
Synthesis and Anti-Neuroinflammatory Activity of 1,7-diphenyl-1,4-heptadien-3-ones in LPS-Stimulated BV2 Microglia Via Inhibiting NF-κB/MAPK Signaling Pathways

Abstract
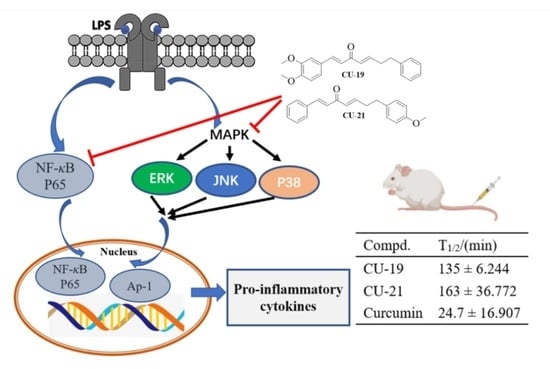
:
1. Introduction
2. Results and Discussions
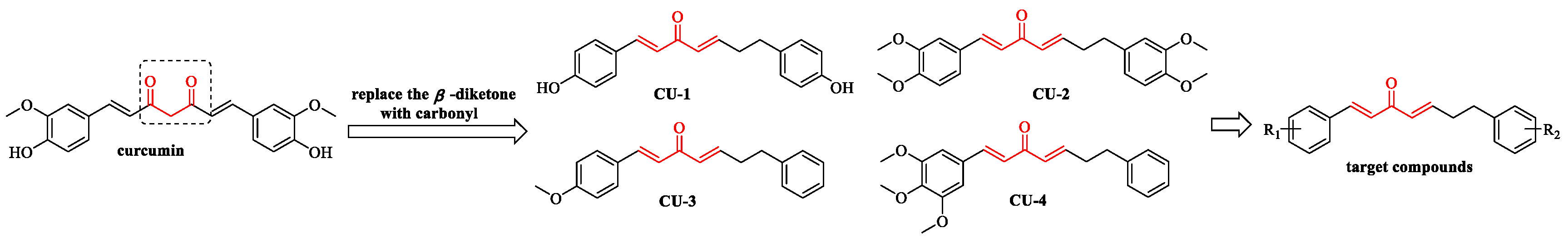
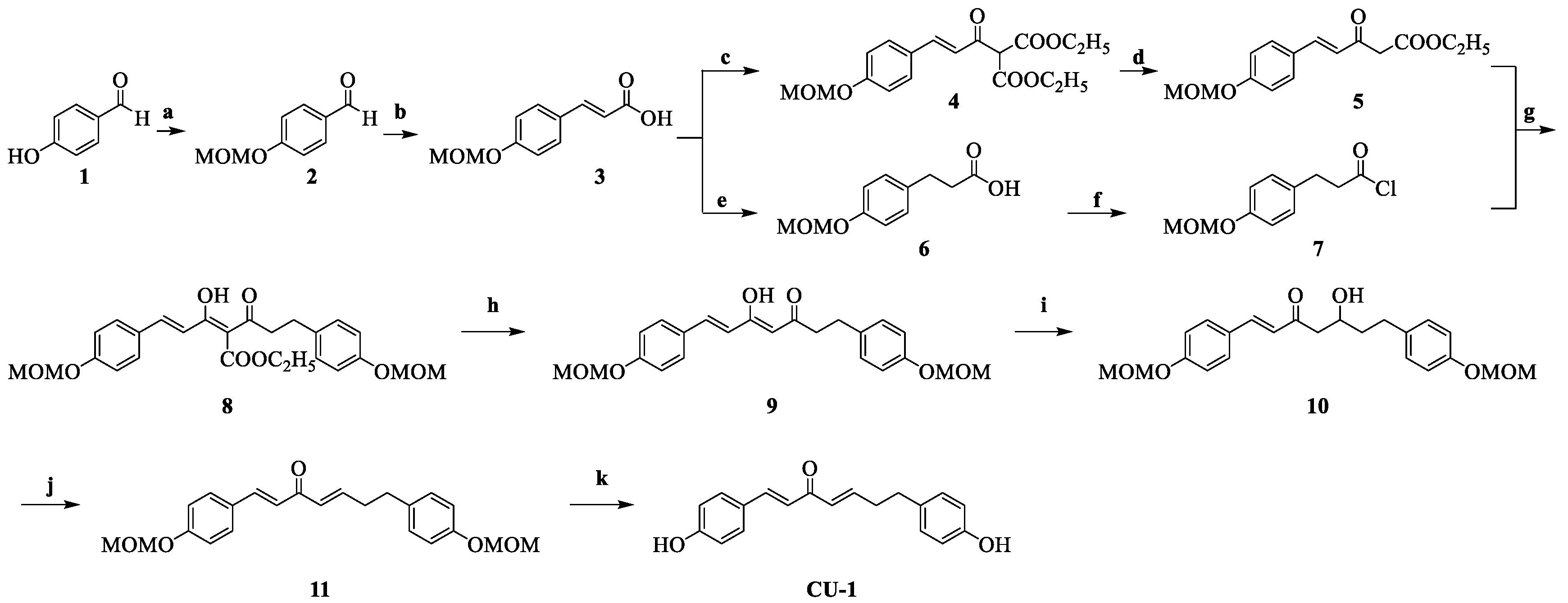
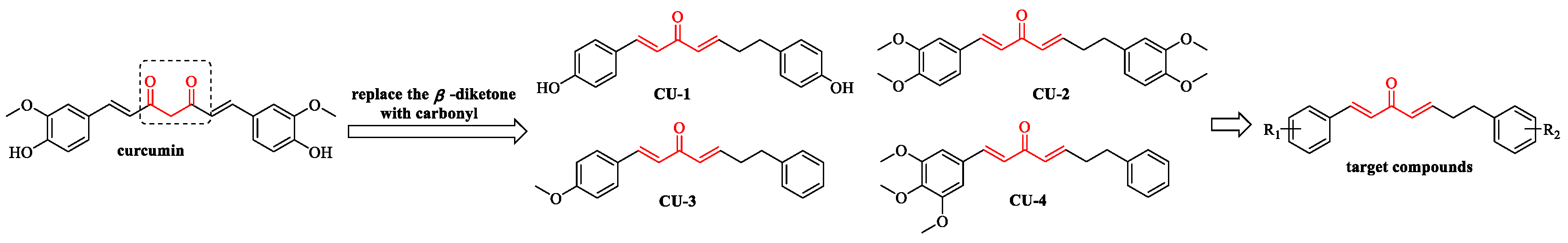
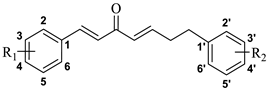
2.1. Chemistry
2.2. Biological Evaluation
2.2.1. Cell Growth Inhibition and Anti-Neuroinflammatory Screening of the Target Compounds CU-1~CU-28
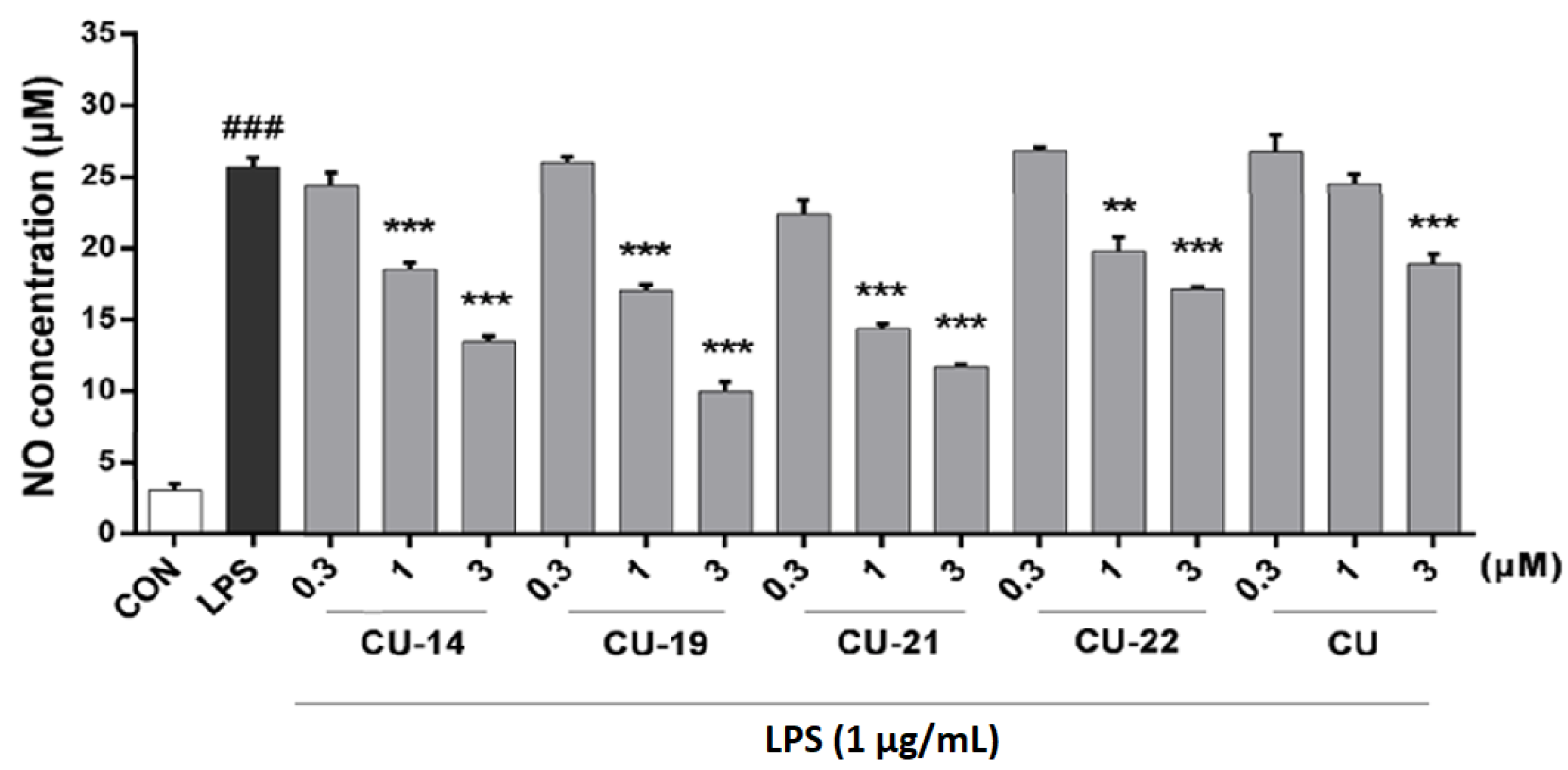
2.2.2. Active Compounds Inhibit LPS-Induced NO Release in a Dose-Dependent Manner
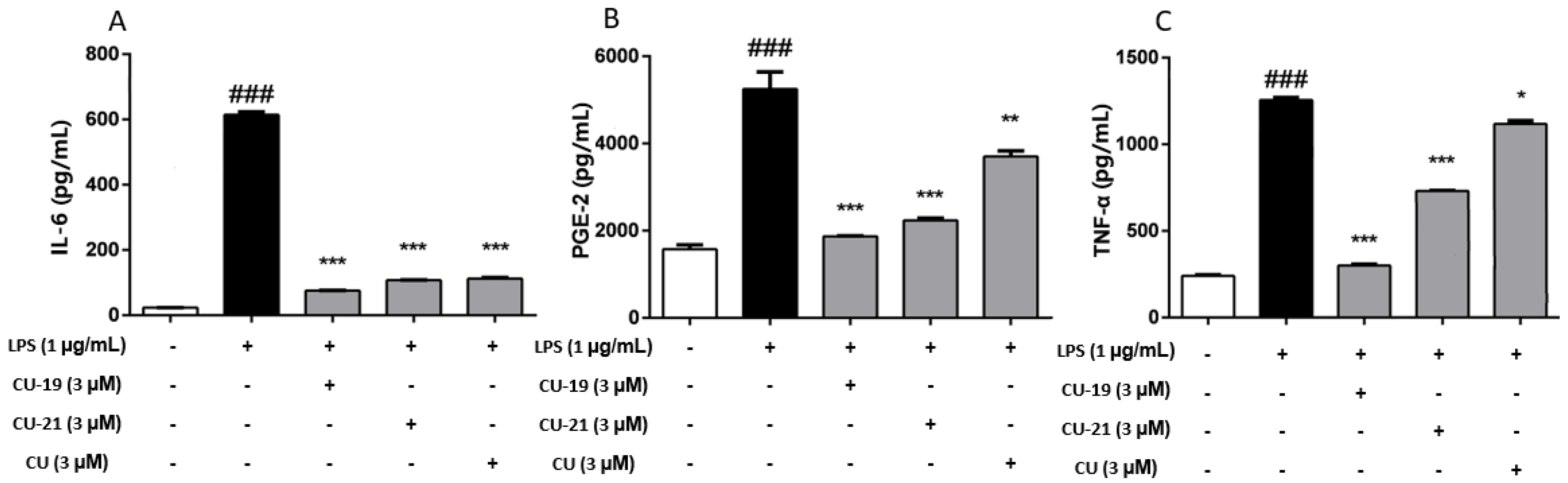
2.2.3. The Ability of CU-19 and CU-21 to Inhibit LPS-Induced Inflammatory Mediator Release
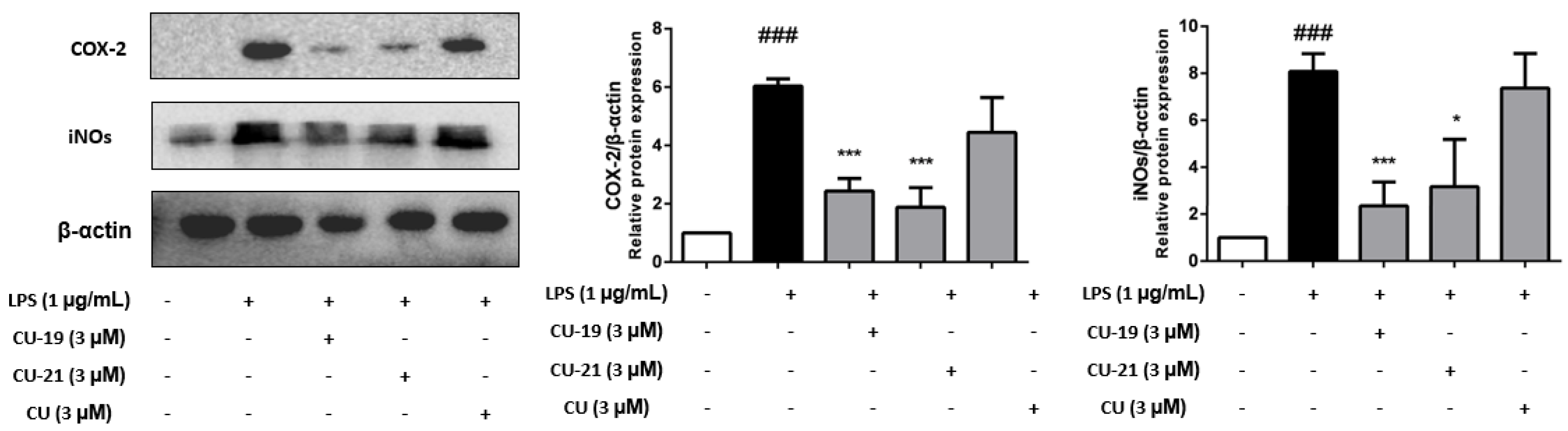
2.2.4. The Ability of CU-19 and CU-21 to Inhibit LPS-Induced iNOS and COX-2 Upregulation
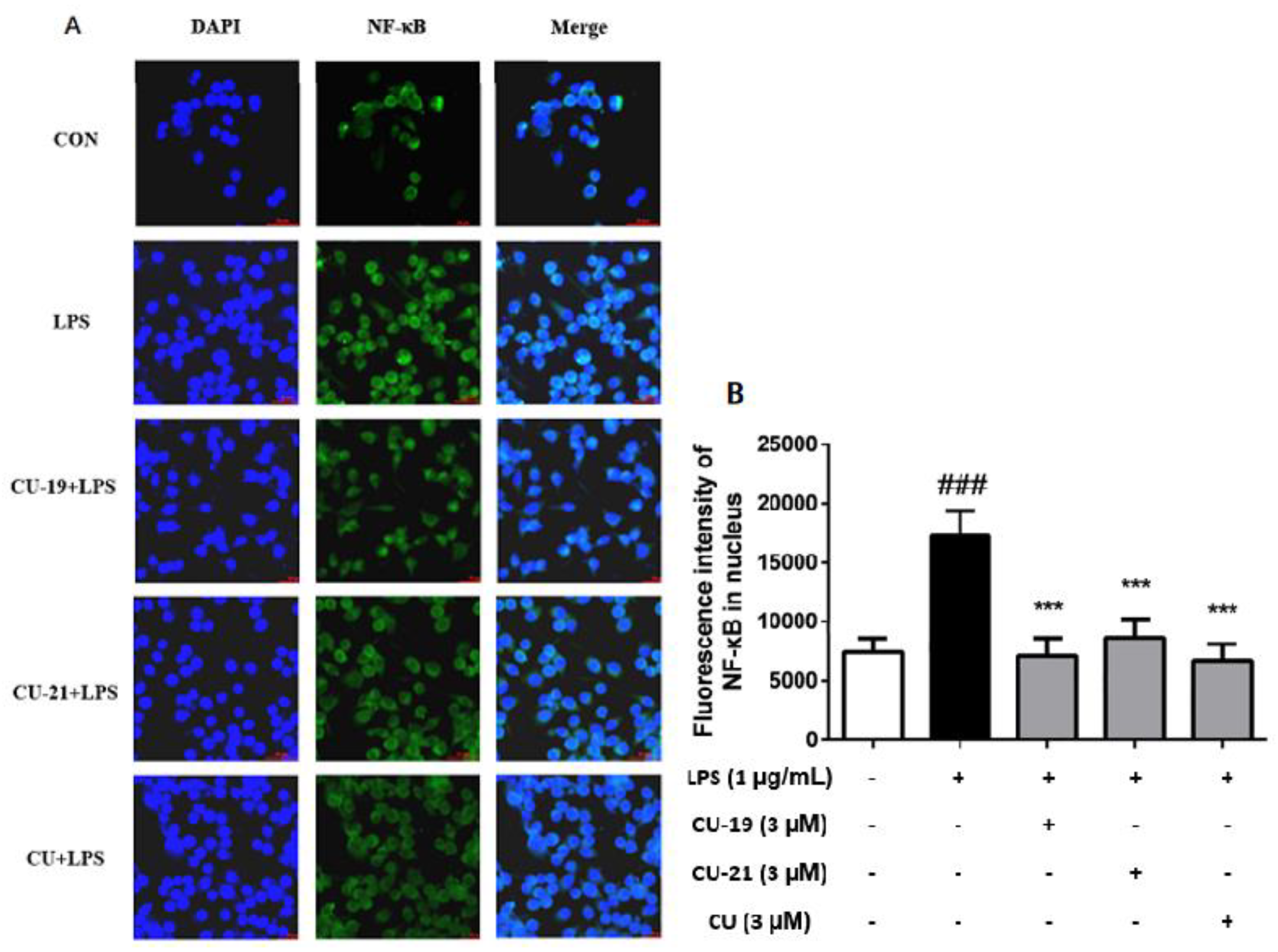
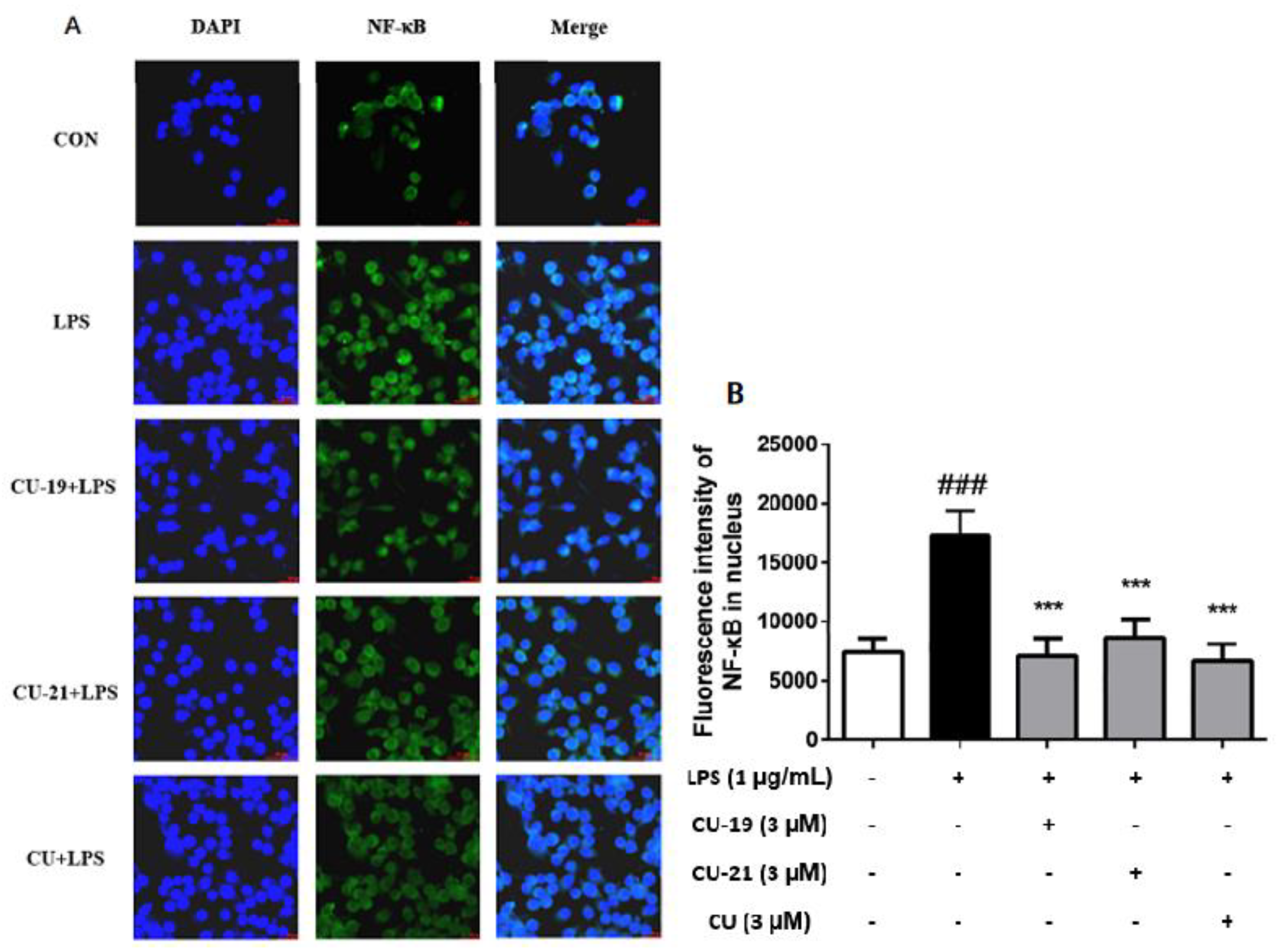
2.2.5. The Ability of CU-19 and CU-21 to Inhibit LPS-Induced NF-κB Activation
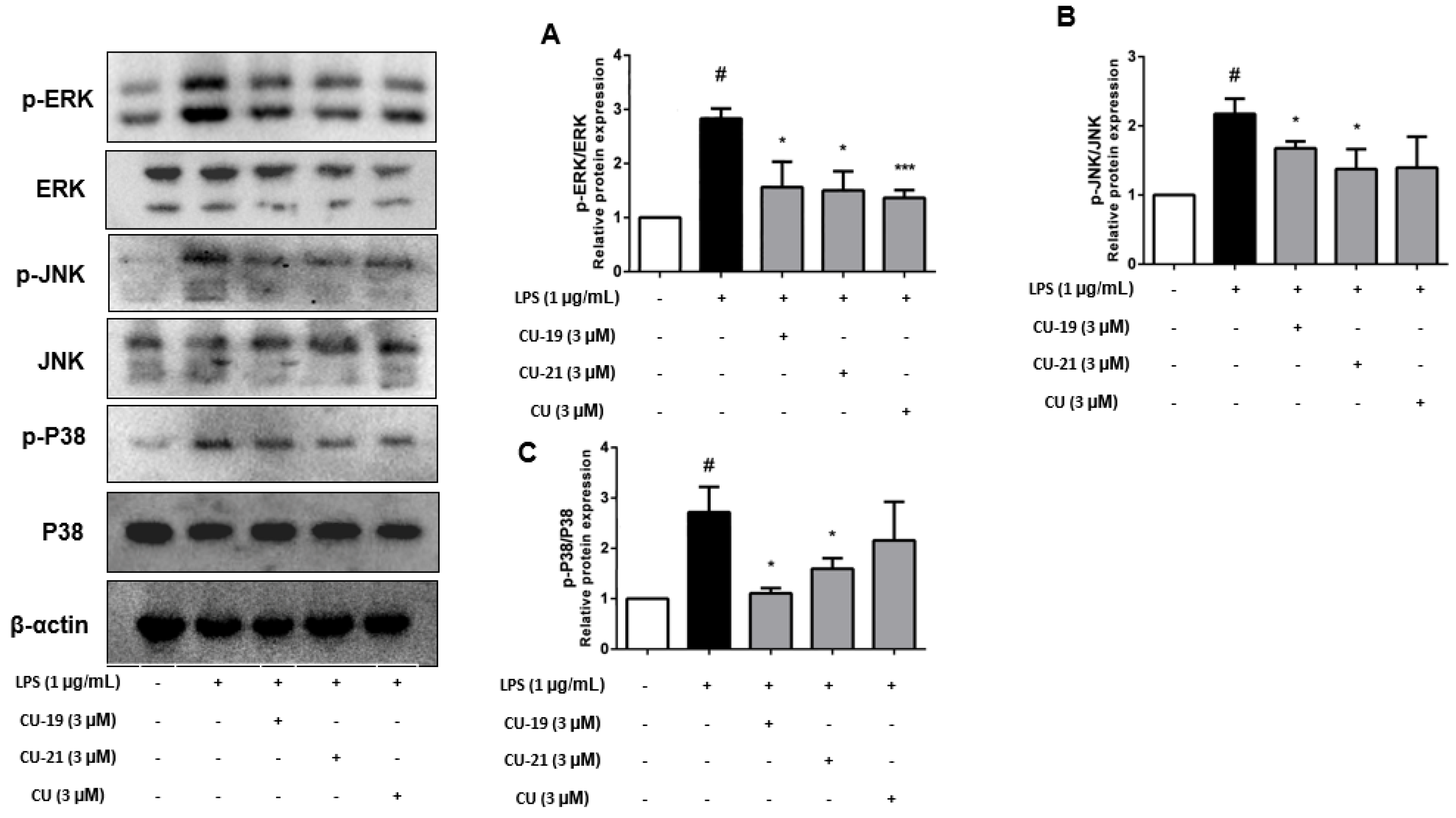
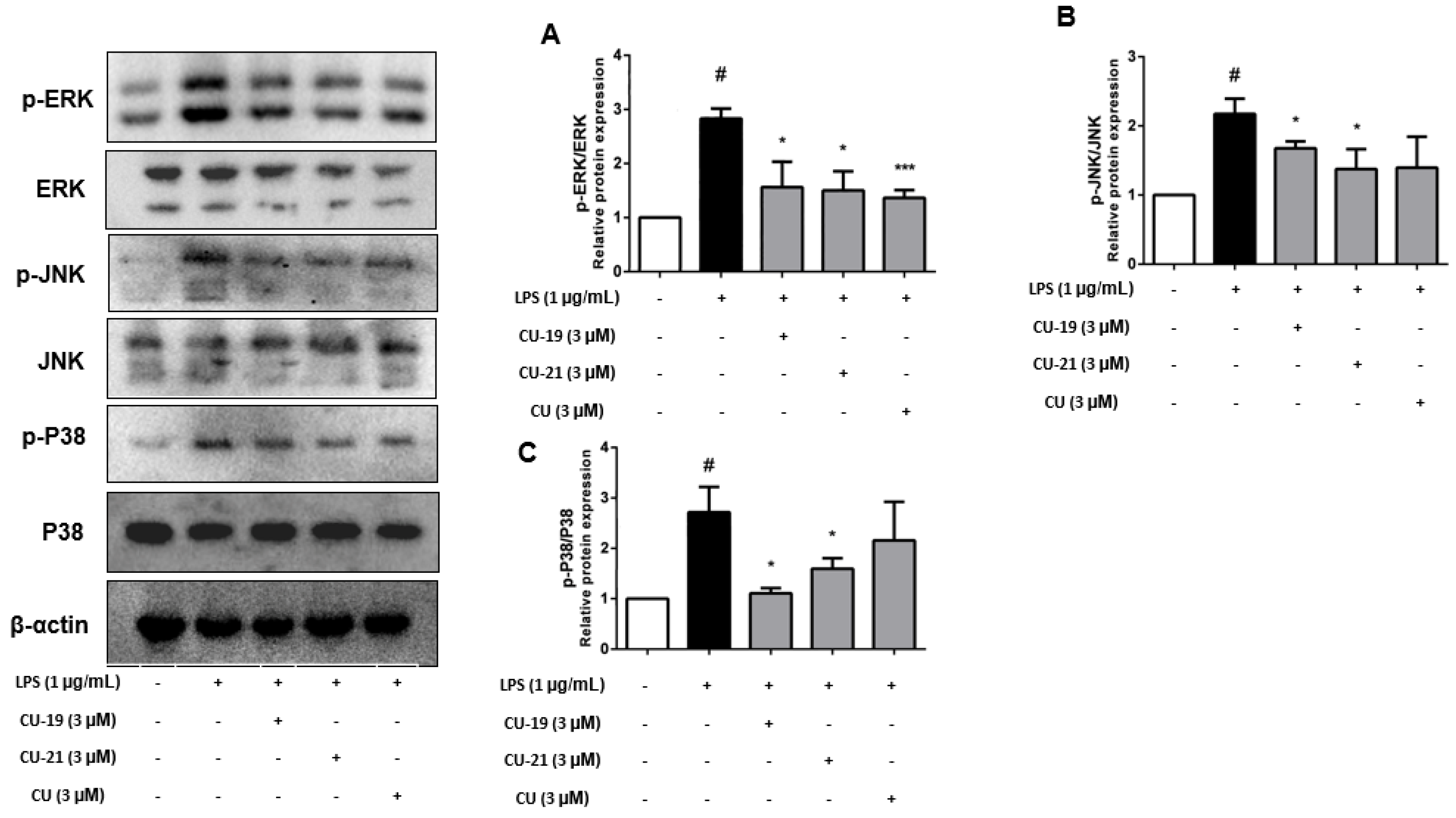
2.2.6. The Ability of CU-19 and CU-21 to Inhibit LPS-Induced MAPK Activation
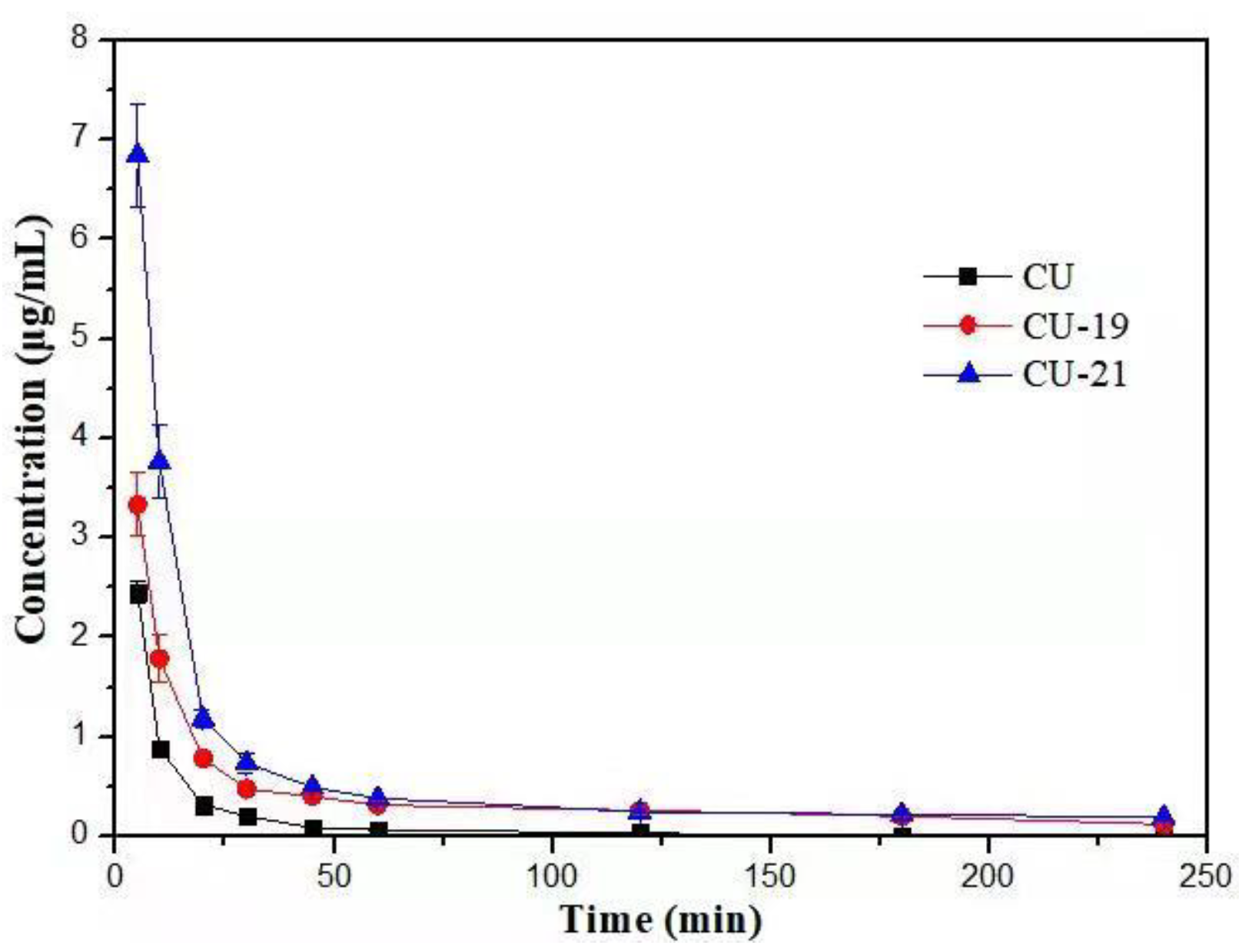
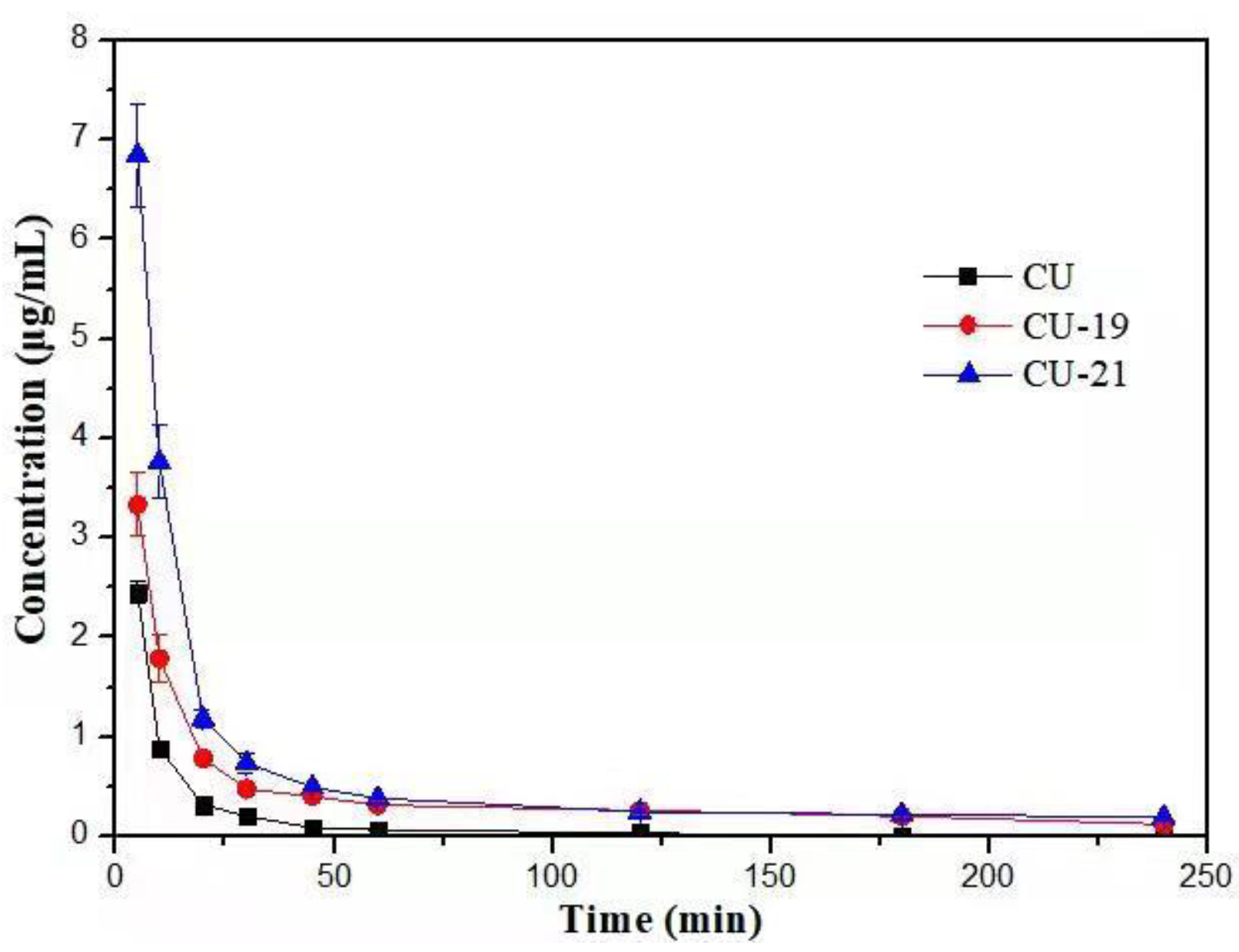
2.3. Pharmacokinetic Study
3. Conclusions
4. Experimental Section
4.1. Chemistry and HPLC Analysis
4.1.1. The Synthesis of 4-(methoxymethoxy)benzaldehyde 2
4.1.2. The Synthesis of (E)-3-(4-(methoxymethoxy)phenyl)acrylic Acid 3
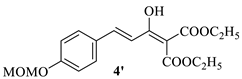
4.1.3. The Synthesis of Diethyl(E)-2-(3-(4-(methoxymethoxy)phenyl)acryloyl)malonate 4 and Diethyl(E)-2-(1-hydroxy-3-(4-(methoxymethoxy)phenyl)allylidene)malonate 4′
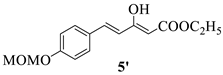
4.1.4. The Synthesis of Ethyl (E)-5-(4-(methoxymethoxy)phenyl)-3-oxo-4-pentenoate 5 and Ethyl (2Z,4E)-3-hydroxy-5-(4-(methoxymethoxy)phenyl)penta-2,4-dienoate 5′
4.1.5. The Synthesis of 3-(4-(methoxymethoxy)phenyl)propionic acid 6
4.1.6. The Synthesis of (1E,3E)-1,7-bis(4-(methoxymethoxy)phenyl)-3-hydroxy-4-(ethoxycarbonyl)-1,3-heptadiene-5-one 8
4.1.7. The Synthesis of (1E,3E)-1,7-bis(4-(methoxymethoxy)phenyl)-3-hydroxy-1,3-heptadiene-5-one 9
4.1.8. The Synthesis of (E)-1,7-bis(4-(methoxymethoxy)phenyl)-5-hydroxy-1-heptene-3-one 10
4.1.9. The Synthesis of (1E,4E)-1,7-bis(4-(methoxymethoxy)phenyl)-1,4-heptadiene-3-one 11
4.1.10. The Synthesis of the Target Compounds
(1E,4E)-1,7-bis(4-hydroxyphenyl)-1,4-heptadien-3-one (CU-1)
(1E,4E)-1,7-bis(3,4-dimethoxyphenyl)-1,4-heptadien-3-one (CU-2)
(1E,4E)-7-phenyl-1-(4-methoxyphenyl)-1,4-heptadien-3-one (CU-3)
(1E,4E)-7-phenyl-1-(3,4,5-trimethoxyphenyl)-1,4-heptadien-3-one (CU-4)
(1E,4E)-1-(2-hydroxyphenyl)-7-phenyl-1,4-heptadien-3-one (CU-5)
(1E,4E)-1-(3-hydroxyphenyl)-7-phenyl-1,4-heptadien-3-one (CU-6)
(1E,4E)-1-(4-hydroxyphenyl)-7-phenyl-1,4-heptadien-3-one (CU-7)
(1E,4E)-1-(3-hydroxy-4-methoxyphenyl)-7-phenyl-1,4-heptadien-3-one (CU-8)
(1E,4E)-1-(4-hydroxy-3-methoxyphenyl)-7-phenyl-1,4-heptadien-3-one (CU-9)
(1E,4E)-1-(4-chlorophenyl)-7-phenyl-1,4-heptadien-3-one (CU-10)
(1E,4E)-1-(4-hydroxyphenyl)-7-(p-tolyl)-1,4-heptadien-3-one (CU-11)
(1E,4E)-7-(4-hydroxy-3-methoxyphenyl)-1-(4-hydroxyphenyl)-1,4-heptadien-3-one (CU-12)
(1E,4E)-1-(4-hydroxyphenyl)-7-(4-methoxyphenyl)-1,4-heptadien-3-one (CU-13)
(1E,4E)-1,7-bis(4-methoxyphenyl)-1,4-heptadien-3-one (CU-14)
(1E,4E)-1,7-diphenyl-1,4-heptadien-3-one (CU-15)
(1E,4E)-7-phenyl-1-(p-tolyl)-1,4- heptadien-3-one (CU-16)
(1E,4E)-1-(4-hydroxy-3-methoxyphenyl)-7-(4-hydroxyphenyl)-1,4-heptadien-3-one (CU-17)
(1E,4E)-1-(3-hydroxy-4-methoxyphenyl)-7-(4-hydroxyphenyl)-1,4-heptadien-3-one (CU-18)
(1E,4E)-1-(3,4-dimethoxyphenyl)-7-phenyl-1,4-heptadien-3-one (CU-19)
(1E,4E)-1,7-bis(4-hydroxy-3-methoxyphenyl)-1,4-heptadien-3-one (CU-20)
(1E,4E)-7-(4-methoxyphenyl)-1-phenyl-1,4-heptadien-3-one (CU-21)
(1E,4E)-1-phenyl-7-(p-tolyl)-1,4-heptadien-3-one (CU-22)
(1E,4E)-7-(3,4-dimethoxyphenyl)-1-phenyl-1,4-heptadien-3-one (CU-23)
(1E,4E)-7-(3-hydroxyphenyl)-1-phenyl-1,4-heptadien-3-one (CU-24)
(1E,4E)-7-(4-hydroxyphenyl)-1-phenyl-1,4-heptadien-3-one (CU-25)
(1E,4E)-7-(4-hydroxy-3-methoxyphenyl)-1-phenyl-1,4-heptadien-3-one (CU-26)
(1E,4E)-1-phenyl-7-(3,4,5-trimethoxyphenyl)-1,4-heptadien-3-one (CU-27)
(1E,4E)-7-(4-chlorophenyl)-1-phenyl-1,4-heptadien-3-one (CU-28)
4.2. Biological Evaluation
4.2.1. Cell Culture
4.2.2. Determination of Cell Viability
4.2.3. Assay for NO Production
4.2.4. Inflammatory Mediator Assays
4.2.5. Western Blot Analysis
4.2.6. Immunofluorescent Staining
4.2.7. Statistical Analysis
4.3. In Vivo Pharmacokinetic Study in Rats
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gao, S.; Cheng, Q.C.; Hu, Y.G.; Tan, Z.Z.; Chen, L.; Liu, S.W.; Kang, Q.Y.; Wei, T. LncRNA AK148321 alleviates neuroinflammation in LPS-stimulated BV2 microglial cell through regulating microRNA-1199-5p/HSPA5 axis. Life Sci. 2021, 266, 118863. [Google Scholar] [CrossRef] [PubMed]
- Qin, Q.; Teng, Z.Q.; Liu, C.M.; Li, Q.; Yin, Y.S.; Tang, Y. TREM2, microglia, and Alzheimer’s disease. Mech. Ageing Dev. 2021, 195, 111438. [Google Scholar] [CrossRef] [PubMed]
- Rapaka, D.; Bitra, V.R.; Challa, S.R.; Adiukwu, P.C. Potentiation of microglia endocannabinoid signaling alleviates neuroinflammation in Alzheimer’s disease. Neuropeptides 2021, 90, 102196. [Google Scholar] [CrossRef]
- Wu, J.Y.; Xu, X.Q.; Zheng, L.; Mo, J.F.; Jin, X.H.; Bao, Y. Nilotinib inhibits microglia-mediated neuroinflammation to protect against dopaminergic neuronal death in Parkinson’s disease models. Int. Immunopharmacol. 2021, 99, 108025. [Google Scholar] [CrossRef]
- Liao, S.; Wu, J.N.; Liu, R.M.; Wang, S.X.; Luo, J.; Yang, Y.; Qin, Y.N.; Li, T.; Zheng, X.H. A novel compound DBZ ameliorates neuroinflammation in LPS-stimulated microglia and ischemic stroke rats: Role of Akt(Ser473)/GSK3β(Ser9)-mediated Nrf2 activation. Redox. Biol. 2020, 36, 101644. [Google Scholar] [CrossRef]
- Tang, M.; Taghibiglou, C. The mechanisms of action of curcumin in Alzheimer’s disease. J. Alzheimer’s Dis. 2017, 58, 1–13. [Google Scholar] [CrossRef]
- Zhang, J.W.; Zheng, Y.L.; Luo, Y.; Du, Y.; Zhang, X.J.; Fu, J.L. Curcumin inhibits LPS-induced neuroinflammation by promoting microglia M2 polarization via TREM2/TLR4/NF-κB pathways in BV2 cells. Mol. Immunol. 2019, 116, 29–37. [Google Scholar] [CrossRef]
- Sun, G.C.; Miao, Z.; Ye, Y.F.; Zhao, P.Z.; Fan, L.; Bao, Z.Y.; Tu, Y.M.; Li, C.; Chao, H.L.; Xu, X.P.; et al. Curcumin alleviates neuroinflammation, enhances hippocampal neurogenesis, and improves spatial memory after traumatic brain injury. Brain Res. Bull. 2020, 162, 84–93. [Google Scholar] [CrossRef]
- Ganesan, P.; Kim, B.; Ramalaingam, P.; Karthivashan, G.; Revuri, V.; Park, S.; Kim, J.S.; Ko, Y.T.; Choi, D.K. Antineuroinflammatory activities and neurotoxicological assessment of curcumin loaded solid lipid nanoparticles on LPS-stimulated BV-2 microglia cell models. Molecules 2019, 24, 1170. [Google Scholar] [CrossRef] [Green Version]
- Schmitt, C.; Lechanteur, A.; Cossais, F.; Bellefroid, C.; Hattermann, K. Liposomal encapsulated curcumin effectively attenuates neuroinflammatory and reactive astrogliosis reactions in glia cells and organotypic brain slices. Int. J. Nanomed. 2020, 15, 3649–3666. [Google Scholar] [CrossRef]
- Azzi, E.; Alberti, D.; Parisotto, S.; Oppedisano, A.; Protti, N.; Altieri, S.; Geninatti-Crich, S.; Deagostino, A. Design, synthesis and preliminary in-vitro studies of novel boronated monocarbonyl analogues of curcumin (BMAC) for antitumor and beta-amiloyd disaggregation activity. Bioorg. Chem. 2019, 93, 103324. [Google Scholar] [CrossRef] [PubMed]
- Liang, G.; Shao, L.; Wang, Y.; Zhao, C.; Chu, Y.; Xiao, J.; Zhao, Y.; Li, X.; Yang, S. Exploration and synthesis of curcumin analogues with improved structural stability both in vitro and in vivo as cytotoxic agents. Bioorg. Med. Chem. 2009, 17, 2623–2631. [Google Scholar] [CrossRef] [PubMed]
- Liang, G.; Zhou, H.; Wang, Y.; Gurley, E.C.; Feng, B.; Chen, L.; Xiao, J.; Yang, S.; Li, X. Inhibition of LPS-induced production of inflammatory factors in the macrophages by mono-carbonyl analogues of curcumin. J. Cell. Mol. Med. 2009, 13, 3370–3379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, C.; Cai, Y.; He, X.; Li, J.; Zhang, L.; Wu, J.; Zhao, Y.; Yang, S.; Li, X.; Li, W.; et al. Synthesis and anti-inflammatory evaluation of novel mono-carbonyl analogues of curcumin in LPS-stimulated RAW 264.7 macrophages. Eur. J. Med. Chem. 2010, 45, 5773–5780. [Google Scholar] [CrossRef]
- Dimmock, J.R.; Padmanilayam, M.P.; Puthucode, R.N.; Nazarali, A.J.; Motaganahalli, N.L.; Zello, G.A.; Quail, J.W.; Oloo, E.O.; Kraatz, H.B.; Prisciak, J.S.; et al. A conformational and structure-activity relationship study of cytotoxic 3,5-bis(arylidene)-4-piperidones and related N-acryloyl analogues. J. Med. Chem. 2001, 44, 586–593. [Google Scholar] [CrossRef]
- Wang, Z.; Mu, W.; Li, P.; Liu, G.; Yang, J. Anti-inflammatory activity of ortho-trifluoromethoxy-substituted 4-piperidione-containing mono-carbonyl curcumin derivatives in vitro and in vivo. Eur. J. Pharm. Sci. 2021, 160, 105756. [Google Scholar] [CrossRef]
- Jirásek, P.; Amslinger, S.; Heilmann, J. Synthesis of Natural and non-natural curcuminoids and their neuroprotective activity against glutamate-induced oxidative stress in HT-22 Cells. J. Nat. Prod. 2014, 77, 2206–2217. [Google Scholar] [CrossRef]
- Zhao, Y.L.; Wang, X.Y.; Sun, L.X.; Fan, R.H.; Bi, K.S.; Yu, Z.G. Cytotoxic constituents of Viscum coloratum. Z. Nat. C 2012, 67, 129–134. [Google Scholar] [CrossRef]
- Chen, C.Y.; Lien, J.C.; Chen, Y.C.; Hung, C.C.; Lin, H.C. Design, synthesis and evaluation of novel derivatives of curcuminoids with cytotoxicity. Int. J. Mol. Sci. 2021, 22, 12171. [Google Scholar] [CrossRef]
- Dong, J.H.; Zhao, X.; Fang, J.Q.; Xia, M.Y.; Zhang, M.H.; Xu, L.Y. Monocarbonyl curcumin analogue for preparation of antitumor drug and its preparation method. CN 113861009, 31 December 2021. [Google Scholar]
- Labib, M.B.; Fayez, A.M.; El-Nahass, E.S.; Awadallah, M.; Halim, P.A. Novel tetrazole-based selective COX-2 inhibitors: Design, synthesis, anti-inflammatory activity, evaluation of PGE2, TNF-alpha, IL-6 and histopathological study. Bioorg. Chem. 2020, 104, 104308. [Google Scholar] [CrossRef]
- Kim, Y.; Lim, H.J.; Jang, H.J.; Lee, S.; Jung, K.; Lee, S.W.; Lee, S.J.; Rho, M.C. Portulaca oleracea extracts and their active compounds ameliorate inflammatory bowel diseases in vitro and in vivo by modulating TNF-alpha, IL-6 and IL-1beta signalling. Food Res. Int. 2018, 106, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Periyasami, G.; Antonisamy, P.; Perumal, K.; Stalin, A.; Rahaman, M.; Alothman, A.A. A competent synthesis and efficient anti-inflammatory responses of isatinimino acridinedione moiety via suppression of in vivo NF-κB, COX-2 and iNOS signaling. Bioorg. Chem. 2019, 90, 103047. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Aranda, M.I.; Gonzalez-Padilla, J.E.; Gomez-Castro, C.Z.; Gomez-Gomez, Y.M.; Rosales-Hernandez, M.C.; Garcia-Baez, E.V.; Franco-Hernandez, M.O.; Castrejon-Flores, J.L.; Martinez, P. Anti-inflammatory effect and inhibition of nitric oxide production by targeting COXs and iNOS enzymes with the 1,2-diphenylbenzimidazole pharmacophore. Bioorg. Med. Chem. 2020, 28, 115427. [Google Scholar] [CrossRef] [PubMed]
- Ge, J.; Guo, K.; Zhang, C.; Talukder, M.; Lv, W.M.; Li, J.Y.; Li, J.L. Comparison of nanoparticle-selenium, selenium-enriched yeast and sodium selenite on the alleviation of cadmium-induced inflammation via NF-κB/IκB pathway in heart. Sci. Total Environ. 2021, 773, 145442. [Google Scholar] [CrossRef]
- Kong, Y.D.; Li, M.; Guo, G.L.; Yu, L.H.; Sun, L.; Yin, Z.; Li, R.M.; Chen, X.M.; Wang, G.Q. Effects of dietary curcumin inhibit deltamethrin-induced oxidative stress, inflammation and cell apoptosis in channa argus via Nrf2 and NF-κB signaling pathways. Aquaculture 2021, 540, 736744. [Google Scholar] [CrossRef]
- Yang, L.; Cao, L.; Li, C.L.; Li, X.B.; Wang, J.S.; Chen, H.P.; He, J.W. Hostaflavone A from Hosta plantaginea (Lam.) Asch. blocked NF-κB/iNOS/COX-2/MAPKs/Akt signaling pathways in LPS-induced RAW 264.7 macrophages. J. Ethnopharmacol. 2021, 282, 114605. [Google Scholar] [CrossRef]
- Jayawardena, T.U.; Kim, H.S.; Sanjeewa, K.A.; Han, E.J.; Jee, Y.; Ahn, G.; Rho, J.R.; Jeon, Y.J. Loliolide, isolated from Sargassum horneri; abate LPS-induced inflammation via TLR mediated NF-κB, MAPK pathways in macrophages. Algal. Res. 2021, 56, 102297. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
|---|---|---|---|---|---|
| Compd. | R1 | R2 | Compd. | R1 | R2 |
| CU-1 | 4-OH | 4′-OH | CU-15 | H | H |
| CU-2 | 3-OCH3, 4- OCH3 | 3′-OCH3, 4′- OCH3 | CU-16 | 4-CH3 | H |
| CU-3 | 4-OCH3 | H | CU-17 | 3-OCH3, 4-OH | 4′-OH |
| CU-4 | 3-OCH3, 4-OCH3, 5-OCH3 | H | CU-18 | 3-OH, 4-OCH3 | 4′-OH |
| CU-5 | 2-OH | H | CU-19 | 3-OCH3, 4-OCH3 | H |
| CU-6 | 3-OH | H | CU-20 | 3-OCH3, 4-OH | 3′-OCH3, 4′-OH |
| CU-7 | 4-OH | H | CU-21 | H | 4′-OCH3 |
| CU-8 | 3-OH, 4-OCH3 | H | CU-22 | H | 4′-CH3 |
| CU-9 | 3-OCH3, 4-OH | H | CU-23 | H | 3′-OCH3, 4′- OCH3 |
| CU-10 | 4-Cl | H | CU-24 | H | 3′-OH |
| CU-11 | 4-OH | 4′-CH3 | CU-25 | H | 4′-OH |
| CU-12 | 4-OH | 3′-OCH3, 4′-OH | CU-26 | H | 3′-OCH3, 4′-OH |
| CU-13 | 4-OH | 4′-OCH3 | CU-27 | H | 3′-OCH3, 4′-OCH3, 5′-OCH3 |
| CU-14 | 4-OCH3 | 4′-OCH3 | CU-28 | H | 4′-Cl |
| Compd. | Cell Viability (%) a | NO Inhibition (%) | |||
|---|---|---|---|---|---|
| 1 µM | 3 µM | 10 µM | 1 µM | 3 µM | |
| CU-1 | 88.07 ± 0.19 | 49.84 ± 0.27 | 23.70 ± 0.38 | 0 | - |
| CU-2 | 76.92 ± 0.19 | 49.16 ± 0.92 | 10.57 ± 1.13 | 0 | - |
| CU-4 | 107.23 ± 0.09 | 73.92 ± 0.30 | 16.61 ± 0.45 | 3.10 | - |
| CU-5 | 95.68 ± 0.11 | 77.39 ± 0.30 | 15.92 ± 0.61 | 0 | - |
| CU-7 | 93.46 ± 0.35 | 52.80 ± 0.50 | 14.26 ± 0.70 | 0 | - |
| CU-12 | 92.03 ± 0.29 | 75.55 ± 0.17 | 12.67 ± 0.59 | 0 | - |
| CU-17 | 99.36 ± 0.01 | 80.72 ± 0.20 | 32.42 ± 0.67 | 0 | - |
| CU-18 | 96.14 ± 0.03 | 46.56 ± 1.16 | 8.63 ± 0.373 | 2.15 | - |
| CU-20 | 97.31 ± 0.03 | 51.82 ± 0.60 | 12.24 ± 0.21 | 16.9 | - |
| CU-3 | 106.72 ± 0.14 | 101.55 ± 0.01 | 98.50 ± 0.01 | - | 35.2 |
| CU-6 | 106.85 ± 0.10 | 100.07 ± 0.01 | 101.56 ± 0.03 | - | 22.1 |
| CU-8 | 101.18 ± 0.04 | 97.40 ± 0.07 | 18.20 ± 0.86 | - | 18.8 |
| CU-9 | 99.38 ± 0.01 | 94.65 ± 0.07 | 14.75 ± 0.75 | - | 7.52 |
| CU-10 | 95.54 ± 0.03 | 95.27 ± 0.16 | 48.66 ± 2.93 | - | 0 |
| CU-11 | 99.17 ± 0.01 | 91.61 ± 0.09 | 11.24 ± 0.56 | - | 0 |
| CU-13 | 109.45 ± 0.27 | 91.40 ± 0.32 | 28.53 ± 1.30 | - | 0 |
| CU-14 | 97.96 ± 0.01 | 97.44 ± 0.03 | 18.06 ± 0.10 | - | 49.7 |
| CU-15 | 104.99 ± 0.03 | 103.11 ± 0.03 | 84.19 ± 0.21 | - | 13.7 |
| CU-16 | 95.00 ± 0.08 | 88.57 ± 0.28 | 33.33 ± 1.20 | - | 15.4 |
| CU-19 | 96.94 ± 0.03 | 96.33 ± 0.02 | 67.91 ± 0.37 | - | 50.3 |
| CU-21 | 96.94 ± 0.01 | 94.55 ± 0.09 | 77.89 ± 0.50 | - | 66.1 |
| CU-22 | 95.14 ± 0.05 | 88.76 ± 0.06 | 46.65 ± 0.59 | - | 48.2 |
| CU-23 | 106.47 ± 0.14 | 100.85 ± 0.01 | 12.35 ± 0.84 | - | 31.4 |
| CU-24 | 103.49 ± 0.02 | 91.48 ± 0.07 | 23.17 ± 0.09 | - | 0 |
| CU-25 | 128.58 ± 0.14 | 96.93 ± 0.01 | 20.30 ± 0.59 | - | 0 |
| CU-26 | 107.17 ± 0.07 | 96.83 ± 0.02 | 12.85 ± 0.14 | - | 37.6 |
| CU-27 | 116.10 ± 0.44 | 101.31 ± 0.03 | 38.93 ± 1.19 | - | 7.02 |
| CU-28 | 95.71 ± 0.07 | 94.75 ± 0.05 | 81.65 ± 0.22 | - | 33.6 |
| CU | 97.57 ± 0.05 | 95.26 ± 0.05 | 28.15 ± 0.19 | - | 37.4 |
| Compd. | CU-14 | CU-19 | CU-21 | CU-22 | CU |
|---|---|---|---|---|---|
| IC50 (μM) | 2.43 ± 0.45 | 1.60 ± 0.31 | 1.74 ± 0.66 | >3 | >3 |
| Parameter | CU | CU-19 | CU-21 |
|---|---|---|---|
| Cmax/(μg·mL−1) | 2.45 ± 0.132 | 3.33 ± 0.393 | 6.85 ± 0.634 |
| T1/2/min | 24.7 ± 16.907 | 135 ± 6.244 | 163 ± 36.772 |
| CL (L/h/Kg) | 0.500 ± 0.069 | 0.150 ± 0.021 | 0.100 ± 0.012 |
| AUC (0-t)/(μg·min·mL−1) | 39.1 ± 4.186 | 106 ± 11.908 | 170 ± 11.121 |
| AUC (0-∞)/(μg·min·mL−1) | 40.7 ± 5.663 | 133 ± 16.995 | 212 ± 24.724 |
| MRT (0-t)/min | 14.1 ± 3.862 | 61.2 ± 3.189 | 45.4 ± 2.659 |
| MRT (0-∞)/min | 20.5 ± 9.253 | 127 ± 3.423 | 139 ± 22.909 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, X.; Fang, J.; Jia, Y.; Wu, Z.; Zhang, M.; Xia, M.; Dong, J. Synthesis and Anti-Neuroinflammatory Activity of 1,7-diphenyl-1,4-heptadien-3-ones in LPS-Stimulated BV2 Microglia Via Inhibiting NF-κB/MAPK Signaling Pathways. Molecules 2022, 27, 3537. https://doi.org/10.3390/molecules27113537
Zhao X, Fang J, Jia Y, Wu Z, Zhang M, Xia M, Dong J. Synthesis and Anti-Neuroinflammatory Activity of 1,7-diphenyl-1,4-heptadien-3-ones in LPS-Stimulated BV2 Microglia Via Inhibiting NF-κB/MAPK Signaling Pathways. Molecules. 2022; 27(11):3537. https://doi.org/10.3390/molecules27113537
Chicago/Turabian StyleZhao, Xuan, Jiqing Fang, Yu Jia, Zi Wu, Meihui Zhang, Mingyu Xia, and Jinhua Dong. 2022. "Synthesis and Anti-Neuroinflammatory Activity of 1,7-diphenyl-1,4-heptadien-3-ones in LPS-Stimulated BV2 Microglia Via Inhibiting NF-κB/MAPK Signaling Pathways" Molecules 27, no. 11: 3537. https://doi.org/10.3390/molecules27113537
APA StyleZhao, X., Fang, J., Jia, Y., Wu, Z., Zhang, M., Xia, M., & Dong, J. (2022). Synthesis and Anti-Neuroinflammatory Activity of 1,7-diphenyl-1,4-heptadien-3-ones in LPS-Stimulated BV2 Microglia Via Inhibiting NF-κB/MAPK Signaling Pathways. Molecules, 27(11), 3537. https://doi.org/10.3390/molecules27113537