2.1. Synthesis, Structure and Stereodynamics of Ligands 1 and 2
Synthesis of the “parent” pyrrolidine-derived ligand was first published in 2004 [
28]. Later, some properties of this compound were revealed [
29].
Starting from (±)-2-(trifluoromethyl)pyrrolidine [
30], we obtained new phenanthroline ligands
1 and
2. Then we started their structural study. Both compounds were characterized by combination of spectral methods and high-resolution mass spectrometry. They are white powders, readily soluble in dichloromethane, chloroform, acetone and moderately soluble in acetonitrile and hexane. In spite of simple structure of prepared diamides
1 and
2, their NMR spectra were surprisingly complicated in CDCl
3 solution at room temperature (
Figures S1 and S2 in Supplementary Materials) even taking into account that mixtures of diastereomers are under study. The
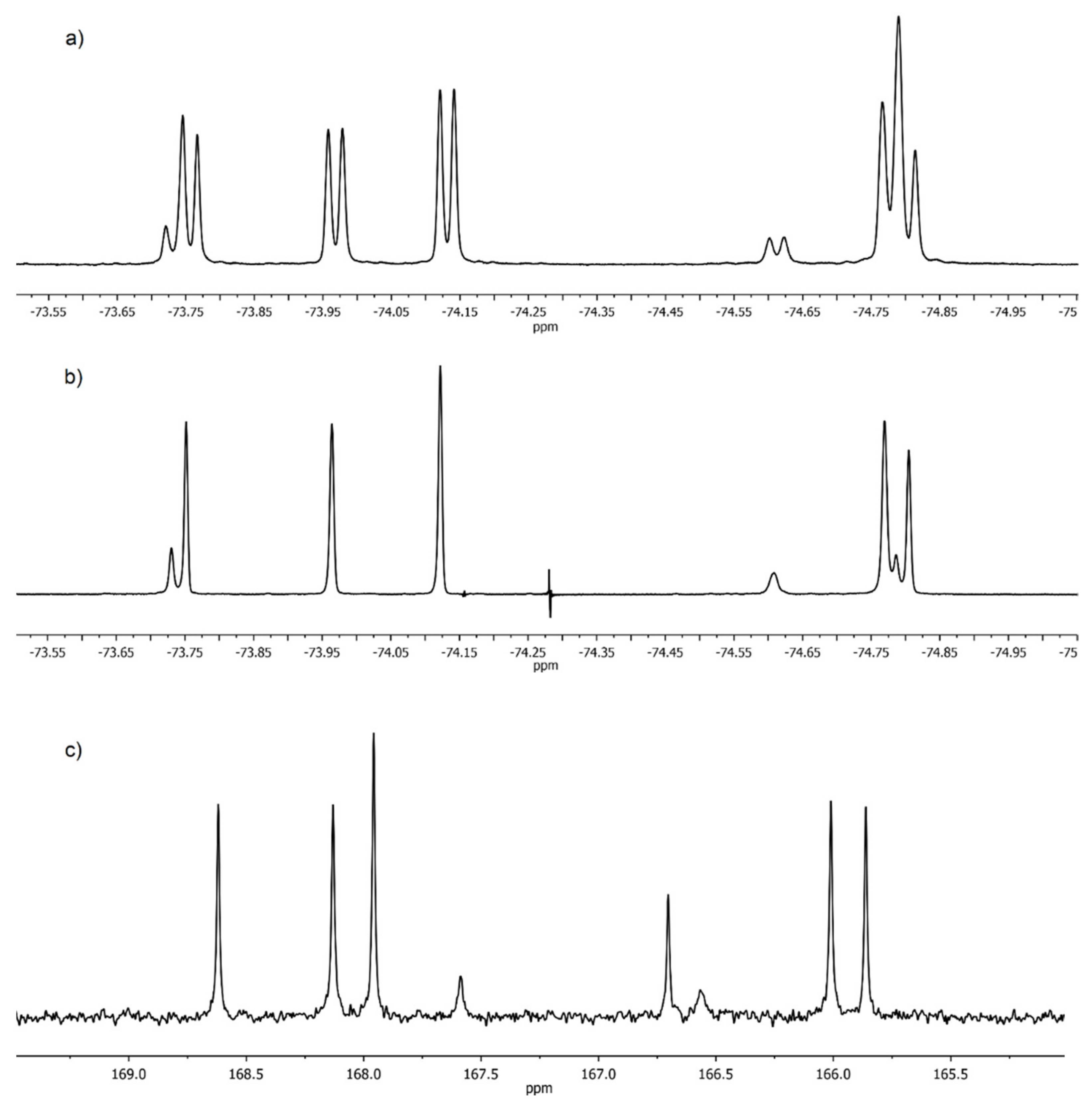
19F-NMR spectrum of
1 (
Figure 2a) contains a set of 8 signals of CF
3 groups the doublet splitting of which is due to the
3J19F,1H spin-spin coupling. Simpler picture is observed in the spectrum of the heteronuclear double resonance
19F-{
1H} (
Figure 2b). Eight singlets of CF
3 groups of different intensities are clearly seen in it. Eight signals of CO-groups are also present in the carbonyl region of the
13C-NMR spectrum of
1 (
Figure 2c).
Such complex
19F and
13C-NMR spectra indicate that the internal rotation along Phen-CO bonds in diamides
1 and
2 in solutions at 23 °C occurs as a fast process in the NMR time scale, while the rotation along the amide bonds N–C=O is completely inhibited. Thus, diamides
1 and
2 in this respect behave similarly to other ligands of this type that we studied earlier [
23]. As a result, three rotamers along the amide bonds (
A–
C) (
Figure 3), differing in the orientation of -CHCF
3 fragments relative to the phenanthroline backbone, which coexist in solutions, give separate signals in the NMR spectra. Note that processes of this type in arylcarboxamides are well documented [
31,
32,
33,
34]. In contrast to symmetric structures
A and
C, the rotamer
B contains two non-equivalent CF
3-groups in the structure. This consideration allows us to explain very well the observed complicated spectra.
To have deeper insight in the equilibrium between rotamers of ligand
1, we decided to separate diastereomers in pure form. The quantitative separation of diastereomers of ligand
1 was carried out by HPLC in the acetonitrile/water system. The chromatogram contains two closely spaced peaks with an intensity ratio of 2:3 (see
Figure S50 in Supplementary Materials). At slow isothermal concentration of the acetonitrile solution of the second fraction, crystals of the racemate of
1 suitable for X-ray were obtained (
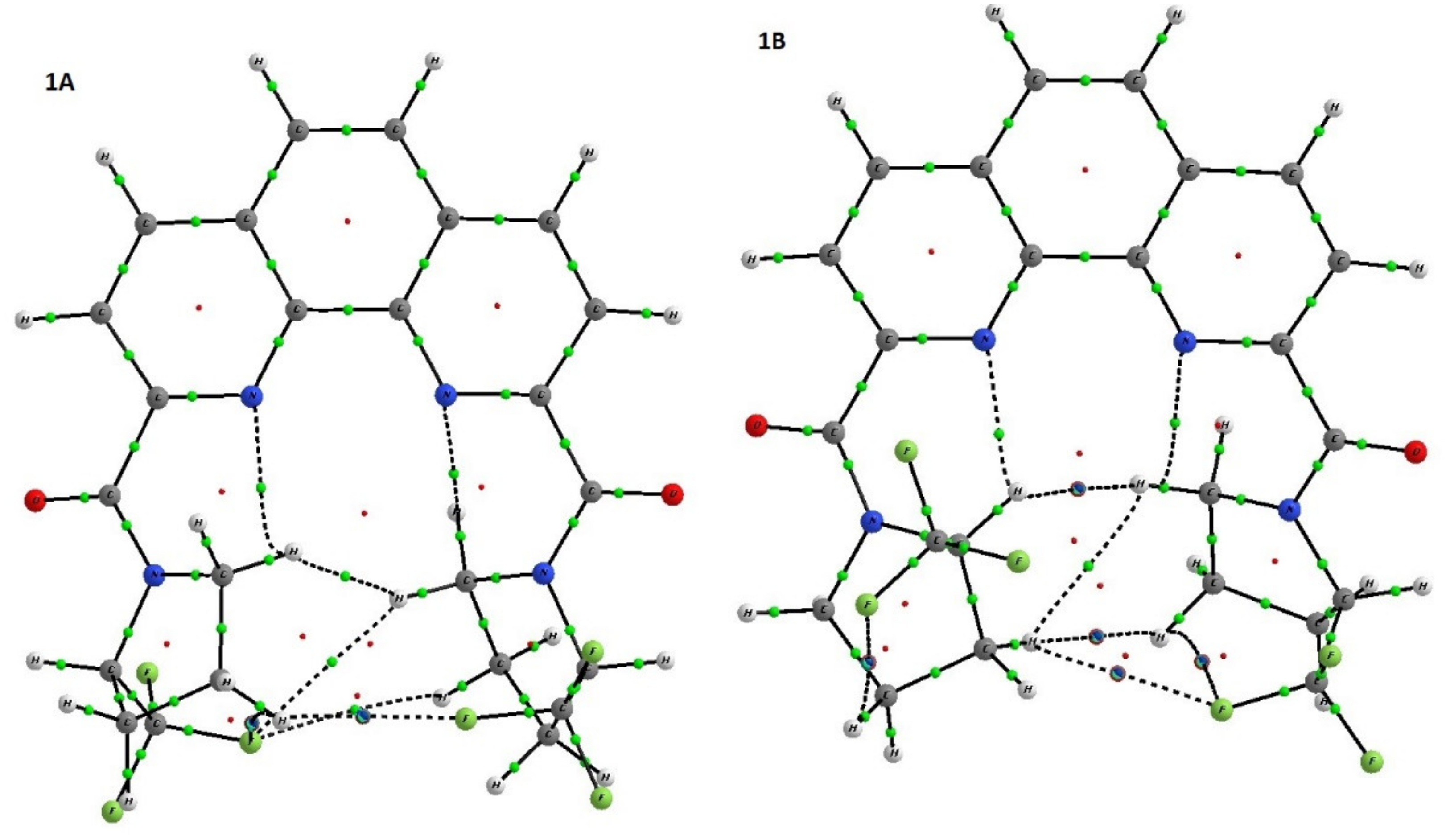
Figure 4). Depending upon the crystal growth procedure, we were able to investigate two racemic crystal forms of ligand
1, which corresponds to isomer
1A and the hydrate of isomer
1B with one water molecule.
The analysis of these two forms clearly shows that they are differ by orientation of pyrrolidine ring in respect to phenanthroline. In
1A and
1B·H
2O the C=O groups are in trans orientation in respect to nitrogen atoms of phenanthroline while the CF
3–C–N–C=O torsion angles in
1A and
1B·H
2O differ. In
1 CF
3–C groups in both pyrrolidine substituents are in syn-periplanar orientation in respect to C=O groups while in
1B·H
2O one pyrrolidine group is syn-periplanar while the other is in the antiperiplanar orientation. The variation of mutual orientation of the trifluoromethylated pyrrolidine almost does not affect the bond lengths distribution in amide fragment (see
Table 1) but leads to different weak interactions between the trifluoromethylated pyrrolidine and nitrogen of phenanthroline ring. As one can see in the case of
1A N(1) and N(10) atoms participate in formation of the weak contacts with CH
2 group of pyrrolidine ring while in
1B·H
2O one of C–H…N contacts is formed by more acidic C-HCF
3 group. Basing on the geometric parameters one can propose that latter contact with H…N distance equal 2.16 Å should be stronger than those (2.36 Å) in
1A and thus such conformation should be more stable.
At the same time, we cannot exclude that stabilization of the conformation in
1B·H
2O is the consequence of crystal packing effects (
Figure 5). Analysis of crystal packing have revealed that both molecules participate in the formation of infinite stacks with comparable interplane separation (3.38 vs. 3.40 Å) but slightly different area of overlap.
Furthermore, the infinite stacks in hydrate are additionally interlinked by O–H…O hydrogen bonds (O…O 2.840(4)-2.859(4) with water molecule which are absent in 1A.
In order to estimate the relative stability of the conformations of the molecule in
1A and
1B·H
2O we performed DFT (PBE/def-2-TZVP) calculations. The geometry of
1A was optimized using the very tight optimization criteria and empirical dispersion corrections on the total energy [
35] with the Becke-Johnson damping (D3) [
36]. The optimization of
1A lead to the geometry that is almost identical to those in
1B·H
2O (see
Table 1). Thus, we can assume that this geometry corresponds to global minimum. In order to estimate the possible way of conformation transformation from
1B·H
2O to the conformation in
1A we check two possible opportunities and performed the relaxed potential energy scan along the Phen–C=O and O=C–N–C–CF
3 torsion angles (step equal to 10°). The barrier to rotation for the first coordinate is equal to 14 kcal/mol while for the other it can be as much as 24 kcal/mol. Upon the relaxed scan we have found the additional minimum which geometry is almost identical to those observed in
1A (see
Table 1). The difference in energy of these two conformers is only 1.04 kcal/mol.
Aiming at estimating the energy of C–H…N contacts in two conformers we have used the topological analysis of the electron density distribution function ρ(
r) within Bader’s quantum theory of “Atoms in Molecule’’ (QTAIM) theory [
37]. Using the AIM formalism, one can distinguish the binding interatomic interactions from all other contacts. When the distribution of ρ(
r) in molecule or crystal is known, one can answer the question whether the bonding interaction is present or not by the search of the bond critical point (3,–1) and predict the energy of weak intermolecular interactions (Econt) with high accuracy on the basis of the potential energy density function v(
r) – the correlation suggested by Espinosa et al. (CEML) [
38]. Recently, the physical interpretation of CEML was suggested [
39].
According to the critical point (CP) search of ρ(
r), CP (3,–1) in conformers in
1A and
1B·H
2O are located not only for all expected bonds but also for weak C–H…N interactions and for series of F…H and H…H interactions between the trifluoromethylated pyrrolidine moieties (
Figure 6). Expectedly, all C–C, C–N, C–H, C–O and C–F bonds are characterized by the negative values of ∇
2ρ(
r) and the negative electron energy density (h
e(
r)) in CP(3,–1) and, therefore, correspond to shared type of interatomic interactions. In contrast all weak H…H, N…H and H…F interactions are characterized by both positive ∇
2ρ(
r) and h
e(
r) in CP (3,–1) and thus correspond to the closed-shell interactions. The energy of H…H and H…F interaction in both conformers is comparable and vary in the range of 0.4–0.9 kcal/mol. In their turn, the energy of C–H…N contacts is higher and equal to ca. 3.2 and 4.6 kcal/mol for CH
2 and CF
3CH group. As one can see, the difference in energy of this interactions is very close to the estimated difference of the conformers’ energy.
Thus, the stabilization of conformation is mainly governed the intramolecular H-bond and, although the difference in energy of two conformers is negligible, the conformation obtained in
1B·H
2O was also observed in the case of meso-
2B, which crystallized without any solvate molecules (
Figure 7).
The interesting feature of
2B is that, due to presence of chlorine substituent, the formation of stacking interaction (with interplane distance ca. 3.3 Å) is accompanied by the Cl…π interaction (
Figure 8).
As expected, the
19F NMR spectrum of racemic
1 (
Figure 9) is simpler, and consists of 4 doublets in the region from –73.9 ppm up to –74.8 ppm. The spectrum of rotamer
B should contain two signals of nonequivalent CF
3 groups of equal intensity, while rotamers
A and
C should have in their
19F NMR spectra by one doublet each since both CF
3 groups of these rotamers are equivalent. These considerations made it possible to distinguish signals from rotamer
B but do not allow the two remaining doublets to be assigned (
Figure 9).
The assignment of signals of two different CF
3-groups of rotamer
B (
B1 and
B2) in the spectrum of racemate was unambiguously confirmed by EXSY
19F NMR (
Figure 10).
The slow exchange of the positions of CF
3-groups is observed due to hindered internal rotation along the amide bonds in EXSY spectrum. As a result, the spectrum contains off-diagonal peaks between signals linked to each other by one exchange act. Such peaks are present only between the
A→C, B1, B2→C doublets since only one rotation around the amide bond is required for the transitions between these rotamers. The EXSY spectrum at short mixing times has no
B1→B2 cross-peaks because two acts of exchange are required for such transition. Consequently, peaks
B1 and
B2 belong to rotamer
B which is realized in the crystal (
Figure 6).
Next, the influence of temperature on the ligand
1 rotamers equilibrium was studied in toluene-d
8. Rising of temperature results in increase of content of rotamers
A and
C (
Table 2) in the equilibrium mixture.
A significant increase in the rate of rotation around the amide bonds is clearly observed above 40 °C. As a result, it is possible to observe in the spectra a gradual broadening and merging of signals from rotamers of each of the diastereomers with increasing of temperature, and two very broad signals are seen at 75 °C (
Figure 11).
The potential energy surfaces of rotamers
A–
C of both diastereomers are rather complex. There are several very close local minima differing in dihedral angles for Phen–CO bonds (the difference in free energies is ±0.5 kcal/mol) near the global minima. The structures corresponding to the global minima are shown in
Figure 12.
As it can be seen, all rotamers
A–C have practically similar stability in the gas phase, but they differ significantly in their dipole moments. Therefore, it is possible to expect that the equilibrium between rotamers can be shifted by solvent polarity change. Indeed, measurement of spectra in four different solvents (toluene, chloroform, acetone and nitrobenzene) confirmed our proposal. Rising the polarity of the solvent leads to an increase in the content of the most polar rotamers
A and
B, while the content of non-polar rotamer
C decreases (
Table 3).
Based on these data, one can assign the signals of rotamers A and C. As a result, we have clear understanding of spectral data for mixture of rotamers A–C. The results obtained for a solution in toluene fall out somewhat from the general dependence. This means that, although the polarity of the solvent is the main factor determining the equilibrium position, other factors, such as the solvent ability to form hydrogen bonds, should be also considered.
We were unable to obtain crystals of the meso-form of
1 suitable for X-ray studies. However, having solved the problem of assigning the signals of the rotamers of the racemic
1, it was possible to make the unambiguous assignment of the signals of rotamers of the meso-form as well. The
19F–{
1H}–NMR spectrum in CDCl
3 is given in
Figure 2b. Two singlets of equal intensity at –73.75 ppm and –74.81 ppm are related to rotamer
B (72% at 21 °C). Two singlets at –73.73 ppm and –74.19 ppm belong to rotamers
A and
C (14% each at 21 °C). The assignment of signals in this spectrum was done according to two-dimensional NMR spectra (see
Figures S9–S13 in Supplementary Materials).
Having established the approach for structure elucidation applied for ligand
1, we were able to determine the structure and composition of diastereoisomers of diamide
2 using a combination of
1H,
13C and
19F NMR spectra (
Table 4).
The
19F NMR spectra of mixture of diastereomers of diamides
1 and
2 are substantially similar (
Figure 2 and
Figure 13). As a result, it possible to distinguish all groups of lines in the spectrum of diamide
2 related to rotamers
A–C for both the meso-form and the racemate. All 8 singlets of CF
3-groups belonging to 6 rotamers are present in the
19F–{
1H}–NMR spectrum in CDCl
3 (
Figure 13). The ratio of two diastereomers of compound
2 is 2:3 according to the integration data. Thus, ligands
1 and
2 have the same ratio of diastereomers.
2.2. Complexation of Ligands 1 and 2 with Ln(III) Nitrates
The prepared amides
1 and
2 are highly attractive ligands for various cations. Based on ligand
1, we obtained a series of complexes with nitrates of La, Nd, Eu, and Lu. It was expected that such interaction can lead to complexes of both 1:1 and 2:1
L:Ln stoichiometry. Using acetonitrile as a solvent for this reaction we were able to synthesize
L·Ln(NO
3)
3 complexes. Complexes of diamide
1 with nitrates La (III), Nd (III), Eu (III), and Lu (III) of 1:1 composition were isolated from solutions in acetonitrile in solid form as light-colored powders. We studied all these complexes using IR and
19F NMR spectroscopy. The coordination of the metal leads to a bathochromic shift of the ν
C=O stretching vibration band in the IR spectrum by 42 cm
−1 in the lanthanum complex. This shift rises with an increase in the atomic number of the cation to reach 49 cm
−1 for Lu complex. Some characteristics of
1·Ln(NO
3)
3 complexes are given in
Table 5.
19F-NMR spectra of
1·Ln(NO
3)
3 complexes have very much in common (
Figure 14). Due to formation of coordination bonds, the amide rotation stops in these cases. This results in two doublet signals are observed in each spectrum which belongs to a complex of
rac- and
meso- forms, respectively.
Having confirmed the possibility of the formation of complexes for the ligands obtained we measured the stability constants by the UV–Vis spectrophotometric titration method. The stability constants were determined by spectrophotometric titration in the UV–visible region (
Table 6,
Figure 15).
Figure 15 represents an example of spectrophotometric data of 1 and Eu(NO
3)
3 complexation in acetonitrile (see
Figures S51–S56 in Supplementary Materials for analogues data for all the titrations). More basic diamide 1 forms complexes of stoichiometry 1:1 and 2:1 with cations of early lanthanoids La(III) and Nd(III) with larger ionic radius. The complexation with lanthanoids of the middle (Eu) and the end of the series (Lu) results in only 1:1 complexes. Less basic diamide 2 contains two electron-withdrawing chlorine atoms at positions 4 and 7 of the phenanthroline nucleus. As a result, this ligand forms only 1:1 complexes with all studied lanthanoids. The stability constants for both ligands increase with an increase in the atomic number of the cation. As expected, the log β
1 for diamide 2 complexes are an order of magnitude lower than the stability constants of diamide 1 complexes.
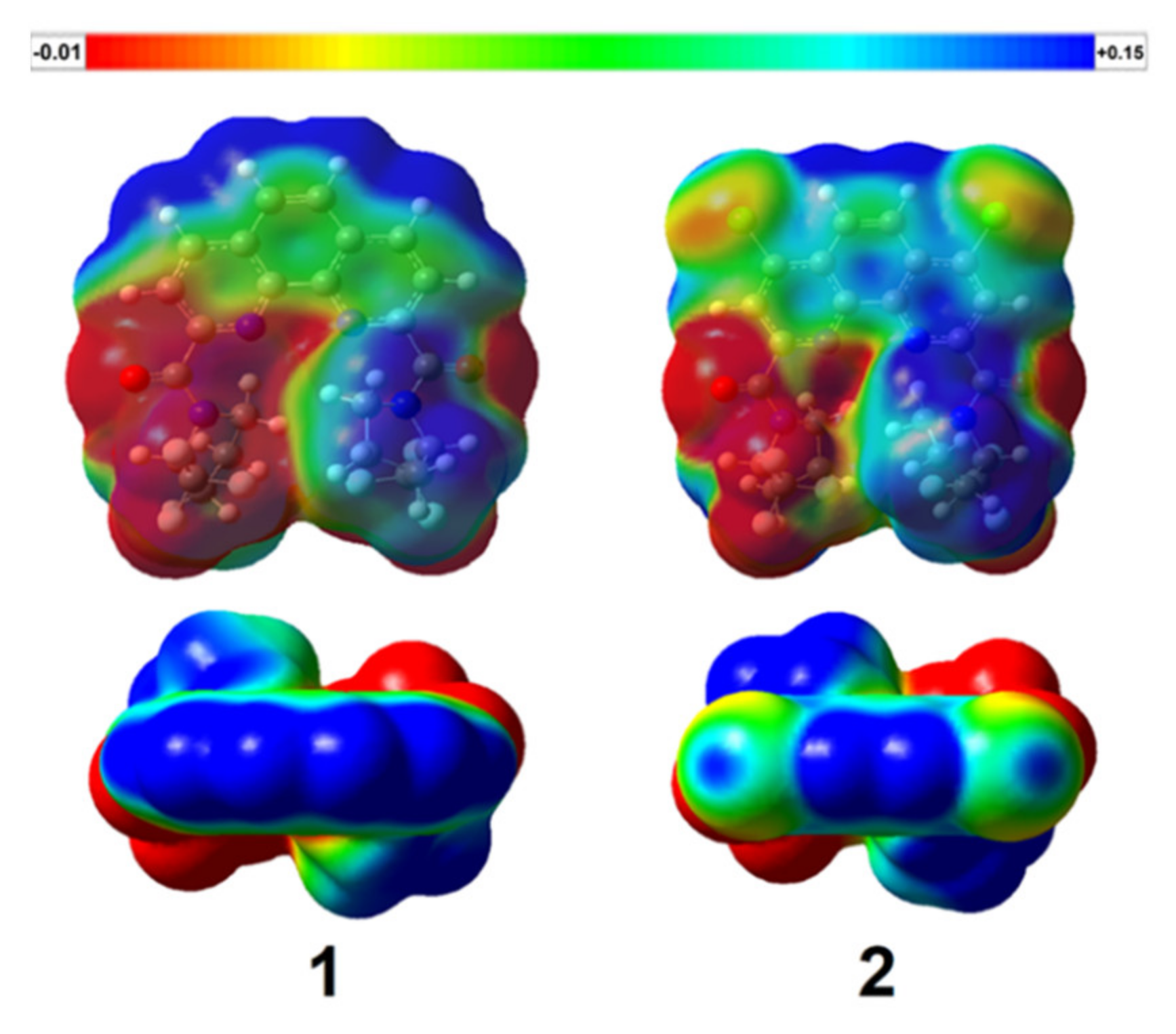
Additionally, we obtained electrostatic potential (ESP) maps at B3LYP/G-31G(d,p) theoretical level (Gaussian 16 [
40]) for both ligands. ESP maps in two projections are given in
Figure 16 (a single potential scale from −0.01 to +0.015 conventional units). The introduction of chlorine in a phenanthroline system leads to a significant change in electron density of the molecules.
Merz-Kollman (ESP) charges of some atoms for optimized geometries of 1 and 2 are shown in
Table 7.
Thus, less effective complexation of 2 with lanthanoids can be associated with the decreased charges at amide oxygens and phenanthroline nitrogens of 2 in comparison to diamide 1, because it leads to weaker ionic component to Ln–O and Ln–N bonds.


 ,
, 

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}