
Use of Passive and Grab Sampling and High-Resolution Mass Spectrometry for Non-Targeted Analysis of Emerging Contaminants and Their Semi-Quantification in Water

Abstract
:1. Introduction
2. Results
2.1. Compound Identification
2.2. Retention Time Prediction
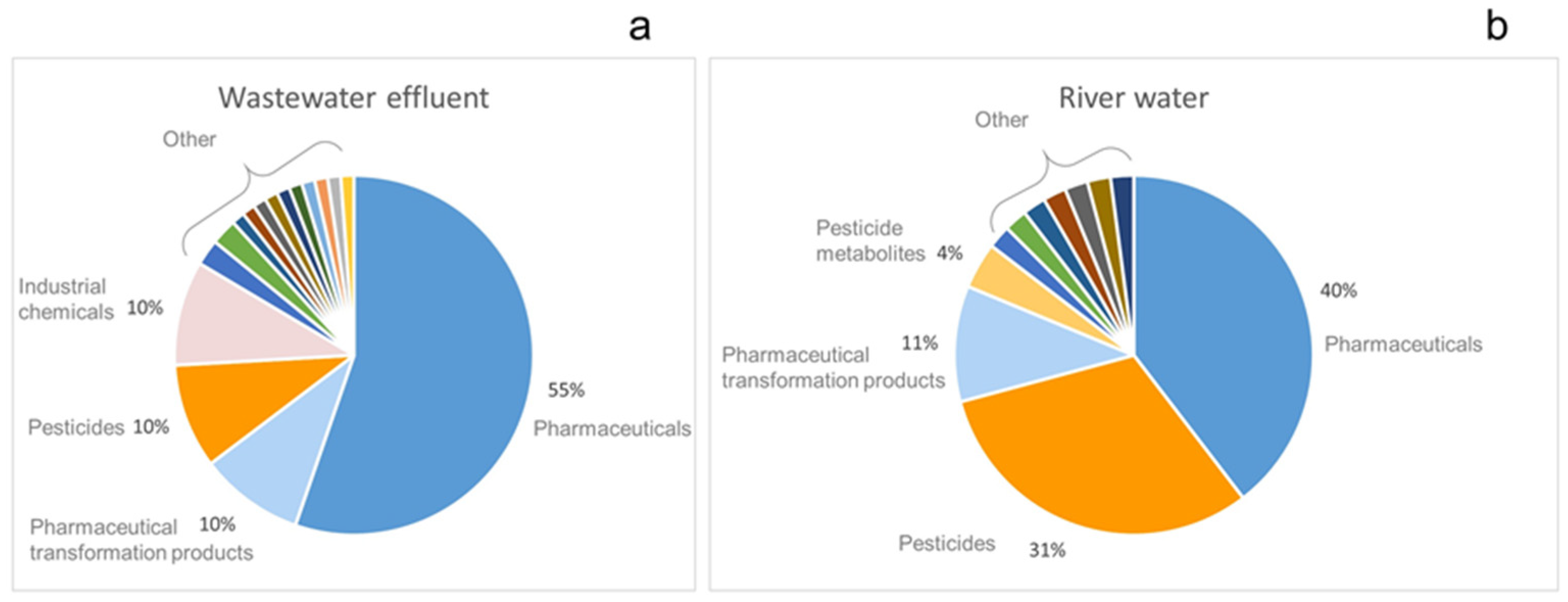
2.3. Contamination Profile
2.4. Quantitative and Semi-Quantitative Analysis
3. Materials and Methods
3.1. Sample Collection and Processing
3.2. Target Analysis
3.3. Non-Target Analysis
3.4. Retention Time Prediction, Semi-Quantification, Data Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Class | Name | RT | Quantification Trace | Recovery Rates in WW (in%) | CAS | Chemical Formula | Log Kow | Analytical LOD (µg/L) | Analytical LOQ (µg/L) | LOQ in Grab Water Sample (ng/L) | LOD in Water Sample (ng/L) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| IS | Simazine D5 | 3.33 | 207.1 > 129 | - | 220621-41-0 | C7H7D5ClN5 | - | - | - | - | - |
| Herbicide | Acetochlor | 7.12 | 224 > 224 | 142 | 34256-82-1 | C14H20ClNO2 | 4.14 | 4.1 | 13.6 | 12.2 | 40.7 |
| Herbicide | Alachlor | 7.06 | 270.1 > 237.9 | 138 | 15972-60-8 | C14H20ClNO2 | 2.97 | 10.0 | 33.2 | 29.9 | 99.7 |
| Herbicide | Atrazine | 3.94 | 216 > 174 | 112 | 1912-24-9 | C8H14ClN5 | 2.50 | 1.9 | 6.4 | 5.8 | 19.3 |
| Herbicide | Atrazine-desethyl (DEA) | 2.72 | 188 > 146 | 116 | 6190-65-4 | C6H10ClN5 | 1.51 | 4.6 | 15.5 | 13.9 | 46.5 |
| Herbicide | Chlortoluron | 3.51 | 213.1 > 72.1 | 129 | 15545-48-9 | C10H13ClN2O | 2.50 | 6.0 | 20.2 | 18.1 | 60.5 |
| Herbicide | DCPMU | 3.33 | 219 > 127.1 | 10 | 3567-62-2 | C8H8Cl2N2O | 2.94 | 5.4 | 17.9 | 16.1 | 53.6 |
| Herbicide | DCPU | 2.9 | 205 > 127 | 0 | 2327-02-8 | C7H6Cl2N2O | 2.35 | 4.1 | 13.5 | 12.2 | 40.6 |
| Herbicide | Terbuthylazine desethyl (DET) | 3.22 | 202.1 >146.1 | 107 | 30125-63-4 | C7H12ClN5 | 2.30 | 1.4 | 4.6 | 4.2 | 13.9 |
| Herbicide | Atrazine desisopropyl (DIA) | 2.54 | 174 > 104 | 154 | 1007-28-9 | C5H8ClN5 | 0.32 | 4.8 | 16.0 | 14.4 | 48.1 |
| Herbicide | Diuron | 3.83 | 233.1 > 72 | 125 | 330-54-1 | C9H10Cl2N2O | 2.87 | 3.7 | 12.5 | 11.2 | 37.4 |
| Herbicide | Flazasulfuron | 3.78 | 408.1 > 182.1 | 7 | 104040-78-0 | C13H12F3N5O5S | −0.06 | 4.5 | 15.1 | 13.6 | 45.3 |
| Herbicide | Isoproturon | 3.67 | 207 > 72 | 124 | 34123-59-6 | C12H18N2O | 2.50 | 3.6 | 12.1 | 10.9 | 36.2 |
| Herbicide | Linuron | 5.21 | 249 > 160 | 122 | 330-55-2 | C9H10Cl2N2O2 | 3.00 | 3.5 | 11.8 | 10.6 | 35.3 |
| Herbicide | Metolachlor | 6.92 | 252 > 252 | 148 | 51218-45-2 | C15H22ClNO2 | 3.40 | 6.9 | 23.0 | 20.7 | 69.0 |
| Herbicide | Oxadixyl | 2.95 | 279.1 > 132.1 | 98 | 77732-09-3 | C14H18N2O4 | 0.65 | 6.4 | 21.2 | 19.1 | 63.7 |
| Herbicide | Propyzamide | 6.06 | 256 > 190 | 133 | 23950-58-5 | C12H11Cl2NO | 3.30 | 4.5 | 14.9 | 13.4 | 44.7 |
| Herbicide | Prosulfocarb | 9.52 | 252 > 91 | 124 | 52888-80-9 | C14H21NOS | 4.48 | 2.8 | 9.3 | 8.4 | 27.9 |
| Herbicide | Simazine | 3.33 | 202 > 124 | 100 | 122-34-9 | C7H12ClN5 | 2.30 | 2.4 | 7.9 | 7.1 | 23.6 |
| Herbicide | Simazine hydroxy | 1.91 | 184 > 114 | 37 | 2599-11-3 | C7H13N5O | 1.67 | 3.8 | 12.8 | 11.5 | 38.4 |
| Herbicide | Terbutylazine | 5.22 | 230.1 > 174 | 124 | 5915-41-3 | C9H16ClN5 | 3.40 | 5.9 | 19.8 | 17.8 | 59.4 |
| Herbicide | Terbutylazine hydroxy | 1.93 | 212.2 > 156 | 0 | 66753-07-9 | C9H17N5O | 1.29 | 2.5 | 8.4 | 7.5 | 25.1 |
| Fungicide | Azoxystrobin | 4.86 | 404 > 344 | 121 | 131860-33-8 | C22H17N3O5 | 2.50 | 3.9 | 13.0 | 11.7 | 39.0 |
| Fungicide | Carbendazim | 2.11 | 192 > 160 | 17 | 10605-21-7 | C9H9N3O2 | 1.48 | 2.8 | 9.2 | 8.3 | 27.5 |
| Fungicide | Dimethomorph | 4.05 | 388 > 301 | 165 | 110488-70-5 | C21H22ClNO4 | 2.63 | 4.3 | 14.4 | 13.0 | 43.3 |
| Fungicide | Epoxiconazole | 5.22 | 330.01 > 121.03 | 157 | 135319-73-2 | C17H13ClFN3O | 3.30 | 3.0 | 10.0 | 9.0 | 29.9 |
| Fungicide | Metalaxyl | 3.67 | 280 > 220 | 125 | 57837-19-1 | C15H21NO4 | 1.65 | 8.8 | 29.3 | 26.4 | 88.0 |
| Fungicide | Penconazole | 6.66 | 284.2 > 159.1 | 145 | 66246-88-6 | C13H15Cl2N3 | 3.72 | 4.2 | 14.1 | 12.7 | 42.4 |
| Fungicide | Prochloraz | 4.59 | 378.04 > 310.18 | 138 | 67747-09-5 | C15H16Cl3N3O2 | 3.50 | 5.0 | 16.7 | 15.0 | 50.0 |
| Fungicide | Pyrimethanil | 4.32 | 200 > 107 | 97 | 53112-28-0 | C12H13N3 | 2.84 | 2.9 | 9.5 | 8.6 | 28.6 |
| Fungicide | Tebuconazole | 5.74 | 308 > 70 | 152 | 107534-96-3 | C16H22ClN3O | 3.49 | 3.0 | 9.9 | 8.9 | 29.6 |
| Fungicide | Tetraconazole | 5.33 | 372 > 159 | 146 | 112281-77-3 | C13H11Cl2F4N3O | 3.60 | 5.0 | 16.7 | 15.0 | 50.0 |
| Insecticide | Imidacloprid | 2.62 | 256.1 > 209.1 | 95 | 138261-41-3 | C9H10ClN5O2 | 0.57 | 4.1 | 13.6 | 12.2 | 40.7 |
References
- Rivera-Utrilla, J.; Sánchez-Polo, M.; Ferro-García, M.Á.; Prados-Joya, G.; Ocampo-Pérez, R. Pharmaceuticals as emerging contaminants and their removal from water. A review. Chemosphere 2013, 93, 1268–1287. [Google Scholar] [CrossRef] [PubMed]
- Bavumiragira, J.P.; Ge, J.; Yin, H. Fate and transport of pharmaceuticals in water systems: A processes review. Sci. Total Environ. 2022, 823, 153635. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, J.L.; Boxall, A.B.A.; Kolpin, D.W.; Leung, K.M.Y.; Lai, R.W.S.; Wong, D.; Ntchantcho, R.; Pizarro, J.; Mart, J.; Echeverr, S.; et al. Pharmaceutical pollution of the world’s rivers. Proc. Natl. Acad. Sci. USA 2022, 119, e2113947119. [Google Scholar] [CrossRef] [PubMed]
- Papagiannaki, D.; Belay, M.H.; Gonçalves, N.P.F.; Robotti, E.; Bianco-Prevot, A.; Binetti, R.; Calza, P. From monitoring to treatment, how to improve water quality: The pharmaceuticals case. Chem. Eng. J. Adv. 2022, 10, 100245. [Google Scholar] [CrossRef]
- Saleh, I.A.; Zouari, N.; Al-Ghouti, M.A. Removal of pesticides from water and wastewater: Chemical, physical and biological treatment approaches. Environ. Technol. Innov. 2020, 19, 101026. [Google Scholar] [CrossRef]
- Campanale, C.; Massarelli, C.; Losacco, D.; Bisaccia, D.; Triozzi, M.; Uricchio, V.F. The monitoring of pesticides in water matrices and the analytical criticalities: A review. TrAC-Trends Anal. Chem. 2021, 144, 116423. [Google Scholar] [CrossRef]
- Sjerps, R.M.A.; Vughs, D.; van Leerdam, J.A.; Laak, T.L.t.; van Wezel, A.P. Data-driven prioritization of chemicals for various water types using suspect screening LC-HRMS. Water Res. 2016, 93, 254–264. [Google Scholar] [CrossRef]
- Blum, K.M.; Andersson, P.L.; Renman, G.; Ahrens, L.; Gros, M.; Wiberg, K.; Haglund, P. Non-target screening and prioritization of potentially persistent, bioaccumulating and toxic domestic wastewater contaminants and their removal in on-site and large-scale sewage treatment plants. Sci. Total Environ. 2017, 575, 265–275. [Google Scholar] [CrossRef] [Green Version]
- Maculewicz, J.; Kowalska, D.; Świacka, K.; Toński, M.; Stepnowski, P.; Białk-Bielińska, A.; Dołżonek, J. Transformation products of pharmaceuticals in the environment: Their fate, (eco)toxicity and bioaccumulation potential. Sci. Total Environ. 2022, 802, 149916. [Google Scholar] [CrossRef]
- Bletsou, A.A.; Jeon, J.; Hollender, J.; Archontaki, E.; Thomaidis, N.S. Targeted and non-targeted liquid chromatography-mass spectrometric workflows for identification of transformation products of emerging pollutants in the aquatic environment. Trends Anal. Chem. 2015, 66, 32–44. [Google Scholar] [CrossRef] [Green Version]
- Puhlmann, N.; Mols, R.; Olsson, O.; Slootweg, J.C.; Kümmerer, K. Towards the design of active pharmaceutical ingredients mineralizing readily in the environment. Green Chem. 2021, 23, 5006–5023. [Google Scholar] [CrossRef]
- Hollender, J.; van Bavel, B.; Dulio, V.; Farmen, E.; Furtmann, K.; Koschorreck, J.; Kunkel, U.; Krauss, M.; Munthe, J.; Schlabach, M.; et al. High resolution mass spectrometry-based non-target screening can support regulatory environmental monitoring and chemicals management. Environ. Sci. Eur. 2019, 31, 42. [Google Scholar] [CrossRef] [Green Version]
- Brack, W.; Hollender, J.; de Alda, M.L.; Müller, C.; Schulze, T.; Schymanski, E.; Slobodnik, J.; Krauss, M. High-resolution mass spectrometry to complement monitoring and track emerging chemicals and pollution trends in European water resources. Environ. Sci. Eur. 2019, 31, 62. [Google Scholar] [CrossRef] [Green Version]
- Gago-Ferrero, P.; Bletsou, A.A.; Damalas, D.E.; Aalizadeh, R.; Alygizakis, N.A.; Singer, H.P.; Hollender, J.; Thomaidis, N.S. Wide-scope target screening of >2000 emerging contaminants in wastewater samples with UPLC-Q-ToF-HRMS/MS and smart evaluation of its performance through the validation of 195 selected representative analytes. J. Hazard. Mater. 2020, 387, 121712. [Google Scholar] [CrossRef] [PubMed]
- Menger, F.; Gago-Ferrero, P.; Wiberg, K.; Ahrens, L. Wide-scope screening of polar contaminants of concern in water: A critical review of liquid chromatography-high resolution mass spectrometry-based strategies. Trends Environ. Anal. Chem. 2020, 28, e00102. [Google Scholar] [CrossRef]
- Schymanski, E.L.; Jeon, J.; Gulde, R.; Fenner, K.; Ruff, M.; Singer, H.P.; Hollender, J. Identifying small molecules via high resolution mass spectrometry: Communicating confidence. Environ. Sci. Technol. 2014, 48, 2097–2098. [Google Scholar] [CrossRef]
- Mathon, B.; Ferreol, M.; Togola, A.; Lardy-Fontan, S.; Dabrin, A.; Allan, I.J.; Staub, P.F.; Mazzella, N.; Miège, C. Polar organic chemical integrative samplers as an effective tool for chemical monitoring of surface waters—Results from one-year monitoring in France. Sci. Total Environ. 2022, 824, 153549. [Google Scholar] [CrossRef]
- González-Gaya, B.; Lopez-herguedas, N.; Santamaria, A.; Mijangos, F.; Etxebarria, N.; Olivares, M.; Prieto, A.; Zuloaga, O. Suspect screening work fl ow comparison for the analysis of organic xenobiotics in environmental water samples. Chemosphere 2021, 274, 129964. [Google Scholar] [CrossRef]
- Bade, R.; Bijlsma, L.; Miller, T.H.; Barron, L.P.; Sancho, J.V.; Hernández, F. Suspect screening of large numbers of emerging contaminants in environmental waters using artificial neural networks for chromatographic retention time prediction and high resolution mass spectrometry data analysis. Sci. Total Environ. 2015, 538, 934–941. [Google Scholar] [CrossRef] [Green Version]
- Aalizadeh, R.; Nika, M.C.; Thomaidis, N.S. Development and application of retention time prediction models in the suspect and non-target screening of emerging contaminants. J. Hazard. Mater. 2019, 363, 277–285. [Google Scholar] [CrossRef]
- Lynn, K.S.; Cheng, M.L.; Chen, Y.R.; Hsu, C.; Chen, A.; Lih, T.M.; Chang, H.Y.; Huang, C.J.; Shiao, M.S.; Pan, W.H.; et al. Metabolite identification for mass spectrometry-based metabolomics using multiple types of correlated ion information. Anal. Chem. 2015, 87, 2143–2151. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.F.; Lu, W.; Rabinowitz, J.D. Avoiding misannotation of in-source fragmentation products as cellular metabolites in liquid chromatography-mass spectrometry-based metabolomics. Anal. Chem. 2015, 87, 2273–2281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Z.; Zhao, L.; Liu, X.; Xu, Y. The application of in-source fragmentation in ultra-high performance liquid chromatography-electrospray ionization -tandem mass spectrometry for pesticide residue analysis. J. Chromatogr. A 2020, 1633, 461637. [Google Scholar] [CrossRef] [PubMed]
- González-Gaya, B.; Lopez-Herguedas, N.; Bilbao, D.; Mijangos, L.; Iker, A.M.; Etxebarria, N.; Irazola, M.; Prieto, A.; Olivares, M.; Zuloaga, O. Suspect and non-target screening: The last frontier in environmental analysis. Anal. Methods 2021, 13, 1876–1904. [Google Scholar] [CrossRef]
- Couto, C.F.; Lange, L.C.; Amaral, M.C.S. Occurrence, fate and removal of pharmaceutically active compounds (PhACs) in water and wastewater treatment plants—A review. J. Water Process Eng. 2019, 32, 100927. [Google Scholar] [CrossRef]
- Pomiès, M.; Choubert, J.M.; Wisniewski, C.; Coquery, M. Modelling of micropollutant removal in biological wastewater treatments: A review. Sci. Total Environ. 2013, 443, 733–748. [Google Scholar] [CrossRef]
- Schollée, J.E.; Schymanski, E.L.; Stravs, M.A.; Gulde, R.; Thomaidis, N.S.; Hollender, J. Similarity of High-Resolution Tandem Mass Spectrometry Spectra of Structurally Related Micropollutants and Transformation Products. J. Am. Soc. Mass Spectrom. 2017, 28, 2692–2704. [Google Scholar] [CrossRef]
- Klaassen, T.; Kasel, D.; Harlfinger, S.; Fuhr, U. Quantification of mephenytoin and its metabolites 4′- hydroxymephenytoin and nirvanol in human urine using a simple sample processing method. Rapid Commun. Mass Spectrom. 2004, 18, 1675–1680. [Google Scholar] [CrossRef]
- Jensen, H.S.; Nichol, K.; Lee, D.; Ebert, B. Clobazam and its active metabolite N-desmethylclobazam display significantly greater affinities for α2- versus α1-GABAa-receptor complexes. PLoS ONE 2014, 9, e88456. [Google Scholar] [CrossRef]
- Gent, L.; Paul, R. The detection of new psychoactive substances in wastewater. A comprehensive review of analytical approaches and global trends. Sci. Total Environ. 2021, 776, 146028. [Google Scholar] [CrossRef]
- Liigand, J.; Wang, T.; Kellogg, J.; Smedsgaard, J.; Cech, N.; Kruve, A. Quantification for non-targeted LC/MS screening without standard substances. Sci. Rep. 2020, 10, 5808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richardson, S.D.; Kimura, S.Y. Water Analysis: Emerging Contaminants and Current Issues. Anal. Chem. 2018, 90, 398–428. [Google Scholar] [CrossRef] [PubMed]
- Loos, R.; Carvalho, R.; António, D.C.; Comero, S.; Locoro, G.; Tavazzi, S.; Paracchini, B.; Ghiani, M.; Lettieri, T.; Blaha, L.; et al. EU-wide monitoring survey on emerging polar organic contaminants in wastewater treatment plant effluents. Water Res. 2013, 47, 6475–6487. [Google Scholar] [CrossRef] [PubMed]
- Kruve, A.; Kiefer, K.; Hollender, J. Benchmarking of the quantification approaches for the non-targeted screening of micropollutants and their transformation products in groundwater. Anal. Bioanal. Chem. 2021, 413, 1549–1559. [Google Scholar] [CrossRef] [PubMed]
- Bengtsson-Palme, J.; Larsson, D.G.J. Concentrations of antibiotics predicted to select for resistant bacteria: Proposed limits for environmental regulation. Environ. Int. 2016, 86, 140–149. [Google Scholar] [CrossRef] [Green Version]
- Branchet, P.; Cadot, E.; Fenet, H.; Sebag, D.; Ngatcha, B.N.; Borrell-Estupina, V.; Ngoupayou, J.R.N.; Kengne, I.; Braun, J.J.; Gonzalez, C. Polar pesticide contamination of an urban and peri-urban tropical watershed affected by agricultural activities (Yaoundé, Center Region, Cameroon). Environ. Sci. Pollut. Res. 2018, 25, 17690–17715. [Google Scholar] [CrossRef]
- Naylor, B.C.; Catrow, J.L.; Maschek, J.A.; Cox, J.E. QSRR automator: A tool for automating retention time prediction in lipidomics and metabolomics. Metabolites 2020, 10, 237. [Google Scholar] [CrossRef]
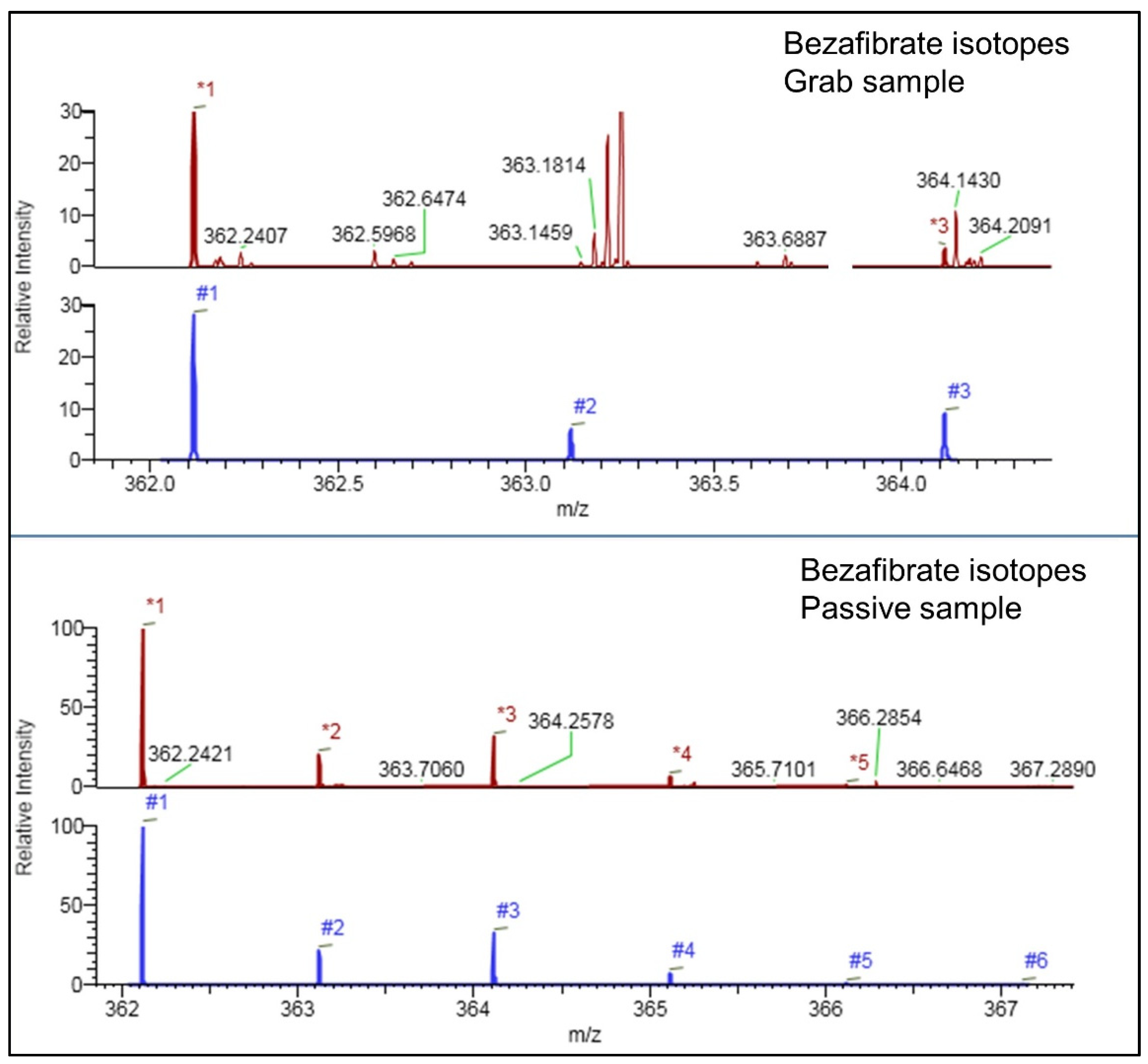
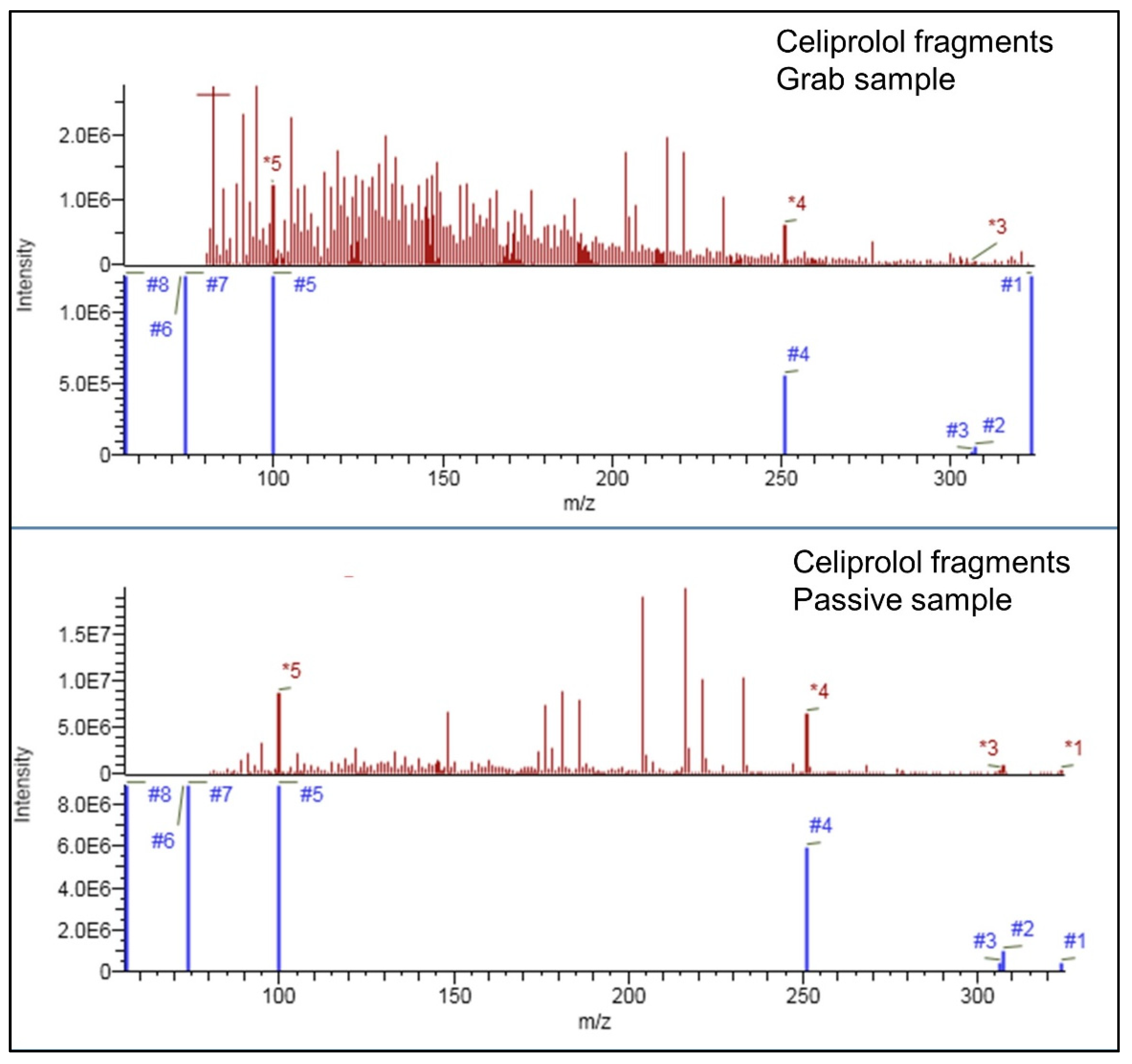
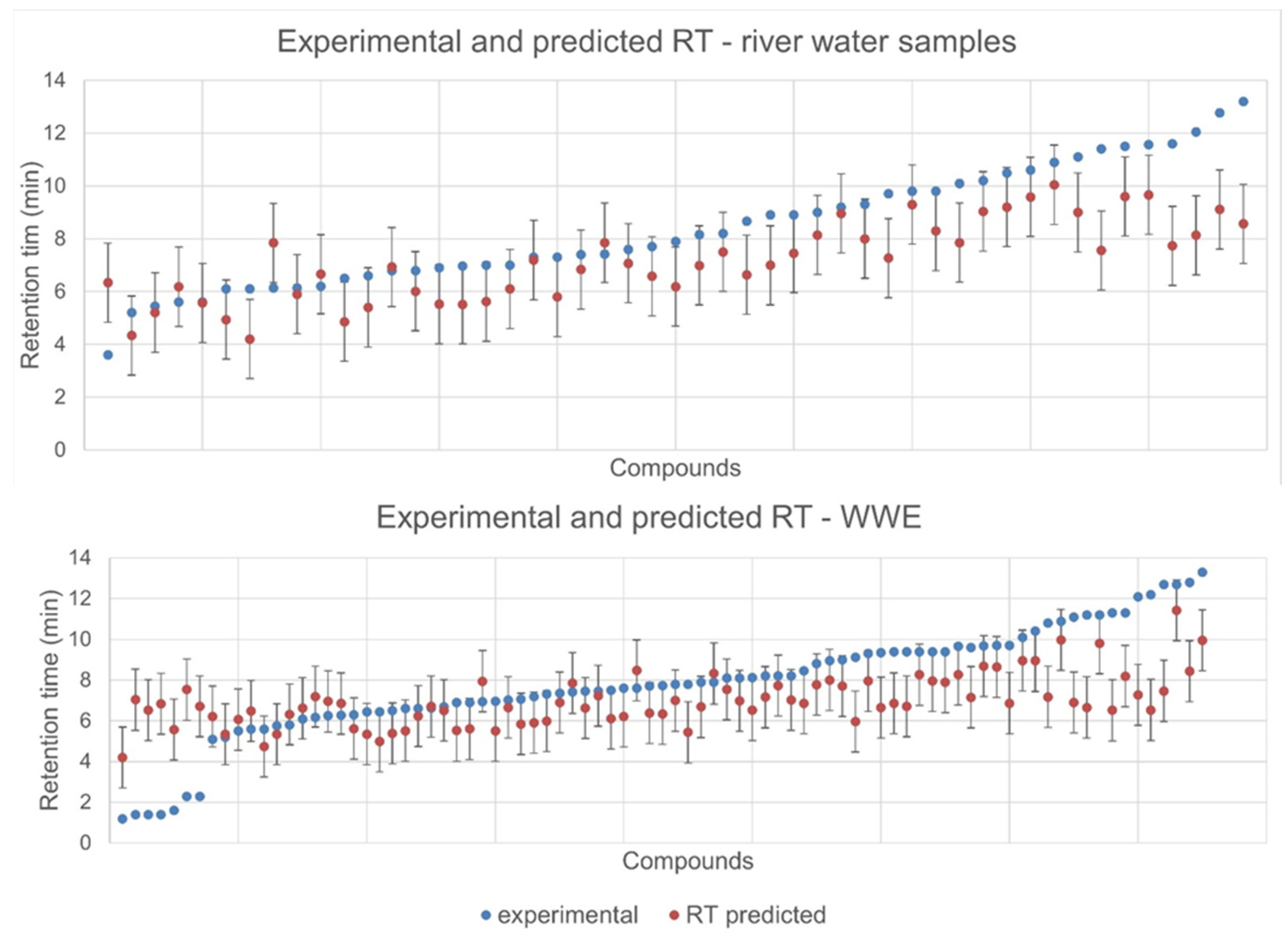
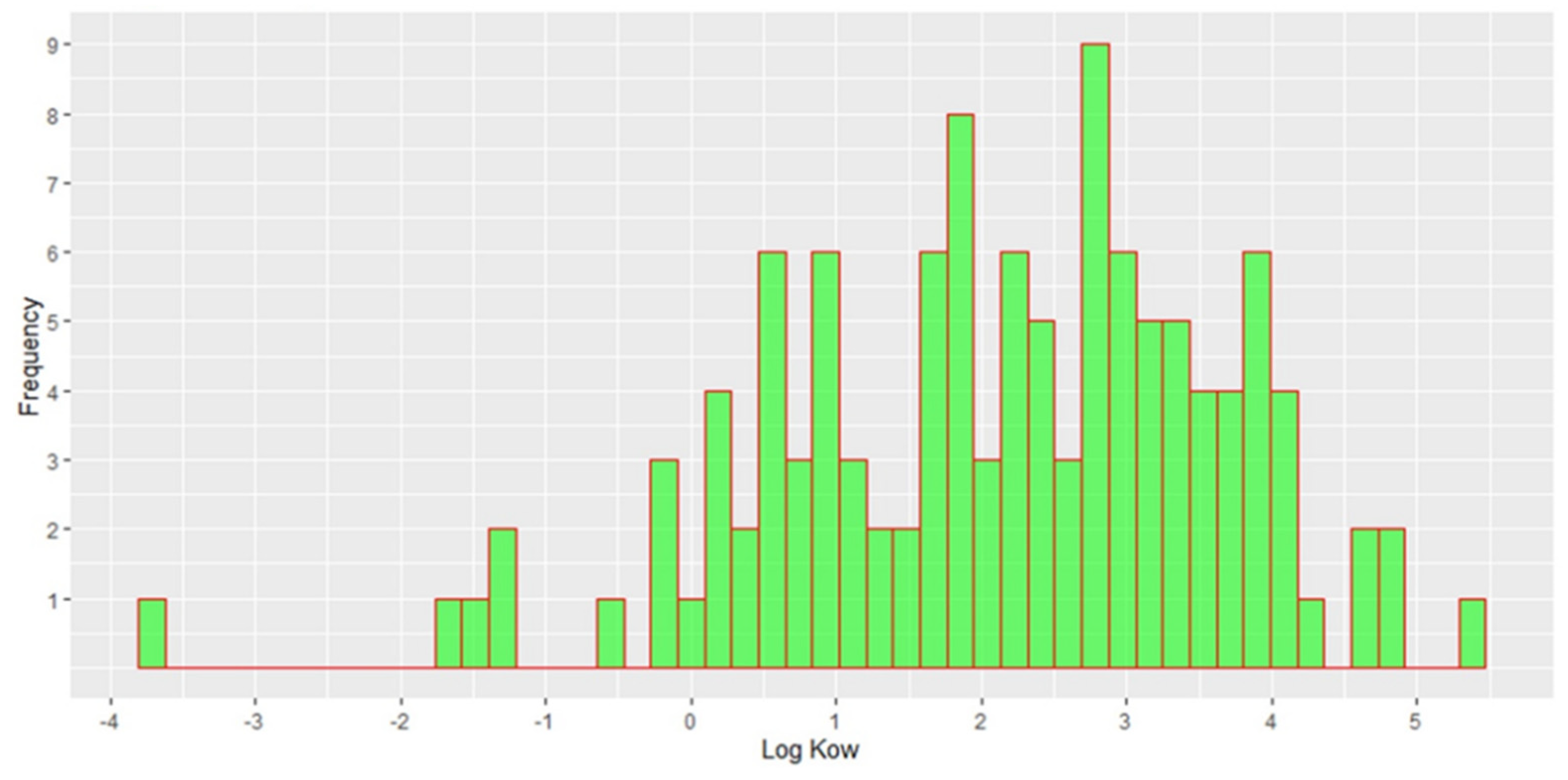
| Compound | Concentration (ng/L) | Compound | Concentration (ng/L) | ||||
|---|---|---|---|---|---|---|---|
| Grab Day 0 | Grab Day 7 | Grab Day 14 | Grab Day 0 | Grab Day 7 | Grab Day 14 | ||
| 1,3-Dicyclohexylurea | 5 (±2.9) | 2.6 (±0.9) | 11.3 (±3.5) | Gemfibrozil | 499 (±260) | 57.9 (±14) | 47.9 (±14.4) |
| 10-Hydroxycarbazepine | 90.9 (±6.7) | 26.6 (±3.8) | 41.1 (±1) | Ibuprofen | 6.8 × 104 (±6.3 × 104) | 2.7 × 103 (±954) | 1.7 × 103 (±894) |
| 2,2,6,6-Tetramethyl-4-piperidinol | 83.8 (±50.6) | 39.6 (±9.3) | 152 (±37.9) | Imidacloprid * | 16.5 (±1.2) | 7.4 (±0.7) | 11.1 (±2.5) |
| 2-Amino-1H-benzimidazole | 49.7 (±27.6) | 31.5 (±12.8) | 154.5 (±64.1) | Irbesartan | 7.2 (±9.1) | 11.9 (±5.8) | 28.3 (±28.4) |
| 2-Hydroxyestrone | 95.9 (±24) | 46.3 (±4.9) | 79.6 (±0.4) | Ketoprofen | 3.6 (±0.09) | 3.3 (±2) | |
| 3,4-Dimethylmethcathinone | 21.2 (±7.8) | 12 (±2.4) | 80.4 (±17.2) | Lacosamide | 2.4 (±0.1) | 1.1 (±0) | 1.2 (±0.2) |
| 3-Acetylindole | 1.5 (±0.1) | 0.7 (±0.1) | 0.7 (±0.2) | Lamotrigine | 1464 (±39) | 626 (±146) | 1263 (±270) |
| Alminoprofen | 2 (±0.7) | 1.6 (±0.4) | 6 (±3.2) | Laurolactam | 27.3 (±2.8) | 12.4 (±3.3) | 49.1 (±5.6) |
| Alprenolol | 2.8 (±2.7) | 2.8 (±1.8) | 25.1 (±16.7) | Lidocaine | 13.9 (±6.1) | 18.5 (±6.2) | 55.9 (±33.1) |
| Aminocarb | 47 (±0.1) | 23.1 (±5.6) | 42.4 (±5.9) | lorazepam | 7.6 (±3.3) | 1.4 (±0.3) | 19.8 (±1.6) |
| Amisulpride N-oxide | Lormetazepam | 8.3 (±2.8) | 2.1 (±0.5) | 10.5 (±1.7) | |||
| Anabasine | 5 (±0.6) | 2.1 (±0.9) | 33.2 (±7) | Metformin | 26.1 (±7.2) | 8.6 (±3.1) | 1.9 (±1) |
| Ancymidol | 1.3 (±0.3) | 1.5 (±0.5) | 3.7 (±0.3) | Mexacarbate | 2 (±0.3) | 1.2 (±0.3) | 14.8 (±0.6) |
| Antipyrine (phenazone) | 2.1 (±1.3) | 0.8 (±0.5) | 11.1 (±1.3) | N-desmethyl mephenytoin | 6 (±4) | 2.8 (±0.3) | |
| Azoxystrobin * | 1.6 (±0.09) | 0.9 (±0.4) | 1.2 (±0.2) | N-desmethyl clobazam | 168 (±76.7) | 29.6 (±6.2) | 552 (±49.8) |
| Benzotriazole | 224 (±3.5) | 166 (±33.2) | 273 (±19) | N-desmethyl venlafaxine | 17.2 (±10.5) | 15 (±7.5) | 60.4 (±31.5) |
| Bezafibrate | 11.1 (±9.7) | 11.8 (±4.5) | 3.2 (±2.6) | Nordazepam | 10.7 (±3.7) | 6.7 (±2.5) | 23.9 (±6.4) |
| Buphedrone | 2.9 (±1.6) | 4 (±1.9) | 1.7 (±0.8) | O-Desmethyl venlafaxine | 1.3 (±1.1) | 0.3 (±0) | 13.5 (±9.5) |
| Bupivacaine | 0.4 (±0.2) | 3 (±1.4) | Picaridin | 5 (±2.1) | 2.2 (±0.6) | 6.8 (±1.5) | |
| Butoxytriglycol | 3.1 × 105 (±1.0 × 105) | 7.2 × 104 (±1.7 × 104) | 3.6 × 104 (±1.8 × 104) | Prazepam | 7.3 (±3.6) | 5.5 (±2.3) | 14 (±5) |
| Caffeine | 616 (±394) | 38.9 (±6.1) | 295 (±39.5) | Propiconazole | 1.4 (±0.5) | 1 (±0.4) | 3 (±1.1) |
| Carbamazepine | 104 (±9.1) | 96.5 (±12) | 119 (±0.7) | Pseudotropine | 11.9 (±5.2) | 10.6 (±4.8) | 45.9 (±13.1) |
| Carbamazepine epoxide | 11.6 (±1.1) | 5 (±1.2) | 10.3 (±0.3) | Pyridostigmine | 127 (±5.5) | 62.5 (±17.4) | 55.5 (±2.9) |
| Celiprolol | 24.4 (±28.5) | 21.6 (±14.2) | 292 (±273) | Pyroquilon | 46.9 (±4.8) | 14.6 (±3.5) | 31.9 (±0.9) |
| Cetirizine | 0.5 (±0.6) | 0.4 (±0.4) | 20.6 (±15.8) | Rosuvastatin | 5.2 (±6.4) | 1.3 (±0.5) | 0.7 (±0.02) |
| Climbazole | 20.1 (±3) | 7.7 (±2.5) | 12.9 (±2.3) | Serotonin | 11.2 (±1.3) | 4.9 (±1.5) | 15.6 (±1) |
| Crotamiton | 9.5 (±5) | 1.1 (±0.3) | 20.1 (±0.9) | Sotalol | 5.8 (±7.1) | 1.1 (±0.6) | 13.3 (±14.5) |
| Cytisine | 5.1 (±1.4) | 3.7 (±2) | 8.7 (±0.9) | Sulfamethoxazole | 63 (±7) | 29.5 (±6.3) | 61.5 (±0.7) |
| Dextromethorphan | 0.4 (±0.2) | Sulpiride | 0.1 (±0.03) | 0.5 (±0.3) | |||
| Diazinon | 0.5 (±0.3) | 0.3 (±0.1) | 1.4 (±0.6) | Terbuthylazin* | 0.2 (±0.09) | 0.2 (±0.2) | 3.9 (±0.4) |
| Dibutyl phthalate | 3870 (±1142) | 998 (±76.8) | 5224 (±872) | Tramadol | 7.7 (±5.5) | 8 (±4.2) | 41.3 (±24.3) |
| Diclofenac | 1 (±0.04) | Tributyl phosphate | 1164 (±151) | 476 (±11.9) | 4739 (±218) | ||
| Diuron * | 3.6 (±0.7) | 2.4 (±1) | 1.9 (±0.9) | Triclosan | 59.6 (±0.5) | ||
| DMACA Reagent | 0.4 (±0.05) | 0.2 (±0) | 0.7 (±0.08) | Triethyl phosphate | 771 (±287) | 190 (±17.1) | 229 (±11.1) |
| Ecgonine | 6.6 (±0.6) | 4.8 (±1.4) | 0.7 (±0.6) | Triisopropanolamine | 3.8 (±2.7) | 1.1 (±0.5) | 11.4 (±8) |
| Estriol | 186 (±13.1) | 62 (±14.2) | 105 (±6) | Trimethoprim | 0.7 (±0.02) | 2.5 (±1.3) | |
| Fexofenadine | 0.3 (±0.5) | 0.5 (±0.4) | 7.9 (±9.9) | Tris (2-butoxyethyl) phosphate | 96.7 (±52.7) | 60.8 (±28.3) | 263 (±87.4) |
| Flecainide | 614 (±520) | 491 (±158) | 1813 (±327) | Tyramine | 99.6 (±21.3) | 39.4 (±8.6) | 68.1 (±2.7) |
| Fluconazole | 33.2 (±9.9) | 17.5 (±5.6) | 37.4 (±6.7) | Valpromide | 518 (±36.9) | 122 (±8.7) | 30.4 (±4) |
| Gabapentin | 195 (±29.4) | 68.1 (±11.5) | 117 (±15.4) | Venlafaxine | 5.3 (±4) | 6.4 (±3.5) | 41.6 (±19.3) |
| Gabapentin lactam | 13.4 (±1.1) | 9.8 (±3.9) | 43.2 (±5.3) | ||||
| Compounds | Concentration (ng/L) | Compounds | Concentration (ng/L) | ||
|---|---|---|---|---|---|
| Grab Day 0 | Grab Day 14 | Grab Day 0 | Grab Day 14 | ||
| Benzophenone | 4 (±1.4) | 15.8 (±0.6) | Trimethoprim | 1.4 (±0.5) | |
| Antipyrine/phenazone | 2.4 (±0.2) | 0.2 (±0.04) | Clobazam | 0.1 (±0.03) | |
| Desethyl sebuthylazine | 11.1 (±0.8) | 3.2 (±0.1) | Ethylmorphine | 18.2 (±2.2) | 29.6 (±1.9) |
| Atraton | 5.3 (±0.5) | 2 (±0.03) | Benalaxyl | 0.8 (±0.1) | |
| Metribuzin | 51.6 (±0.8) | Tiapride | 0.2 (±0.2) | 0.1 (±0.02) | |
| Prometon | 8.8 (±0.8) | 0.2 (±0.002) | Retrorsine | 2.3 (±0.07) | |
| Picaridin | 1.1 (±1.5) | 7.4 (±2.3) | Amisulpride | 2.4 (±0.1) | |
| Norfentanyl | 65.9 (±66.7) | 65.9 (±26.5) | Fluopyram | 2.1 (±0.3) | |
| Lidocaine | 0.09 (±0.01) | Azoxystrobin * | 11.7 (±0.4) | ||
| Prometryn | 1.1 (±0.01) | 0.07 (±0.04) | Flecainide | 0.3 (±0.03) | |
| Meperidine | 0.05 (±0.009) | 0.5 (±0.02) | Sulpiride | 9.8 (±1.7) | 17.8 (±1.5) |
| Lamotrigine | 4 (±0.7) | 167.2 (±1.6) | Tramadol | 4 (±0.1) | 3.6 (±0.01) |
| Napropamide | 8.2 (±0.8) | Simazin* | 5.7 (±0.07) | ||
| Metolachlor * | 4.2 (±0.3) | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tadić, Đ.; Manasfi, R.; Bertrand, M.; Sauvêtre, A.; Chiron, S. Use of Passive and Grab Sampling and High-Resolution Mass Spectrometry for Non-Targeted Analysis of Emerging Contaminants and Their Semi-Quantification in Water. Molecules 2022, 27, 3167. https://doi.org/10.3390/molecules27103167
Tadić Đ, Manasfi R, Bertrand M, Sauvêtre A, Chiron S. Use of Passive and Grab Sampling and High-Resolution Mass Spectrometry for Non-Targeted Analysis of Emerging Contaminants and Their Semi-Quantification in Water. Molecules. 2022; 27(10):3167. https://doi.org/10.3390/molecules27103167
Chicago/Turabian StyleTadić, Đorđe, Rayana Manasfi, Marine Bertrand, Andrés Sauvêtre, and Serge Chiron. 2022. "Use of Passive and Grab Sampling and High-Resolution Mass Spectrometry for Non-Targeted Analysis of Emerging Contaminants and Their Semi-Quantification in Water" Molecules 27, no. 10: 3167. https://doi.org/10.3390/molecules27103167
APA StyleTadić, Đ., Manasfi, R., Bertrand, M., Sauvêtre, A., & Chiron, S. (2022). Use of Passive and Grab Sampling and High-Resolution Mass Spectrometry for Non-Targeted Analysis of Emerging Contaminants and Their Semi-Quantification in Water. Molecules, 27(10), 3167. https://doi.org/10.3390/molecules27103167