conventional Cigarettes, Electronic Ciga-rettes, and Heat-Not-Burn Tobacco Products. Chem. Res. Toxicol. 2019, 32, 1289–1298. [Google Scholar] [CrossRef] [PubMed]
- Bergstrom, J. Tobacco smoking and chronic destructive periodontal disease. Odontology 2004, 92, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Qiu, F.; Liang, C.L.; Liu, H.; Zeng, Y.Q.; Hou, S.; Huang, S.; Lai, X.; Dai, Z. Impacts of cigarette smoking on immune responsiveness: Up and down or upside down? Oncotarget 2017, 8, 268–284. [Google Scholar] [CrossRef] [PubMed]
- Heyn, J.; Luchting, B.; Azad, S.C. Smoking Associated T-Cell Imbalance in Patients with Chronic Pain. Nicotine Tob. Res. 2020, 22, 111–117. [Google Scholar] [CrossRef]
- Andersen, A.M.; Lei, M.K.; Beach, S.R.H.; Philibert, R.A.; Sinha, S.; Colgan, J.D. Cigarette and Cannabis Smoking Effects on GPR15+ Helper T Cell Levels in Peripheral Blood: Relationships with Epigenetic Biomarkers. Genes 2020, 11, 149. [Google Scholar] [CrossRef]
- Hartmann, T.; Marino, F.; Duffield, R. Tobacco smoking and acute exercise on immune-inflammatory responses among relative short and longer smoking histories. Cytokine 2019, 123, 154754. [Google Scholar] [CrossRef]
- Jamil, A.; Rashid, A.; Majeed, A. Correlation between Genotoxicity and Interleukin-6 in Smokers: A Rodent Model. J. Coll. Physicians Surg. Pak. 2018, 28, 821–823. [Google Scholar] [CrossRef]
- Gonzalez-Quintela, A.; Alende, R.; Gude, F.; Campos, J.; Rey, J.; Meijide, L.M.; Fernandez-Merino, C.; Vidal, C. Serum levels of immunoglobulins (IgG, IgA, IgM) in a general adult population and their relationship with alcohol consumption, smoking and common metabolic abnormalities. Clin. Exp. Immunol. 2008, 151, 42–50. [Google Scholar] [CrossRef]
- Yao, L.K.; Liu, G.N.; Huang, S.M.; Li, W.T.; Li, Y. Relationship between expression of HDAC2, IL-8, TNF-alpha in lung adenocarcinoma tissues and smoking. Natl. Med. J. China 2016, 96, 1410–1413. [Google Scholar]
- Pace, E.; Di Vincenzo, S.; Di Salvo, E.; Genovese, S.; Dino, P.; Sangiorgi, C.; Ferraro, M.; Gangemi, S. MiR-21 upregulation increases IL-8 expression and tumorigenesis program in airway epithelial cells exposed to cigarette smoke. J. Cell. Physiol. 2019, 234, 22183–22194. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Li, J.; Wang, L.; Zhang, Q. Association between IL-1beta, IL-8, and IL-10 polymorphisms and risk of acute pancreatitis. Genet. Mol. Res. 2015, 14, 6635–6641. [Google Scholar] [CrossRef] [PubMed]
- Griffith, J.W.; Sokol, C.L.; Luster, A.D. Chemokines and chemokine receptors: Positioning cells for host defense and immunity. Annu. Rev. Immunol. 2014, 32, 659–702. [Google Scholar] [CrossRef] [PubMed]
- Romagnani, P.; Rotondi, M.; Lazzeri, E.; Lasagni, L.; Francalanci, M.; Buonamano, A.; Milani, S.; Vitti, P.; Chiovato, L.; Tonacchera, M.; et al. Expression of IP-10/CXCL10 and MIG/CXCL9 in the thyroid and increased levels of IP-10/CXCL10 in the serum of patients with recent-onset Graves’ disease. Am. J. Pathol. 2002, 161, 195–206. [Google Scholar] [CrossRef]
- Yun, J.J.; Fischbein, M.P.; Whiting, D.; Irie, Y.; Fishbein, M.C.; Burdick, M.D.; Belperio, J.; Strieter, R.M.; Laks, H.; Berliner, J.A.; et al. The role of MIG/CXCL9 in cardiac allograft vasculopathy. Am. J. Pathol. 2002, 161, 1307–1313. [Google Scholar] [CrossRef][Green Version]
- Maruoka, H.; Inoue, D.; Takiuchi, Y.; Nagano, S.; Arima, H.; Tabata, S.; Matsushita, A.; Ishikawa, T.; Oita, T.; Takahashi, T. IP-10/CXCL10 and MIG/CXCL9 as novel markers for the diagnosis of lymphoma-associated hemophagocytic syndrome. Ann. Hematol. 2014, 93, 393–401. [Google Scholar] [CrossRef]
- Semba, T.; Nishimura, M.; Nishimura, S.; Ohara, O.; Ishige, T.; Ohno, S.; Nonaka, K.; Sogawa, K.; Satoh, M.; Sawai, S.; et al. The FLS (fatty liver Shionogi) mouse reveals local expressions of lipocalin-2, CXCL1 and CXCL9 in the liver with non-alcoholic steatohepatitis. BMC Gastroenterol. 2013, 13, 120. [Google Scholar] [CrossRef]
- Onor, I.O.; Stirling, D.L.; Williams, S.R.; Bediako, D.; Borghol, A.; Harris, M.B.; Darensburg, T.B.; Clay, S.D.; Okpechi, S.C.; Sarpong, D.F. Clinical Effects of Cigarette Smoking: Epidemiologic Impact and Review of Pharmacotherapy Options. Int. J. Environ. Res. Public Health 2017, 14, 1147. [Google Scholar] [CrossRef]
- Shiels, M.S.; Katki, H.A.; Freedman, N.D.; Purdue, M.P.; Wentzensen, N.; Trabert, B.; Kitahara, C.M.; Furr, M.; Li, Y.; Kemp, T.J.; et al. Cigarette smoking and variations in systemic immune and inflammation markers. J. Natl. Cancer. Inst. 2014, 106, dju294. [Google Scholar] [CrossRef]
- Chahal, N.; McLain, A.C.; Ghassabian, A.; Michels, K.A.; Bell, E.M.; Lawrence, D.A.; Yeung, E.H. Maternal Smoking and Newborn Cytokine and Immunoglobulin Levels. Nicotine Tob. Res. 2017, 19, 789–796. [Google Scholar] [CrossRef] [PubMed]
- Schett, G. Physiological effects of modulating the interleukin-6 axis. Rheumatology 2018, 57 (Suppl. 2), ii43–ii50. [Google Scholar] [CrossRef] [PubMed]
- Rose-John, S. The Soluble Interleukin 6 Receptor: Advanced Therapeutic Options in Inflammation. Clin. Pharmacol. Ther. 2017, 102, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Karatayli, E.; Hall, R.A.; Weber, S.N.; Dooley, S.; Lammert, F. Effect of alcohol on the interleukin 6-mediated inflammatory response in a new mouse model of acute-on-chronic liver injury. Biochim. Biophys. Acta. Mol. Basis. Dis. 2019, 1865, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Speyer, C.B.; Costenbader, K.H. Cigarette smoking and the pathogenesis of systemic lupus erythematosus. Expert Rev. Clin. Immunol. 2018, 14, 481–487. [Google Scholar] [CrossRef]
- Xian-jin, Z.; Dong-hui, S. I-TAC: A Novel Chemokine. Int. J. Med. Microbiol. 2006, 29, 330–334. [Google Scholar]
- Albuquerque, R.J. The newest member of the VEGF family. Blood 2013, 121, 4015–4016. [Google Scholar] [CrossRef]
- Kanefendt, F.; Lindauer, A.; Mross, K.; Fuhr, U.; Jaehde, U. Determination of soluble vascular endothelial growth factor receptor 3 (sVEGFR-3) in plasma as pharmacodynamic biomarker. J. Pharm. Biomed. Anal. 2012, 70, 485–491. [Google Scholar] [CrossRef]
- Takahashi, K.; Mizukami, H.; Saga, Y.; Takei, Y.; Urabe, M.; Kume, A.; Machida, S.; Fujiwara, H.; Suzuki, M.; Ozawa, K. Suppression of lymph node and lung metastases of endometrial cancer by muscle-mediated expression of soluble vascular endothelial growth factor receptor-3. Cancer Sci. 2013, 104, 1107–1111. [Google Scholar] [CrossRef]
- Su, S.B.; Gong, W.; Gao, J.L.; Shen, W.; Murphy, P.M.; Oppenheim, J.J.; Wang, J.M. A seven-transmembrane, G pro-tein-coupled receptor, FPRL1, mediates the chemotactic activity of serum amyloid A for human phagocytic cells. J. Exp. Med. 1999, 189, 395–402. [Google Scholar] [CrossRef]
- He, R.; Sang, H.; Ye, R.D. Serum amyloid A induces IL-8 secretion through a G protein-coupled receptor, FPRL1/LXA4R. Blood 2003, 101, 1572–1581. [Google Scholar] [CrossRef] [PubMed]
- Bozinovski, S.; Uddin, M.; Vlahos, R.; Thompson, M.; McQualter, J.L.; Merritt, A.S.; Wark, P.A.; Hutchinson, A.; Irving, L.B.; Levy, B.D.; et al. Serum amyloid A opposes lipoxin A4 to mediate glucocorticoid refractory lung inflammation in chronic obstructive pulmonary disease. Proc. Natl. Acad. Sci. USA 2012, 109, 935–940. [Google Scholar] [CrossRef] [PubMed]
- Paffenbarger, R.S., Jr.; Hyde, R.T.; Wing, A.L.; Hsieh, C.C. Physical activity, all-cause mortality, and longevity of college alumni. N. Engl. J. Med. 1986, 314, 605–613. [Google Scholar] [CrossRef]
- Bendlin, B.B.; Carlsson, C.M.; Gleason, C.E.; Johnson, S.C.; Sodhi, A.; Gallagher, C.L.; Puglielli, L.; Engelman, C.D.; Ries, M.L.; Xu, G.; et al. Midlife predictors of Alzheimer’s disease. Maturitas 2010, 65, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Krintus, M.; Kozinski, M.; Kubica, J.; Sypniewska, G. Critical appraisal of inflammatory markers in cardiovascular risk stratification. Crit. Rev. Clin. Lab. Sci. 2014, 51, 263–279. [Google Scholar] [CrossRef]
- Arnson, Y.; Shoenfeld, Y.; Amital, H. Effects of tobacco smoke on immunity, inflammation and autoimmunity. J. Autoimmun. 2010, 34, J258–J265. [Google Scholar] [CrossRef]
- House, I.G.; Savas, P.; Lai, J.; Chen, A.X.Y.; Oliver, A.J.; Teo, Z.L.; Todd, K.L.; Henderson, M.A.; Giuffrida, L.; Petley, E.V.; et al. Macrophage-Derived CXCL9 and CXCL10 Are Required for Antitumor Immune Responses Following Immune Checkpoint Blockade. Clin. Cancer Res. 2020, 26, 487–504. [Google Scholar] [CrossRef]
- Koper, O.M.; Kaminska, J.; Sawicki, K.; Kemona, H. CXCL9, CXCL10, CXCL11, and their receptor (CXCR3) in neuroinflammation and neurodegeneration. Adv. Clin. Exp. Med. 2018, 27, 849–856. [Google Scholar] [CrossRef]
- Bonecchi, R.; Galliera, E.; Borroni, E.M.; Corsi, M.M.; Locati, M.; Mantovani, A. Chemokines and chemokine receptors: An overview. Front. Biosci. 2009, 14, 540–551. [Google Scholar] [CrossRef]
- Pekarek, V.; Srinivas, S.; Eskdale, J.; Gallagher, G. Interferon lambda-1 (IFN-lambda1/IL-29) induces ELR(-) CXC chem-okine mRNA in human peripheral blood mononuclear cells, in an IFN-gamma-independent manner. Genes Immun. 2007, 8, 177–180. [Google Scholar] [CrossRef]
- Sheppard, P.; Kindsvogel, W.; Xu, W.; Henderson, K.; Schlutsmeyer, S.; Whitmore, T.E.; Kuestner, R.; Garrigues, U.; Birks, C.; Roraback, J.; et al. IL-28, IL-29 and their class II cytokine receptor IL-28R. Nat. Immunol. 2003, 4, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Honda, M.; Yamamoto, S.; Cheng, M.; Yasukawa, K.; Suzuki, H.; Saito, T.; Osugi, Y.; Tokunaga, T.; Kishimoto, T. Human soluble IL-6 receptor: Its detection and enhanced release by HIV infection. J. Immunol. 1992, 148, 2175–2180. [Google Scholar] [PubMed]
- Hurst, S.M.; Wilkinson, T.S.; McLoughlin, R.M.; Jones, S.; Horiuchi, S.; Yamamoto, N.; Rose-John, S.; Fuller, G.M.; Topley, N.; Jones, S.A. Il-6 and its soluble receptor orchestrate a temporal switch in the pattern of leukocyte recruitment seen during acute inflammation. Immunity 2001, 14, 705–714. [Google Scholar] [CrossRef]
- Richards, P.J.; Nowell, M.A.; Horiuchi, S.; McLoughlin, R.M.; Fielding, C.A.; Grau, S.; Yamamoto, N.; Ehrmann, M.; Rose-John, S.; Williams, A.S.; et al. Functional characterization of a soluble gp130 isoform and its therapeutic capacity in an experimental model of inflammatory arthritis. Arthritis Rheum. 2006, 54, 1662–1672. [Google Scholar] [CrossRef]
- Heinz, D.; Peters, M.; Prange, R.; Gerken, G.; Rose-John, S. Possible role of human interleukin-6 and soluble interleukin-6 receptor in hepatitis B virus infection. J. Viral. Hepat. 2001, 8, 186–193. [Google Scholar] [CrossRef]
- Salvi, M.; Pedrazzoni, M.; Girasole, G.; Giuliani, N.; Minelli, R.; Wall, J.R.; Roti, E. Serum concentrations of proin-flammatory cytokines in Graves’ disease: Effect of treatment, thyroid function, ophthalmopathy and cigarette smoking. Eur. J. Endocrinol. 2000, 143, 197–202. [Google Scholar] [CrossRef]
- Ritter, M.; Göggel, R.; Chaudhary, N.; Wiedenmann, A.; Jung, B.; Weith, A.; Seither, P. Elevated expression of TARC (CCL17) and MDC (CCL22) in models of cigarette smoke-induced pulmonary inflammation. Biochem. Biophys. Res. Commun. 2005, 334, 254–262. [Google Scholar] [CrossRef]
- Aldaham, S.; Foote, J.A.; Chow, H.H.; Hakim, I.A. Smoking Status Effect on Inflammatory Markers in a Randomized Trial of Current and Former Heavy Smokers. Int. J. Inflam. 2015, 2015, 439396. [Google Scholar] [CrossRef]
- Shiels, M.S.; Shu, X.O.; Chaturvedi, A.K.; Gao, Y.T.; Xiang, Y.B.; Cai, Q.; Hu, W.; Shelton, G.; Ji, B.T.; Pinto, L.A.; et al. A prospective study of immune and inflammation markers and risk of lung cancer among female never smokers in Shanghai. Carcinogenesis 2017, 38, 1004–1010. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Kozaki, K.; Karpanen, T.; Koshikawa, K.; Yla-Herttuala, S.; Takahashi, T.; Alitalo, K. Suppression of tumor lymphangiogenesis and lymph node metastasis by blocking vascular endothelial growth factor receptor 3 signaling. J. Natl. Cancer Inst 2002, 94, 819–825. [Google Scholar] [CrossRef]
- Ting-ting, L.; Zhong-lan, Y.; Ze-wei, Y. Carotid ultrasound screening and related behavioral factors in high risk population of cerebral hemorrhage. China J. Mod. Med. 2015, 25, 64–68. [Google Scholar]
- Papaioannou, A.I.; Koutsokera, A.; Tanou, K.; Kiropoulos, T.S.; Tsilioni, I.; Oikonomidi, S.; Liadaki, K.; Pournaras, S.; Gourgoulianis, K.I.; Kostikas, K. The acute effect of smoking in healthy and asthmatic smokers. Eur. J. Clin. Investig. 2010, 40, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Kotani, K.; Satoh-Asahara, N.; Kato, Y.; Araki, R.; Himeno, A.; Yamakage, H.; Koyama, K.; Tanabe, M.; Oishi, M.; Okajima, T.; et al. Serum amyloid A low-density lipoprotein levels and smoking status in obese Japanese patients. J. Int. Med. Res. 2011, 39, 1917–1922. [Google Scholar] [CrossRef]
- Higham, A.; Bostock, D.; Booth, G.; Dungwa, J.V.; Singh, D. The effect of electronic cigarette and tobacco smoke exposure on COPD bronchial epithelial cell inflammatory responses. Int. J. Chron. Obstruct. Pulmon. Dis. 2018, 13, 989–1000. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.Y.; Lee, Y.S.; Min, K.H.; Hur, G.Y.; Lee, S.Y.; Kang, K.H.; Rhee, C.K.; Park, S.J.; Shim, J.J. Difference in systemic inflammation and predictors of acute exacerbation between smoking-associated COPD and tuberculosis-associated COPD. Int. J. Chron. Obstruct. Pulmon. Dis. 2018, 13, 3381–3387. [Google Scholar] [CrossRef]
- Goldbach-Mansky, R. Immunology in clinic review series; focus on autoinflammatory diseases: Update on monogenic autoinflammatory diseases: The role of interleukin (IL)-1 and an emerging role for cytokines beyond IL-1. Clin. Exp. Immunol. 2012, 167, 391–404. [Google Scholar] [CrossRef]
- Gao, C.; Li, S.; Zhao, T.; Chen, J.; Ren, H.; Zhang, H.; Wang, X.; Lang, M.; Liu, J.; Gao, S.; et al. SCF, regulated by HIF-1alpha, promotes pancreatic ductal adenocarcinoma cell progression. PLoS ONE 2015, 10, e0121338. [Google Scholar]
- Wang, Y.; Ma, H.; Tao, X.; Luo, Y.; Wang, H.; He, J.; Fang, Q.; Guo, S.; Song, C. SCF promotes the production of IL-13 via the MEK-ERK-CREB signaling pathway in mast cells. Exp. Ther. Med. 2019, 18, 2491–2496. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Group | Frequency | Age (Median; Min/Max) | Sex | Race |
|---|---|---|---|---|
| non-smokers | 30 | 36.6 yr; 19/57 yr | Male | Han |
| current-smokers | 30 | 35.5 yr; 24/49 yr | Male | Han |
| ex-smokers | 18 | 42.2 yr; 26/56 yr | Male | Han |
| Total | 78 | 37.5 yr; 19/57 yr | Male | Han |
| Expression Trends | Cytokine | Non-Smokers | Current-Smokers | Ex-Smokers | p-Value |
|---|---|---|---|---|---|
| Continuous increase | CXCL9/MIG | 1320.65 | 1493.70 | 1928.06 | 0.03/0.03 |
| sIL-6R | 57,248.93 | 70,286.72 | 76,155.75 | 2.96 × 10−3/6.84 × 10−3 | |
| Recovery after smoking cessation | TARC | 19.47 | 25.47 | 22.39 | 1.28 × 10−4/1.61 × 10−2 |
| ITAC | 48.09 | 105.34 | 84.09 | 0.07 × 10−4/3.89 × 10−2 | |
| sVEGFR3 | 20,058.64 | 27,610.30 | 26,732.49 | 0.06 × 10−4/2.53 × 10−2 | |
| SAA | 8833.60 | 4771.36 | 7609.59 | 0.03 × 10−4/9.37 × 10−3 | |
| No significant difference | IL-1β | 12.35 | 22.21 | 38.24 | 0.35/0.43 |
| BCA-1 | 24.92 | 39.37 | 27.55 | 0.84/0.35 | |
| TNF-α | 11.21 | 13.95 | 10.63 | 0.99/0.191 | |
| CRP | 9055.50 | 7058.28 | 11,859.6 | 0.918/0.30 | |
| ENA-78 | 547.60 | 457.60 | 649.28 | 0.51/0.34 | |
| MDC | 408.08 | 430.63 | 396.81 | 0.47/0.34 | |
| TNFRII | 14,775.96 | 15,543.39 | 13,188.03 | 0.94/0.17 | |
| Not detected | IL-1α | — | — | — | / |
| IL-6 | — | — | — | / | |
| IL-8 | — | — | — | / | |
| SCF | — | — | — | / |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, H.; Chen, H.; Fu, Y.; Liu, M.; Zhang, J.; Han, S.; Tian, Y.; Hou, H.; Hu, Q. Effects of Smoking on Inflammatory-Related Cytokine Levels in Human Serum. Molecules 2022, 27, 3715. https://doi.org/10.3390/molecules27123715
Wang H, Chen H, Fu Y, Liu M, Zhang J, Han S, Tian Y, Hou H, Hu Q. Effects of Smoking on Inflammatory-Related Cytokine Levels in Human Serum. Molecules. 2022; 27(12):3715. https://doi.org/10.3390/molecules27123715
Chicago/Turabian StyleWang, Hongjuan, Huan Chen, Yaning Fu, Min Liu, Jingni Zhang, Shulei Han, Yushan Tian, Hongwei Hou, and Qingyuan Hu. 2022. "Effects of Smoking on Inflammatory-Related Cytokine Levels in Human Serum" Molecules 27, no. 12: 3715. https://doi.org/10.3390/molecules27123715
APA StyleWang, H., Chen, H., Fu, Y., Liu, M., Zhang, J., Han, S., Tian, Y., Hou, H., & Hu, Q. (2022). Effects of Smoking on Inflammatory-Related Cytokine Levels in Human Serum. Molecules, 27(12), 3715. https://doi.org/10.3390/molecules27123715