
Identification of Human Dihydroorotate Dehydrogenase Inhibitor by a Pharmacophore-Based Virtual Screening Study

 , , , ,
, , , , 

Abstract
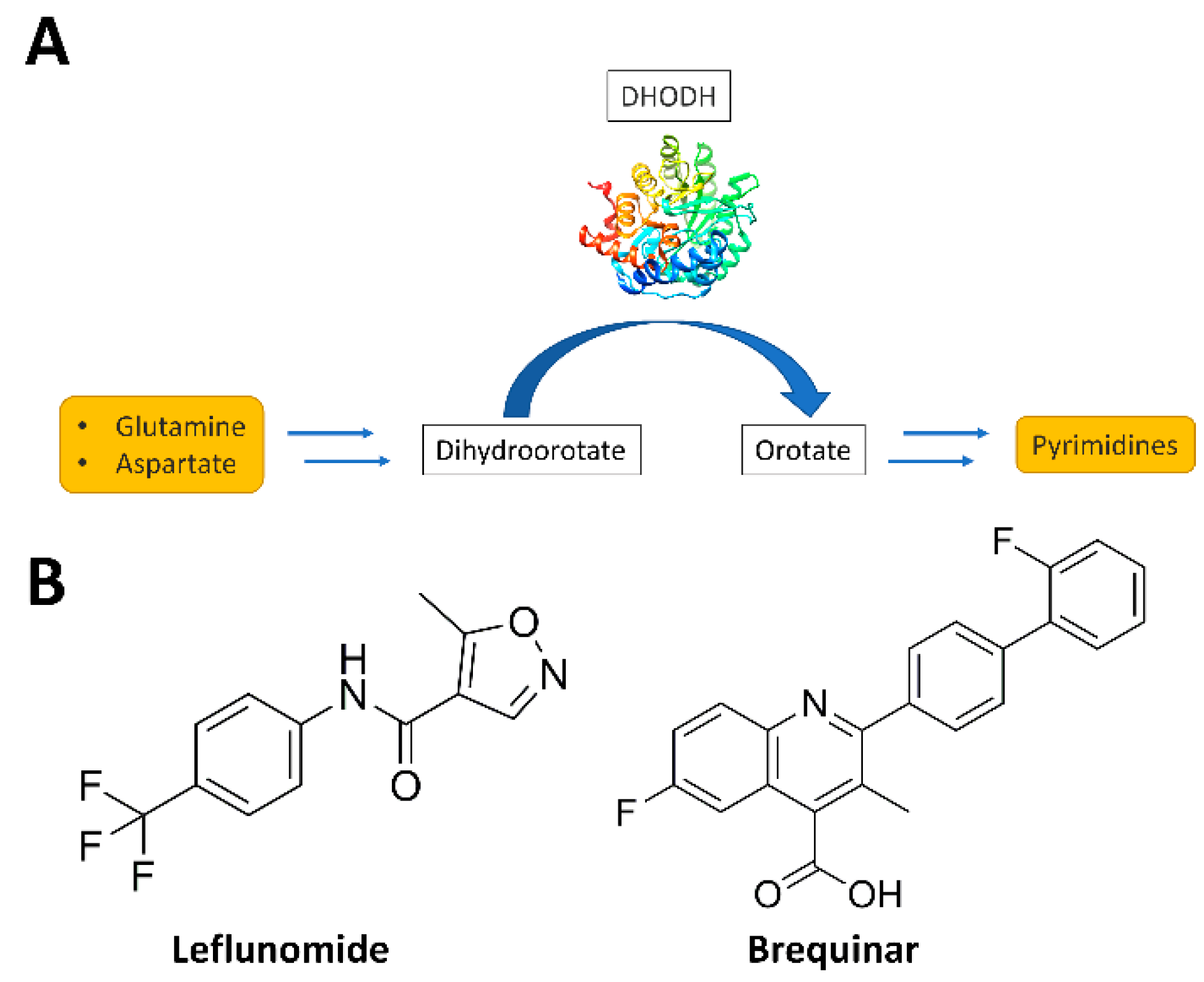
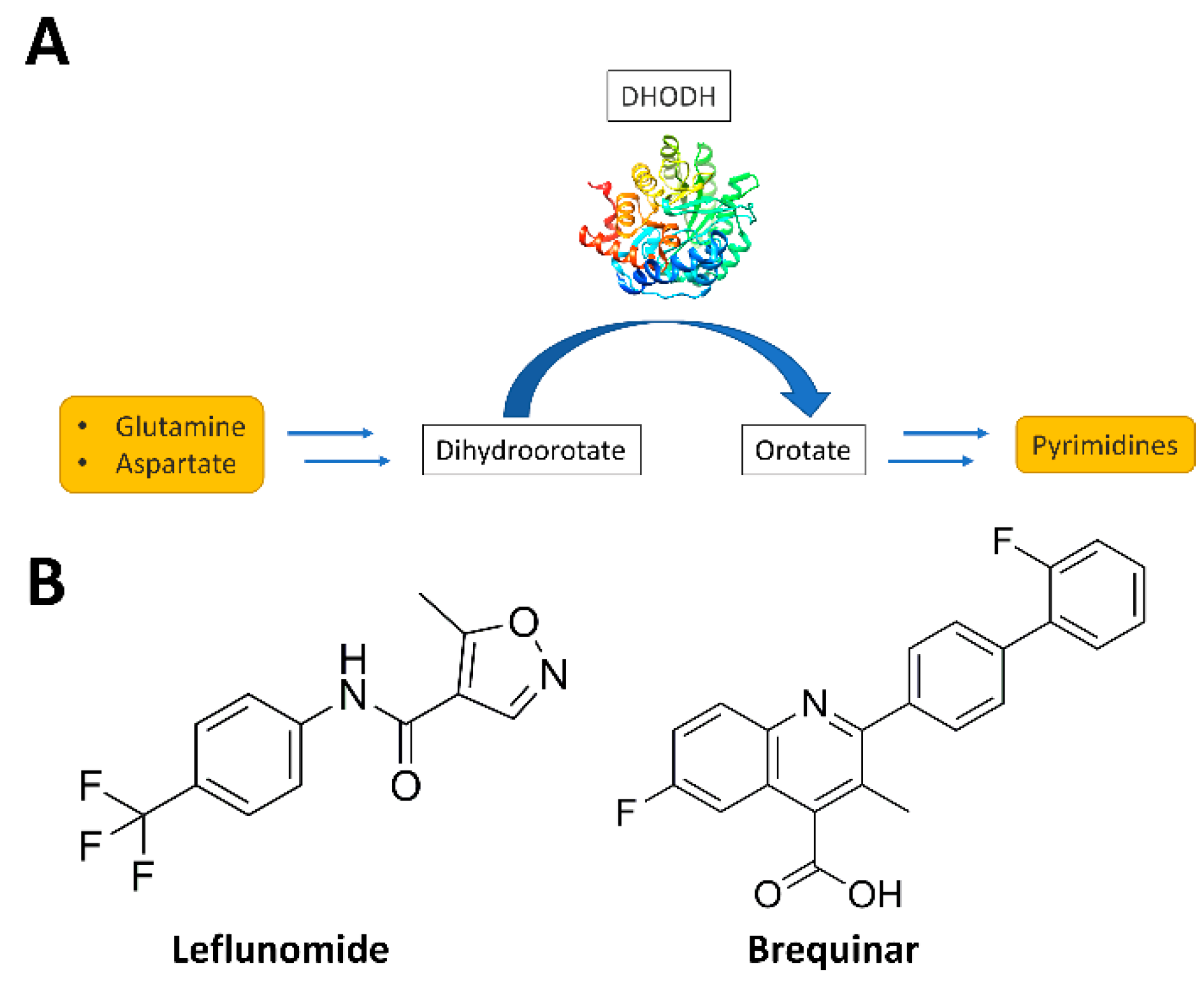
:1. Introduction
2. Materials and Methods
2.1. Pharmacophore Model Creation
2.2. Database Generation and Pharmacophore Screening
2.3. Active Ligands Dataset Generation
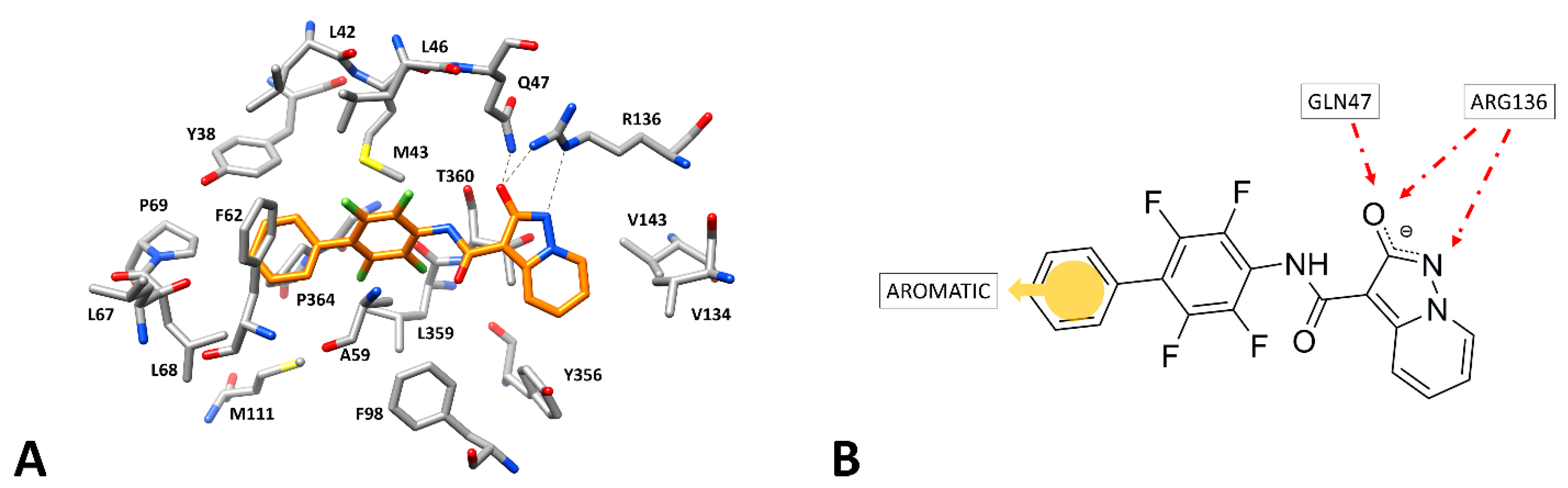
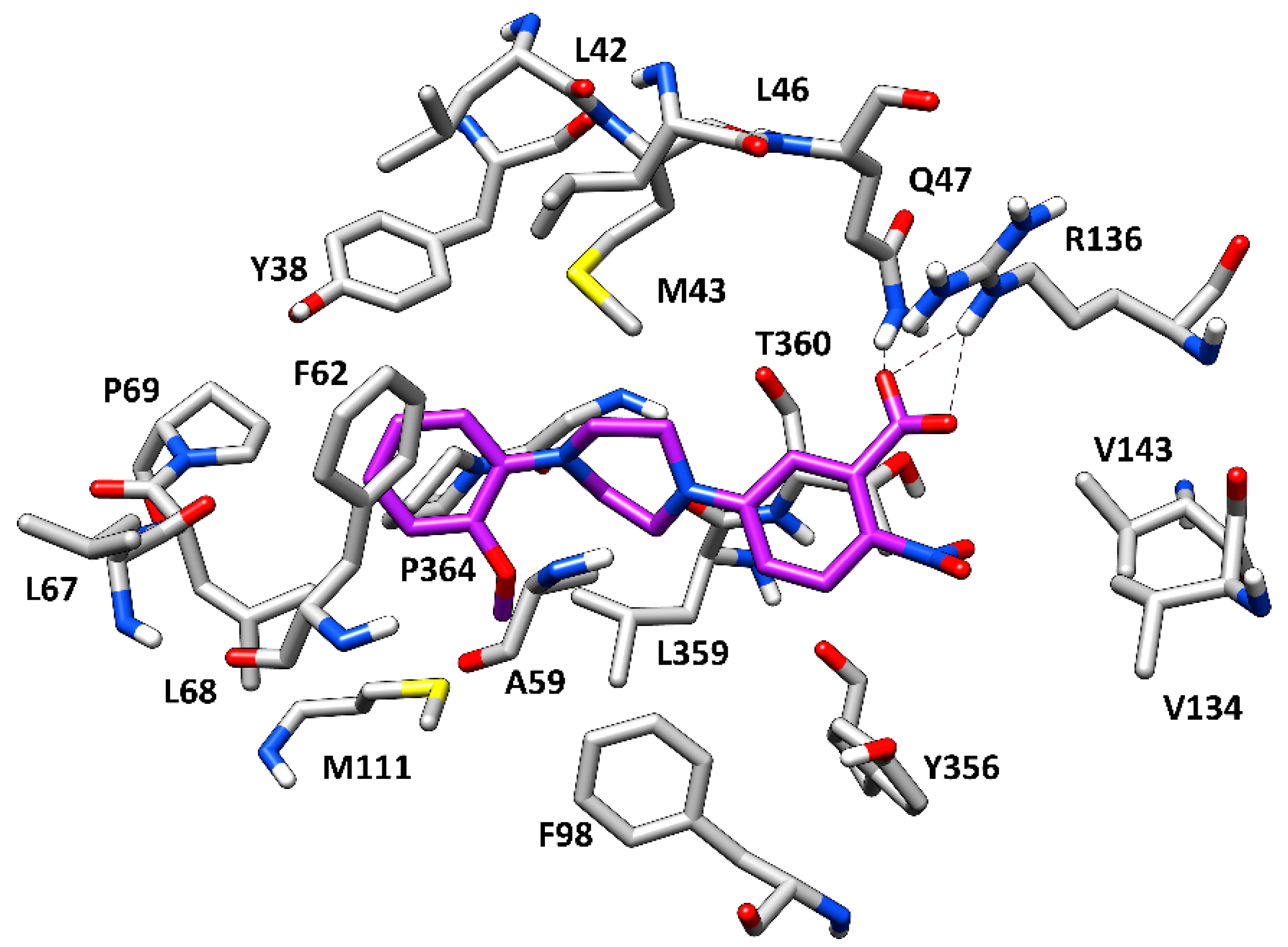
2.4. Molecular Docking Studies
2.5. Consensus Docking Evaluation
2.6. Molecular Dynamics Simulations
2.7. hDHODH Inhibition Assay
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Evans, D.R.; Guy, H.I. Mammalian Pyrimidine Biosynthesis: Fresh Insights into an Ancient Pathway. J. Biol. Chem. 2004, 279, 33035–33038. [Google Scholar] [CrossRef] [Green Version]
- Barnes, T.; Parry, P.; Hart, I.; Jones, C.; Minet, M.; Patterson, D. Regional mapping of the gene encoding dihydroorotate dehydrogenase, an enzyme involved in UMP synthesis, electron transport, and superoxide generation, to human chromosome region 16q22. Somat. Cell Mol. Genet. 1993, 19, 405–411. [Google Scholar] [CrossRef]
- Sørensen, G.; Dandanell, G. A new type of dihydroorotate dehydrogenase, type 1S, from the thermoacidophilic archaeon Sulfolobus solfataricus. Extremophiles 2002, 6, 245–251. [Google Scholar] [CrossRef]
- Mohamad Fairus, A.K.; Choudhary, B.; Hosahalli, S.; Kavitha, N.; Shatrah, O. Dihydroorotate dehydrogenase (DHODH) inhibitors affect ATP depletion, endogenous ROS and mediate S-phase arrest in breast cancer cells. Biochimie 2017, 135, 154–163. [Google Scholar] [CrossRef]
- Reis, R.A.G.; Calil, F.A.; Feliciano, P.R.; Pinheiro, M.P.; Nonato, M.C. The dihydroorotate dehydrogenases: Past and present. Arch. Biochem. Biophys. 2017, 632, 175–191. [Google Scholar] [CrossRef]
- Boschi, D.; Pippione, A.C.; Sainas, S.; Lolli, M.L. Dihydroorotate dehydrogenase inhibitors in anti-infective drug research. Eur. J. Med. Chem. 2019, 183, 111681. [Google Scholar] [CrossRef]
- Lolli, M.L.; Sainas, S.; Pippione, A.C.; Giorgis, M.; Boschi, D.; Dosio, F. Use of human Dihydroorotate Dehydrogenase (hDHODH) Inhibitors in Autoimmune Diseases and New Perspectives in Cancer Therapy. Recent Pat. Anticancer. Drug Discov. 2018, 13, 86–105. [Google Scholar] [CrossRef]
- Lewis, T.A.; Sykes, D.B.; Law, J.M.; Muñoz, B.; Rustiguel, J.K.; Nonato, M.C.; Scadden, D.T.; Schreiber, S.L. Development of ML390: A Human DHODH Inhibitor That Induces Differentiation in Acute Myeloid Leukemia. ACS Med. Chem. Lett. 2016, 7, 1112–1117. [Google Scholar] [CrossRef] [Green Version]
- Sykes, D.B.; Kfoury, Y.S.; Mercier, F.E.; Wawer, M.J.; Law, J.M.; Haynes, M.K.; Lewis, T.A.; Schajnovitz, A.; Jain, E.; Lee, D.; et al. Inhibition of Dihydroorotate Dehydrogenase Overcomes Differentiation Blockade in Acute Myeloid Leukemia. Cell 2016, 167, 171–186.e15. [Google Scholar] [CrossRef] [Green Version]
- Rosenzweig, M.; Palmer, J.; Tsai, N.-C.; Synold, T.; Wu, X.; Tao, S.; Hammond, S.N.; Buettner, R.; Duarte, L.; Htut, M.; et al. Repurposing leflunomide for relapsed/refractory multiple myeloma: A phase 1 study. Leuk. Lymphoma 2020, 61, 1669–1677. [Google Scholar] [CrossRef]
- Cody, R.; Stewart, D.; DeForni, M.; Moore, M.; Dallaire, B.; Azarnia, N.; Gyves, J. Multicenter phase II study of brequinar sodium in patients with advanced breast cancer. Am. J. Clin. Oncol. 1993, 16, 526–528. [Google Scholar] [CrossRef]
- Dodion, P.F.; Wagener, T.; Stoter, G.; Drozd, A.; Lev, L.M.; Skovsgaard, T.; Renard, J.; Cavalli, F. Phase II trial with Brequinar (DUP-785, NSC 368390) in patients with metastatic colorectal cancer: A study of the Early Clinical Trials Group of the EORTC. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 1990, 1, 79–80. [Google Scholar] [CrossRef] [PubMed]
- Maroun, J.; Ruckdeschel, J.; Natale, R.; Morgan, R.; Dallaire, B.; Sisk, R.; Gyves, J. Multicenter phase II study of brequinar sodium in patients with advanced lung cancer. Cancer Chemother. Pharmacol. 1993, 32, 64–66. [Google Scholar] [CrossRef] [Green Version]
- Natale, R.; Wheeler, R.; Moore, M.; Dallaire, B.; Lynch, W.; Carlson, R.; Grillo-Lopez, A.; Gyves, J. Multicenter phase II trial of brequinar sodium in patients with advanced melanoma. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 1992, 3, 659–660. [Google Scholar] [CrossRef] [PubMed]
- Wolber, G.; Langer, T. LigandScout: 3-D Pharmacophores Derived from Protein-Bound Ligands and Their Use as Virtual Screening Filters. J. Chem. Inf. Model. 2005, 45, 160–169. [Google Scholar] [CrossRef]
- Sainas, S.; Pippione, A.C.; Lupino, E.; Giorgis, M.; Circosta, P.; Gaidano, V.; Goyal, P.; Bonanni, D.; Rolando, B.; Cignetti, A.; et al. Targeting Myeloid Differentiation Using Potent 2-Hydroxypyrazolo[1,5-a]pyridine Scaffold-Based Human Dihydroorotate Dehydrogenase Inhibitors. J. Med. Chem. 2018, 61, 6034–6055. [Google Scholar] [CrossRef]
- Landrum, G. RDKit: Open-Source Cheminformatics. 2006. Available online: https://www.rdkit.org (accessed on 1 June 2022).
- Poli, G.; Seidel, T.; Langer, T. Conformational Sampling of Small Molecules With iCon: Performance Assessment in Comparison With OMEGA. Front. Chem. 2018, 6, 229. [Google Scholar] [CrossRef]
- Pini, E.; Poli, G.; Tuccinardi, T.; Chiarelli, L.; Mori, M.; Gelain, A.; Costantino, L.; Villa, S.; Meneghetti, F.; Barlocco, D. New Chromane-Based Derivatives as Inhibitors of Mycobacterium tuberculosis Salicylate Synthase (MbtI): Preliminary Biological Evaluation and Molecular Modeling Studies. Molecules 2018, 23, 1506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuccinardi, T.; Poli, G.; Romboli, V.; Giordano, A.; Martinelli, A. Extensive consensus docking evaluation for ligand pose prediction and virtual screening studies. J. Chem. Inf. Model. 2014, 54, 2980–2986. [Google Scholar] [CrossRef] [PubMed]
- Giorgis, M.; Lolli, M.L.; Rolando, B.; Rao, A.; Tosco, P.; Chaurasia, S.; Marabello, D.; Fruttero, R.; Gasco, A. 1,2,5-Oxadiazole analogues of leflunomide and related compounds. Eur. J. Med. Chem. 2011, 46, 383–392. [Google Scholar] [CrossRef]
- Sainas, S.; Giorgis, M.; Circosta, P.; Gaidano, V.; Bonanni, D.; Pippione, A.C.; Bagnati, R.; Passoni, A.; Qiu, Y.; Cojocaru, C.F.; et al. Targeting Acute Myelogenous Leukemia Using Potent Human Dihydroorotate Dehydrogenase Inhibitors Based on the 2-Hydroxypyrazolo[1,5-a]pyridine Scaffold: SAR of the Biphenyl Moiety. J. Med. Chem. 2021, 64, 5404–5428. [Google Scholar] [CrossRef] [PubMed]
- Calistri, A.; Luganini, A.; Mognetti, B.; Elder, E.; Sibille, G.; Conciatori, V.; Del Vecchio, C.; Sainas, S.; Boschi, D.; Montserrat, N.; et al. The New Generation hDHODH Inhibitor MEDS433 Hinders the In Vitro Replication of SARS-CoV-2 and Other Human Coronaviruses. Microorganisms 2021, 9, 1731. [Google Scholar] [CrossRef]
- Poli, G.; Martinelli, A.; Tuccinardi, T. Reliability analysis and optimization of the consensus docking approach for the development of virtual screening studies. J. Enzyme Inhib. Med. Chem. 2016, 31, 167–173. [Google Scholar] [CrossRef]
- Russo Spena, C.; De Stefano, L.; Poli, G.; Granchi, C.; El Boustani, M.; Ecca, F.; Grassi, G.; Grassi, M.; Canzonieri, V.; Giordano, A.; et al. Virtual screening identifies a PIN1 inhibitor with possible antiovarian cancer effects. J. Cell. Physiol. 2019, 234, 15708–15716. [Google Scholar] [CrossRef]
- Chiarelli, L.R.; Mori, M.; Barlocco, D.; Beretta, G.; Gelain, A.; Pini, E.; Porcino, M.; Mori, G.; Stelitano, G.; Costantino, L.; et al. Discovery and development of novel salicylate synthase (MbtI) furanic inhibitors as antitubercular agents. Eur. J. Med. Chem. 2018, 155, 754–763. [Google Scholar] [CrossRef]
- Poli, G.; Giuntini, N.; Martinelli, A.; Tuccinardi, T. Application of a FLAP-consensus docking mixed strategy for the identification of new fatty acid amide hydrolase inhibitors. J. Chem. Inf. Model. 2015, 55, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Lapillo, M.; Salis, B.; Palazzolo, S.; Poli, G.; Granchi, C.; Minutolo, F.; Rotondo, R.; Caligiuri, I.; Canzonieri, V.; Tuccinardi, T.; et al. First-of-its-kind STARD 3 Inhibitor: In Silico Identification and Biological Evaluation as Anticancer Agent. ACS Med. Chem. Lett. 2019, 10, 475–480. [Google Scholar] [CrossRef] [PubMed]
- Madak, J.T.; Cuthbertson, C.R.; Miyata, Y.; Tamura, S.; Petrunak, E.M.; Stuckey, J.A.; Han, Y.; He, M.; Sun, D.; Showalter, H.D.; et al. Design, Synthesis, and Biological Evaluation of 4-Quinoline Carboxylic Acids as Inhibitors of Dihydroorotate Dehydrogenase. J. Med. Chem. 2018, 61, 5162–5186. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
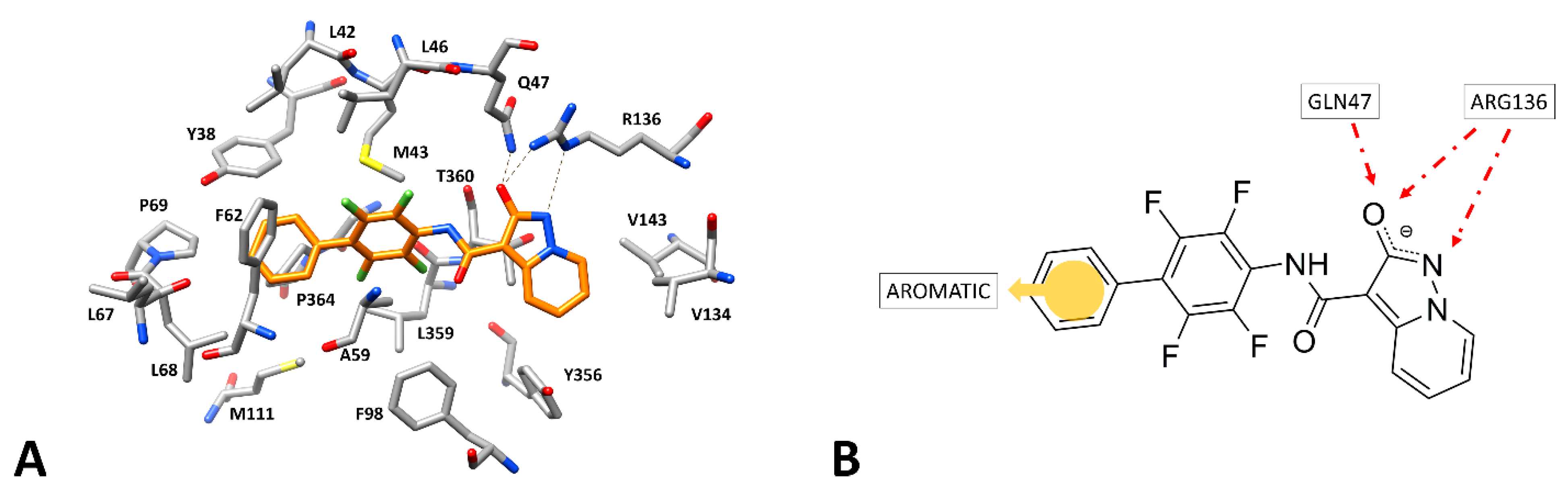{kind=link}
{kind=link}
| Docking Software | RMSD (Å) |
|---|---|
| rDock | 1.2 |
| Gold PLP | 0.3 |
| Gold Chemscore | 0.7 |
| Gold ASP | 0.4 |
| Gold Goldscore | 0.6 |
| Plants | 0.4 |
| Glide SP | 0.2 |
| Glide XP | 0.2 |
| Fred | 0.9 |
| Dock 6 | 0.2 |
| Autodock | 0.6 |
| Glamdock | 0.5 |
| Vina | 0.4 |
| Consensus Level | No. of Compounds |
|---|---|
| 13 | 0 |
| 12 | 0 |
| 11 | 0 |
| 10 | 7 |
| 9 | 11 |
| 8 | 22 |
| 7 | 37 |
| 6 | 47 |
| 5 | 76 |
| 4 | 79 |
| 3 | 320 |
| 2 | 721 |
| 1 | 166 |
| Structure | Compounds ID | % Inhibition (100 μM) | IC50 (μM) |
|---|---|---|---|
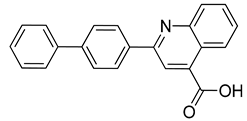 | CPD1 | n.d | 0.051 ± 0.027 a |
 | CPD2 | 8% | n.d |
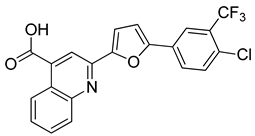 | CPD3 | 48% | n.d |
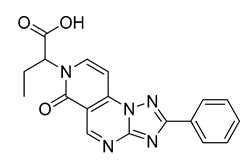 | CPD4 | 16% | n.d |
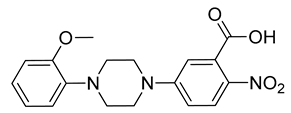 | CPD5 | 72% | 48 ± 8 |
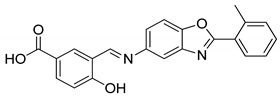 | CPD6 | 0% | n.d |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Galati, S.; Sainas, S.; Giorgis, M.; Boschi, D.; Lolli, M.L.; Ortore, G.; Poli, G.; Tuccinardi, T. Identification of Human Dihydroorotate Dehydrogenase Inhibitor by a Pharmacophore-Based Virtual Screening Study. Molecules 2022, 27, 3660. https://doi.org/10.3390/molecules27123660
Galati S, Sainas S, Giorgis M, Boschi D, Lolli ML, Ortore G, Poli G, Tuccinardi T. Identification of Human Dihydroorotate Dehydrogenase Inhibitor by a Pharmacophore-Based Virtual Screening Study. Molecules. 2022; 27(12):3660. https://doi.org/10.3390/molecules27123660
Chicago/Turabian StyleGalati, Salvatore, Stefano Sainas, Marta Giorgis, Donatella Boschi, Marco L. Lolli, Gabriella Ortore, Giulio Poli, and Tiziano Tuccinardi. 2022. "Identification of Human Dihydroorotate Dehydrogenase Inhibitor by a Pharmacophore-Based Virtual Screening Study" Molecules 27, no. 12: 3660. https://doi.org/10.3390/molecules27123660
APA StyleGalati, S., Sainas, S., Giorgis, M., Boschi, D., Lolli, M. L., Ortore, G., Poli, G., & Tuccinardi, T. (2022). Identification of Human Dihydroorotate Dehydrogenase Inhibitor by a Pharmacophore-Based Virtual Screening Study. Molecules, 27(12), 3660. https://doi.org/10.3390/molecules27123660