
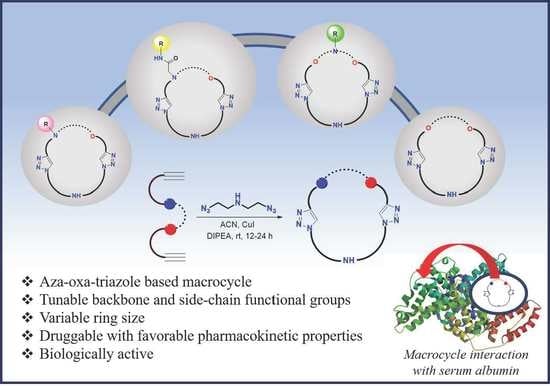
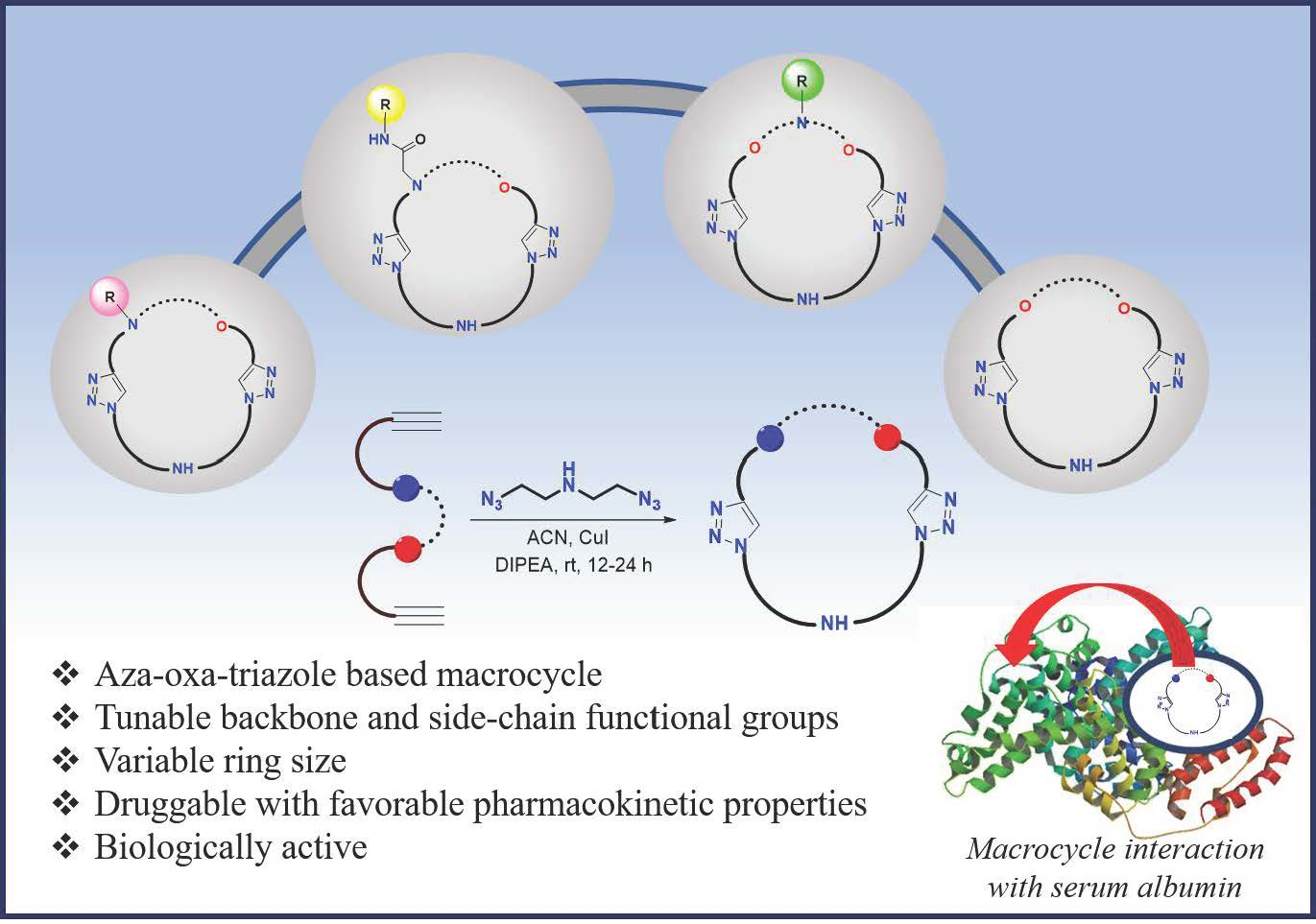
Aza-Oxa-Triazole Based Macrocycles with Tunable Properties: Design, Synthesis, and Bioactivity



Abstract
:
1. Introduction
2. Results and Discussion
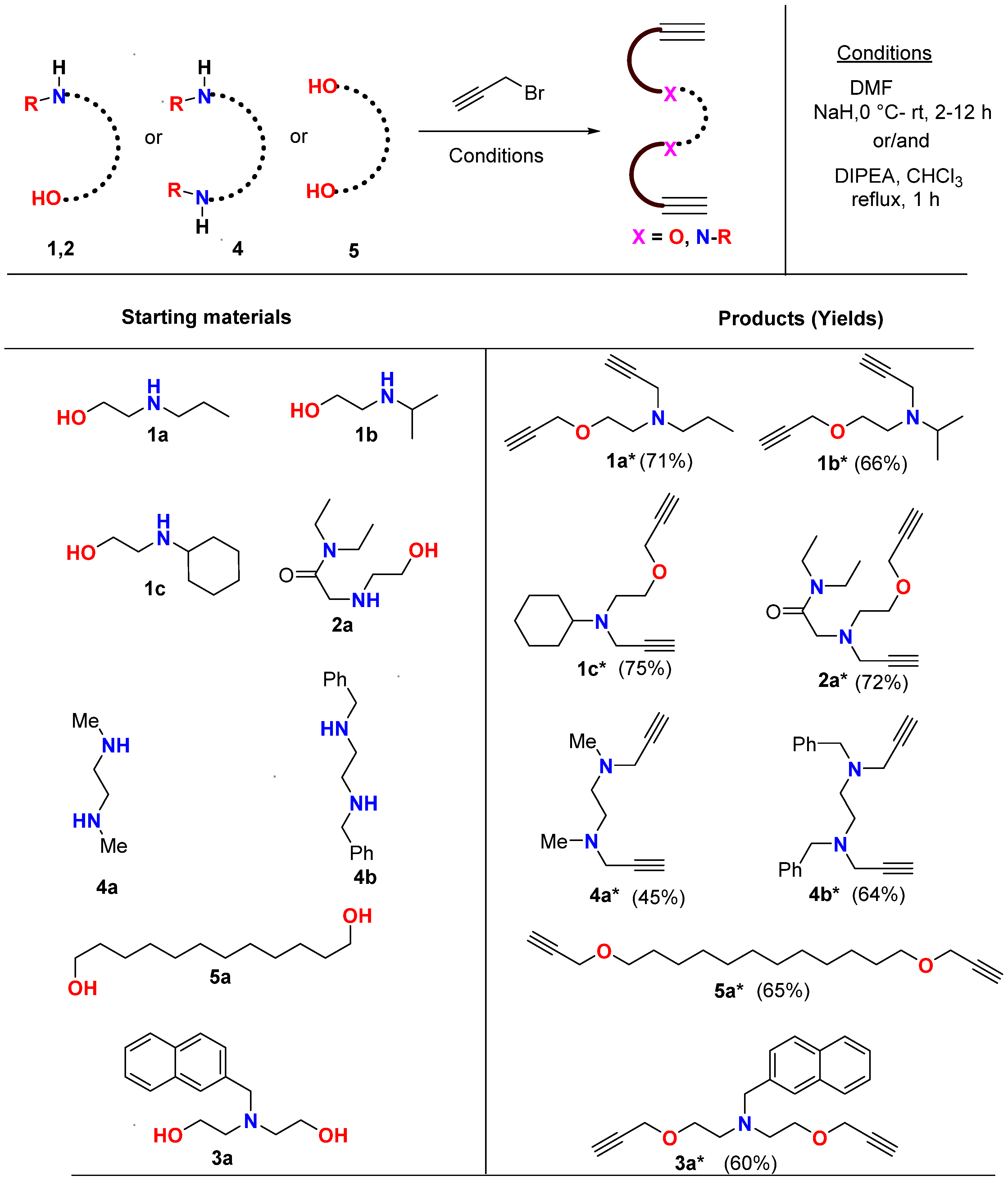
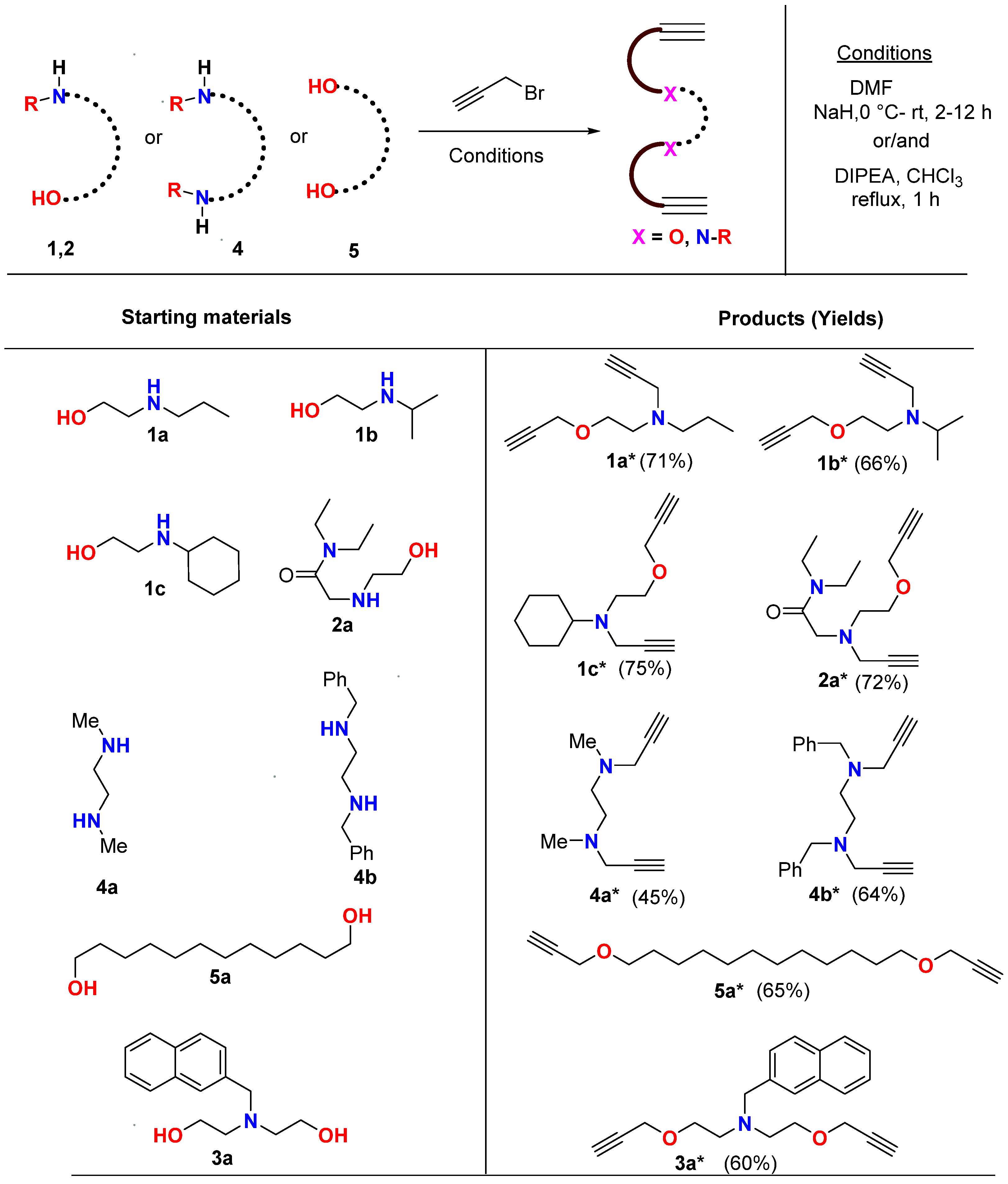
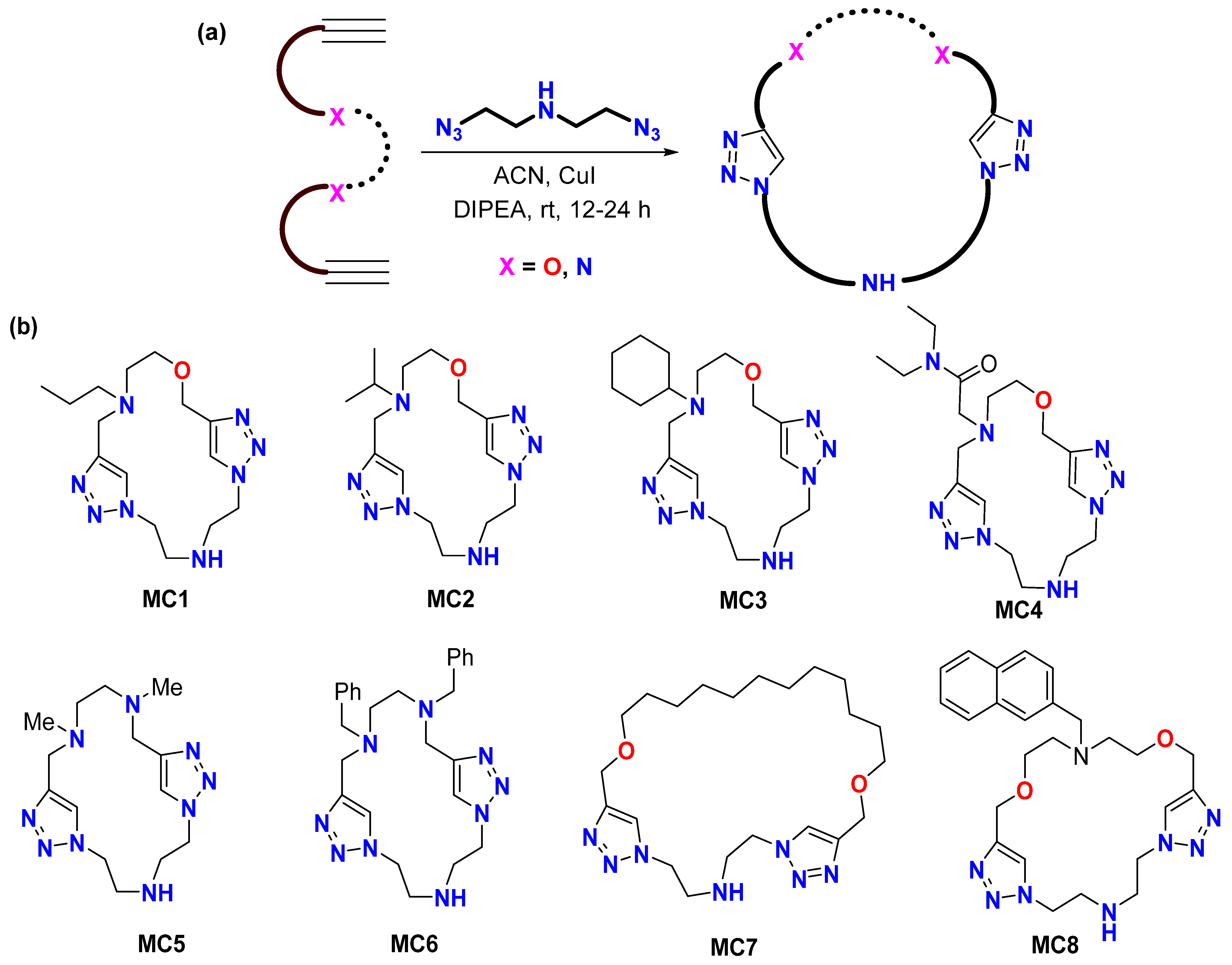
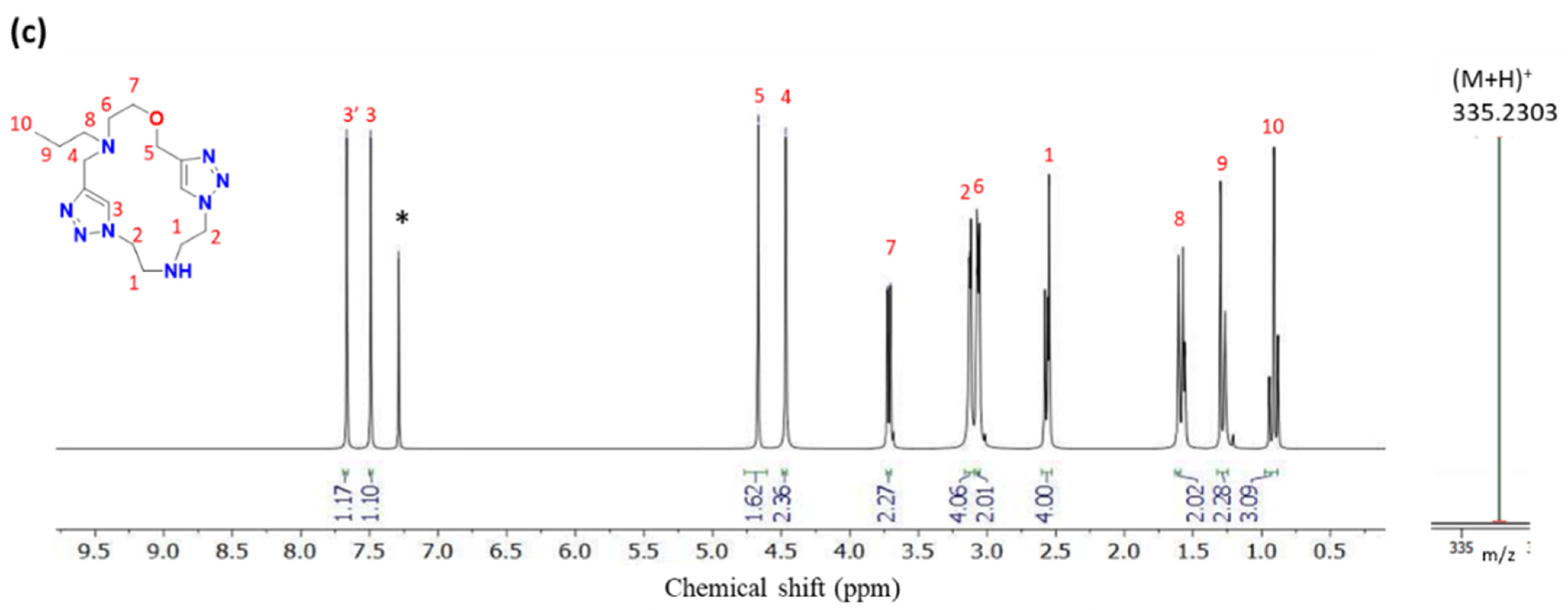
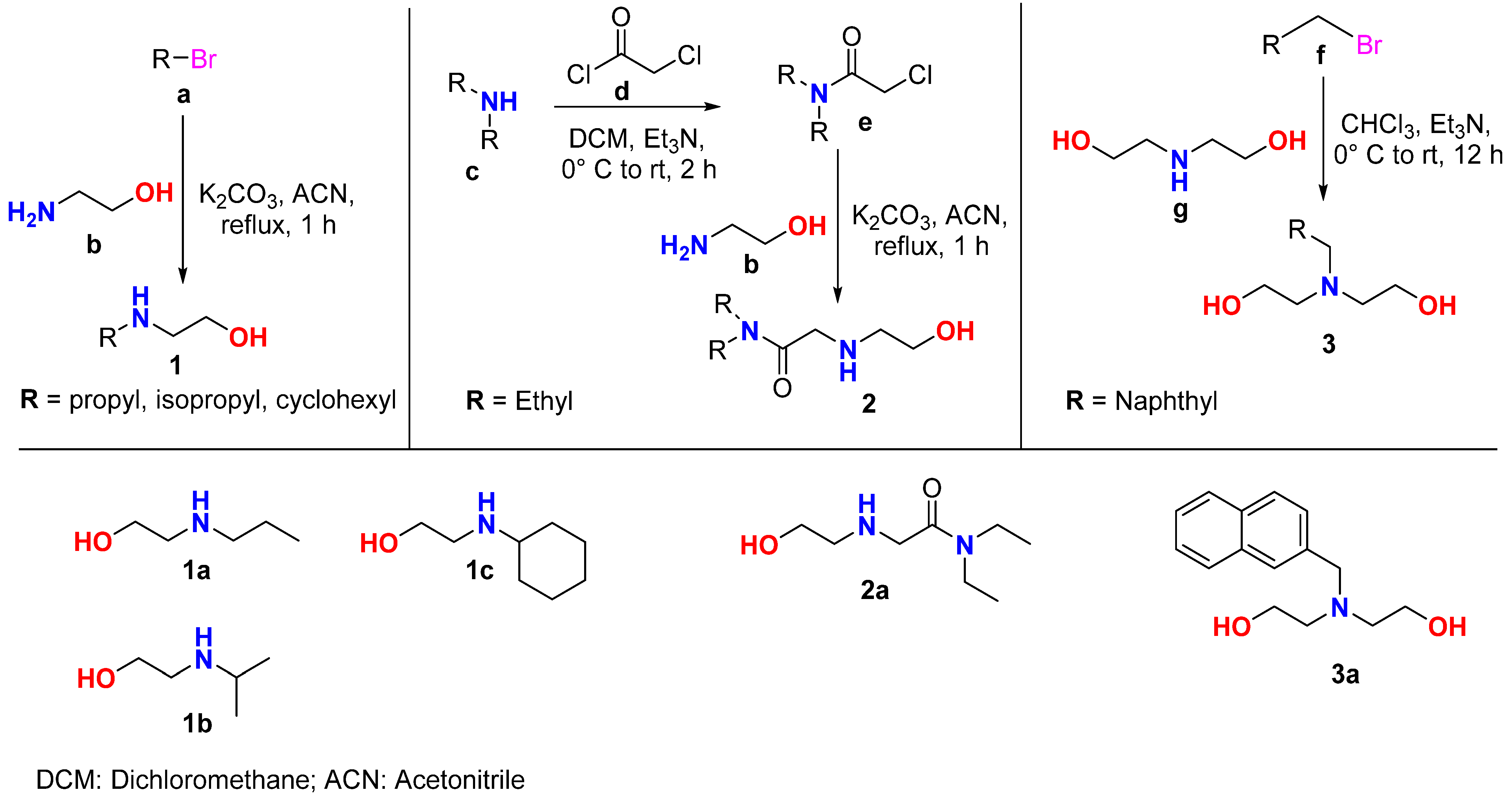
2.1. Synthesis and Characterization
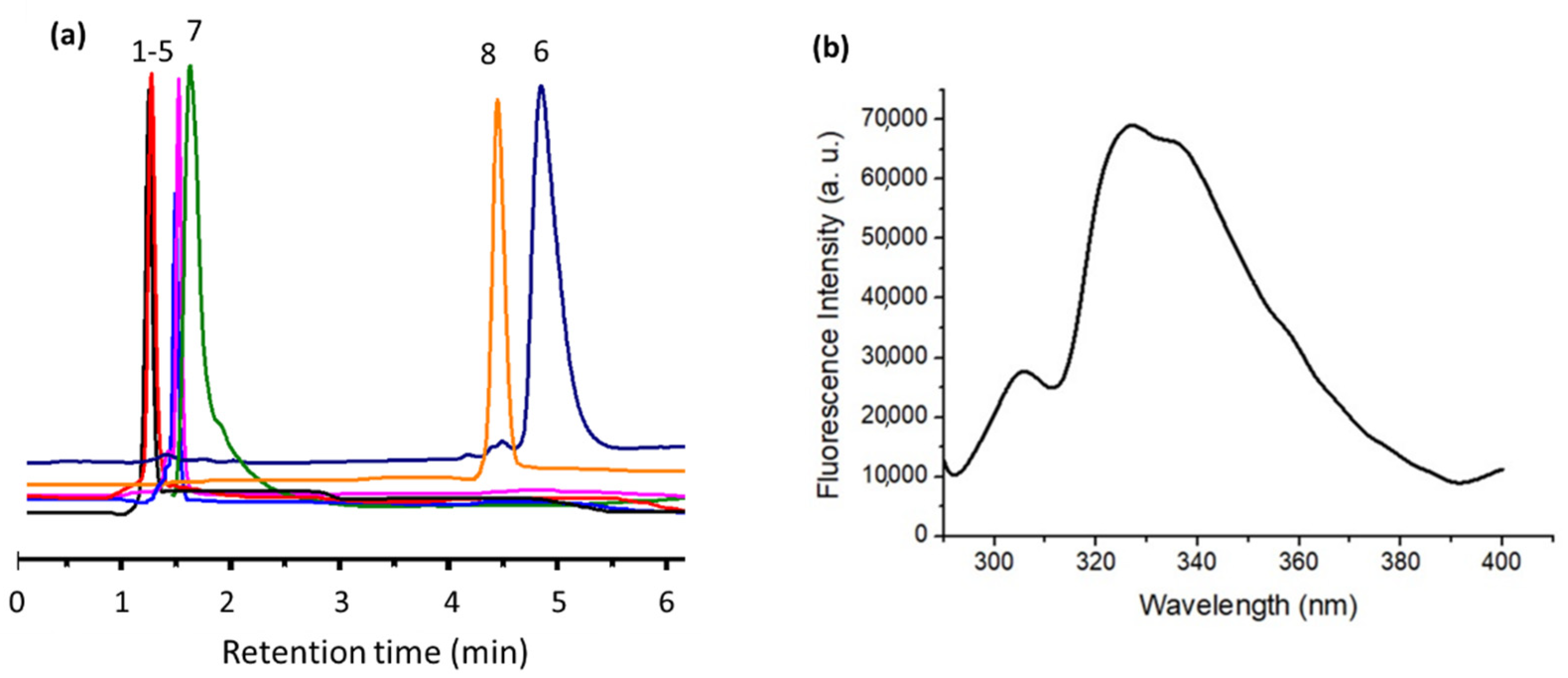
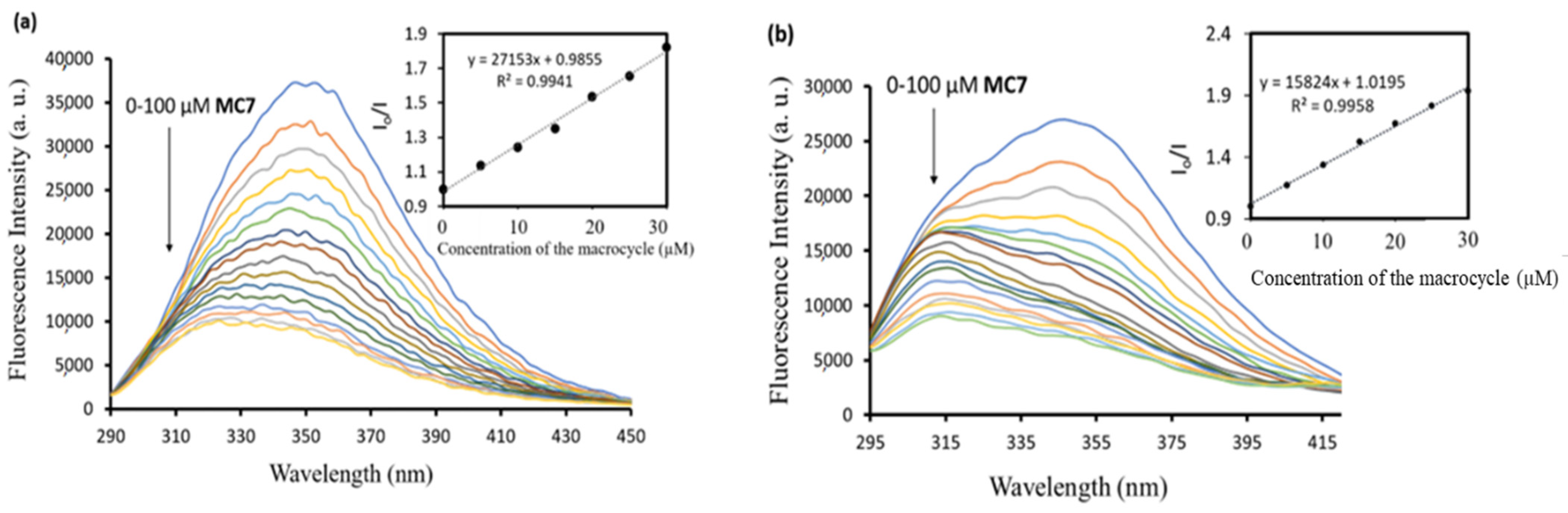
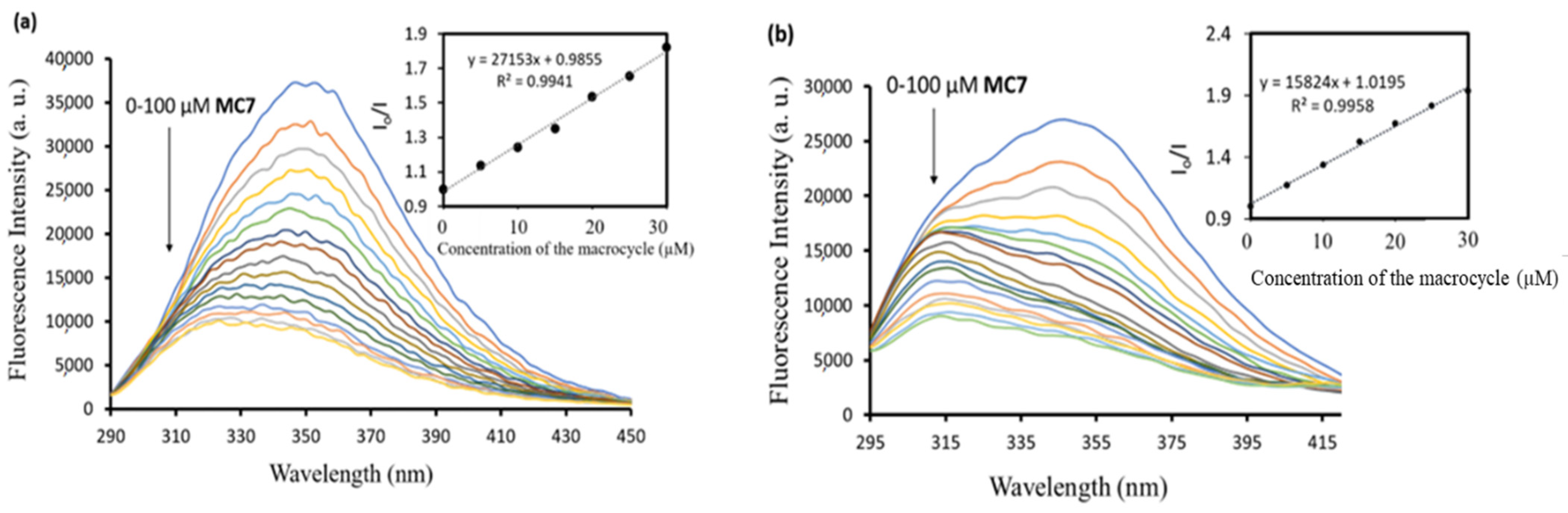
2.2. Bioactive Properties of the Macrocycles
3. Materials and Methods
3.1. Synthesis of Bis-Azide
3.2. Synthesis of Alkyne Precursors (Amino-Alcohols)
3.3. Preparation of Chloro-Amides
3.4. Preparation of Mono/di-Alkynes: General Procedure for N-Propargylation
3.5. Preparation of Bis-Alkynes: General Procedure for O-Propargylation
3.6. Synthesis of Triazole-based Macrocycles: General Procedure for Azide-Alkyne Click (CuAAC) Cyclization
3.7. Protein Interaction Studies by Fluorescence Spectroscopy
3.8. LC-MS, HRMS, and NMR
3.9. Molecular Docking Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Yu, J.; Qi, D.; Li, J. Design, synthesis and applications of responsive macrocycles. Commun. Chem. 2020, 3, 1–14. [Google Scholar] [CrossRef]
- Marti-Centelles, V.; Pandey, M.D.; Burguete, M.I.; Luis, S.V. Macrocyclization Reactions: The Importance of Conformational, Configurational, and Template-Induced Preorganization. Chem. Rev. 2015, 115, 8736–8834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mallinson, J.; Collins, I. Macrocycles in new drug discovery. Futur. Med. Chem. 2012, 4, 1409–1438. [Google Scholar] [CrossRef]
- Yu, X.; Sun, D. Macrocyclic Drugs and Synthetic Methodologies toward Macrocycles. Molecules 2013, 18, 6230–6268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frank, A.T.; Farina, N.S.; Sawwan, N.; Wauchope, O.R.; Qi, M.; Brzostowska, E.M.; Chan, W.; Grasso, F.W.; Haberfield, P.; Greer, A. Natural macrocyclic molecules have a possible limited structural diversity. Mol. Divers. 2007, 11, 115–118. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, K.T.; Osberger, T.J.; King, T.A.; Sore, H.F.; Spring, D.R. Strategies for the Diversity-Oriented Synthesis of Macro-cycles. Chem. Rev. 2019, 119, 10288–10317. [Google Scholar] [CrossRef]
- Chen, C.-F.; Han, Y. Triptycene-Derived Macrocyclic Arenes: From Calixarenes to Helicarenes. Accounts Chem. Res. 2018, 51, 2093–2106. [Google Scholar] [CrossRef]
- Park, C.; Burgess, K. Facile Macrocyclizations to β-Turn Mimics with Diverse Structural, Physical, and Conformational Prop-erties. J. Comb. Chem. 2001, 3, 257–266. [Google Scholar] [CrossRef]
- Gradillas, A.; Pérez-Castells, J. Macrocyclization by Ring-closing Metathesis in the Total Synthesis of Natural Products: Reaction Conditions and Limitations. Angew. Chem. Int. Ed. 2006, 45, 6086–6101. [Google Scholar] [CrossRef]
- Rivera, D.G.; Ojeda-Carralero, G.M.; Reguera, L.; van der Eycken, E.V. Peptide Macrocyclization by Transition Metal Catalysis. Chem. Soc. Rev. 2020, 49, 2039–2059. [Google Scholar] [CrossRef]
- Parenty, A.; Moreau, X.; Campagne, J.-M. Macrolactonizations in the Total Synthesis of Natural Products. Chem. Rev. 2006, 106, 911–939. [Google Scholar] [CrossRef] [PubMed]
- Pasini, D. The Click Reaction as an Efficient Tool for the Construction of Macrocyclic Structures. Molecules 2013, 18, 9512–9530. [Google Scholar] [CrossRef] [PubMed]
- White, C.J.; Yudin, A.K. Contemporary strategies for peptide macrocyclization. Nat. Chem. 2011, 3, 509–524. [Google Scholar] [CrossRef]
- Aimetti, A.A.; Shoemaker, R.K.; Lin, C.-C.; Anseth, K.S. On-resin peptide macrocyclization using thiol–ene click chemistry. Chem. Commun. 2010, 46, 4061–4063. [Google Scholar] [CrossRef] [Green Version]
- Fitzgerald, M.E.; Mulrooney, C.A.; Duvall, J.R.; Wei, J.; Suh, B.-C.; Akella, L.B.; Vrcic, A.; Marcaurelle, L.A. Build/Couple/Pair Strategy for the Synthesis of Stereochemically Diverse Macrolactams via Head-to-Tail Cyclization. ACS Comb. Sci. 2012, 14, 89–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolaou, K.C.; Bulger, P.G.; Sarlah, D. Palladium-Catalyzed Cross-Coupling Reactions in Total Synthesis. Angew. Chem. Int. Ed. 2005, 44, 4442–4489. [Google Scholar] [CrossRef] [PubMed]
- Larsen, B.J.; Sun, Z.; Nagorny, P. Synthesis of Eukaryotic Translation Elongation Inhibitor Lactimidomycin via Zn(II)-Mediated Horner–Wadsworth–Emmons Macrocyclization. Org. Lett. 2013, 15, 2998–3001. [Google Scholar] [CrossRef]
- Breazzano, S.P.; Poudel, Y.B.; Boger, D.L. A Pd(0)-Mediated Indole (Macro)cyclization Reaction. J. Am. Chem. Soc. 2013, 135, 1600–1606. [Google Scholar] [CrossRef] [Green Version]
- Nishikawa, K.; Yoshimi, Y.; Maeda, K.; Morita, T.; Takahashi, I.; Itou, T.; Inagaki, S.; Hatanaka, M. Radical Photocyclization Route for Macrocyclic Lactone Ring Expansion and Conversion to Macrocyclic Lactams and Ketones. J. Org. Chem. 2012, 78, 582–589. [Google Scholar] [CrossRef]
- Abdelraheem, E.M.M.; Khaksar, S.; Kurpiewska, K.; Kalinowska-Tłuścik, J.; Shaabani, S.; Dömling, A. Two-Step Macrocycle Synthesis by Classical Ugi Reaction. J. Org. Chem. 2018, 83, 1441–1447. [Google Scholar] [CrossRef] [Green Version]
- Zapf, C.W.; Harrison, B.A.; Drahl, C.; Sorensen, E.J. A Diels-Alder Macrocyclization Enables an Efficient Asymmetric Synthesis of the Antibacterial Natural Product Abyssomicin C. Angew. Chem. 2005, 117, 6691–6695. [Google Scholar] [CrossRef]
- Thurakkal, L.; Nanjan, P.; Porel, M. Design, Synthesis and Bioactive Properties of a Class of Macrocycles with Tunable Func-tional Groups and Ring Size. Sci. Rep. 2022, 12, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Madhavachary, R.; Abdelraheem, E.M.M.; Rossetti, A.; Twarda-Clapa, A.; Musielak, B.; Kurpiewska, K.; Kalinowska-Tłuścik, J.; Holak, T.A.; Dömling, A. Two-Step Synthesis of Complex Artificial Macrocyclic Compounds. Angew. Chem. Int. Ed. 2017, 56, 10725–10729. [Google Scholar] [CrossRef] [PubMed]
- Krakowiak, K.E.; Bradshaw, J.S.; Zamecka-Krakowiak, D.J. Synthesis of aza-crown ethers. Chem. Rev. 1989, 89, 929–972. [Google Scholar] [CrossRef]
- Hänni, K.D.; Leigh, D.A. The Application of CuAAC ‘Click’Chemistry to Catenane and Rotaxane Synthesis. Chem. Soc. Rev. 2010, 39, 1240–1251. [Google Scholar] [CrossRef]
- Tron, G.C.; Pirali, T.; Billington, R.; Canonico, P.L.; Sorba, G.; Genazzani, A. Click chemistry reactions in medicinal chemistry: Applications of the 1,3-dipolar cycloaddition between azides and alkynes. Med. Res. Rev. 2007, 28, 278–308. [Google Scholar] [CrossRef]
- Agrahari, A.K.; Bose, P.; Jaiswal, M.K.; Rajkhowa, S.; Singh, A.S.; Hotha, S.; Mishra, N.; Tiwari, V.K. Cu(I)-Catalyzed Click Chemistry in Glycoscience and Their Diverse Applications. Chem. Rev. 2021, 121, 7638–7956. [Google Scholar] [CrossRef]
- García, F.; Torres, M.R.; Matesanz, E.; Sánchez, L. Open aryl triazole receptors: Planar sheets, spheres and anion binding. Chem. Commun. 2011, 47, 5016–5018. [Google Scholar] [CrossRef]
- Wang, Y.; Bie, F.; Jiang, H. Controlling Binding Affinities for Anions by a Photoswitchable Foldamer. Org. Lett. 2010, 12, 3630–3633. [Google Scholar] [CrossRef]
- Kumar, A.; Pandey, P.S. Anion Recognition by 1, 2, 3-Triazolium Receptors: Application of Click Chemistry in Anion Recog-nition. Org. Lett. 2008, 10, 165–168. [Google Scholar] [CrossRef]
- Lau, Y.H.; Rutledge, P.J.; Watkinson, M.; Todd, M.H. Chemical sensors that incorporate click-derived triazoles. Chem. Soc. Rev. 2011, 40, 2848–2866. [Google Scholar] [CrossRef] [PubMed]
- Campo, V.L.; Carvalho, I.; Da Silva, C.H.T.P.; Schenkman, S.; Hill, L.; Nepogodiev, S.A.; Field, R.A. Cyclooligomerisation of azido-alkyne-functionalised sugars: Synthesis of 1,6-linked cyclic pseudo-galactooligosaccharides and assessment of their sialylation by Trypanosoma cruzi trans-sialidase. Chem. Sci. 2010, 1, 507–514. [Google Scholar] [CrossRef]
- Romański, J.; Jaworski, P. Synthesis of the novel crown and lariat ethers with integrated 1,2,3-triazole ring. Phosphorus Sulfur Silicon Relat. Elem. 2016, 192, 231–234. [Google Scholar] [CrossRef]
- Nanjan, P.; Jose, A.; Thurakkal, L.; Porel, M. Sequence-Defined Dithiocarbamate Oligomers via a Scalable, Support-Free, It-erative Strategy. Macromolecules 2020, 53, 11019–11026. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Peters, T., Jr. Serum Albumin. Adv. Clin. Chem. 1970, 13, 37–111. [Google Scholar]
- Kumaran, R.; Ramamurthy, P. Photophysical studies on the interaction of amides with Bovine Serum Albumin (BSA) in aqueous solution: Fluorescence quenching and protein unfolding. J. Lumin. 2014, 148, 277–284. [Google Scholar] [CrossRef]
- Steinhardt, J.; Krijn, J.; Leidy, J.G. Differences between bovine and human serum albumins. Binding isotherms, optical rotatory dispersion, viscosity, hydrogen ion titration, and fluorescence effects. Biochemistry 1971, 10, 4005–4015. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ranjith, D. Molecular Docking Studies of Aloe Vera for Their Potential Antibacterial Activity Using Argus Lab 4.0. 1. Pharma Innov. J. 2019, 8, 481–487. [Google Scholar]
- Biovia, D.S. Discovery Visualizer Studio; Dassault Systèmes: San Diego, CA, USA, 2019. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Macrocycle | Binding Constant (×103 M−1) | |
|---|---|---|
| BSA | HSA | |
| MC1 | 4.6 | 4.2 |
| MC2 | 4.0 | 4.4 |
| MC3 | 4.2 | 3.5 |
| MC4 | 9.1 | 8.4 |
| MC5 | 1.4 | 3.2 |
| MC6 | 0.8 | 0.93 |
| MC7 | 27.1 | 15.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheekatla, S.R.; Thurakkal, L.; Jose, A.; Barik, D.; Porel, M. Aza-Oxa-Triazole Based Macrocycles with Tunable Properties: Design, Synthesis, and Bioactivity. Molecules 2022, 27, 3409. https://doi.org/10.3390/molecules27113409
Cheekatla SR, Thurakkal L, Jose A, Barik D, Porel M. Aza-Oxa-Triazole Based Macrocycles with Tunable Properties: Design, Synthesis, and Bioactivity. Molecules. 2022; 27(11):3409. https://doi.org/10.3390/molecules27113409
Chicago/Turabian StyleCheekatla, Subba Rao, Liya Thurakkal, Anna Jose, Debashis Barik, and Mintu Porel. 2022. "Aza-Oxa-Triazole Based Macrocycles with Tunable Properties: Design, Synthesis, and Bioactivity" Molecules 27, no. 11: 3409. https://doi.org/10.3390/molecules27113409
APA StyleCheekatla, S. R., Thurakkal, L., Jose, A., Barik, D., & Porel, M. (2022). Aza-Oxa-Triazole Based Macrocycles with Tunable Properties: Design, Synthesis, and Bioactivity. Molecules, 27(11), 3409. https://doi.org/10.3390/molecules27113409