
Chrysosporazines Revisited: Regioisomeric Phenylpropanoid Piperazine P-Glycoprotein Inhibitors from Australian Marine Fish-Derived Fungi





Abstract
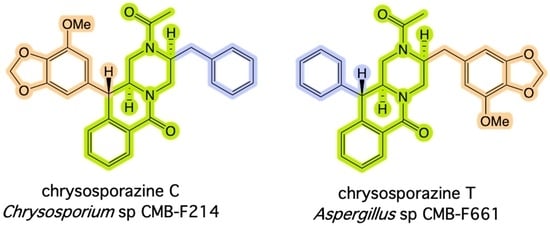
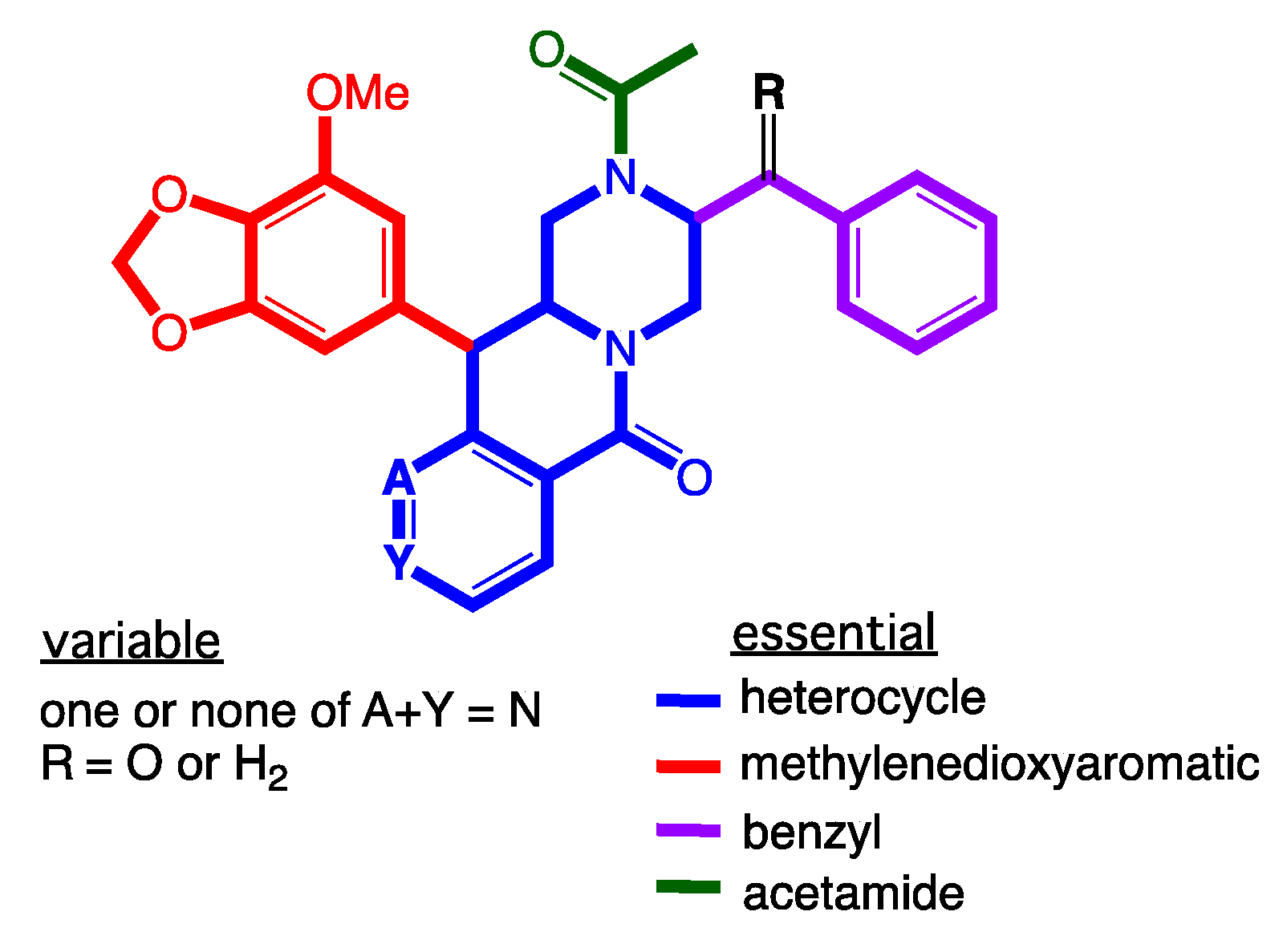
1. Introduction
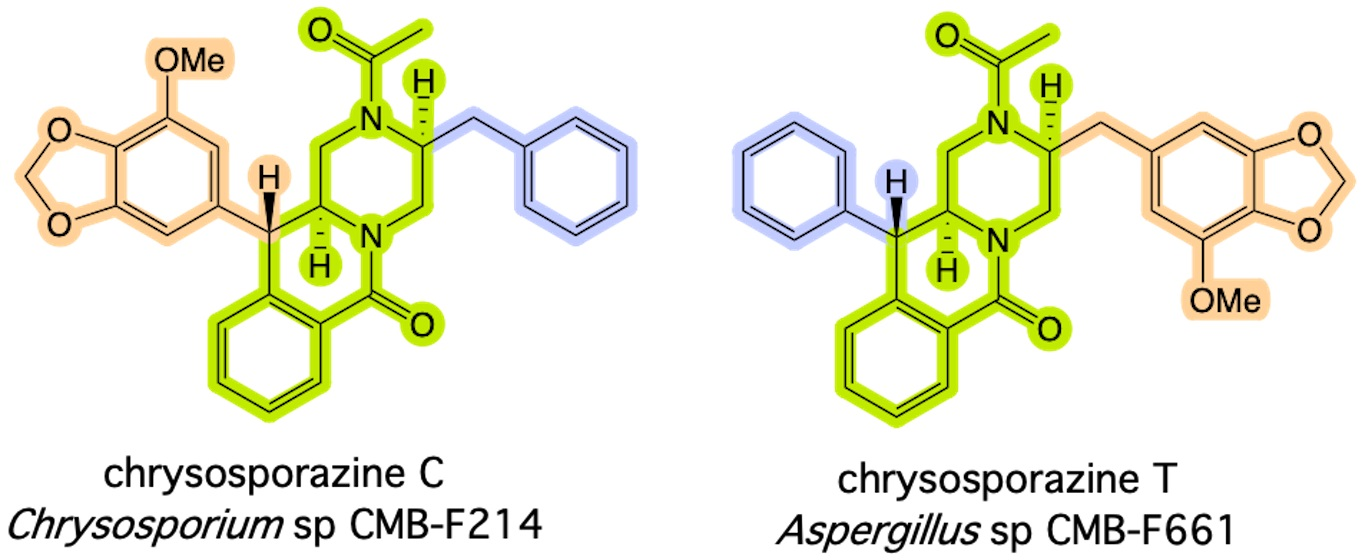
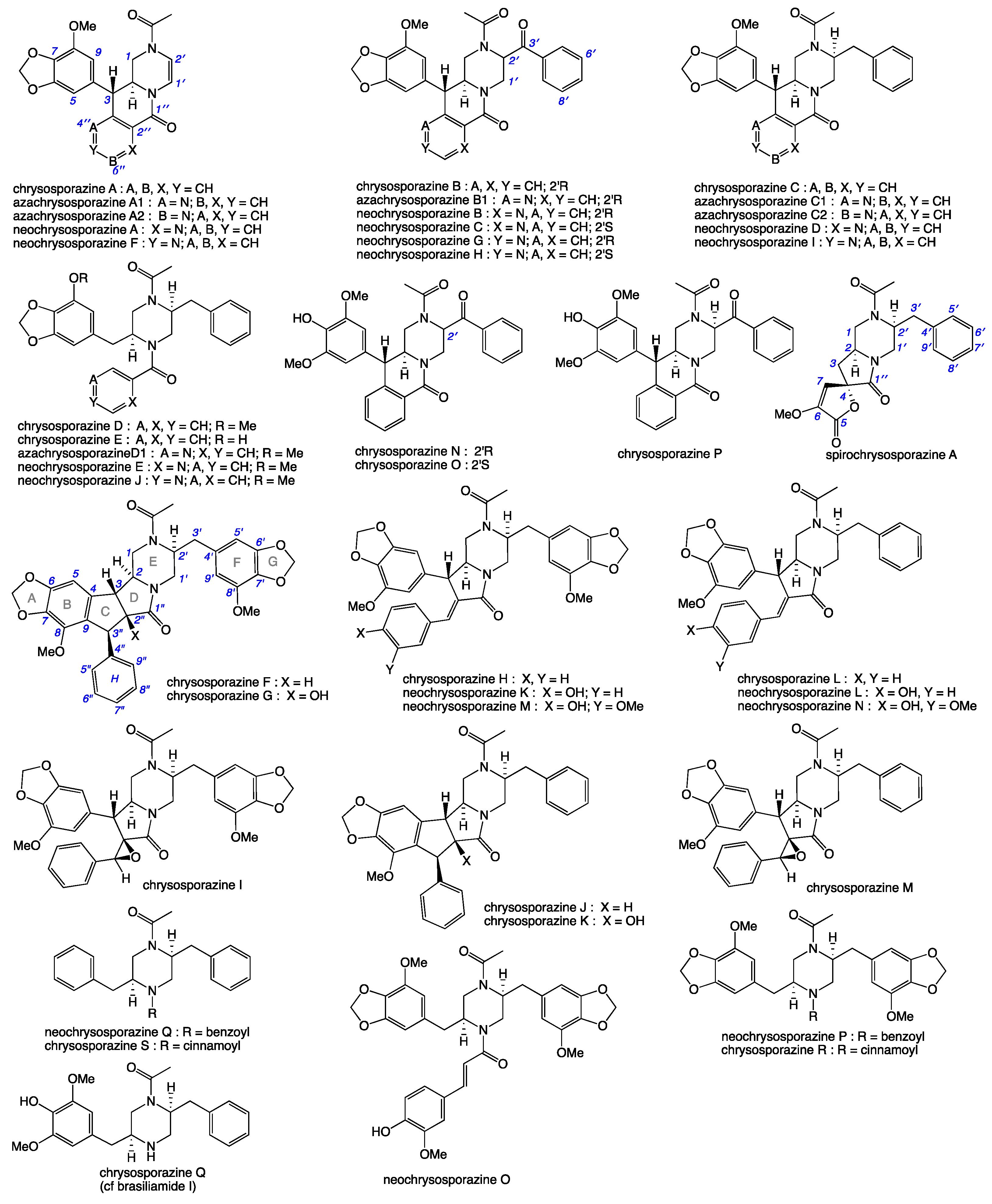
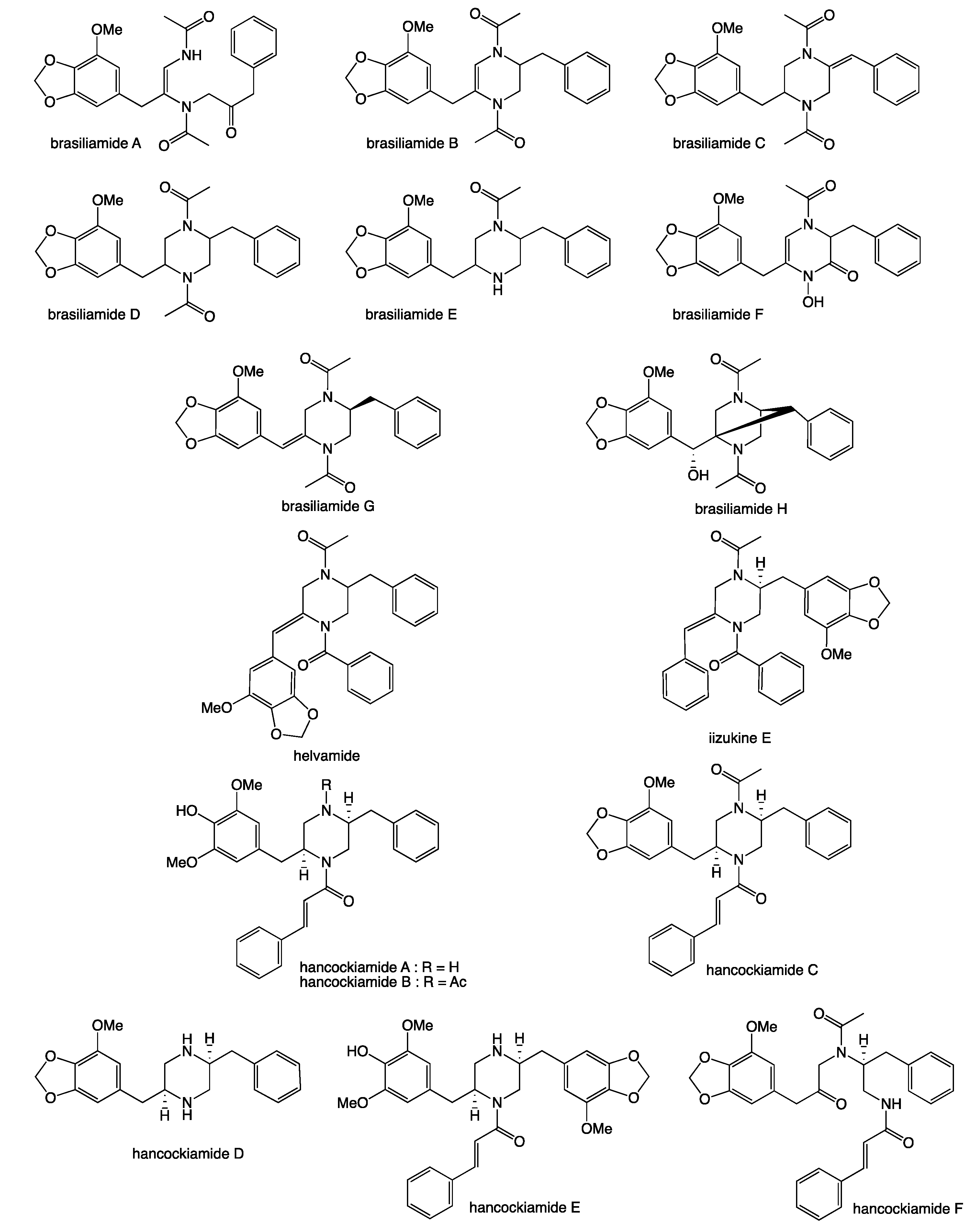
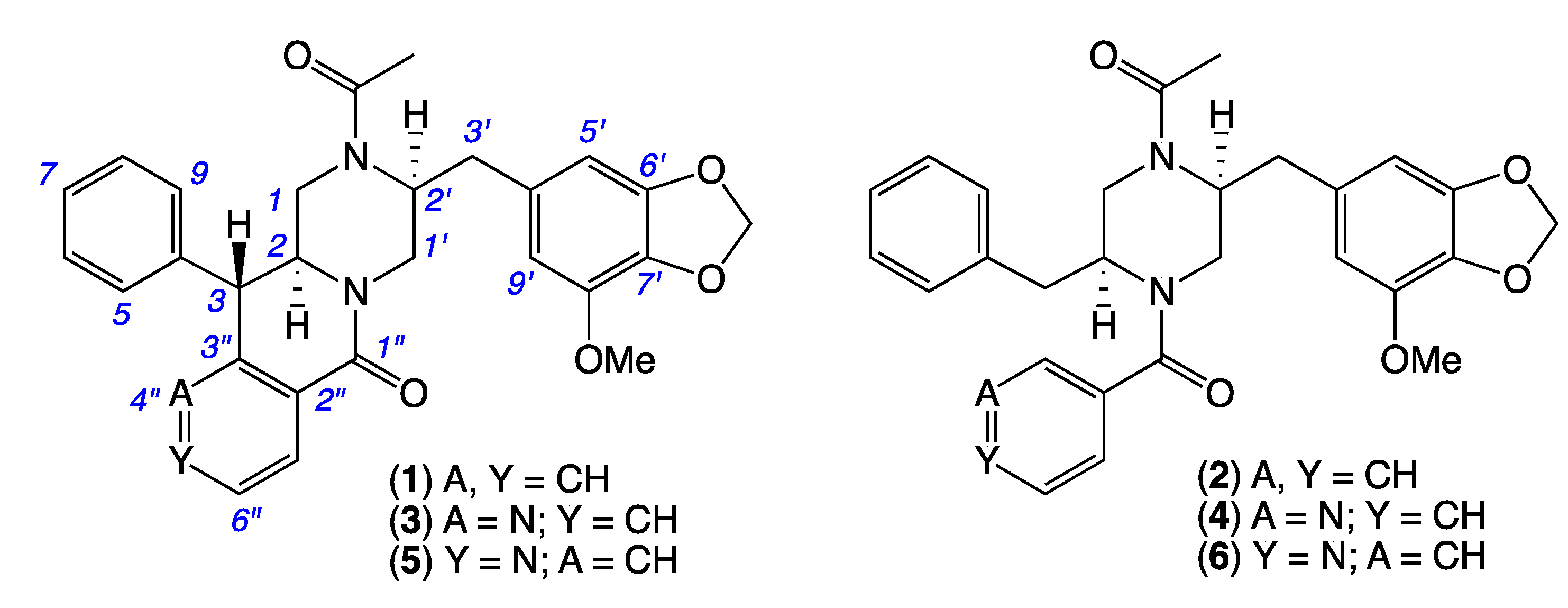
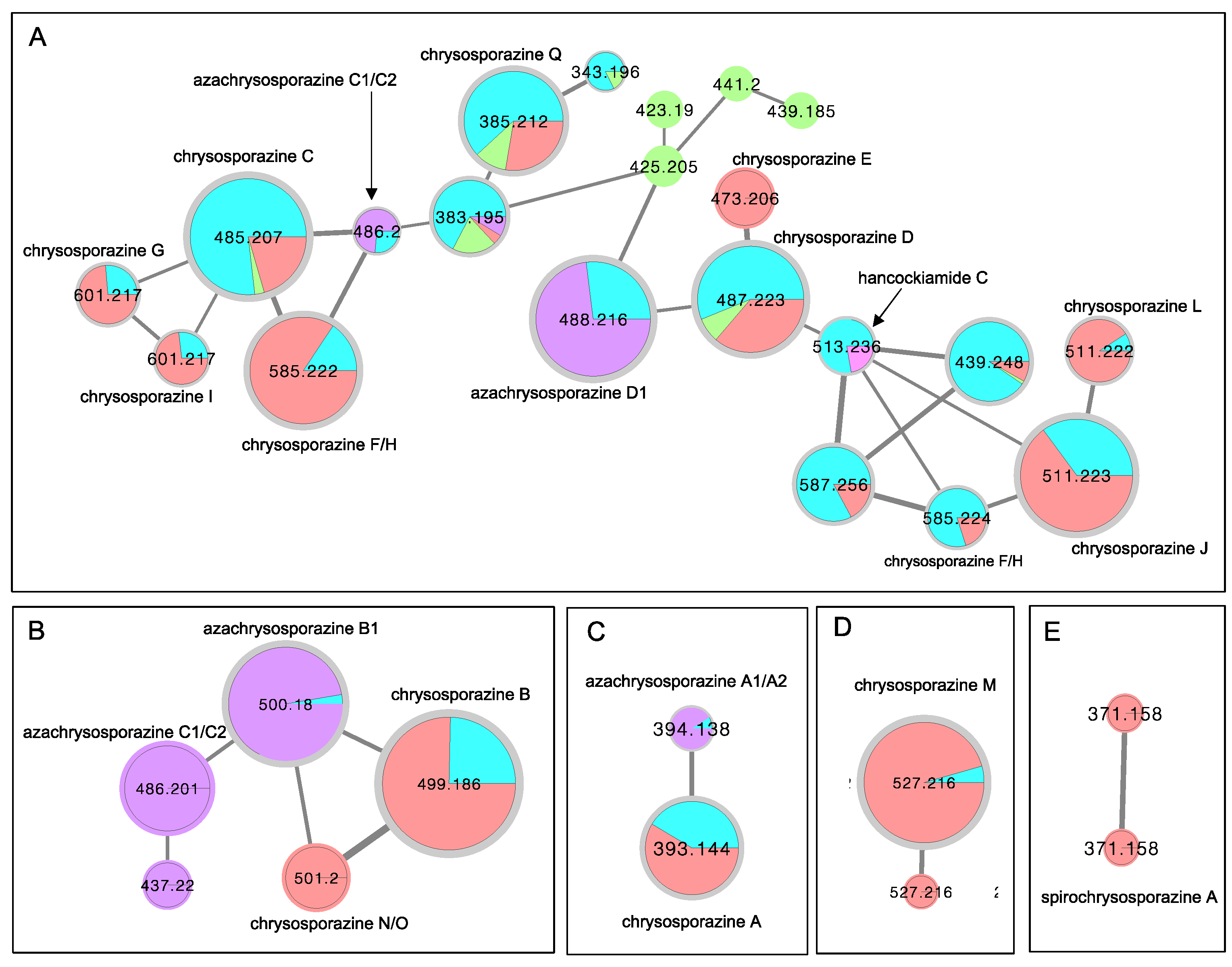
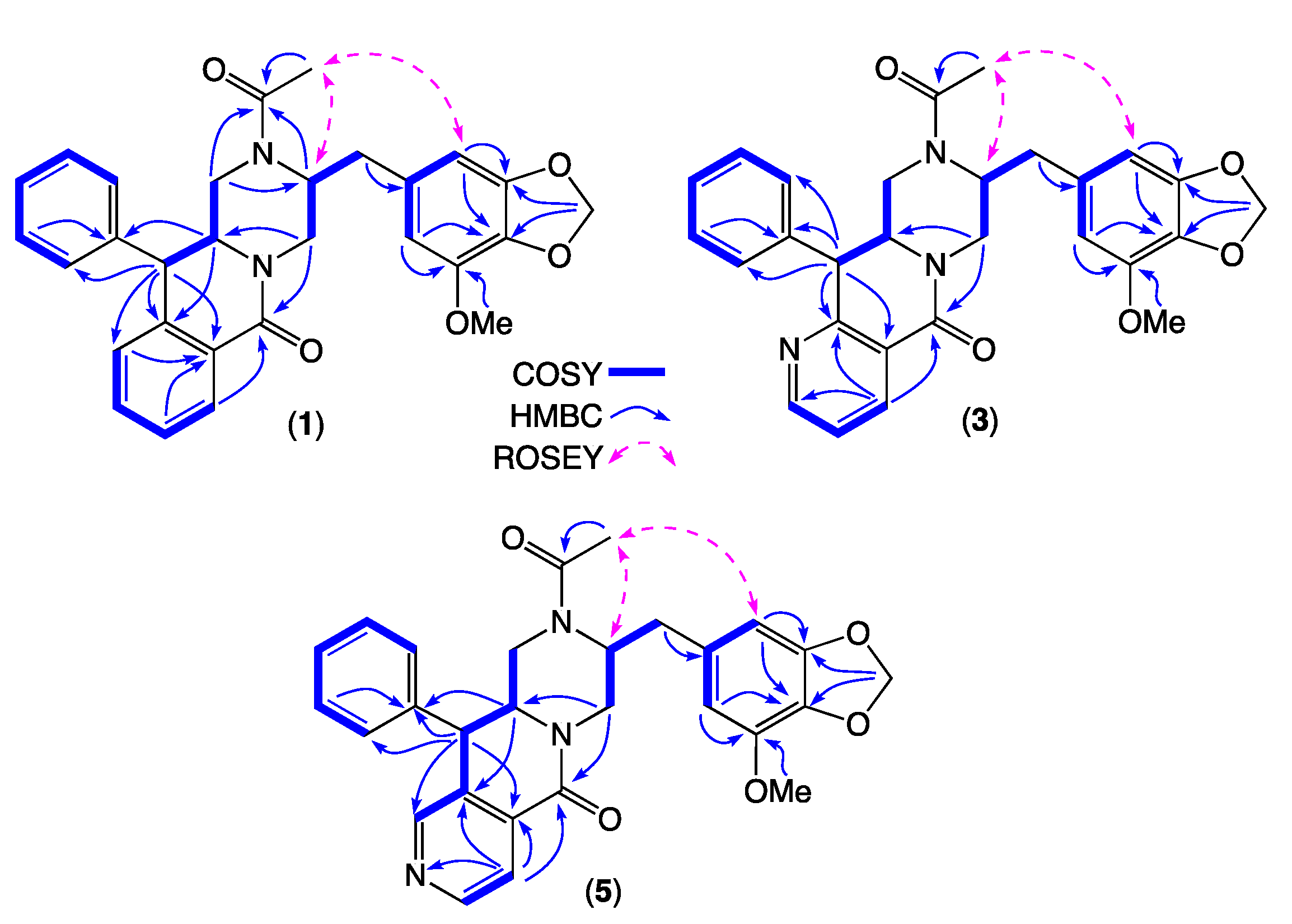
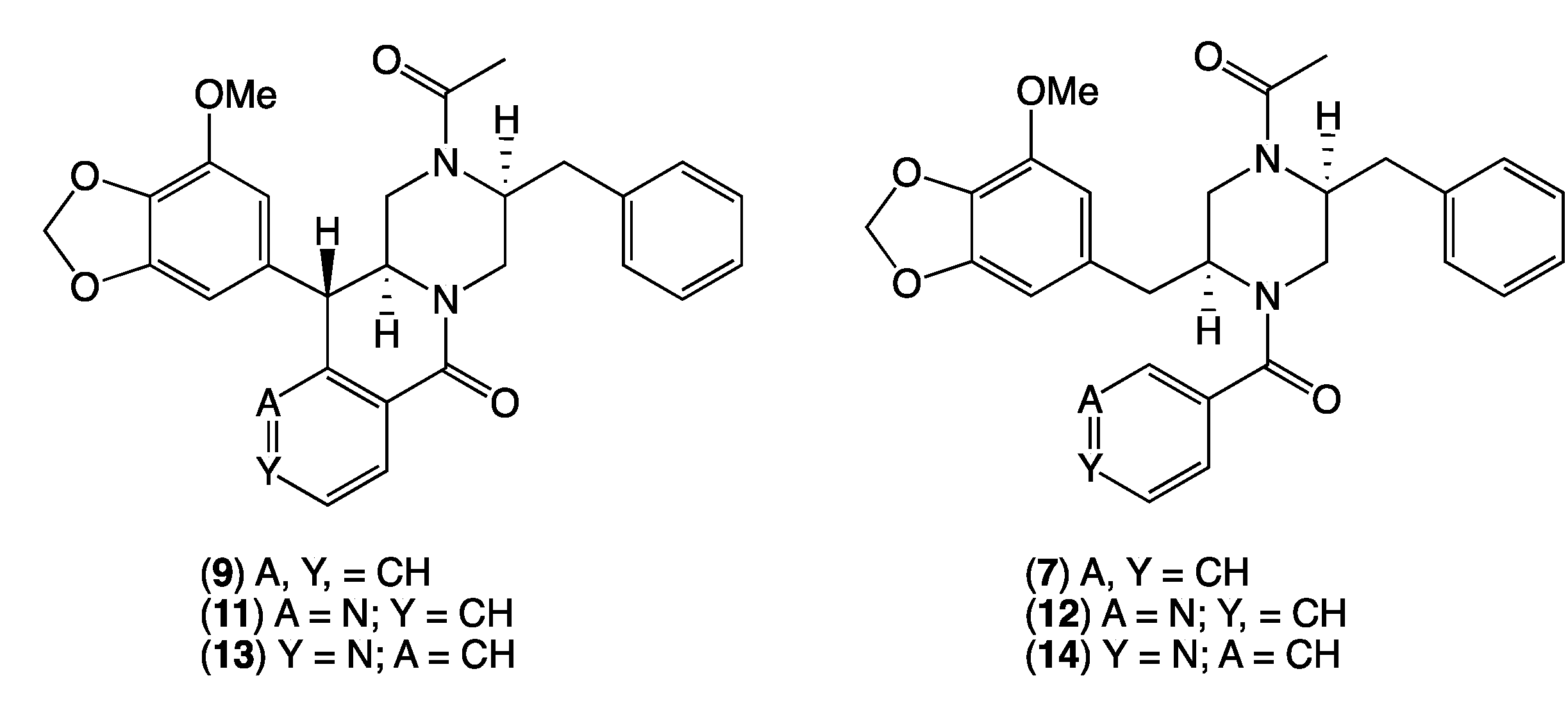
2. Results and Discussion
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Fungal Isolation
3.3. Fungal Taxonomy
3.4. Global Natural Product Social (GNPS) Molecular Networking
3.5. Fractionation of a Scaled Up PDA Culture of Aspergillus sp. CMB-F661
3.6. Media MATRIX Cultivation Profiling of Aspergillus sp. CMB-F661
3.7. Precursor-Directed Biosynthesis Cultivation Profiling of Aspergillus sp. CMB-F661
3.8. Scaled-Up Cultivation of Aspergillus sp. CMB-F661 on PDA with Sodium Nicotinate
3.9. Scaled-Up Cultivation of Aspergillus sp. CMB-F661 on PDA with Sodium Isonicotinate
3.10. Fractionation of a Scaled Up M1 Culture of Spiromastix sp. CMB-F455
3.11. Media MATRIX Cultivation Profiling of Spiromastix sp. CMB-F455
3.12. Precursor-Directed Biosynthesis Cultivation Profiling of Spiromastix sp. CMB-F455
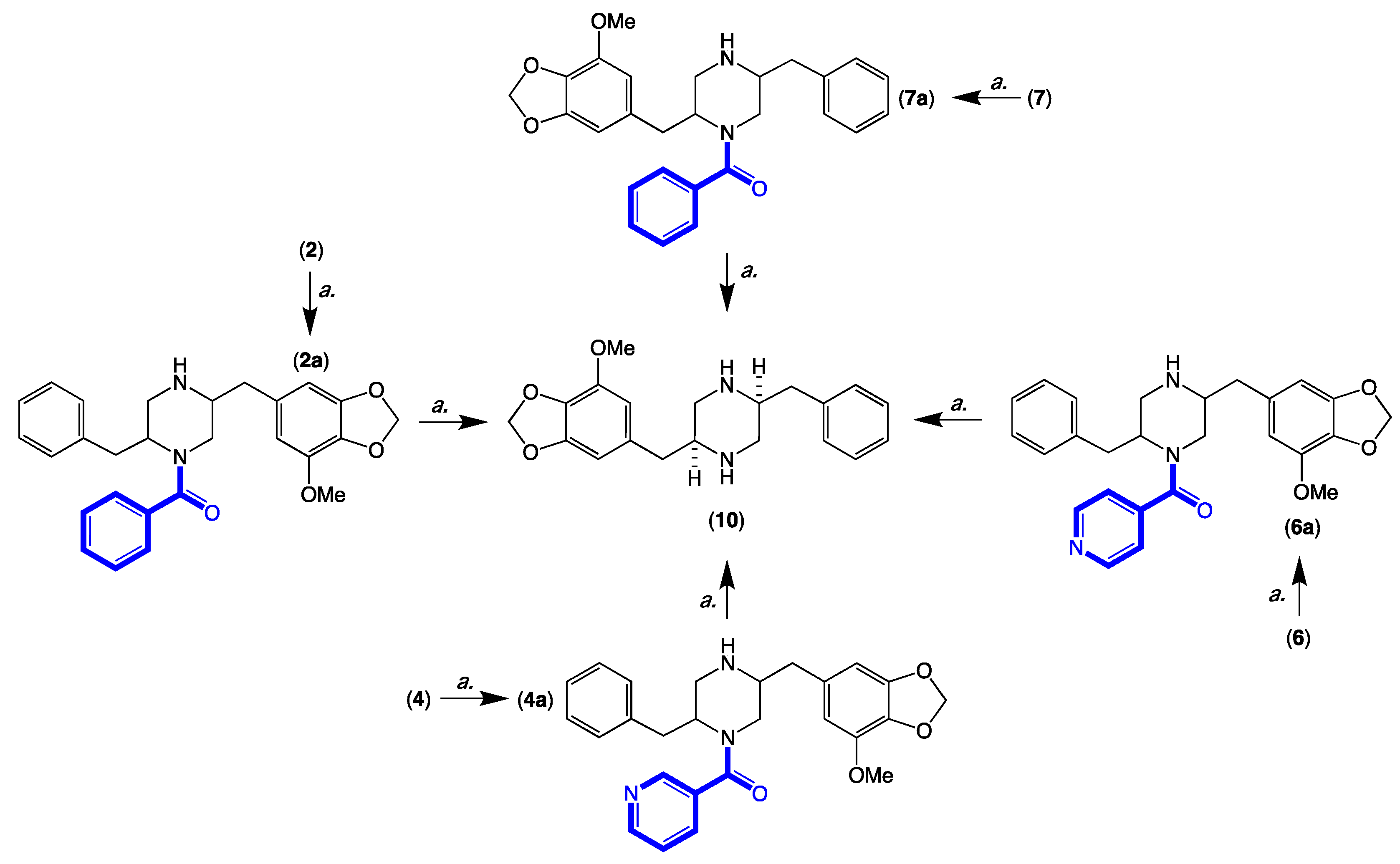
3.13. Acid Hydrolysis and Chemical Correlation of 2, 4, 6 and 7 to the Common Product 10
3.14. Antibacterial Assay
3.15. Antifungal Assay
3.16. Cytotoxicity (MTT) Assay
3.17. P-Glycoprotein Mediated MDR Reversal Assay
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Elbanna, A.H.; Khalil, Z.G.; Bernhardt, P.V.; Capon, R.J. Scopularides revisited: Molecular networking guided exploration of lipodepsipeptides in Australian marine fish gastrointestinal tract-derived fungi. Mar. Drugs 2019, 17, 475. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, O.G.; Khalil, Z.G.; Capon, R.J. Prolinimines: N-Amino-l-Pro-methyl ester (hydrazine) Schiff bases from a fish gastrointestinal tract-derived fungus, Trichoderma sp. CMB-F563. Org. Lett. 2018, 20, 377–380. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, O.G.; Khalil, Z.G.; Capon, R.J. N-Amino-l-Pro-methyl ester from an Australian fish gut-derived fungus: Challenging the distinction between natural product and artifact. Mar. Drugs 2021, 19, 151. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, O.G.; Khalil, Z.G.; Santiago, V.; Capon, R.J. Metarhizides A–C and metarhizosides A–B: PKS-NRPS macrolides and aromatic glycosides from an Australian fish gut-derived fungus, Metarhizium sp. CMB-F624. Tetrahedron 2022, 113, 132759. [Google Scholar] [CrossRef]
- Elbanna, A.H.; Khalil, Z.G.; Bernhardt, P.V.; Capon, R.J. Chrysosporazines A-E: P-Glycoprotein inhibitory piperazines from an Australian marine fish gastrointestinal tract-derived fungus, Chrysosporium sp. CMB-F214. Org. Lett. 2019, 21, 8097–8100. [Google Scholar] [CrossRef]
- Mohamed, O.G.; Salim, A.A.; Khalil, Z.G.; Elbanna, A.H.; Bernhardt, P.V.; Capon, R.J. Chrysosporazines F-M: P-Glycoprotein inhibitory phenylpropanoid piperazines from an Australian marine fish derived fungus, Chrysosporium sp. CMB-F294. J. Nat. Prod. 2020, 83, 497–504. [Google Scholar] [CrossRef]
- Elbanna, A.H.; Agampodi Dewa, A.; Khalil, Z.G.; Capon, R.J. Precursor-directed biosynthesis mediated amplification of minor aza phenylpropanoid piperazines in an Australian marine fish-gut-derived fungus, Chrysosporium sp. CMB-F214. Mar. Drugs 2021, 19, 478. [Google Scholar] [CrossRef]
- Dewa, A.A.; Elbanna, A.H.; Khalil, Z.G.; Capon, R.J. Neochrysosporazines: Precursor-directed biosynthesis defines a marine-derived fungal natural product P-Glycoprotein inhibitory pharmacophore. J. Med. Chem. 2022, 65, 2610–2622. [Google Scholar] [CrossRef]
- Li, H.; Lacey, A.E.; Shu, S.; Kalaitzis, J.A.; Vuong, D.; Crombie, A.; Hu, J.; Gilchrist, C.L.M.; Lacey, E.; Piggott, A.M.; et al. Hancockiamides: Phenylpropanoid piperazines from Aspergillus hancockii are biosynthesised by a versatile dual single-module NRPS pathway. Org. Biomol. Chem. 2021, 19, 587–595. [Google Scholar] [CrossRef]
- Fujita, T.; Makishima, D.; Akiyama, K.; Hayashi, H. New convulsive compounds, brasiliamides A and B, from Penicillium brasilianum batista JV-379. Biosci. Biotechnol. Biochem. 2002, 66, 1697–1705. [Google Scholar] [CrossRef]
- Fujita, T.; Hayashi, H. New brasiliamide congeners, brasiliamides C, D and E, from Penicillium brasilianum Batista JV-379. Biosci. Biotechnol. Biochem. 2004, 68, 820–826. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Fill, T.P.; Geris dos Santos, R.M.; Barisson, A.; Rodrigues-Filho, E.; Souza, A.Q. Co-production of bisphenylpropanoid amides and meroterpenes by an endophytic Penicillium brasilianum found in the root bark of Melia azedarach. Z Nat. C J. Biosci. 2009, 64, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Fill, T.P.; Silva, B.F.D.; Rodrigues-Fo, E. Biosynthesis of phenylpropanoid amides by an endophytic Penicillium brasilianum found in root bark of Melia azedarach. J. Microbiol. Biotechnol. 2010, 20, 622–629. [Google Scholar] [PubMed]
- Liu, Y.; Mandi, A.; Li, X.M.; Meng, L.H.; Kurtan, T.; Wang, B.G. Peniciadametizine A, a dithiodiketopiperazine with a unique spiro[furan-2,7’-pyrazino[1,2-b][1,2]oxazine] skeleton, and a related analogue, peniciadametizine b, from the marine sponge-derived fungus Penicillium adametzioides. Mar. Drugs 2015, 13, 3640–3652. [Google Scholar] [CrossRef] [PubMed]
- Paluka, J.; Kanokmedhakul, K.; Soytong, M.; Soytong, K.; Yahuafai, J.; Siripong, P.; Kanokmedhakul, S. Meroterpenoid pyrones, alkaloid and bicyclic brasiliamide from the fungus Neosartorya hiratsukae. Fitoterapia 2020, 142, 104485. [Google Scholar] [CrossRef]
- Eamvijarn, A.; Kijjoa, A.; Bruyere, C.; Mathieu, V.; Manoch, L.; Lefranc, F.; Silva, A.; Kiss, R.; Herz, W. Secondary metabolites from a culture of the fungus Neosartorya pseudofischeri and their in vitro cytostatic activity in human cancer cells. Planta Med. 2012, 78, 1767–1776. [Google Scholar] [CrossRef]
- Liu, Z.G.; Bao, L.; Liu, H.W.; Ren, J.W.; Wang, W.Z.; Wang, L.; Li, W.; Yin, W.B. Chemical diversity from the Tibetan Plateau fungi Penicillium kongii and P. brasilianum. Mycology 2018, 9, 10–19. [Google Scholar] [CrossRef]
- Yuan, B.; Liu, D.; Guan, X.; Yan, Y.; Zhang, J.; Zhang, Y.; Yang, D.; Ma, M.; Lin, W. Piperazine ring formation by a single-module NRPS and cleavage by an alpha-KG-dependent nonheme iron dioxygenase in brasiliamide biosynthesis. Appl. Microbiol. Biotechnol. 2020, 104, 6149–6159. [Google Scholar] [CrossRef]
- Kang, H.H.; Zhong, M.J.; Ma, L.Y.; Rong, X.G.; Liu, D.S.; Liu, W.Z. Iizukines C-E from a saline soil fungus Aspergillus iizukae. Bioorg. Chem. 2019, 91, 103167. [Google Scholar] [CrossRef]
- Fukuda, T.; Furukawa, T.; Kobayashi, K.; Nagai, K.; Uchida, R.; Tomoda, H. Helvamide, a new inhibitor of sterol O-acyltransferase produced by the fungus Aspergillus nidulans BF-0142. J. Antibiot. 2019, 72, 8–14. [Google Scholar] [CrossRef]
- Wang, M.; Carver, J.J.; Phelan, V.V.; Sanchez, L.M.; Garg, N.; Peng, Y.; Nguyen, D.D.; Watrous, J.; Kapono, C.A.; Luzzatto-Knaan, T.; et al. Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nat. Biotechnol. 2016, 34, 828–837. [Google Scholar] [CrossRef] [PubMed]
- Salim, A.A.; Khalil, Z.G.; Elbanna, A.H.; Wu, T.; Capon, R.J. Methods in microbial biodiscovery. Mar. Drugs 2021, 19, 503. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | (1) δH, Multi (J in Hz) | (3) δH, Multi (J in Hz) | (5) δH, Multi (J in Hz) |
|---|---|---|---|
| 1 | a. 4.14, dd (13.8, 3.9) | a. 4.25, dd (13.3, 3.8) | a. 4.19, dd (14.5, 4.0) |
| b. 2.94, m | b. 3.03, m | b. 2.98, m | |
| 2 | 3.84, ddd (10.6, 10.6, 3.9) | 3.87, ddd (11.2, 8.8, 3.8) | 3.92, ddd (14.5, 10.3, 3.7) |
| 3 | 4.47, d (10.6) | 4.56, d (8.8) | 4.56, d (10.3) |
| 5/9 | 7.35, m | 7.26, m | 7.40, m |
| 6/8 | 7.44, m | 7.35, m | 7.46, m |
| 7 | 7.37, m | 7.29, m | 7.40, m |
| 1′ | a. 4.57, dd (13.3, 1.2) | a. 4.56, m | a. 4.55, dd (13.4, 1.3) |
| b. 2.95, m | b. 3.01, m | b. 2.99, m | |
| 2′ | 4.21, m | 4.21, m | 4.23, m |
| 3′ | a. 2.90, dd (13.5, 8.1) | a. 2.88, dd (13.5, 8.3) | a. 2.90, dd (13.4, 8.6) |
| b. 2.86, dd (13.5, 6.6) | b. 2.83, dd (13.5, 6.3) | b. 2.85, dd (13.4, 6.4) | |
| 4′ | - | - | - |
| 5′ | 6.54, d (1.2) | 6.54, d (1.3) | 6.54, d (1.4) |
| 9′ | 6.55, d (1.2) | 6.55, d (1.3) | 6.55, d (1.4) |
| 4″ | 6.60, d (7.8) | - | 7.89, s |
| 5″ | 7.44, m | 8.57, dd (4.7, 1.8) | - |
| 6″ | 7.39, m | 7.44, ddd (7.8, 4.7) | 8.64, d (4.7) |
| 7″ | 8.04, dd (7.7, 1.4) | 8.34, dd (7.8, 1.8) | 7.88, d (4.7) |
| 1-NCOCH3 | - | - | - |
| 1-NCOCH3 | 1.70, s | 1.68, s | 1.70, s |
| 6′-OCH2 | 5.94/5.93, ABq | 5.95/5.93, ABq | 5.95/5.94, ABq |
| 8′-OCH3 | 3.79, s | 3.78, s | 3.78, s |
| Position | (1) δC, Type | (3) δC, Type | (5) δC, Type |
|---|---|---|---|
| 1 | 40.1, CH2 | 40.4, CH2 | 40.1, CH2 |
| 2 | 57.8, CH | 58.6, CH | 58.2, CH |
| 3 | 46.3, CH | 48.8, CH | 43.7, CH |
| 4 | 140.4 A, C | 140.7, C | 139.4, C |
| 5/9 | 129.3, CH | 129.3, CH | 129.2A, CH |
| 6/8 | 129.1, CH | 128.6, CH | 129.1A, CH |
| 7 | 127.6, CH | 127.1, CH | 127.9, CH |
| 1′ | 44.6, CH2 | 45.0, CH2 | 44.8, CH2 |
| 2′ | 54.6, CH | 54.9, CH | 54.6, CH |
| 3′ | 34.9, CH2 | 35.0, CH2 | 34.8, CH2 |
| 4′ | 132.5, C | 132.6, C | 132.6, C |
| 5′ | 103.3, CH | 103.4, CH | 103.3, CH |
| 6′ | 148.3, C | 148.2, C | 148.3, C |
| 7′ | 133.0, C | 133.4, C | 133.3, C |
| 8′ | 143.0, C | 143.1, C | 143.0, C |
| 9′ | 109.0, CH | 109.1, CH | 109.0, CH |
| 1″ | 163.9, C | 163.2, C | 162.3, C |
| 2″ | 127.4, C | 122.9 A, C | 134.4, C |
| 3″ | 140.3 A, C | 158.4, C | 133.7, C |
| 4″ | 126.9 B, CH | - | 148.6, CH |
| 5″ | 132.3, CH | 152.6, CH | - |
| 6″ | 127.0 B, CH | 122.8 A, CH | 148.3, CH |
| 7″ | 127.6, CH | 135.6, CH | 120.2, CH |
| 1-NCOCH3 | 168.4, C | 168.6, C | 168.5, C |
| 1-NCOCH3 | 20.8, CH3 | 20.8, CH3 | 20.8, CH3 |
| 6′-OCH2 | 101.0, CH2 | 101.0, CH2 | 101.0, CH2 |
| 8′-OCH3 | 56.2, CH3 | 56.3, CH3 | 56.2, CH3 |
| SW620 Ad300 | ||||
|---|---|---|---|---|
| Treatment | IC50 (μM) | FR | GS | FI |
| doxorubicin | 5.75 | 57.5 | 1.0 | 0.12 |
| verapamil | >30 | ND | ND | - |
| + verapamil (2.5 μM) | 0.71 | 7.1 | 8.1 | 1.00 |
| + chrysosporazine T (1) | 0.97 | 9.7 | 5.9 | 0.61 |
| + chrysosporazine C (9) | 0.31 | 3.1 | 18.5 | 2.28 |
| + chrysosporazine U (2) | 2.76 | 27.6 | 2.0 | 0.25 |
| + chrysosporazine D (7) | 4.36 | 43.6 | 1.32 | 0.16 |
| + azachrysosporazine T1 (3) | 0.89 | 8.9 | 6.4 | 0.80 |
| + azachrysosporazine C1 (11) | 0.27 | 2.7 | 21.3 | 2.63 |
| + azachrysosporazine U1 (4) | 2.78 | 27.8 | 2.0 | 0.25 |
| + azachrysosporazine D1 (12) | 3.55 | 35.5 | 1.6 | 0.20 |
| + neochrysosporazine R (5) | 0.58 | 5.8 | 9.9 | 1.22 |
| + neochrysosporazine I (13) | 1.01 | 10.1 | 5.7 | 0.70 |
| + neochrysosporazine S (6) | 1.95 | 19.5 | 2.9 | 0.36 |
| + neochrysosporazine J (14) | 6.18 | 61.8 | 0.9 | 0.11 |
| + brasiliamide A (8) | 5.27 | 52.7 | 1.1 | 0.13 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Agampodi Dewa, A.; Khalil, Z.G.; Elbanna, A.H.; Capon, R.J. Chrysosporazines Revisited: Regioisomeric Phenylpropanoid Piperazine P-Glycoprotein Inhibitors from Australian Marine Fish-Derived Fungi. Molecules 2022, 27, 3172. https://doi.org/10.3390/molecules27103172
Agampodi Dewa A, Khalil ZG, Elbanna AH, Capon RJ. Chrysosporazines Revisited: Regioisomeric Phenylpropanoid Piperazine P-Glycoprotein Inhibitors from Australian Marine Fish-Derived Fungi. Molecules. 2022; 27(10):3172. https://doi.org/10.3390/molecules27103172
Chicago/Turabian StyleAgampodi Dewa, Amila, Zeinab G. Khalil, Ahmed H. Elbanna, and Robert J. Capon. 2022. "Chrysosporazines Revisited: Regioisomeric Phenylpropanoid Piperazine P-Glycoprotein Inhibitors from Australian Marine Fish-Derived Fungi" Molecules 27, no. 10: 3172. https://doi.org/10.3390/molecules27103172
APA StyleAgampodi Dewa, A., Khalil, Z. G., Elbanna, A. H., & Capon, R. J. (2022). Chrysosporazines Revisited: Regioisomeric Phenylpropanoid Piperazine P-Glycoprotein Inhibitors from Australian Marine Fish-Derived Fungi. Molecules, 27(10), 3172. https://doi.org/10.3390/molecules27103172