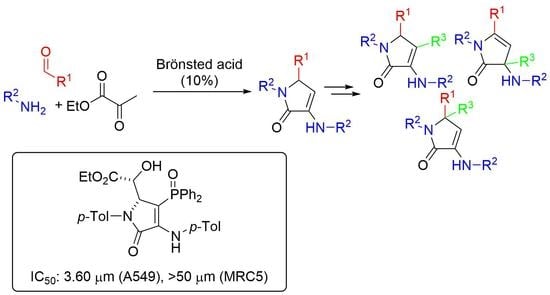
3.1.3. Experimental Procedures and Characterization Data for Compounds 13, 15, 16, and 23
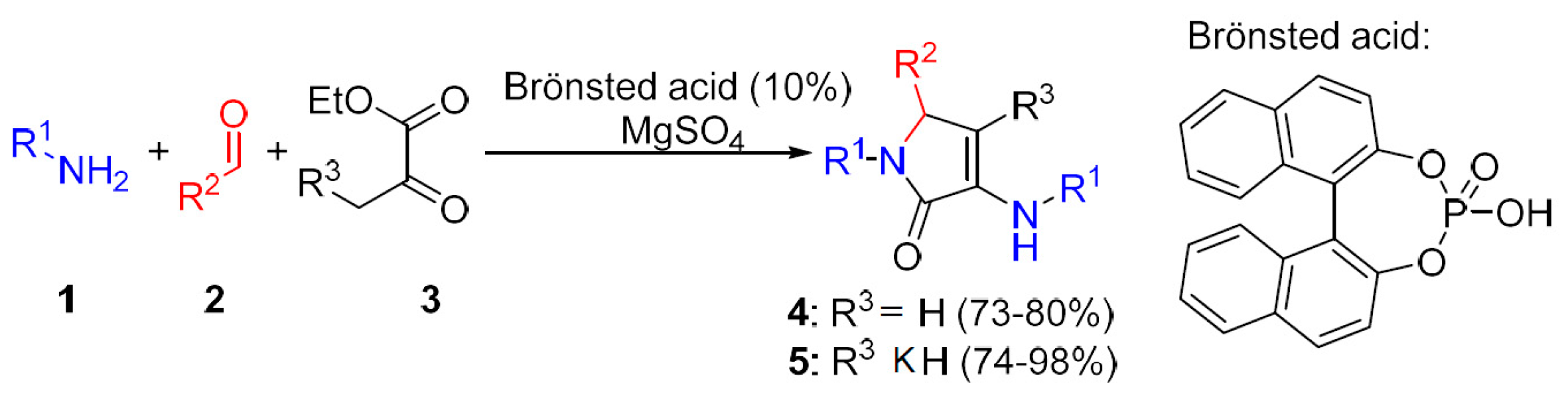
General Procedure for the Multicomponent Synthesis of γ-Lactams 4 and 5
Following a known procedure [
11], a solution of amine
1 (10 mmol), aldehyde
2 (5 mmol), ethyl pyruvate
3 (1.74 g, 15 mmol), and 1,1′-binaphthyl-2,2′-diyl hydrogen phosphate (174 mg, 10 mol%) was stirred in Et
2O (25 mL) at room temperature or MTBE (25 mL) at 55 °C (heating plate with Heat-On) in the presence of anhydrous MgSO
4 (2.5 g) for 48 h. The volatiles were distilled off at reduced pressure and the crude residue was purified by chromatography (AcOEt/hexanes 9:1) to afford pure γ-lactams
4 and
5.
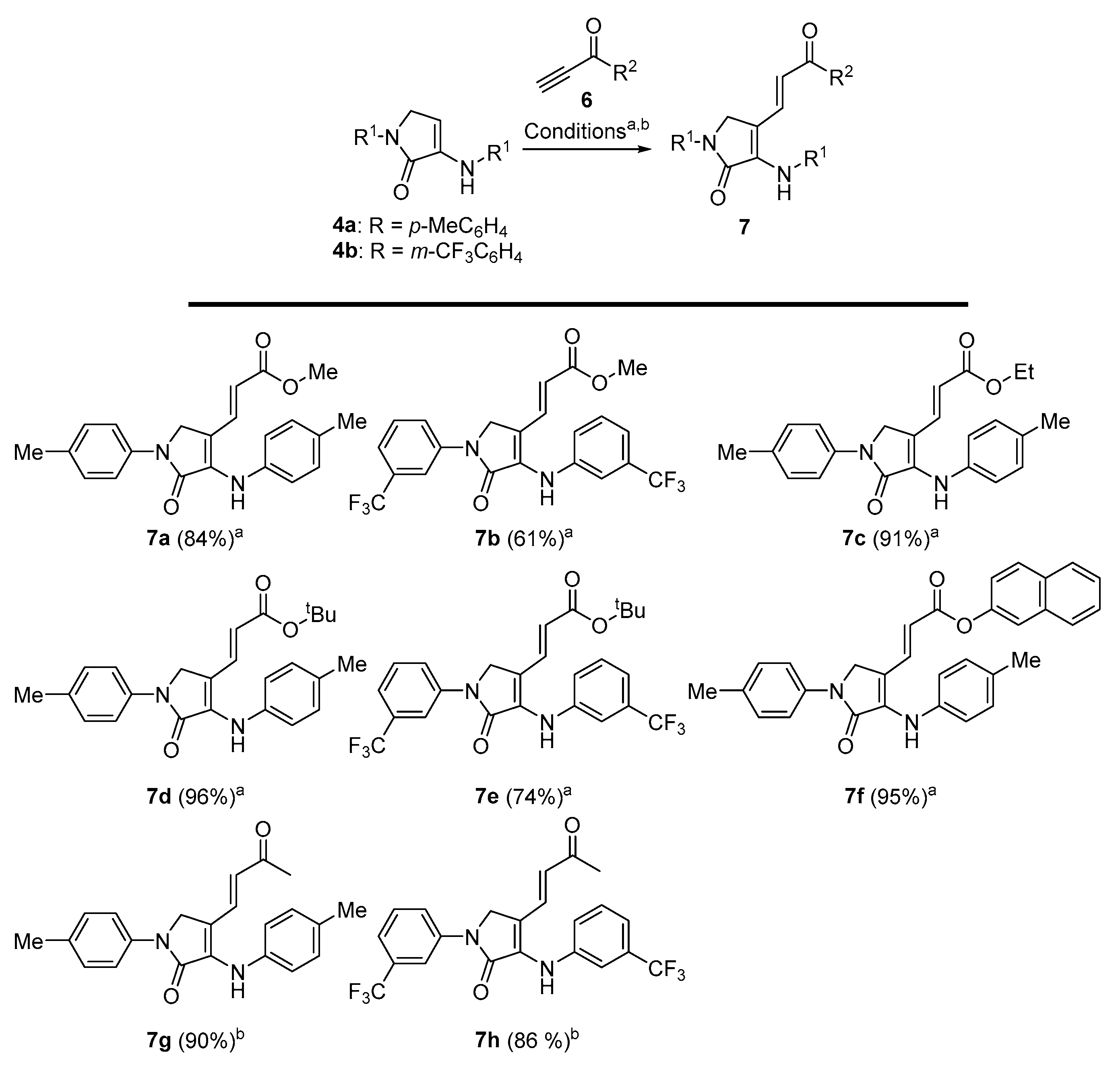
General Procedure for the Nucleophilic Conjugate Addition of γ-Lactams 4 to Activated Alkynes
Procedure A (Esters). The corresponding γ-lactam 4 (1 mmol) was added to a freshly prepared solution of LDA (1.2 mmol) in THF (3 mL) at −78 °C under N2 atmosphere. After 1 h, the selected alkyl propiolate (1.2 mmol) was added and the reaction was stirred at room temperature until the disappearance of the starting materials (monitored by TLC and/or 1H RMN, ~4 h). The reaction was acidified with a 0.5 M aqueous solution of HCl (10 mL), and extracted with dichloromethane (2 × 10 mL). The combined organic layers were washed with water (2 × 10 mL), dried over MgSO4, and concentrated under vacuum. The crude residue was purified by crystallization, affording pure functionalized γ-lactam derivatives 6. In some cases, a previous purification by chromatography was necessary as detailed for each compound.
Procedure B (Ketones). A solution of the corresponding γ-lactam 4 (1 mmol), 3-butn-2-one (0.097 mL, 1.2 mmol) and Yb(OTf)3 (1 mmol, 620 mg) in dichloromethane (3 mL) was stirred at room temperature until the disappearance of the starting (monitored by TLC, 4 to 20 h). The reaction mixture was filtered through celite and concentrated under vacuum and the crude residue was purified by crystallization, affording pure functionalized γ-lactam derivatives 7. In some cases, previous purification by chromatography column was necessary as detailed for each compound.
Methyl (E)-3-(5-oxo-1-(p-tolyl)-4-(p-tolylamino)-2,5-dihydro-1H-pyrrol-3-yl)acrylate (7a). The general procedure A was followed using 1-(p-tolyl)-3-(p-tolylamino)-1,5-dihydro-2H-pyrrol-2-one 4a (278 mg, 1 mmol) and methyl propiolate (107 μL, 1.2 mmol), affording 304 mg (84%) of 7a as yellow crystals after crystallization (Dichloromethane/Hexanes 1:3). M.p. (Dichloromethane/Hexanes) = 189–191 °C. 1H NMR (400 MHz, CDCl3) δ 7.66 (d, 3JHH = 8.6 Hz, 2H, 2 × CHAr), 7.56 (d, 3JHH = 15.7 Hz, 1H, CH=), 7.22 (d, 3JHH = 8.6 Hz, 2H, 2 × CHAr), 7.19 (d, 3JHH = 8.4 Hz, 2H, 2 × CHAr), 7.11 (d, 3JHH = 8.4 Hz, 2H, 2 × CHAr), 6.74 (s, 1H, NH), 5.63 (d, 3JHH = 15.7 Hz, 1H, CH=), 4.41 (s, 2H, CH2), 3.68 (s, 3H, OCH3), 2.37 (s, 3H, CH3Tol), 2.36 (s, 3H, CH3Tol) ppm. 13C NMR {1H} (101 MHz, CDCl3) δ 167.4 (C=O), 165.6 (C=O), 136.6 (Cquat), 136.4 (Cquat), 136.3 (Cquat), 135.5 (CH=), 135.1 (Cquat), 134.8 (Cquat), 130.2 (2 × CHAr), 129.9 (2 × CHAr), 122.5 (2 × CHAr), 119.1 (2 × CHAr), 113.2 (CH=), 107.5 (Cquat), 51.6 (OCH3), 49.2 (CH2), 21.1 (CH3Tol), 21.0 (CH3Tol) ppm. FTIR (neat) νmax: 3321 (NH st), 3059 (=CH st), 1701 (C=O st), 1676 (C=O st), 1611 (C=C st), cm−1. HRMS (ESI-TOF) m/z: [M+H]+ calcd for C22H23N2O3 363.1709, Found 363.1708.
Methyl (E)-3-(5-oxo-1-(3-(trifluoromethyl)phenyl)-4-((3-(trifluoromethyl)phenyl)amino)- 2,5-dihydro-1H-pyrrol-3-yl)acrylate (7b). The general procedure A was followed using 1-(3-(trifluoromethyl)phenyl)-3-((3-(trifluoromethyl)phenyl)amino)-1,5-dihydro-2H-pyrrol-2-one 4b (386 mg, 1 mmol) and methyl propiolate 107 μL, 1.2 mmol), affording 285 mg (61%) of 7b as a red oil after chromatography (Hexane/AcOEt 9:1). 1H NMR (400 MHz, CDCl3) δ 8.10–7.99 (m, 2H, 2 × CHAr), 7.59–7.44 (m, 3H, 3 × CHAr), 7.54 (d, 3JHH = 15.8 Hz, 1H, CH=), 7.40 (d, 3JHH = 7.4 Hz, 2H, 2 × CHAr), 7.33 (d, 3JHH = 7.4 Hz, 1H, CHAr), 6.82 (s, 1H, NH), 5.80 (d, 3JHH = 15.8 Hz, 1H, CH=), 4.54 (s, 2H, CH2), 3.71 (s, 3H, CH3) ppm. 13C NMR {1H} (101 MHz, Acetone d6) δ 167.0 (C=O), 166.6 (C=O), 143.0 (Cquat), 141.0 (Cquat), 136.0 (Cquat), 134.6 (CH=), 131.6 (q, 2JCF = 32.3 Hz, Cquat), 131.6 (q, 2JCF = 31.9 Hz, Cquat), 130.9 (2 × CHar), 125.2 (q, 1JCF = 271.6 Hz, CF3), 125.1 (q, 1JCF = 271.3 Hz, CF3), 124.2 (CHar), 122.4 (CHar), 121.2 (q, 3JFC = 4.0 Hz, CHAr), 119.8 (q, 3JFC = 3.8 Hz, CHAr), 117.9 (CH=), 117.7 (Cquat), 117.2 (q, 3JFC = 3.9 Hz, CHAr), 115.6 (q, 3JFC = 4.1 Hz, CHAr), 51.7 (CH3), 49.6 (CH2) ppm. 19F-NMR (282 MHz, CDCl3) δ −63.1, −63.5 ppm. FTIR (neat) νmax: 3309 (NH st), 3062 (=CH st), 1706 (C=O st), 1679 (C=O st), 1609 (C=C st) cm−1. HRMS (ESI-TOF) m/z: [M+H]+ calcd for C22H17F6N2O3 471.1143, Found 471.1140.
Ethyl (E)-3-(5-oxo-1-(p-tolyl)-4-(p-tolylamino)-2,5-dihydro-1H-pyrrol-3-yl)acrylate (7c). The general procedure A was followed using 1-(p-tolyl)-3-(p-tolylamino)-1,5-dihydro-2H-pyrrol-2-one 4a (278 mg, 1 mmol) and ethyl propiolate (118 μL, 1.2 mmol), affording 345 mg (91%) of 7c as brown crystals after crystallization (Dichloromethane/Hexanes 1:3). M.p. (Dichloromethane/Hexanes) = 211–212 °C. 1H NMR (400 MHz, CDCl3) δ 7.65 (d, 3JHH = 8.4 Hz, 2H, 2 × CHAr), 7.54 (d, 3JHH = 15.7 Hz, 1H, CH=), 7.22 (d, 3JHH = 8.4 Hz, 2H, 2 × CHAr), 7.19 (d, 3JHH = 8.3 Hz, 2H, 2 × CHAr), 7.11 (d, 3JHH = 8.3 Hz, 2H, 2 × CHAr), 6.75 (s, 1H, NH), 5.62 (d, 3JHH = 15.7 Hz, 1H, CH=), 4.40 (s, 2H, CH2), 4.12 (q, 3JHH = 7.1 Hz, 2H, CH2CH3), 2.35 (s, 6H, 2 × CH3Tol), 1.23 (t, 3JHH = 7.1 Hz, 3H, CH2CH3) ppm. 13C NMR {1H} (101 MHz, CDCl3) δ 166.9 (C=O), 165.6 (C=O), 136.6 (Cquat), 136.3 (Cquat), 136.3 (Cquat), 135.2 (Cquat), 135.2 (Cquat), 134.8 (CH=), 130.2 (2 × CHAr), 129.9 (2 × CHAr), 122.8 (2 × CHAr), 119.0 (2 × CHAr), 113.6 (CH=), 107.4 (Cquat), 60.3 (CH2CH3), 49.2 (CH2), 21.1 (CH3Tol), 21.0 (CH3Tol), 14.3 (CH2CH3) ppm. FTIR (neat) νmax: 3381 (NH st), 3066 (=CH st), 1698 (C=O st), 1676 (C=O st), 1605 (C=C st) cm−1. HRMS (ESI-TOF) m/z: [M+H]+ calcd for C23H25N2O3 377.1865, Found 377.1863.
Tert-butyl (E)-3-(5-oxo-1-(p-tolyl)-4-(p-tolylamino)-2,5-dihydro-1H-pyrrol-3-yl)acrylate (7d). The general procedure A was followed using 1-(p-tolyl)-3-(p-tolylamino)-1,5-dihydro-2H-pyrrol-2-one 4a (278 mg, 1 mmol) and tert-butyl propiolate (166 μL, 1.2 mmol), affording 388 mg (96%) of 7d as orange crystals after crystallization (Dichloromethane/Hexanes 1:3). M.p. (Dichloromethane/Hexanes) = 186–187 °C. 1H NMR (400 MHz, CDCl3) δ 7.66 (d, 3JHH = 8.6 Hz, 2H, 2 × CHAr), 7.44 (d, 3JHH = 15.7 Hz, 1H, CH=), 7.22 (d, 3JHH = 8.6 Hz, 2H, 2 × CHAr), 7.18 (d, 3JHH = 8.3 Hz, 2H, 2 × CHAr), 7.11 (d, 3JHH = 8.3 Hz, 2H, 2 × CHAr), 6.68 (s, 1H, NH), 5.55 (d, 3JHH = 15.7 Hz, 1H, CH=), 4.40 (s, 2H, CH2), 2.36 (s, 3H, CH3Tol), 2.34 (s, 3H, CH3Tol), 1.41 (s, 9H, 3 × CH3tBu) ppm. 13C NMR {1H} (101 MHz, CDCl3) δ 166.1 (C=O), 165.8 (C=O), 136.7 (Cquat), 136.4 (Cquat), 135.8 (Cquat), 134.9 (Cquat), 134.7 (Cquat), 134.2 (CH=), 130.1 (2 × CHAr), 129.9 (2 × CHAr), 122.8 (2 × CHAr), 119.0 (2 × CHAr), 116.0 (CH=), 107.7 (Cquat), 80.1 (CquattBu), 49.2 (CH2), 28.24 (3 × CH3tBu), 21.1 (CH3Tol), 21.0 (CH3Tol) ppm. FTIR (neat) νmax: 3402 (NH st), 3041 (=CH st), 1694 (C=O st), 1679 (C=O st), 1615 (C=C st) cm−1. HRMS (ESI-TOF) m/z: [M+H]+ calcd for C25H29N2O3 405.2178, Found 405.2179.
Tert-butyl (E)-3-(5-oxo-1-(3-(trifluoromethyl)phenyl)-4-((3-(trifluoromethyl)phenyl) amino)-2,5-dihydro-1H-pyrrol-3-yl)acrylate (7e). The general procedure A was followed using 1-(3-(trifluoromethyl)phenyl)-3-((3-(trifluoromethyl)phenyl)amino)-1,5-dihydro- 2H-pyrrol-2-one 4b (386 mg, 1 mmol) and tert-butyl propiolate (166 μL, 1.2 mmol), affording 380 mg (74%) of 7e as a yellow oil after chromatography (Hexanes/AcOEt 9:1). 1H NMR (400 MHz, CDCl3) δ 8.10–7.99 (m, 2H, 2 × CHar), 7.59–7.43 (m, 3H, 3 × CHar), 7.42–7.28 (m, 3H), 7.38 (d, 3JHH = 15.8 Hz, 1H), 6.78 (s, 1H, NH), 5.73 (d, 3JHH = 15.8 Hz, 1H, CH=), 4.52 (s, 2H, CH2), 1.42 (s, 9H, 3 × CH3tBu) ppm. 13C NMR {1H} (75 MHz, CDCl3) δ 166.1 (C=O), 165.4 (C=O), 140.4 (Cquat), 139.3 (Cquat), 134.1 (CH=), 132.9 (Cquat), 132.2 (q, 2JFC = 32.3 Hz, Cquat), 132.0 (q, 2JFC = 32.7 Hz, Cquat), 130.2 (2 × CHAr), 124.0 (q, 1JFC = 272.4 Hz), 123.9 (q, 1JFC = 272.7 Hz), 124.1 (CHAr), 121.9 (CHAr), 121.6 (q, 3JFC = 3.9 Hz, CHAr), 120.9 (q, 3JFC = 3.9 Hz, CHAr), 119.4 (CH=), 117.5 (q, 3JFC = 3.9 Hz, CHAr), 115.3 (q, 3JFC = 3.9 Hz, CHAr), 112.1 (Cquat), 81.0 (CquattBu), 49.2 (CH2), 28.22 (3 × CH3tBu) ppm. 19F-NMR (282 MHz, CDCl3) δ −63.1, −63.3 ppm. FTIR (neat) νmax: 3301 (NH st), 3051 (=CH st), 1708 (C=O st), 1675 (C=O st), 1607 (C=C st) cm−1. HRMS (ESI-TOF) m/z: [M+H]+ calcd for C25H23F6N2O3 513.1613, Found 513.1610.
Naphthalen-2-yl (E)-3-(5-oxo-1-(p-tolyl)-4-(p-tolylamino)-2,5-dihydro-1H-pyrrol-3-yl) acrylate (7f). The general procedure A was followed using 1-(p-tolyl)-3-(p-tolylamino)-1,5-dihydro-2H-pyrrol-2-one 4a (278 mg, 1 mmol) and naphthalen-2-yl prop-2-ynoate (235 mg, 1.2 mmol), affording 450 mg (95%) of 7f as brown crystals after crystallization (Dichloromethane/Hexanes 1:3). M.p. (Dichloromethane/Hexanes) = 228–230 °C. (dec.). 1H NMR (400 MHz, CDCl3) δ 7.84 (d, 3JHH = 8.4 Hz, 2H, 2 × CHAr), 7.75 (d, 3JHH = 15.6 Hz, 1H, CH=), 7.70 (d, 3JHH = 8.6 Hz, 2H, 2 × CHAr), 7.58–7.35 (m, 5H, 5 × CHAr), 7.25–7.02 (m, 6H, 6 × CHAr), 6.86 (s, 1H, NH), 5.84 (d, 3JHH = 15.6 Hz, 1H, CH=), 4.50 (s, 2H, CH2), 2.38 (s, 3H, CH3Tol), 2.30 (s, 3H, CH3Tol) ppm. 13C NMR {1H} (101 MHz, CDCl3) δ 165.5 (C=O), 165.4 (C=O), 145.7 (Cquat), 137.4 (Cquat), 137.2 (CH=), 136.2 (Cquat), 135.8 (Cquat), 135.0 (Cquat), 134.8 (Cquat), 133.9 (Cquat), 131.5 (Cquat), 130.3 (2 × CHAr), 130.0 (2 × CHAr), 127.9 (CHAr), 127.7 (CHAr), 126.6 (CHAr), 126.5 (CHAr), 125.7 (CHAr), 123.2 (2 × CHAr), 121.3 (CHAr), 119.2 (2 × CHAr), 118.5 (CHAr), 112.0 (CH=), 106.8 (Cquat), 49.3 (CH2), 21.2 (CH3Tol), 21.1 (CH3Tol) ppm. FTIR (neat) νmax: 3284 (NH st), 3056 (=CH st), 1714 (C=O st), 1676 (C=O st), 1603 (=CH st) cm−1. HRMS (ESI-TOF) m/z: [M+H]+ calcd for C31H26N2O3 475.2022, Found 475.2024.
(E)-4-(3-oxobut-1-en-1-yl)-1-(p-tolyl)-3-(p-tolylamino)-1,5-dihydro-2H-pyrrol-2-one (7g). The general procedure B was followed using 1-(p-tolyl)-3-(p-tolylamino)-1,5-dihydro-2H-pyrrol-2-one (278 mg, 1 mmol) 4a, affording 312 mg (90%) of 7g as yellow crystals after crystallization (Et2O/pentane 1:3). M.p. (Et2O /Pentane) = 195–196 °C. 1H NMR (300 MHz, CDCl3) δ 7.66 (d, 3JHH = 8.5 Hz, 2H, 2 × CHAr), 7.36–7.10 (m, 7H, 6 × CHAr + CH=), 6.81 (s, 1H, NH), 5.86 (d, 3JHH = 16.1 Hz, 1H, CH=), 4.41 (s, 2H, CH2), 2.38 (s, 3H, CH3Tol), 2.36 (s, 3H, CH3Tol), 2.01 (d, 4JHH = 1.5 Hz, 3H, COCH3) ppm. 13C NMR {1H} (75 MHz, CDCl3) δ 198.1 (C=O), 165.3 (C=O), 137.6 (Cquat), 136.6 (Cquat), 136.2 (Cquat), 135.8 (Cquat), 134.9 (Cquat), 134.7 (CH=), 130.2 (2 × CHAr), 129.9 (2 × CHAr), 123.5 (2 × CHAr), 119.1 (2 × CHAr), 119.1 (CH=), 107.3 (Cquat), 49.1 (CH2), 26.0 (COCH3), 21.1 (CH3Tol), 21.1 (CH3Tol) ppm. FTIR (neat) νmax: 3281 (NH st), 3059 (=CH st), 1713 (C=O st), 1679 (C=O st), 1603 (C=C st) cm−1. HRMS (ESI-TOF) m/z: [M+H]+ calcd for C22H23N2O2 347.1760, Found 347.1757.
(E)-4-(3-oxobut-1-en-1-yl)-1-(3-(trifluoromethyl)phenyl)-3-((3-(trifluoromethyl)phenyl) amino)-1,5-dihydro-2H-pyrrol-2-one (7h). The general procedure B was followed using 1-(3-(trifluoromethyl)phenyl)-3-((3-(trifluoromethyl)phenyl)amino)-1,5-dihydro-2H-pyrrol-2-one 4b (386 mg, 1 mmol), affording 391 mg (86%) of 7h as a pale yellow oil after chromatography (Hexanes/AcOEt 8:2). 1H NMR (400 MHz, Acetone d6) δ 8.42 (m, 1H, CHAr), 8.22 (m, 1H, CHAr), 8.05 (s, 1H, NH), 7.67 (m, 1H, CHAr), 7.62–7.49 (m, 4H, 4 × CHAr), 7.45 (d, 3JHH = 16.1 Hz, 1H, CH=), 7.39 (m, 1H, CHAr), 6.3 (d, 3JHH = 16.1 Hz, 1H, CH=), 4.83 (s, 2H, CH2), 2.10 (s, 3H, COCH3) ppm. 13C NMR {1H} (101 MHz, Acetone d6) δ 197.1 (C=O), 165.5 (C=O), 143.0 (Cquat), 141.0 (Cquat), 136.6 (Cquat), 133.1 (CH=), 131.7 (q, 2JFC = 32.1 Hz, Cquat), 131.6 (q, 2JFC = 32.2 Hz, Cquat), 130.9 (2 × CHAr), 127.1 (CHAr), 125.2 (q, 1JFC = 271.4 Hz, Cquat), 125.2 (q, 1JFC = 271.6 Hz, Cquat), 124.5 (CHAr), 122.4 (CH=), 121.2 (q, 3JFC = 3.8 Hz, CHAr), 119.9 (q, 3JFC = 3.7 Hz, CHAr), 117.7 (Cquat), 117.4 (q, 3JFC = 4.2 Hz, CHar), 115.6 (q, 3JFC = 3.8 Hz, CHAr), 49.6 (CH2), 26.8 (COCH3) ppm. 19F-NMR (282 MHz, CDCl3) δ −63.1, −63.5 ppm. FTIR (neat) νmax: 3297 (NH st), 3054 (=CH st), 1715 (C=O st), 1677 (C=O st), 1608 (C=C st) cm−1. HRMS (ESI-TOF) m/z: [M+H]+ calcd for C22H17F6N2O2 455.1194, Found 455.1187.
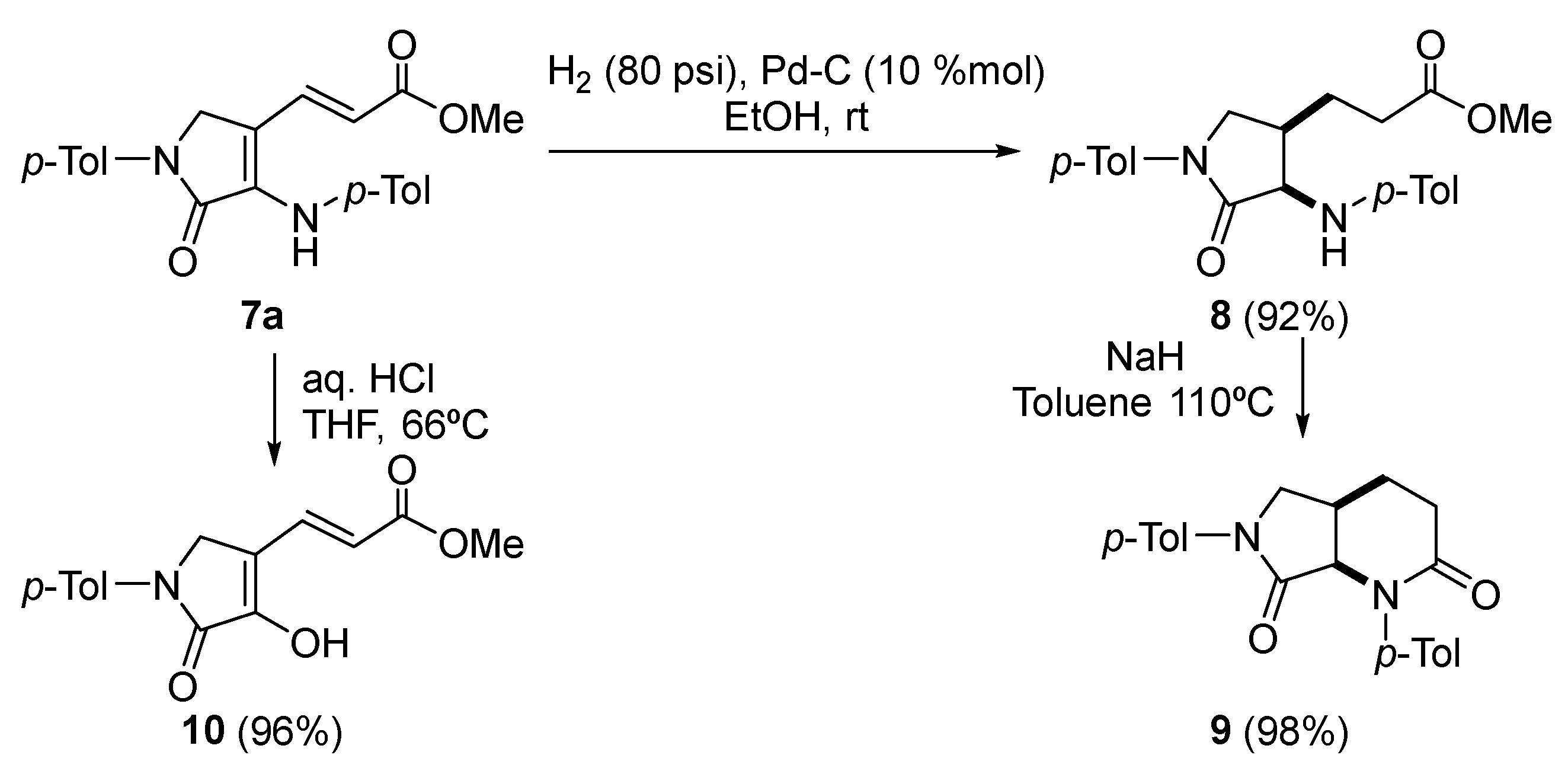
Procedure for the Catalytic Hydrogenation of 7a
A solution of (E)-3-(5-oxo-1-(p-tolyl)-4-(p-tolylamino)-2,5-dihydro-1H-pyrrol-3-yl) acrylate (7a) (363 mg, 1mmol) in MeOH (5 mL) was stirred for 2 days under H2 pressure (80 psi) in the presence of palladium on carbon (10%) (213 mg, 10% mmol). The reaction mixture was filtered through celite and concentrated under vacuum. The crude residue was purified by crystallization (Dichloromethane/Hexanes 1:3), affording pure saturated γ-lactam 7.
Methyl 3-((3R*,4R*)-5-oxo-1-(p-tolyl)-4-(p-tolylamino)pyrrolidin-3-yl)propanoate (8). The procedure was followed, affording 327 mg (90%) of 8 as white crystals M.p. (Dichloromethane/Hexanes) = 158–160 °C. 1H NMR (400 MHz, CDCl3) δ 7.54 (d, 3JHH = 8.5 Hz, 2H, 2 × CHAr), 7.21 (d, 3JHH = 8.5 Hz, 2H, 2 × CHAr), 7.04 (d, 3JHH = 8.4 Hz, 2H, 2 × CHAr), 6.63 (d, 3JHH = 8.4 Hz, 2H, 2 × CHAr), 4.47 (s, 1H, NH), 4.13 (dd, 3JHH = 7.2 Hz, 4JHH = 2.0 Hz, 1H, NCH), 4.00 (dd, 2JHH = 10.2 Hz, 3JHH = 5.8 Hz, 1H, NCHAHB), 3.59 (s, 3H, OCH3), 2.91 (m, 1H, CH), 2.43–2.29 (m, 5H, CH3Tol + COCH2), 2.27 (s, 3H, CH3Tol), 1.96 (m, 1H, CH), 1.54 (m, 1H, CHAHB) ppm. 13C NMR {1H} (101 MHz, CDCl3) δ 173.5 (C=O), 171.4 (C=O), 145.4 (Cquat), 136.7 (Cquat), 134.9 (Cquat), 130.0 (2 × CHAr), 129.7 (2 × CHAr), 127.8 (Cquat), 119.8 (2 × CHAr), 113.5 (2 × CHAr), 60.0 (CH), 51.7 (OCH3), 50.1 (CH2), 37.1 (CH), 31.7 (CH2), 23.0 (CH2), 21.0 (CH3Tol), 20.5 (CH3Tol) ppm. FTIR (neat) νmax: 3325 (NH st), 1733 (C=O st), 1698 (C=O st) cm−1. HRMS (ESI-TOF) m/z: [M+H]+ calcd for C22H27N2O3 367.2022, Found 367.2011.
Procedure for the Intramolecular Cyclization of 8
To a suspension of NaH (29 mg, 1.2 mmol) in toluene (3 mL) was added neat 3-((3R*,4R*)-5-oxo-1-(p-tolyl)-4-(p-tolylamino)pyrrolidin-3-yl)propanoate (8) (366 mg, 1mmol). The reaction was stirred under reflux overnight and the resulting mixture was then quenched with water (10 mL) and extracted with dichloromethane (2 × 10 mL). The combined organic layers were dried over MgSO4 and concentrated under vacuum and the crude residue was purified by chromatography (Hexanes/AcOEt 1:1), affording pure bicyclic γ-lactam 9.
(4aR*,7aR*)-1,6-Di-p-tolylhexahydro-1H-pyrrolo[3,4-b]pyridine-2,7-dione (9). The procedure was followed, affording 330 mg (98%) of 9 as a pale yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.51 (d, 3JHH = 8.6 Hz, 2H, 2 × CHAr), 7.37 (d, 3JHH = 8.4 Hz, 2H, 2 × CHAr), 7.19 (d, 3JHH = 8.4 Hz, 2H, 2 × CHAr), 7.17 (d, 3JHH = 8.6 Hz, 2H, 2 × CHAr), 4.65 (d, 3JHH = 8.0 Hz, 1H, NCH), 4.04 (dd, 2JHH = 10.1 Hz, 3JHH = 6.4 Hz, 1H, NCHACHB), 3.56 (d, 2JHH = 10.1 Hz, 1H, NCHACHB), 3.00 (p, 3JHH = 7.0 Hz, 1H, CH), 2.54 (t, 3JHH = 6.7 Hz, 2H, COCH2), 2.34 (s, 3H, CH3Tol), 2.33 (s, 3H, CH3Tol), 2.16–1.92 (m, 2H, COCH2CH2) ppm. 13C NMR {1H} (101 MHz, CDCl3) δ 170.4 (C=O), 169.7 (C=O), 139.7 (Cquat), 136.9 (Cquat), 136.4 (Cquat), 134.9 (Cquat), 129.6 (2 × CHAr), 129.5 (2 × CHAr), 127.3 (2 × CHAr), 119.5 (2 × CHAr), 64.3 (CH), 51.7 (CH2), 31.5 (CH2), 30.3 (CH), 25.7 (CH2), 21.2 (CH3Tol), 20.9 (CH3Tol) ppm. FTIR (neat) νmax: 1698 (C=O st), 1673 (C=O st) cm−1. HRMS (ESI-TOF) m/z: [M+H]+ calcd for C21H23N2O2 335.1760, Found 335.1757.
Procedure for the Acidic Hydrolysis of 7a
A solution of (E)-3-(5-oxo-1-(p-tolyl)-4-(p-tolylamino)-2,5-dihydro-1H-pyrrol-3-yl) acrylate (7a) (0.363, 1 mmol) in toluene (2 mL) and 37% aqueous HCl (2 mL) was stirred at room temperature overnight. The mixture was diluted with water and extracted with dichloromethane (2 × 10 mL). The combined organic layers were washed with water (2 × 10 mL), dried over MgSO4, and concentrated under vacuum. The crude residue was purified by chromatography (Hexanes/AcOEt 4:1), affording pure enol-derived γ-lactam 10 after crystallization (Et2O /Pentane 1:3).
Methyl (E)-3-(4-hydroxy-5-oxo-1-(p-tolyl)-2,5-dihydro-1H-pyrrol-3-yl)acrylate (10). The procedure was followed, affording 262 mg (96%) of 9 as white crystals. M.p. (Et2O /Pentane) = 239–241 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.33 (s, 1H, OH), 7.72 (d, 3JHH = 8.7 Hz, 2H, 2 × CHAr), 7.66 (d, 3JHH = 15.9 Hz, 1H, CH=), 7.22 (d, 3JHH = 8.7 Hz, 2H, 2 × CHAr), 6.11 (d, 3JHH = 15.9 Hz, 1H, CH=), 4.49 (s, 2H, CH2), 3.71 (s, 3H, OCH3), 2.29 (s, 3H, CH3Tol) ppm. 13C NMR {1H} (101 MHz, DMSO-d6) δ 167.1 (C=O), 164.6 (C=O), 149.6 (Cquat), 137.2 (Cquat), 133.9 (CH=), 133.8 (Cquat), 129.8 (2 × CHAr), 118.8 (2 × CHAr), 116.9 (CH=), 114.9 (Cquat), 52.0 (OCH3), 47.2 (CH2), 20.9 (CH3Tol) ppm. FTIR (neat) νmax: 3460 (OH st), 3046 (=CH st), 1701 (C=O st), 1679 (C=O st), 1612 (C=C st) cm−1. HRMS (ESI-TOF) m/z: [M+H]+ calcd for C15H16NO4 274.1079, Found 274.1076.
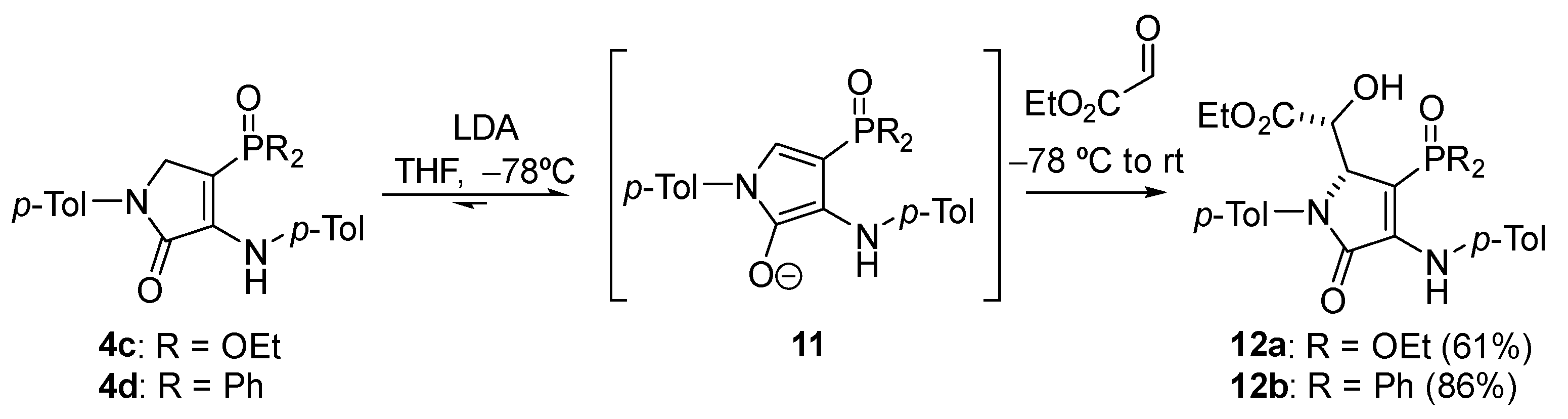
General Procedure for the Nucleophilic Addition of γ-Lactams 4c–d to Ethyl Glyoxalate
A solution of diethyl (5-oxo-1-(p-tolyl)-4-(p-tolylamino)-2,5-dihydro-1H-pyrrol-3-yl) phosphonate 4c (414 mg, 1 mmol) in THF (1 mL) was added to a freshly prepared solution of LDA (1.2 Eq) in THF (2 mL) at −78 °C. After 1 h, a 1.6 M solution of ethyl glyoxalate in toluene (1.2 mmol, 750 μL) was added and the reaction was left to warm to rt overnight. The reaction was quenched with a 0.5 M aqueous solution of HCl and the resulting mixture was extracted with dichloromethane (2 × 10 mL). The combined organic layers were washed with water (2 × 10 mL), dried over MgSO4, and concentrated under vacuum. The crude residue was purified by chromatography (Hexanes/AcOEt 6:4), affording pure γ-lactam derivatives 12.
Ethyl (R*)-2-((R*)-3-(diethoxyphosphoryl)-5-oxo-1-(p-tolyl)-4-(p-tolylamino)-2,5-dihydro-1H-pyrrol-2-yl)-2-hydroxyacetate (12a). The general procedure was followed using diethyl (5-oxo-1-(p-tolyl)-4-(p-tolylamino)-2,5-dihydro-1H-pyrrol-3-yl)phosphonate 4c (414 mg, 1 mmol) affording 315 mg (61%) of 12a as a pale yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.45 (s, 1H, NH), 7.34 (d, 3JHH = 7.3 Hz, 2H, 2 × CHAr), 7.20 (d, 3JHH = 8.0 Hz, 2H, 2 × CHAr), 7.14–7.03 (m, 4H, 4 × CHAr), 5.16 (t, 3JHH = 1.5 Hz, 1H, CH), 4.47 (d, 3JPH = 7.9 Hz, 1H, CH), 4.42 (broad s, 1H, OH), 4.03 (q, 3JHH = 7.2 Hz, 4H, 2 × CH2), 3.94 (q, 3JHH = 7.2 Hz, 2H, CH2), 2.35 (s, 3H, CH3Tol), 2.32 (s, 3H, CH3Tol), 1.47–0.98 (m, 9H, 3 × CH3) ppm. 13C NMR {1H} (101 MHz, CDCl3) δ 171.5 (C=O), 164.1 (d, 2JCP = 19.4 Hz, Cquat), 146.2 (d, 3JCP = 7.2 Hz, C=O), 137.0 (Cquat), 136.5 (Cquat), 135.0 (Cquat), 133.1 (Cquat), 129.8 (2 × CHAr), 129.3 (2 × CHAr), 124.4 (2 × CHAr), 123.8 (2 × CHAr), 96.4 (d, 1JCP = 211.3 Hz, Cquat), 70.2 (CH), 63.9 (d, 2JCP = 17.4 Hz, CH), 62.5 (d, 2JCP = 5.5 Hz, CH2), 62.4 (d, 2JCP = 5.4 Hz, CH2), 61.8 (CH2), 21.2 (CH3Tol), 21.1 (CH3Tol), 16.3 (d, 3JCP = 6.7 Hz, CH3), 16.3 (d, 3JCP = 6.9 Hz, CH3), 14.1 (CH3) ppm. 31P NMR (162 MHz, CDCl3) δ 19.1 ppm. FTIR (neat) νmax: 3430 (OH), 3338 (NH), 1714 (C=O st), 1694 (C=O st), 1609 (C=C), 1213 (P=O) cm−1. HRMS (ESI-TOF) m/z: [M+H]+ calcd for C26H34N2O7P 517.2104, Found 517.2100.
Ethyl (R*)-2-((R*)-3-(diphenylphosphoryl)-5-oxo-1-(p-tolyl)-4-(p-tolylamino)-2,5-dihydro-1H-pyrrol-2-yl)-2-hydroxyacetate (12b). The general procedure was followed using 4-(diphenylphosphoryl)-1-(p-tolyl)-3-(p-tolylamino)-1,5-dihydro-2H-pyrrol-2-one 4d (478 mg, 1 mmol), affording 498 mg (86%) of 12b as yellow crystals. M.p. (Dichloromethane/Hexanes) = 211–213 °C. 1H NMR (400 MHz, CDCl3) δ 7.87–7.70 (m, 2H, 2 × CHAr), 7.61–7.43 (m, 5H, 5 × CHAr), 7.33 (d, 3JHH = 8.2 Hz, 2H, 2 × CHAr), 7.31–7.23 (m, 3H, 3 × CHAr), 7.17 (d, 3JHH = 8.2 Hz, 2H, 2 × CHAr), 6.82 (d, 3JHH = 8.2 Hz, 2H, 2 × CHAr), 6.72 (d, 3JHH = 8.2 Hz, 2H, 2 × CHAr), 6.64 (s, 1H, NH), 5.28 (broad s, 1H, OH), 5.09 (t, 3JHH = 2.8 Hz, 1H, CH), 4.38 (dd, 3JPH = 9.3 Hz, 3JHH = 2.8 Hz, 1H, CH), 3.91 (q, 3JHH = 7.1 Hz, 2H, CH2), 2.33 (s, 3H, CH3Tol), 2.21 (s, 3H, CH3Tol), 1.12 (t, 3JHH = 7.1 Hz, 3H, CH3) ppm. 13C NMR {1H} (101 MHz, CDCl3) δ 171.0 (C=O), 164.5 (d, 3JCP = 14.5 Hz, C=O), 144.9 (d, 2JCP = 3.9 Hz, Cquat), 137.2 (Cquat), 136.7 (d, 4JCP = 1.2 Hz, Cquat), 133.8 (Cquat), 133.3 (Cquat), 132.9 (d, 1JCP = 109.0 Hz, Cquat), 132.1 (d, 4JCP = 2.5 Hz, CHAr), 131.9 (d, 4JCP = 2.9 Hz, CHAr), 131.3 (d, 2JCP = 9.6 Hz, 2 × CHAr), 131.2 (d, 2JCP = 10.3 Hz, 2 × CHAr), 131.0 (Cquat), 129.7 (2 × CHAr), 129.5 (2 × CHAr), 129.0 (d, 3JCP = 12.4 Hz, 2 × CHAr), 128.6 (d, 3JCP = 12.7 Hz, 2 × CHAr), 123.7 (2 × CHAr), 121.5 (2 × CHAr), 102.5 (d, 1JCP = 113.1 Hz, Cquat), 70.6 (CH), 64.7 (d, 2JCP = 12.7 Hz, CH), 61.6 (CH2), 21.2 (CH3Tol), 20.9 (CH3Tol), 14.0 (CH3) ppm. 31P NMR (121 MHz, CDCl3) δ 19.4 ppm. FTIR (neat) νmax: 3417 (OH), 3262 (NH), 1720 (C=O), 1679 (C=O), 1606 (C=C), 1175 (P=O). HRMS (ESI-TOF) m/z: [M+H]+ calcd for C34H34N2O5P 581.2205, Found 581.2202.
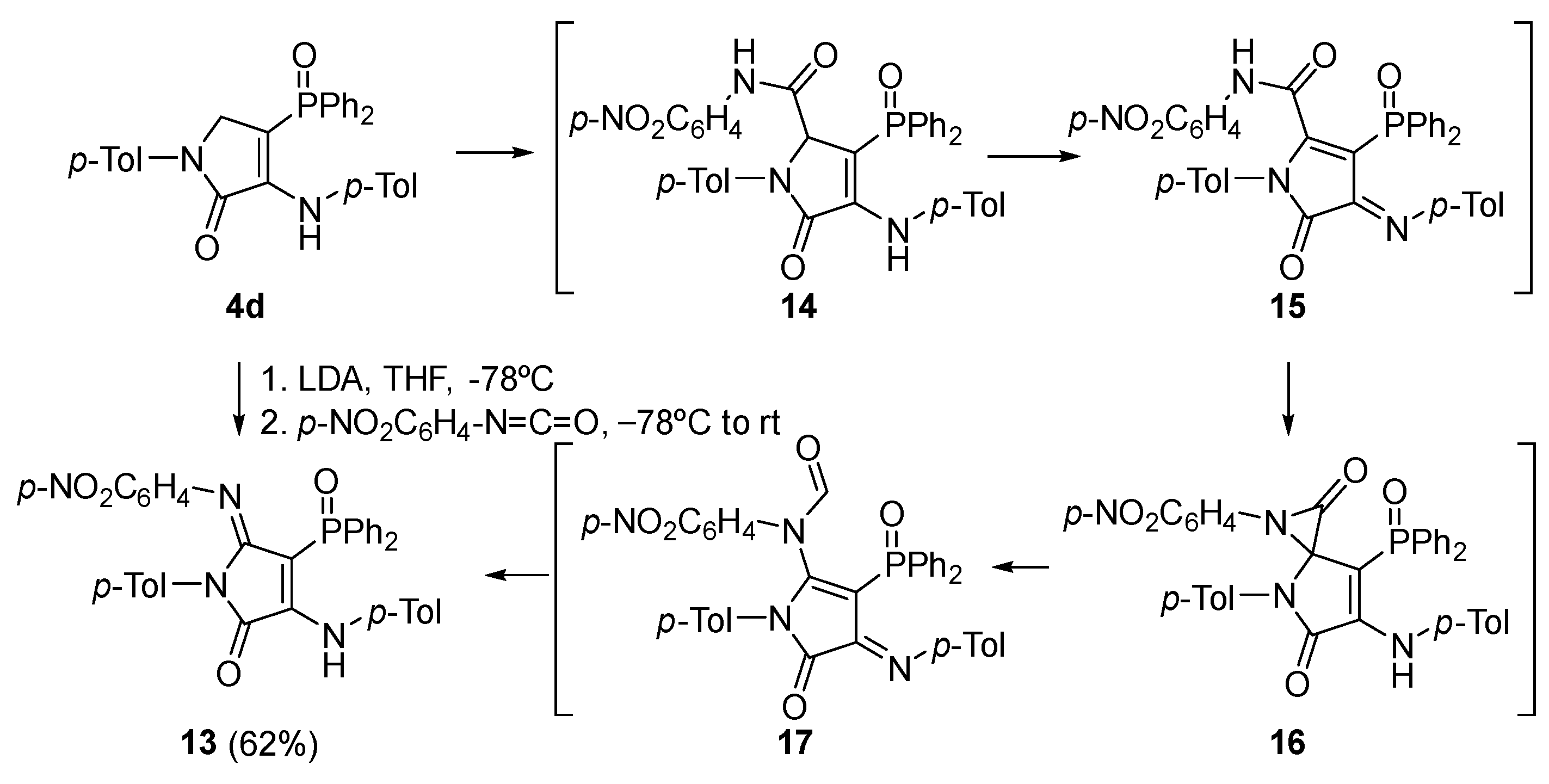
Procedure for the Synthesis of γ-Lactam Derivative 13
A solution of 4-(diphenylphosphoryl)-1-(p-tolyl)-3-(p-tolylamino)-1,5-dihydro-2H-pyrrol-2-one (4d) (478 mg, 1 mmol) in THF (1 mL) was added to a freshly prepared solution of LDA (1.2 Eq) in THF (2 mL) at −78 °C. After 1 h, a solution of 1-isocyanato-4-nitrobenzene (197 mg, 1.2 mmol) in THF (1 mL) was added and the reaction was left to warm to rt overnight. The reaction was quenched with a 0.5 M aqueous solution of HCl and the resulting mixture was extracted with dichloromethane (2 × 10 mL). The combined organic layers were washed with water (2 × 10 mL), dried over MgSO4, and concentrated under vacuum. The crude residue was purified by chromatography (Hexanes/AcOEt 4:1), affording pure γ-lactam derivative 13.
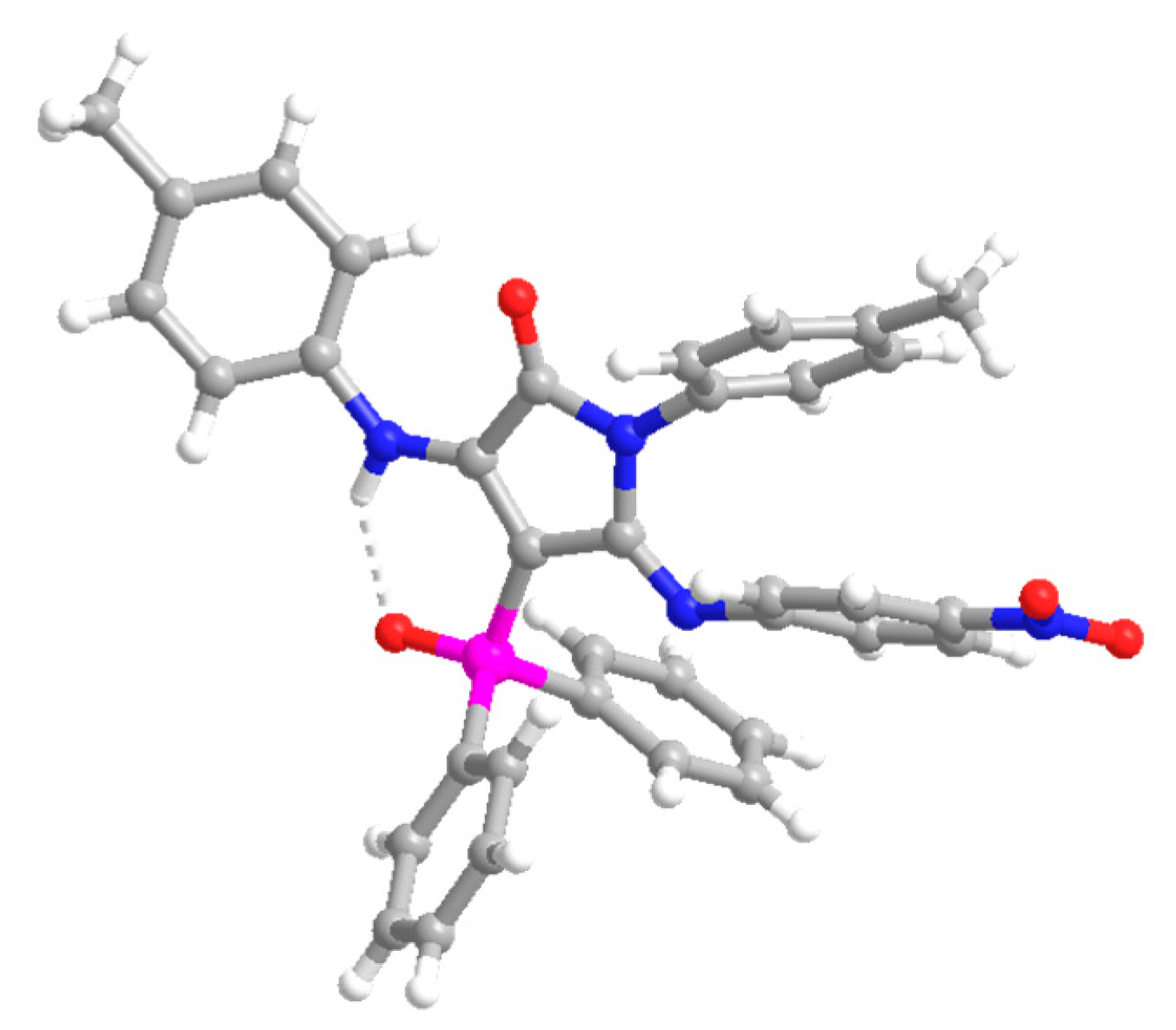
4-(Diphenylphosphoryl)-5-((4-nitrophenyl)imino)-1-(p-tolyl)-3-(p-tolylamino)-1,5-dihydro-2H-pyrrol-2-one (13). The procedure was followed, affording 380 mg (62%) of 13 as an orange oil. 1H NMR (400 MHz, CDCl3) δ 10.54 (s, 1H, NH), 8.03–7.91 (m, 4H, 4 × CHAr), 7.66 (d, 3JHH = 8.9 Hz, 2H, 2 × CHAr), 7.63–7.56 (m, 2H, 2 × CHAr), 7.54–7.47 (m, 4H, 4 × CHAr), 7.18 (d, 3JHH = 8.3 Hz, 2H, 2 × CHAr), 7.11 (d, 3JHH = 8.2 Hz, 2H, 2 × CHAr), 6.76 (d, 3JHH = 8.2 Hz, 2H, 2 × CHAr), 6.66 (d, 3JHH = 8.3 Hz, 2H, 2 × CHAr), 6.27 (d, 3JHH = 8.9 Hz, 2H, 2 × CHAr), 2.32 (s, 3H, CH3Tol), 2.10 (s, 3H, CH3Tol) ppm. 13C NMR {1H} (101 MHz, CDCl3) δ 164.7 (d, 2JCP = 14.4 Hz, C=N), 153.2 (Cquat), 152.1 (d, 3JCP = 3.7 Hz, C=O), 148.7 (Cquat), 148.6 (Cquat), 142.5 (Cquat), 138.7 (Cquat), 136.4 (Cquat), 134.7 (Cquat), 133.4 (Cquat), 132.5 (d, 4JPC = 2.7 Hz, 2 × CHAr), 132.2 (2 × CHAr), 132.1 (2 × CHAr), 130.7 (Cquat), 129.4 (2 × CHAr), 129.3 (2 × CHAr), 128.5 (2 × CHAr), 128.4 (2 × CHAr), 128.1 (2 × CHAr), 124.6 (2 × CHAr), 123.8 (2 × CHAr), 120.6 (2 × CHAr), 93.7 (d, 1JCP = 116.4 Hz, Cquat), 21.2 (CH3Tol), 21.1 (CH3Tol) ppm. 31P NMR (162 MHz, CDCl3) δ 30.9 ppm. FTIR (neat) νmax: 3330 (NH), 1667 (C=O st), 1606 (C=C), 1204 (P=O) cm−1. HRMS (ESI-TOF) m/z: [M+H]+ calcd for C36H30N4O4P 613.2005, Found 613.2025.
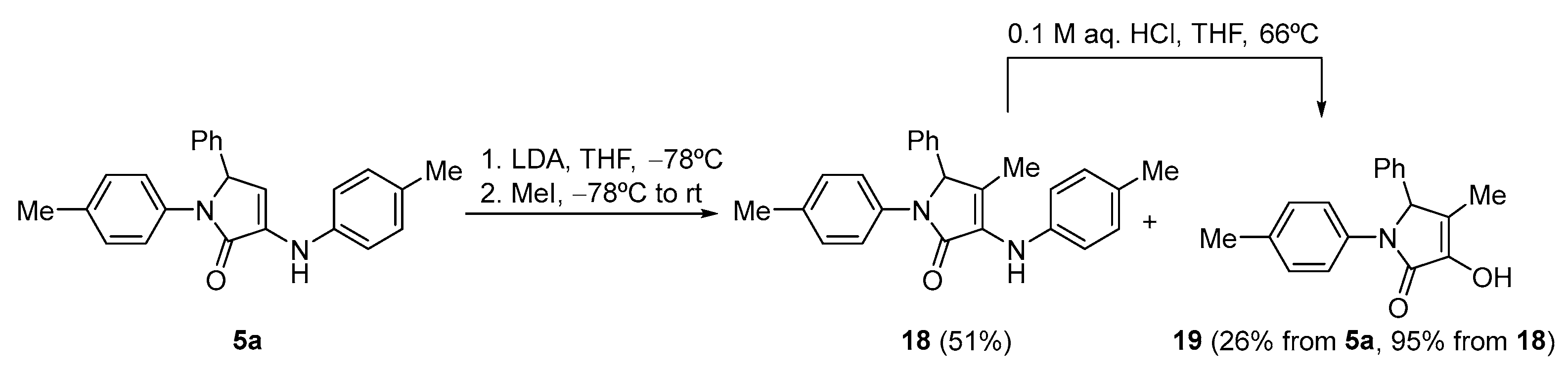
Procedure for the Functionalization of γ-Lactam 5a with Methyl Iodide
A solution of 5-phenyl-1-(p-tolyl)-3-(p-tolylamino)-1,5-dihydro-2H-pyrrol-2-one (5a) (354 mg, 1mmol) in THF (1 mL) was added to a solution of freshly prepared LDA (1.2 mmol) in tetrahydrofuran (2 mL) at −78 °C. After 1 h, an excess of methyl iodide (93 μL, 1.5 mmol) was added. The reaction mixture was stirred overnight at −78 °C and was then quenched with a 0.5 M aqueous solution of HCl (10 mL). The reaction mixture was extracted with dichloromethane (2 × 10 mL) and the combined organic layers were washed with water (2 × 10 mL), dried with MgSO4, and concentrated under vacuum. The crude residue was purified by chromatography (Hexanes), affording a 3:1 mixture of 18 and 19 that was isolated as pure after chromatography followed by crystallization (Et2O/Pentane).
4-methyl-5-phenyl-1-(p-tolyl)-3-(p-tolylamino)-1,5-dihydro-2H-pyrrol-2-one (18). The procedure was followed, affording 0.188 g (51%) of 18 as white crystals. M.p. (Et2O/Pentane) = 191–192 °C. 1H NMR (400 MHz, CDCl3) δ 7.40 (d, 3JHH = 8.5 Hz, 2H, 2 × CHAr), 7.35–7.20 (m, 5H, 5 × CHAr), 7.05 (d, 3JHH = 8.5 Hz, 2H, 2 × CHAr), 7.05 (d, 3JHH = 8.5 Hz, 2H, 2 × CHAr), 6.78 (d, 3JHH = 8.5 Hz, 2H, 2 × CHAr), 5.94 (s, 1H, NH), 5.33 (s, 1H, CH), 2.28 (s, 3H, CH3Tol), 2.24 (s, 3H, CH3Tol), 1.60 (s, 3H, CH3) ppm. 13C NMR {1H} (101 MHz, CDCl3) δ 168.2 (C=O), 139.7 (Cquat), 137.3 (Cquat), 135.2 (Cquat), 134.2 (Cquat), 130.8 (Cquat), 129.7 (Cquat), 129.6 (2 × CHAr), 129.5 (2 × CHAr), 129.1 (2 × CHAr), 128.4 (CHAr), 127.2 (2 × CHAr), 125.7 (Cquat), 121.4 (2 × CHAr), 118.8 (2 × CHAr), 67.6 (CH), 21.0 (CH3Tol), 20.8 (CH3Tol), 13.6 (CH3) ppm. FTIR (neat) νmax: 3306 (NH st), 1688 (C=O st), 1608 (C=C st) cm−1. HRMS (ESI-TOF) m/z: [M+H]+ calcd for C25H25N2O 369.1967, Found 369.1973.
3-Hydroxy-4-methyl-5-phenyl-1-(p-tolyl)-1,5-dihydro-2H-pyrrol-2-one (19). The procedure was followed, affording 76 mg (26%) of 19 of as a pale yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.34 (d, 3JHH = 8.5 Hz, 2H, 2 × CHAr), 7.33–7.24 (m, 3H, 3 × CHAr), 7.16 (d, 3JHH = 8.3 Hz, 2H, 2 × CHAr), 7.04 (d, 3JHH = 8.5 Hz, 2H, 2 × CHAr), 6.15 (s, 1H, OH), 5.26 (s, 1H, CH), 2.23 (s, 3H, CH3Tol), 1.73 (s, 3H, CH3) ppm. 13C NMR {1H} (101 MHz, CDCl3) δ 166.9 (C=O), 140.7 (Cquat), 136.2 (Cquat), 134.8 (Cquat), 134.6 (Cquat), 129.6 (2 × CHAr), 129.1 (2 × CHAr), 128.5 (CHAr), 127.3 (2 × CHAr), 122.1 (Cquat), 121.3 (2 × CHAr), 66.0 (CH), 21.0 (CH3Tol), 9.5 (CH3) ppm. FTIR (neat) νmax: 3501 (OH st), 1681 (C=O st), 1607 (C=C st) cm−1. HRMS (ESI-TOF) m/z: [M+H]+ calcd for C18H18N2O 280.1338, Found 280.1343. Additionally, substrate 18 can be obtained in 95% yield from 17 by refluxing in a mixture of THF 1 (mL) and a 0.1 M aqueous solution of HCl (1 mL) for 1 h.
Procedure for the Vinylogous Mannich Reaction of γ-Lactam 5b with Ethyl Glyoxalate
A solution of ethyl 5-oxo-1-(p-tolyl)-4-(p-tolylamino)-2,5-dihydro-1H-pyrrole-2-carboxylate (5b)(350 mg, 1 mmol), N,N-dimethylmethyleneiminium iodide (370 mg, 2 mmol) and triethylamine (350 μL, 2.5 mmol, in chloroform (3 mL) was refluxed overnight. The reaction mixture was then acidified with a 0.5 M aqueous solution of HCl (10 mL) and extracted with dichloromethane (2 × 10 mL). The combined organic layers were washed with water (2 × 10 mL), dried over MgSO4, and concentrated under vacuum. The crude residue was purified by crystallization from a mixture of dichloromethane/hexanes (1:3), affording the pure functionalized γ-lactam derivative 20.
Ethyl 2-((dimethylamino)methyl)-5-oxo-1-(p-tolyl)-4-(p-tolylamino)-2,5-dihydro-1H- pyrrole-2-carboxylate (20). The procedure was followed, affording 321 mg (68%) of 20 as yellow crystals. M.p. (Dichloromethane/Hexanes) = 170–172 °C. 1H NMR (400 MHz, CDCl3) δ 7.25 (d, 3JHH = 8.5 Hz, 2H, 2 × CHAr), 7.20 (d, 3JHH = 8.2 Hz, 2H, 2 × CHAr), 7.13 (d, 3JHH = 8.2 Hz, 2H, 2 × CHAr), 7.00 (d, 3JHH = 8.5 Hz, 2H, 2 × CHAr), 6.65 (s, 1H, NH), 5.95 (s, 1H, CH), 4.30–4.09 (m, 2H, CH2), 3.17 (d, 2JHH = 14.0 Hz, 1H, CHACHB), 2.74 (d, 2JHH = 14.0 Hz, 1H, CHACHB), 2.36 (s, 3H, CH3Tol), 2.31 (s, 3H, CH3Tol), 2.16 (s, 6H, 2 × NCH3), 1.24 (t, 3JHH = 7.1 Hz, 3H, CH3) ppm. 13C NMR {1H} (101 MHz, CDCl3) δ 171.1 (C=O), 168.3 (C=O), 138.8 (Cquat), 136.6 (Cquat), 134.1 (Cquat), 133.7 (Cquat), 130.1 (Cquat), 130.0 (2 × CHAr), 129.7 (2 × CHAr), 125.6 (2 × CHAr), 116.9 (2 × CHAr), 104.9 (CHAr), 72.7 (CH), 62.1 (CH2O), 60.9 (CH2), 47.8 (2 × NCH3), 21.2 (CH3, Tol), 20.8 (CH3, Tol), 14.2 (CH3, OEt) ppm. FTIR (neat) νmax: 3312 (=CH st), 1691 (C=O st), 1615 (C=C st) cm−1. HRMS (ESI-TOF) m/z: [M+H]+-Me2N calcd for C25H23N2O 367.1801, Found 367.1806.
Procedure for the Hydrogenation Reaction of γ-Lactam 5b
A solution of ethyl 5-oxo-1-(p-tolyl)-4-(p-tolylamino)-2,5-dihydro-1H-pyrrole-2- carboxylate (5b) (350 mg, 1mmol) in EtOH (5 mL) was stirred for 2 days under H2 pressure (80 psi) in the presence of palladium on carbon (10%) (320 mg, 10% mmol). The reaction mixture was filtered through celite and concentrated under vacuum. The crude residue was purified by crystallization from a mixture of Et2O/Pentane 1:3, affording pure 21 as white crystals
Ethyl (2S*,4S*)-5-oxo-1-(p-tolyl)-4-(p-tolylamino)pyrrolidine-2-carboxylate (21). The procedure was followed, affording 326 mg (93%) of 21 as white crystals M.p. (Et2O/Pentane) = 142–144 °C. 1H NMR (400 MHz, CDCl3) δ 7.35 (d, 3JHH = 8.5 Hz, 2H, 2 × CHAr), 7.18 (d, 3JHH = 8.2 Hz, 2H, 2 × CHAr), 7.03 (d, 3JHH = 8.2 Hz, 2H, 2 × CHAr), 6.62 (d, 3JHH = 8.5 Hz, 2H, 2 × CHAr), 4.78 (dd, 3JHH = 7.7 Hz, 3JHH = 7.7 Hz, 1H, CH), 4.16 (dd, 3JHH = 8.3 Hz, 3JHH = 8.3 Hz, 1H, CH), 4.10 (m, 2H, CH2O), 3.09 (m, 1H, CH2), 2.33 (s, 3H, CH3Tol), 2.26 (s, 3H, CH3Tol), 2.08 (m,, 1H, CH2), 1.12 (m, 3H, CH3 OEt) ppm. 13C NMR {1H} (101 MHz, CDCl3) δ 172.4 (C=O), 171.0 (C=O), 144.7 (Cquat), 136.1 (Cquat), 135.2 (Cquat), 130.0 (2 × CHAr), 129.7 (2 × CHAr), 128.1 (Cquat), 122.1 (2 × CHAr), 114.0 (2 × CHAr), 61.9 (CH2), 59.1 (CH), 55.4 (CH), 33.1 (CH2), 21.1 (CH3Tol), 20.5 (CH3Tol), 14.1 (CH3) ppm. FTIR (neat) νmax: 3306 (N-H), 1748 (C=O), 1686 (C=O) cm−1. HRMS (ESI-TOF) m/z: [M+H]+ calcd for C21H24NO3 353.1865, Found 353.1862.
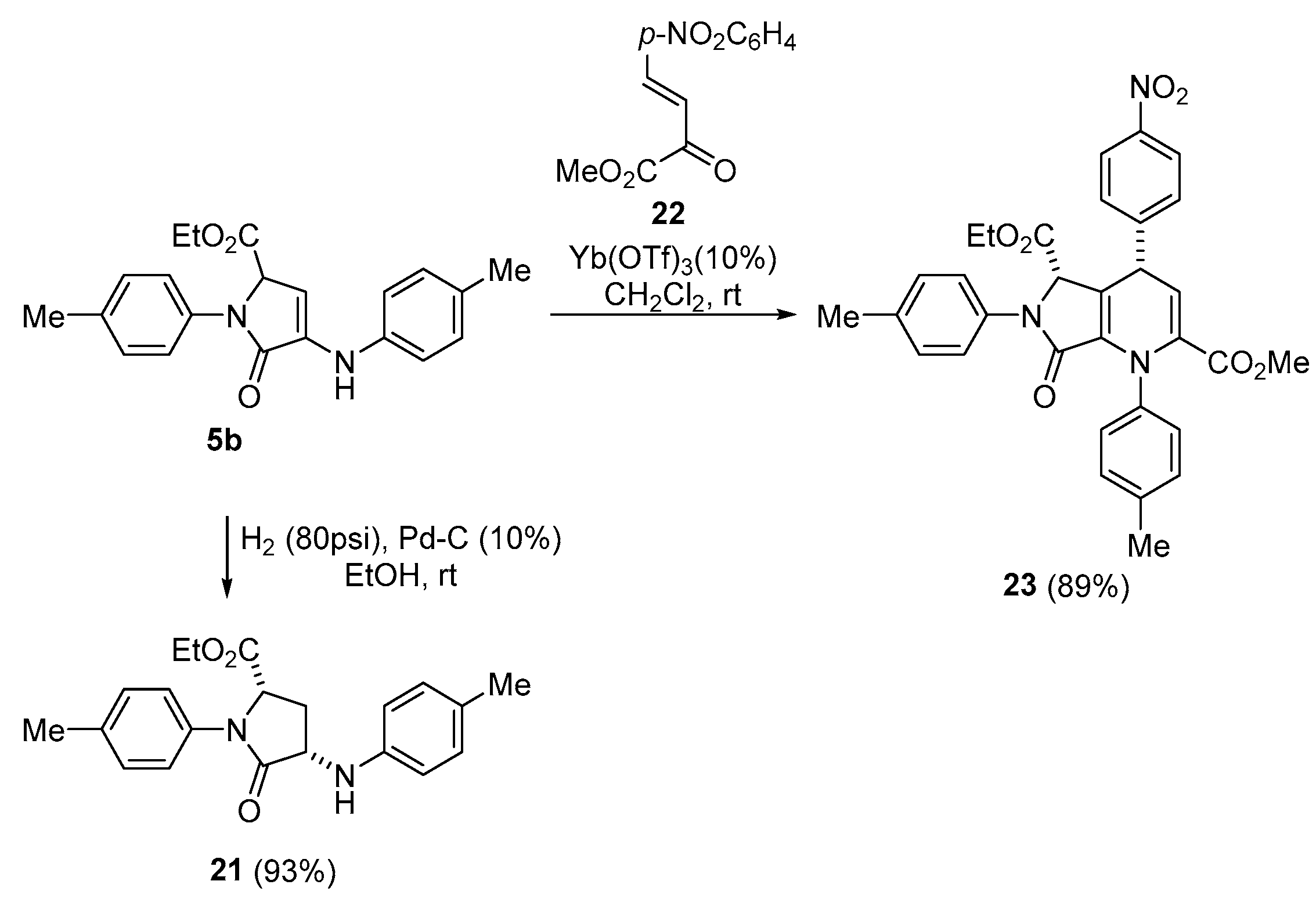
Procedure for the [3+3] Annulation Reaction of γ-Lactam 5b and β,γ-Unsaturated Ketoester 22
A solution of 5-oxo-1-(p-tolyl)-4-(p-tolylamino)-2,5-dihydro-1H-pyrrole- 2-carboxylate (5b) (350 mg, 1 mmol), Yb(OTf)3 (62.0 mg, 0.1 mmol) and β,γ--unsaturated ketoester 22 (282 mg, 1.2 mmol) was stirred in CH2Cl2 at room temperature for 6 h. The resulting mixture was filtered through celite and concentrated under vacuum. The crude residue was purified by column chromatography (Hexanes/AcOEt (8:2), affording pure 23 as a white solid.
5-Ethyl 2-methyl (4S*,5S*)-4-(4-nitrophenyl)-7-oxo-1,6-di-p-tolyl-4,5,6,7-tetrahydro- 1H-pyrrolo[3,4-b]pyridine-2,5-dicarboxylate (23). The procedure was followed, affording 504 mg (89%) of 23 as a white solid. 1H NMR (400 MHz, CDCl3) δ 8.23 (d, 3JHH = 8.7 Hz, 2H, 2 × CHAr), 7.52 (d, 3JHH = 8.7 Hz, 2H, 2 × CHAr), 7.34 (d, 3JHH = 8.2 Hz, 2H, 2 × CHAr), 7.27 (d, 3JHH = 8.0 Hz, 2H, 2 × CHAr), 7.18 (d, 3JHH = 8.0 Hz, 2H, 2 × CHAr), 7.06 (d, 3JHH = 8.2 Hz, 2H, 2 × CHAr), 5.76 (d, 3JHH = 4.7 Hz, 1H, CH), 5.03 (s, 1H, CH), 4.91 (d, 3JHH = 4.7 Hz, 1H, CH), 3.49 (s, 3H, OCH3), 3.34 (m, 1H, CH2O), 3.11 (m, 1H, CH2O), 2.36 (s, 3H, CH3Tol), 2.25 (s, 3H, CH3Tol), 0.79 (dd, 3JHH = 7.1 Hz, 3JHH = 7.1 Hz, 3H, CH3 OEt) ppm. 13C NMR {1H} (101 MHz, CDCl3) δ 167.8 (C=O), 163.6 (C=O), 163.0 (C=O), 149.8 (Cquat), 147.4 (Cquat), 138.2 (Cquat), 137.9 (Cquat), 136.1 (Cquat), 135.8 (Cquat), 135.2 (Cquat), 134.6 (Cquat), 129.7 (2 × CHAr), 129.2 (2 × CHAr), 129.2 (2 × CHAr), 128.6 (2 × CHAr), 124.1 (2 × CHAr), 121.0 (2 × CHAr), 119.6 (Cquat), 113.4 (CH), 63.1 (CH), 61.9 (CH2), 52.3 (OCH3), 41.0 (CH), 21.4 (CH3Tol), 20.8 (CH3Tol), 13.6 (CH3) ppm. FTIR (neat) νmax: 3053 (=CH st), 1739 (C=O), 1723 (C=O), 1676 (C=O), 1622 (C=C) cm−1. HRMS (ESI-TOF) m/z: [M+H]+ calcd for C32H29N33O7 568.2084, Found 568.2078.
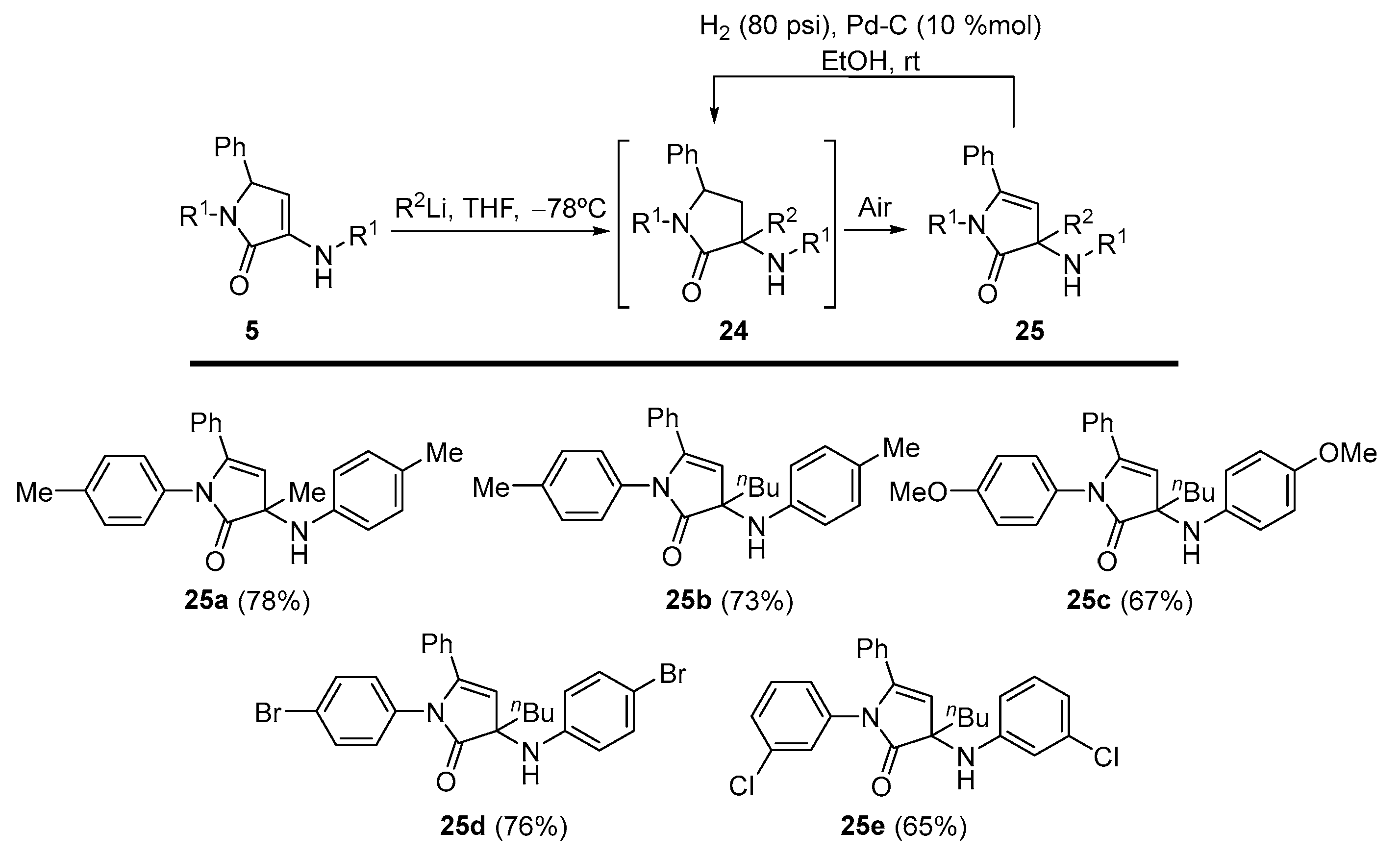
General Procedure for the Nucleophilic Addition of Organolithium Reagents to γ-Lactams 5
A 1.6 M solution of the corresponding organolithium reagent in hexanes (1.25 mL, 2 mmol) was added to a solution of the corresponding γ-lactam 5 (1 mmol) in tetrahydrofuran (3 mL) at −78 °C under N2 atmosphere. The reaction was warmed to rt overnight. The reaction crude was quenched with a 0.5 M aqueous solution of HCl (5 mL), and extracted with dichloromethane (2 × 10 mL). The combined organic layers were washed with water, dried over MgSO4, and concentrated under reduced pressure. The crude residue was purified by crystallization, providing the pure functionalized γ-lactam derivatives 25. In some cases, a previous purification by chromatography was necessary as detailed for each compound.
3-Methyl-5-phenyl-1-(p-tolyl)-3-(p-tolylamino)-1,3-dihydro-2H-pyrrol-2-one (25a). The general procedure was followed, using 5-phenyl-1-(p-tolyl)-3-(p-tolylamino)-1,5-dihydro- 2H-pyrrol-2-one (354 mg, 1 mmol) (5a) and a 1.6 M solution of methyllithium in hexanes (1.25 mL, 2 mmol), affording 288 mg (78%) of 25a as yellow crystals after chromatography (Hexanes/AcOEt 8:2) and subsequent crystallization (Et2O /Pentane 1:3). M.p. (Et2O /Pentane) = 183–185 °C. 1H NMR (400 MHz, CDCl3) δ 7.31–7.25 (m, 4H, 4 × CHAr), 7.24–7.20 (m, 2H, 2 × CHAr), 7.11 (d, 3JHH = 8.1 Hz, 2H, 2 × CHAr), 7.03–6.95 (m, 6H, 5 × CHAr + NH), 5.63 (s, 1H, =CH), 2.33 (s, 3H, CH3Tol), 2.28 (s, 3H, CH3Tol), 1.45 (s, 3H, CH3) ppm. 13C NMR {1H} (101 MHz, CDCl3) δ 175.4 (Cquat), 165.6 (C=O), 148.9 (Cquat), 137.1 (Cquat), 135.6 (Cquat), 133.1 (Cquat), 131.9 (Cquat), 130.0 (CHAr), 129.6 (2 × CHAr), 129.4 (2 × CHAr), 128.6 (2 × CHAr), 128.3 (2 × CHAr), 127.3 (2 × CHAr), 121.8 (2 × CHAr), 94.8 (=CH), 92.8 (Cquat), 25.3 (CH3), 21.1 (CH3Tol), 21.1 (CH3Tol) ppm. FTIR (neat) νmax: 3403 (NH st), 3053 (=CH st), 1674 (C=O st) cm−1. HRMS (ESI-TOF) m/z: [M+H]+ calcd for C25H25N2O 369.1967, Found 369.1972.
3-Butyl-5-phenyl-1-(p-tolyl)-3-(p-tolylamino)-1,3-dihydro-2H-pyrrol-2-one (25b). The general procedure was followed, using 5-phenyl-1-(p-tolyl)-3-(p-tolylamino)-1,5-dihydro- 2H-pyrrol-2-one (0.354 g, 1 mmol) (5a) and a 1.6 M solution of n-butyllithium in hexanes (1.25 mL, 2 mmol), affording 300 mg (73%) of 25b as an orange oil after chromatography (Hexanes/AcOEt 8:2). 1H NMR (400 MHz, CDCl3) δ 7.34–7.16 (m, 6H, 6 × CHAr), 7.11 (d, 3JHH = 8.3 Hz, 2H, 2 × CHAr), 7.05–6.95 (m, 6H, 5 × CHAr + NH), 5.61 (s, 1H, =CH), 2.33 (s, 3H, CH3Tol), 2.29 (s, 3H, CH3Tol), 2.12 (m, 1H, CHAHB), 1.85 (m, 1H, CHAHB), 1.36–1.00 (m, 4H, 2 × CH2Bu), 0.76 (t, 3JHH = 6.9 Hz, 3H, CH3Bu) ppm. 13C NMR {1H} (101 MHz, CDCl3) δ 175.0 (Cquat), 166.6 (C=O), 148.6 (Cquat), 136.2 (Cquat), 135.2 (Cquat), 133.0 (Cquat), 131.9 (Cquat), 129.9 (CHAr), 129.5 (2 × CHAr), 129.2 (2 × CHAr), 128.6 (2 × CHAr), 128.2 (2 × CHAr), 127.0 (2 × CHAr), 121.9 (2 × CHAr), 95.5 (=CH), 95.0 (Cquat), 38.0 (CH2Bu), 25.0 (CH2Bu), 22.6 (CH2Bu), 21.1 (CH3Tol), 21.0 (CH3Tol), 14.1 (CH3Bu) ppm. FTIR (neat) νmax: 3411 (NH st), 3053 (=CH st), 1703 (C=O st) cm−1. HRMS (ESI-TOF) m/z: [M+H]+ calcd for C28H31N2O 411.2436, Found 411.2442.
3-Butyl-1-(4-methoxyphenyl)-3-((4-methoxyphenyl)amino)-5-phenyl-1,3-dihydro-2H-pyrrol-2-one (25c). The general procedure was followed, using 1-(4-methoxyphenyl)-3-((4-methoxyphenyl)amino)-5-phenyl-1,5-dihydro-2H-pyrrol-2-one (386 mg, 1 mmol) (5c) and a 1.6 M solution of n-butyllithium in hexanes (1.25 mL, 2 mmol), affording 296 mg (67%) of 25c as an orange oil after chromatography (Hexanes/AcOEt 7:3). 1H NMR (400 MHz, CDCl3) δ 7.34–7.20 (m, 6H, 5 × CHAr + NH), 7.06 (d, 3JHH = 8.8 Hz, 2H, 2 × CHAr), 7.05 (d, 3JHH = 8.8 Hz, 2H, 2 × CHAr), 6.87 (d, 3JHH = 8.8 Hz, 2H, 2 × CHAr), 6.74 (d, 3JHH = 8.8 Hz, 2H, 2 × CHAr), 5.63 (s, 1H, =CH), 3.79 (s, 3H, OCH3), 3.77 (s, 3H, OCH3), 2.08 (m, 1H, CHAHB), 1.82 (m, 1H, CHAHB), 1.31–1.07 (m, 4H, 2 × CH2Bu), 0.77 (t, 3JHH = 6.8 Hz, 3H, CH3Bu) ppm. 13C NMR {1H} (101 MHz, CDCl3) δ 174.4 (Cquat), 167.4 (C=O), 157.7 (Cquat), 156.4 (Cquat), 143.6 (Cquat), 131.5 (Cquat), 131.4 (Cquat), 130.2 (CHAr), 128.7 (2 × CHAr), 128.7 (2 × CHAr), 128.4 (2 × CHAr), 123.1 (2 × CHAr), 114.3 (2 × CHAr), 113.9 (2 × CHAr), 95.1 (Cquat), 94.8 (=CH), 55.6 (OCH3), 55.5 (OCH3), 37.9 (CH2Bu), 25.1 (CH2Bu), 22.7 (CH2Bu), 14.1 (CH3Bu) ppm. FTIR (neat) νmax: 3391 (NH st), 3047 (=CH st), 1693 (C=O st) cm−1. HRMS (ESI-TOF) m/z: [M+H]+ calcd for C28H31N2O3 443.2335, Found 443.2334.
1-(4-Bromophenyl)-3-((4-bromophenyl)amino)-3-butyl-5-phenyl-1,3-dihydro-2H-pyrrol-2-one (25d). The general procedure was followed, using 1-(4-bromophenyl)-3-((4-bromophenyl)amino)-5-phenyl-1,5-dihydro-2H-pyrrol-2-one (5d) (484 mg, 1 mmol) and a 1.6 M solution of n-butyllithium in hexanes (1.25 mL, 2 mmol), affording 411 mg (76%) of 25d as yellow crystals after chromatography (Hexanes/AcOEt 85:15) followed by crystallization (Et2O /Pentane 1:3). M.p. (Et2O/Pentane) = 129–130 °C. 1H NMR (400 MHz, CDCl3) δ 7.42 (d, 3JHH = 8.7 Hz, 2H, 2 × CHAr), 7.34–7.10 (m, 10H, 9 × CHAr + NH), 6.94 (d, 3JHH = 8.7 Hz, 2H, 2 × CHAr), 5.55 (s, 1H, =CH), 1.37–1.03 (m, 5H, 5 × CHBu), 0.87 (m, 1H, CHBu), 0.74 (t, 3JHH = 6.9 Hz, 3H, CH3Bu) ppm. 13C NMR {1H} (75 MHz, CDCl3) δ 175.3 (Cquat), 167.5 (C=O), 150.6 (Cquat), 138.8 (Cquat), 132.1 (2 × CHAr), 131.5 (Cquat), 130.5 (CHAr), 128.8 (2 × CHAr), 128.7 (2 × CHAr), 128.6 (2 × CHAr), 127.3 (2 × CHAr), 123.7 (2 × CHAr), 116.8 (Cquat), 108.2 (Cquat), 95.5 (Cquat), 94.9 (=CH), 37.9 (CH2Bu), 25.1 (CH2Bu), 22.7 (CH2Bu), 14.0 (CH3Bu) ppm. FTIR (neat) νmax: 3397 (NH st), 3056 (=CH st), 1692 (C=O st) cm−1. HRMS (ESI-TOF) m/z: [M+H]+-Br, calcd for C26H26BrN2O 463.1208, Found 463.1215.
3-Butyl-1-(3-chlorophenyl)-3-((3-chlorophenyl)amino)-5-phenyl-1,3-dihydro-2H-pyrrol-2-one (25e). The general procedure was followed, using 1-(3-chlorophenyl)-3-((3-chlorophenyl)amino)-5-phenyl-1,5-dihydro-2H-pyrrol-2-one (5e) (395 mg, 1 mmol) and a 1.6 M solution of n-butyllithium in hexanes (1.25 mL, 2 mmol), affording 338 mg (75%) of 25e as yellow crystals after chromatography (Hexanes/AcOEt 85:15) followed by crystallization (Dichloromethane/Hexanes 1:3). M.p. (Dichloromethane/Hexanes) = 50–51 °C. 1H NMR (400 MHz, CDCl3) δ 7.39–7.21 (m, 7H, 7 × CHAr), 7.13–7.09 (m, 2H, 2 × CHAr), 7.08–7.02 (m, 2H, 2 × CHAr), 6.94–6.87 (m, 2H, 2 × CHAr), 5.57 (s, 1H, =CH), 2.05 (m, 1H, CHAHB), 1.86 (m, 1H, CHAHB), 1.32–1.02 (m, 4H, 2 × CH2Bu), 0.75 (t, 3JHH = 7.0 Hz, 3H, CH3Bu) ppm. 13C NMR {1H} (101 MHz, CDCl3) δ 175.2 (Cquat), 166.9 (C=O), 152.8 (Cquat), 140.1 (Cquat), 134.6 (Cquat), 134.3 (Cquat), 131.1 (Cquat), 130.6 (CHAr), 130.2 (CHAr), 129.4 (CHAr), 128.6 (2 × CHAr), 128.5 (2 × CHAr), 126.7 (CHAr), 125.9 (CHAr), 125.1 (CHAr), 123.9 (CHAr), 121.9 (CHAr), 120.2 (CHAr), 96.3 (Cquat), 94.8 (=CH), 37.7 (CH2Bu), 24.9 (CH2Bu), 22.6 (CH2Bu), 13.9 (CH3Bu) ppm. FTIR (neat) νmax: 3391 (NH st), 3069 (=CH st), 1705 (C=O st) cm−1. HRMS (ESI-TOF) m/z: [M+H]+ calcd for C26H25Cl2N2O 451.1344, Found 451.1352.


 ,
, 
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
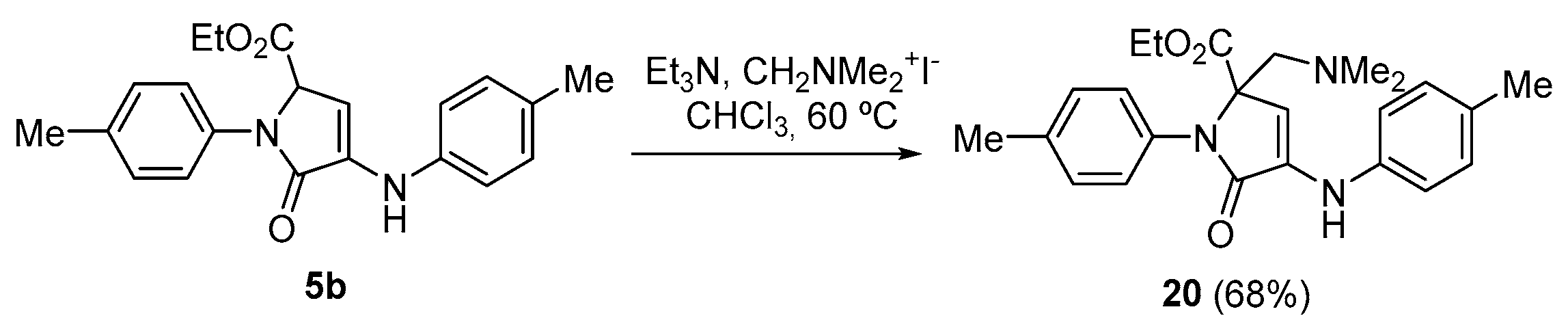{kind=link}
{kind=link}
{kind=link}
