Determination of 18 Intact Glucosinolates in Brassicaceae Vegetables by UHPLC-MS/MS: Comparing Tissue Disruption Methods for Sample Preparation



Abstract
:
1. Introduction
2. Results and Discussion
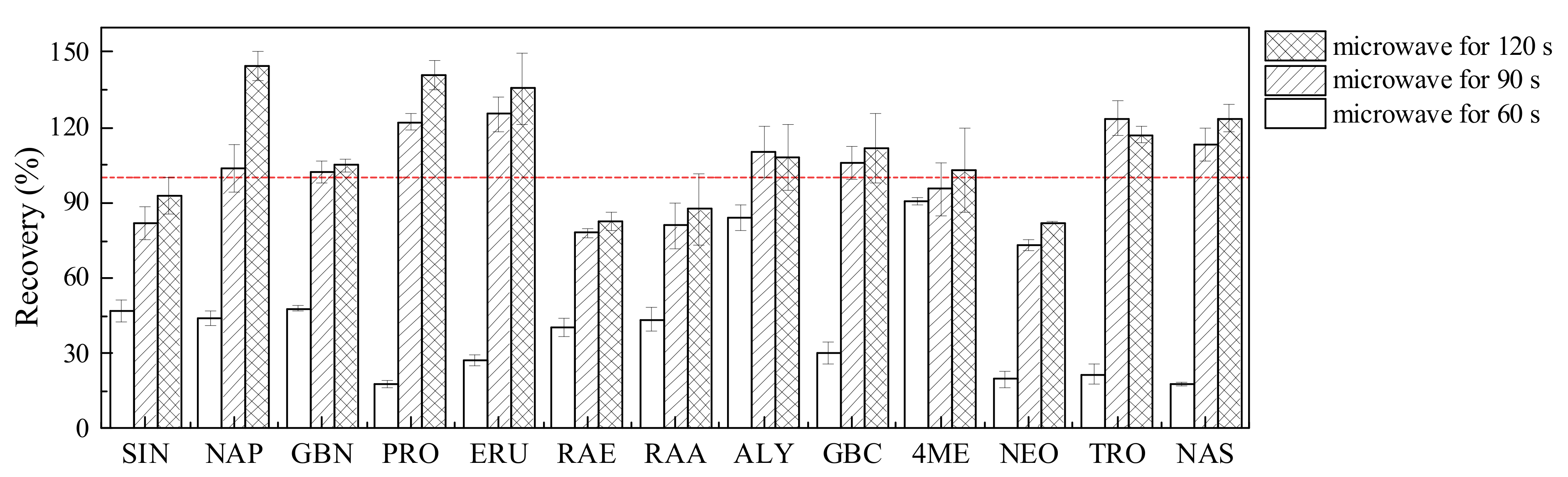
2.1. Pretreatment Optimization
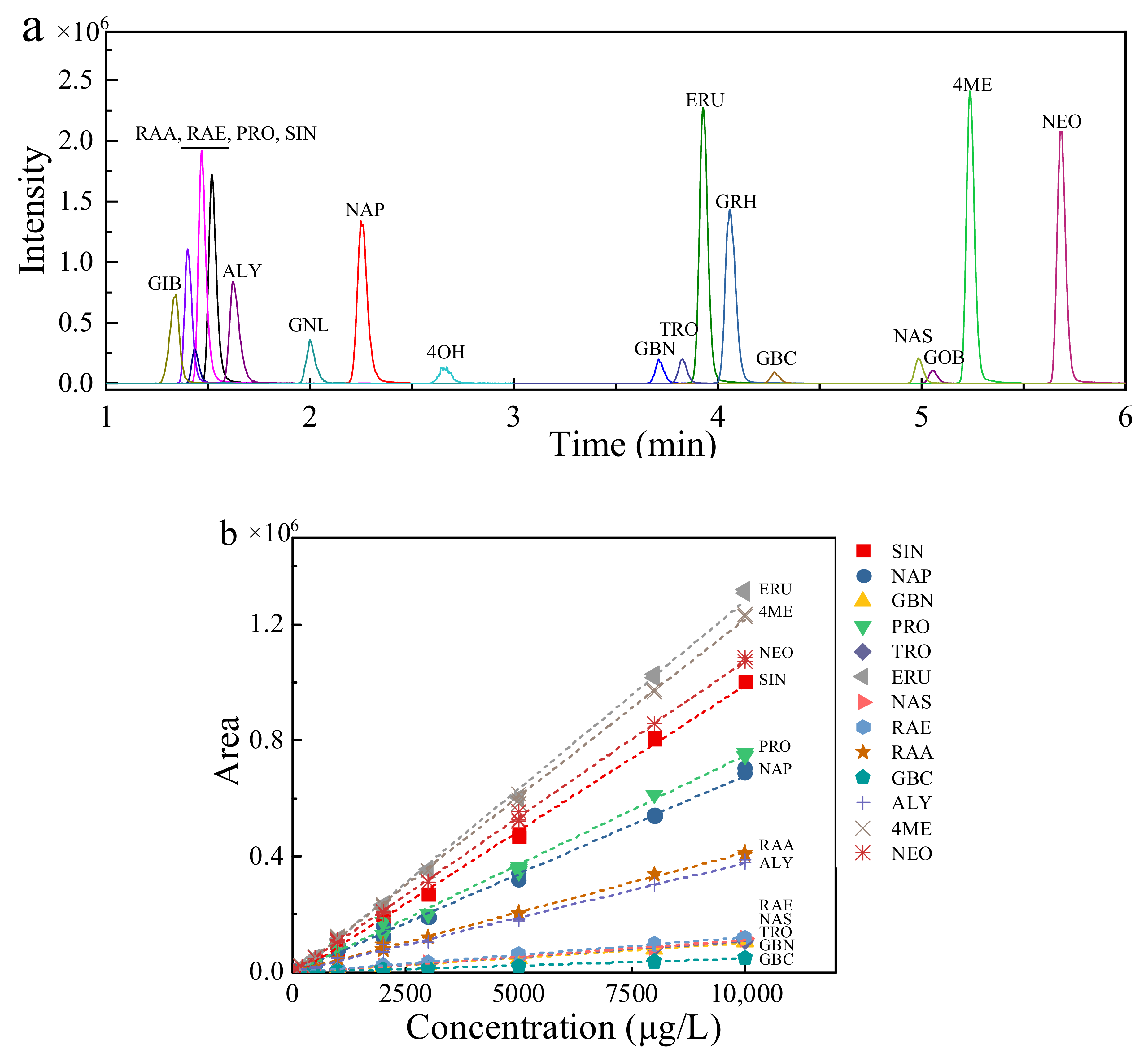
2.2. Analytical Method Validation
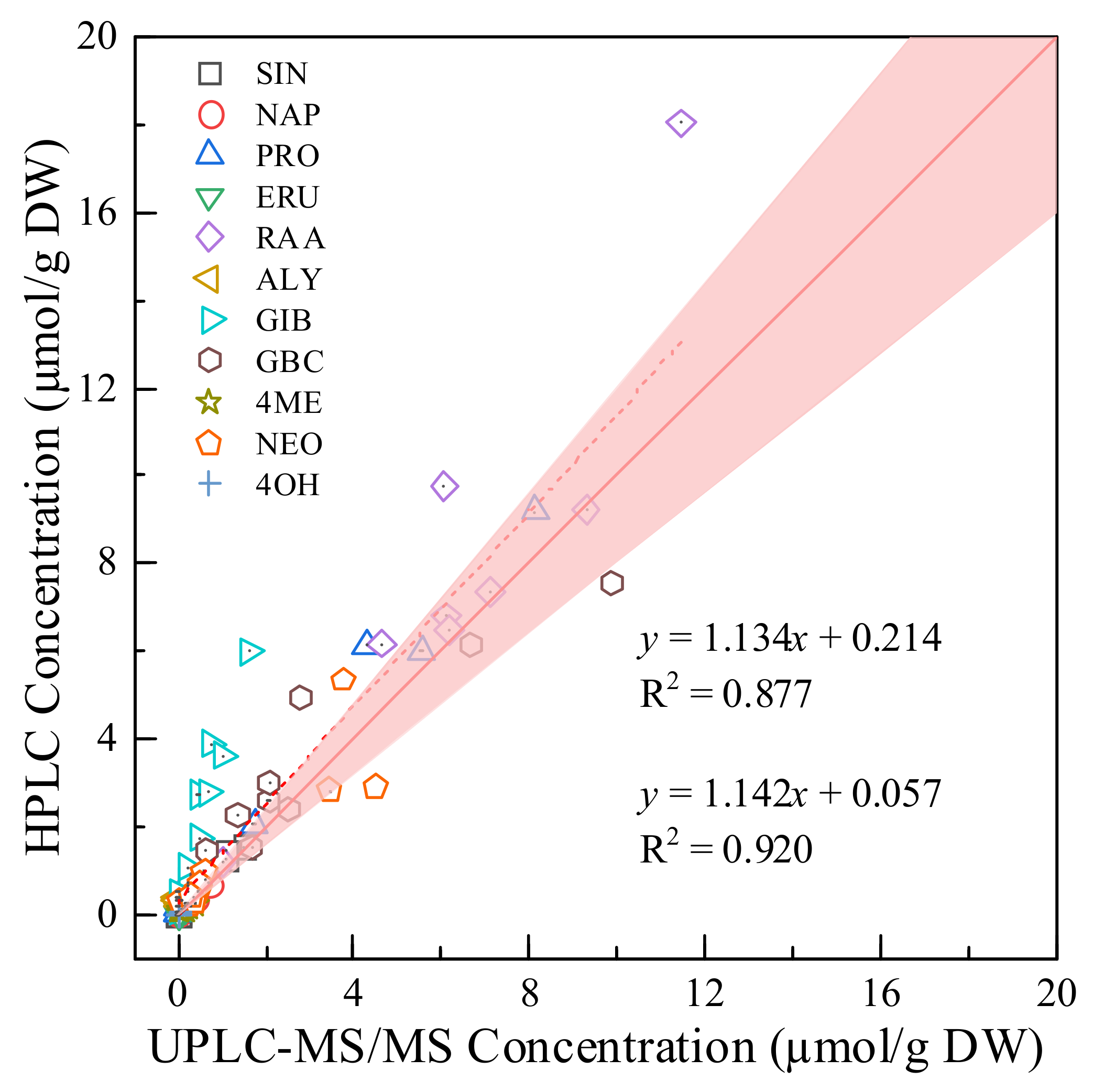
2.3. Comparison of the UHPLC-MS/MS Method with the HPLC Method
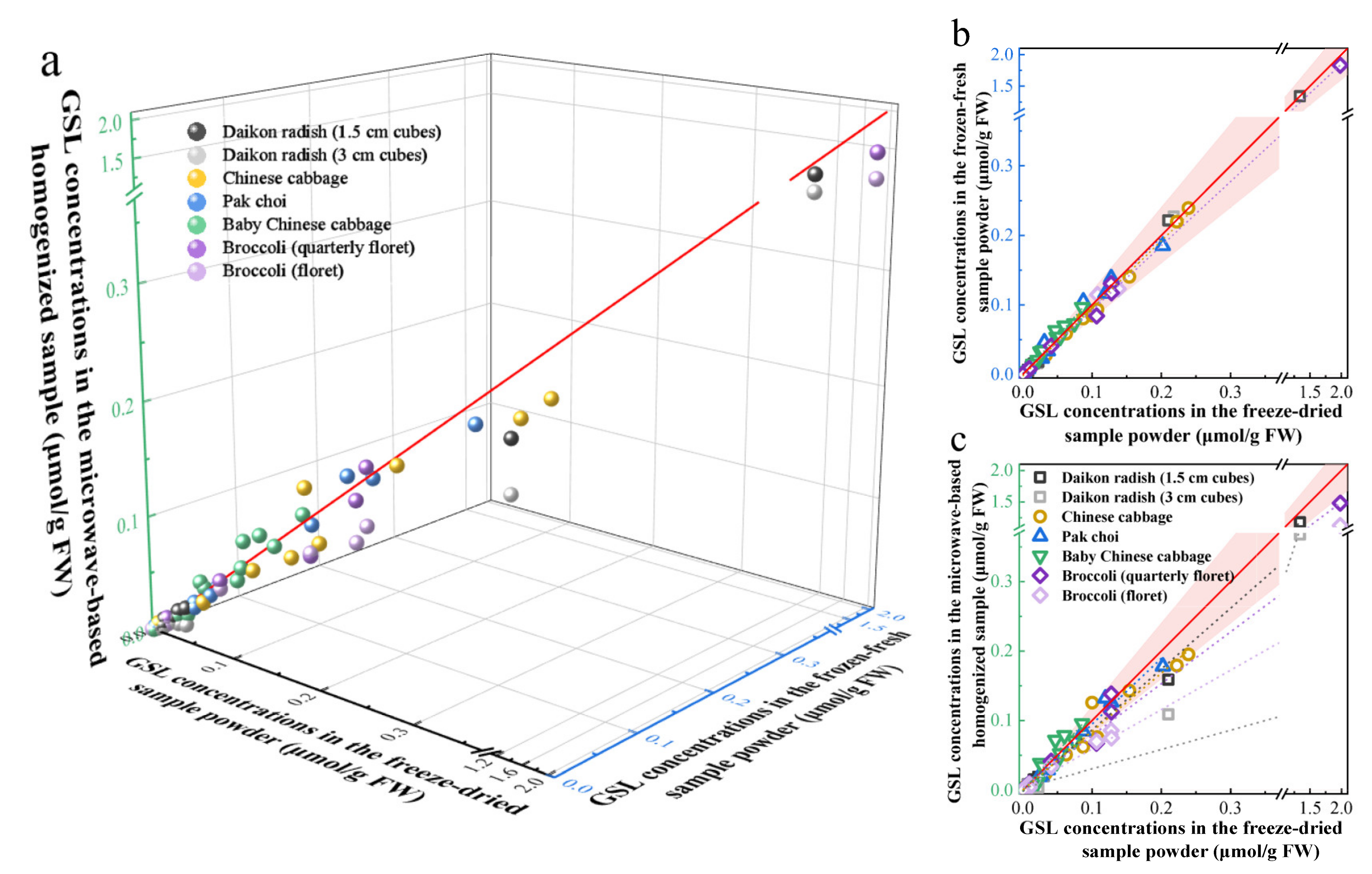
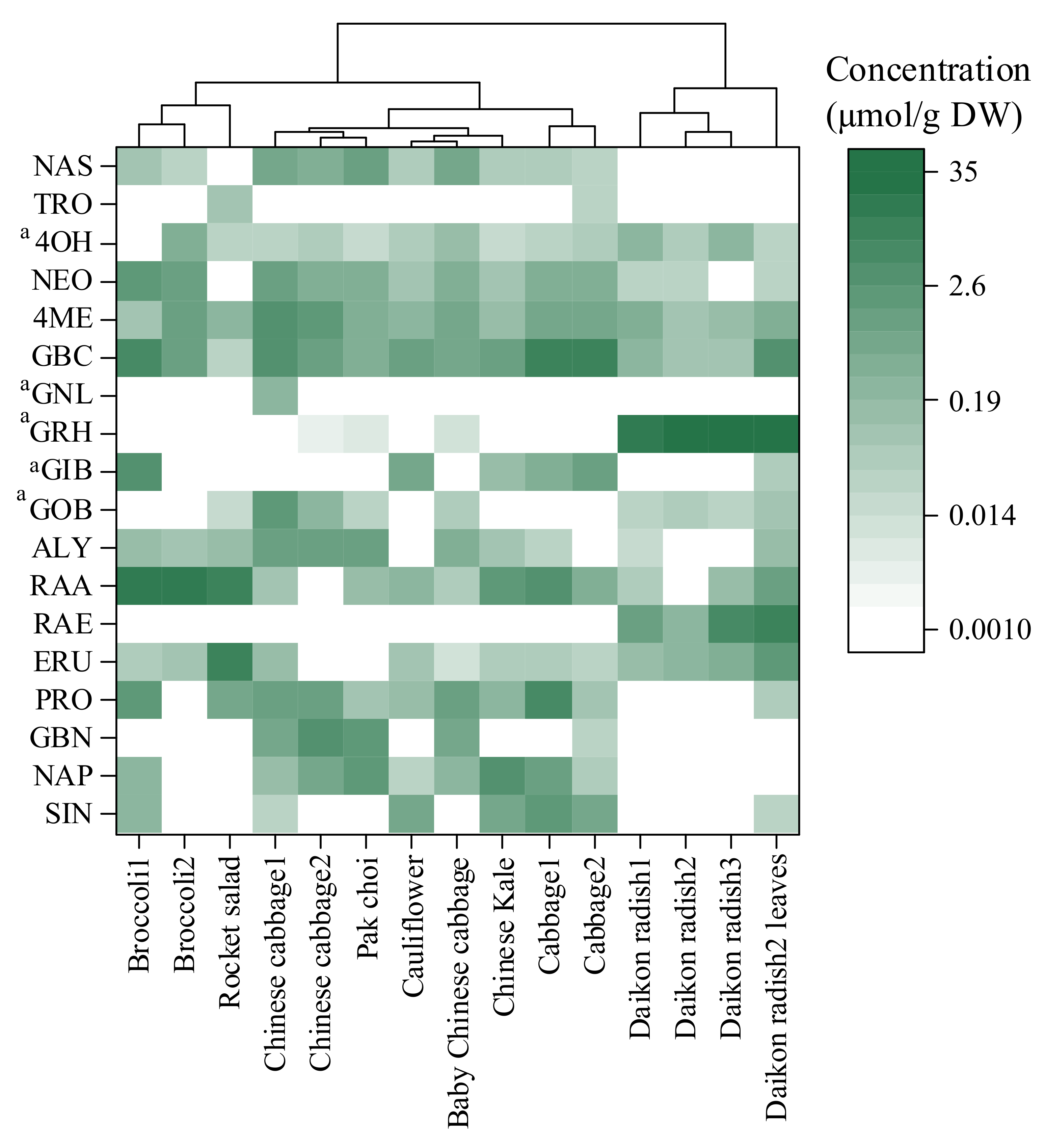
2.4. Applications of the Developed Method
3. Materials and Methods
3.1. Standards and Reagents
3.2. Sample Collection and Tissue Disruption
3.3. Intact Glucosinolate Extraction
3.4. UHPLC-MS/MS Analysis
3.5. Quantitative Analysis of Desulfo-Glucosinolates
3.6. Quantification and Method Performance Validation
3.7. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
References
- Sikorska-Zimny, K.; Beneduce, L. The glucosinolates and their bioactive derivatives in Brassica: A review on classification, biosynthesis and content in plant tissues, fate during and after processing, effect on the human organism and interaction with the gut microbiota. Crit. Rev. Food Sci. 2021, 61, 2544–2571. [Google Scholar] [CrossRef] [PubMed]
- Hanschen, F.S.; Kühn, C.; Nickel, M.; Rohn, S.; Dekker, M. Leaching and degradation kinetics of glucosinolates during boiling of Brassica oleracea vegetables and the formation of their breakdown products. Food Chem. 2018, 263, 240–250. [Google Scholar] [CrossRef] [PubMed]
- Mithen, R. Glucosinolates-biochemistry, genetics and biological activity. Plant Growth Regul. 2001, 34, 91–103. [Google Scholar] [CrossRef]
- Vo, Q.V.; Trenerry, C.; Rochfort, S.; Wadeson, J.; Leyton, C.; Hughes, A.B. Synthesis and anti-inflammatory activity of aromatic glucosinolates. Bioorgan. Med. Chem. 2013, 21, 5945–5954. [Google Scholar] [CrossRef]
- Lee, Y.; Chen, M.; Lee, J.D.; Zhang, J.; Lin, S.; Fu, T.; Chen, H.; Ishikawa, T.; Chiang, S.; Katon, J.; et al. Reactivation of pten tumor suppressor for cancer treatment through inhibition of a myc-wwp1 inhibitory pathway. Science 2019, 364, u159. [Google Scholar] [CrossRef]
- Li, Z.; Zheng, S.; Liu, Y.; Fang, Z.; Yang, L.; Zhuang, M.; Zhang, Y.; Lv, H.; Wang, Y.; Xu, D. Characterization of glucosinolates in 80 broccoli genotypes and different organs using UHPLC-Triple-TOF-MS method. Food Chem. 2021, 334, 127519. [Google Scholar] [CrossRef]
- Franklin, S.J.; Dickinson, S.E.; Karlage, K.L.; Bowden, G.T.; Myrdal, P.B. Stability of sulforaphane for topical formulation. Drug Dev. Ind. Pharm. 2013, 40, 494–502. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, L.; Xu, K.; Zhu, Y.; Wang, F.; Xiao, J.; Guo, L. Calcium affects glucoraphanin metabolism in broccoli sprouts under ZnSO4 stress. Food Chem. 2021, 334, 127520. [Google Scholar] [CrossRef] [PubMed]
- Blažević, I.; Montaut, S.; Burčul, F.; Olsen, C.E.; Burow, M.; Rollin, P.; Agerbirk, N. Glucosinolate structural diversity, identification, chemical synthesis and metabolism in plants. Phytochemistry 2020, 169, 112100. [Google Scholar] [CrossRef]
- Doheny-Adams, T.; Redeker, K.; Kittipol, V.; Bancroft, I.; Hartley, S.E. Development of an efficient glucosinolate extraction method. Plant Methods 2017, 13, 17. [Google Scholar] [CrossRef] [Green Version]
- Maldini, M.; Foddai, M.; Natella, F.; Petretto, G.L.; Rourke, J.P.; Chessa, M.; Pintore, G. Identification and quantification of glucosinolates in different tissues of Raphanus raphanistrum by liquid chromatography tandem-mass spectrometry. J. Food Compos. Anal. 2017, 61, 20–27. [Google Scholar] [CrossRef]
- Sookjitsumran, W.; Devahastin, S.; Mujumdar, A.S.; Chiewchan, N. Comparative evaluation of microwave-assisted extraction and preheated solvent extraction of bioactive compounds from a plant material: A case study with cabbages. Int. J. Food Sci. Technol. 2016, 51, 2440–2449. [Google Scholar] [CrossRef]
- López-Berenguer, C.; Carvajal, M.; Moreno, D.A.; García-Viguera, C. Effects of microwave cooking conditions on bioactive compounds present in broccoli inflorescences. J. Agric. Food Chem. 2007, 55, 10001–10007. [Google Scholar] [CrossRef]
- Verkerk, R.; Dekker, M. Glucosinolates and myrosinase activity in red cabbage (Brassica oleracea L. Var. Capitata f. rubra DC.) after various microwave treatments. J. Agric. Food Chem. 2004, 52, 7318–7323. [Google Scholar]
- Hwang, I.M.; Park, B.; Dang, Y.M.; Kim, S.; Seo, H.Y. Simultaneous direct determination of 15 glucosinolates in eight Brassica species by UHPLC-Q-Orbitrap-MS. Food Chem. 2019, 282, 127–133. [Google Scholar] [CrossRef]
- Verkerk, R.; Dekker, M.; Jongen, W.M.F. Post-harvest increase of indolyl glucosinolates in response to chopping and storage ofbrassica vegetables. J. Sci. Food Agric. 2001, 81, 953–958. [Google Scholar] [CrossRef]
- Grosser, K.; van Dam, N.M. A straightforward method for glucosinolate extraction and analysis with high-pressure liquid chromatography (HPLC). J. Vis. Exp. 2017, 121, e55425. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.G.; Bonnema, G.; Zhang, N.; Kwak, J.H.; de Vos, R.C.H.; Beekwilder, J. Evaluation of glucosinolate variation in a collection of turnip (Brassica rapa) germplasm by the analysis of intact and desulfo glucosinolates. J. Agric. Food Chem. 2013, 61, 3984–3993. [Google Scholar] [CrossRef]
- Kim, H.J.; Lee, M.J.; Jeong, M.H.; Kim, J.E. Identification and quantification of glucosinolates in kimchi by liquid chromatography-electrospray tandem mass spectrometry. Int. J. Anal. Chem. 2017, 2017, 6753481. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Cheung, C.; Ma, W.; Au, W.; Zhang, X.Y.; Lee, A. Determination of two intact glucosinolates in vegetables and Chinese herbs. Anal. Bioanal. Chem. 2004, 378, 827–833. [Google Scholar] [CrossRef]
- Piekarska, A.; Kusznierewicz, B.; Meller, M.; Dziedziul, K.; Namieśnik, J.; Bartoszek, A. Myrosinase activity in different plant samples; Optimisation of measurement conditions for spectrophotometric and pH-stat methods. Ind. Crop. Prod. 2013, 50, 58–67. [Google Scholar] [CrossRef]
- Liang, H.; Yuan, Q.P.; Dong, H.R.; Liu, Y.M. Determination of sulforaphane in broccoli and cabbage by high-performance liquid chromatography. J. Food Compos. Anal. 2006, 19, 473–476. [Google Scholar] [CrossRef]
- Francisco, M.; Moreno, D.A.; Cartea, M.E.; Ferreres, F.; García-Viguera, C.; Velasco, P. Simultaneous identification of glucosinolates and phenolic compounds in a representative collection of vegetable Brassica rapa. J. Chromatogr. A 2009, 1216, 6611–6619. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Zhao, Y.; Sun, J.; Yu, L.L.; Chen, P. Chemical profiling of glucosinolates in cruciferous vegetables-based dietary supplements using ultra-high performance liquid chromatography coupled to tandem high resolution mass spectrometry. J. Food Compos. Anal. 2017, 61, 67–72. [Google Scholar] [CrossRef]
- Campos, D.; Aguilar Galvez, A.; García Ríos, D.; Chirinos, R.; Limaymanta, E.; Pedreschi, R. Postharvest storage and cooking techniques affect the stability of glucosinolates and myrosinase activity of andean mashua tubers (Tropaeolum tuberosum). Int. J. Food Sci. Technol. 2019, 54, 2387–2395. [Google Scholar] [CrossRef]
- Hart, D.J.; Scott, K.J. Development and evaluation of an HPLC method for the analysis of carotenoids in foods, and the measurement of the carotenoid content of vegetables and fruits commonly consumed in the UK. Food Chem. 1995, 54, 101–111. [Google Scholar] [CrossRef]
- Liang, X.; Lee, H.W.; Li, Z.; Lu, Y.; Zou, L.; Ong, C.N. Simultaneous quantification of 22 glucosinolates in 12 Brassicaceae vegetables by hydrophilic interaction chromatography-tandem mass spectrometry. ACS Omega 2018, 3, 15546–15553. [Google Scholar] [CrossRef]
- Chan, C.; Yusoff, R.; Ngoh, G.; Kung, F.W. Microwave-assisted extractions of active ingredients from plants. J. Chromatogr. A 2011, 1218, 6213–6225. [Google Scholar] [CrossRef]
- Kuljarachanan, T.; Chiewchan, N.; Devahastin, S. Profiles of major glucosinolates in different parts of white cabbage and their evolutions during processing into vegetable powder by various methods. Int. Food Res. J. 2019, 26, 1763–1772. [Google Scholar]
- Jimenez-Garcia, S.N.; Vazquez-Cruz, M.A.; Ramirez-Gomez, X.S.; Beltran-Campos, V.; Contreras-Medina, L.M.; Garcia-Trejo, J.F.; Feregrino-Pérez, A.A. Changes in the content of phenolic compounds and biological activity in traditional mexican herbal infusions with different drying methods. Molecules 2020, 25, 1601. [Google Scholar] [CrossRef] [Green Version]
- Alolga, R.N.; Osae, R.; Essilfie, G.; Saalia, F.K.; Akaba, S.; Chikari, F. Sonication, osmosonication and vacuum-assisted osmosonication pretreatment of ghanaian garlic slices: Effect on physicochemical properties and quality characteristics. Food Chem. 2021, 343, 128535. [Google Scholar] [CrossRef] [PubMed]
- Chiu, Y.; Matak, K.; Ku, K. Methyl jasmonate treatment of broccoli enhanced glucosinolate concentration, which was retained after boiling, steaming, or microwaving. Foods 2020, 9, 758. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.; Jayaprakasha, G.K.; Patil, B.S. Rapid and efficient desulfonation method for the analysis of glucosinolates by high-resolution liquid chromatography coupled with quadrupole time-of-flight mass spectrometry. J. Agric. Food Chem. 2017, 65, 11100–11108. [Google Scholar] [CrossRef]
- Ares, A.M.; Nozal, M.J.; Bernal, J.L.; Bernal, J. Analysis of intact glucosinolates in beeswax by liquid chromatography tandem mass spectrometry. Food Anal. Method. 2016, 9, 795–804. [Google Scholar] [CrossRef]
- Andini, S.; Araya-Cloutier, C.; Sanders, M.; Vincken, J. Simultaneous analysis of glucosinolates and isothiocyanates by reversed-phase ultra-high-performance liquid chromatography-electron spray ionization-tandem mass spectrometry. J. Agric. Food Chem. 2020, 68, 3121–3131. [Google Scholar] [CrossRef] [Green Version]
- ISO 9167:2019(E). Rapeseed and Rapeseed Meals—Determination of Glucosinolates Content-Method Using High-Performance Liquid Chromatography; ISO Technical Committees: Geneva, Switzerland, 2019. [Google Scholar]
- Förster, N.; Ulrichs, C.; Schreiner, M.; Müller, C.T.; Mewis, I. Development of a reliable extraction and quantification method for glucosinolates in moringa oleifera. Food Chem. 2015, 166, 456–464. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Liu, H.; Pu, C.; Chen, J.; Sun, Y.; Hu, L. Determination of multiple antibiotics in leafy vegetables using QuEChERS-UHPLC-MS/MS. J. Sep. Sci. 2018, 41, 713–722. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chemical Group | Compound | Abbreviation | Chemical Formula | Retention Time (min) | Parent (m/z) | Cone (V) | Daughter (m/z) | Collision (V) | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Aliphatic | Sinigrin | SIN | C10H17NO9S2 | 1.52 | 357.86 | 4 | 96.62 | 161.89 | 194.93 | 18 | 18 | 20 |
| Gluconapin | NAP | C11H19NO9S2 | 2.24 | 371.83 | 4 | 96.56 | 194.81 | 258.94 | 22 | 16 | 18 | |
| Glucobrassicanapin | GBN | C12H21NO9S2 | 3.69 | 385.91 | 4 | 194.92 | 258.92 | 274.94 | 16 | 20 | 20 | |
| Progoitrin | PRO | C11H19NO10S2 | 1.46 | 387.89 | 4 | 96.75 | 194.91 | 258.91 | 20 | 18 | 16 | |
| Glucoerucin | ERU | C12H23NO9S3 | 3.90 | 419.96 | 10 | 96.68 | 258.98 | 195.00 | 22 | 18 | 20 | |
| Glucoraphenin | RAE | C12H21NO10S3 | 1.43 | 433.87 | 4 | 96.50 | 258.98 | 419.01 | 22 | 20 | 20 | |
| Glucoraphanin | RAA | C12H23NO10S3 | 1.40 | 435.89 | 2 | 96.69 | 259.01 | 371.93 | 24 | 20 | 18 | |
| Glucoalyssin | ALY | C13H25NO10S3 | 1.62 | 449.90 | 6 | 191.92 | 274.94 | 386.02 | 24 | 20 | 20 | |
| Glucoberteroin a | GOB | C13H24NO9S3 | 5.06 | 434.06 | 10 | 354.11 | 258.98 | 195.03 | 20 | 20 | 18 | |
| Glucoiberin a | GIB | C11H21NO10S3 | 1.35 | 422.02 | 12 | 358.00 | 258.98 | 195.00 | 26 | 15 | 20 | |
| Glucoraphasatin a | GRH | C12H21NO9S3 | 4.08 | 418.03 | 10 | 338.00 | 258.98 | 175.00 | 18 | 20 | 18 | |
| Gluconapoleiferin a | GNL | C12H20NO10S2 | 2.00 | 402.05 | 10 | 332.00 | 258.98 | 195.00 | 25 | 22 | 20 | |
| Indolic | Glucobrassicin | GBC | C16H20N2O9S2 | 4.26 | 446.90 | 18 | 194.97 | 258.95 | 274.94 | 24 | 18 | 22 |
| 4-methoxyglucobrassicin | 4ME | C17H22N2O10S2 | 5.23 | 476.91 | 10 | 96.62 | 194.93 | 258.90 | 22 | 18 | 20 | |
| Neoglucobrassicin | NEO | C17H22N2O10S2 | 5.67 | 477.00 | 10 | 96.96 | 258.98 | 446.00 | 22 | 18 | 14 | |
| 4-hydroxyglucobrassicin a | 4OH | C16H20N2O10S2 | 2.64 | 463.05 | 10 | 285.00 | 267.00 | 259.01 | 18 | 20 | 26 | |
| Aromatic | Glucotropaeolin | TRO | C14H19NO9S2 | 3.80 | 407.86 | 2 | 165.93 | 194.93 | 258.91 | 24 | 20 | 20 |
| Gluconasturtiin | NAS | C15H21NO9S2 | 4.96 | 421.91 | 12 | 179.95 | 194.93 | 258.96 | 22 | 16 | 20 | |
| Compound | Freeze-Dried Sample Powder | Frozen-Fresh Sample Powder | Calibration Curve Equation | R2 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Concentration | Recovery | RSD | LOD | LOQ | Concentration | Recovery | RSD | LOD | LOQ | |||
| (nmol/g DW) | (%) | (%) | (nmol/g DW) | (nmol/g DW) | (nmol/g FW) | (%) | (%) | (nmol/g FW) | (nmol/g FW) | |||
| Sinigrin | 50 | 107 | 5 | 3.23 | 9.69 | 5 | 94 | 1 | 0.37 | 1.11 | y = 94.9167x − 810.16 | 0.997 |
| 250 | 110 | 3 | 25 | 80 | 7 | |||||||
| 1000 | 90 | 5 | 100 | 90 | 12 | |||||||
| Gluconapin | 50 | 92 | 6 | 2.52 | 7.56 | 5 | 91 | 8 | 0.33 | 0.98 | y = 67.5645x − 191.872 | 0.993 |
| 250 | 92 | 3 | 25 | 81 | 7 | |||||||
| 1000 | 77 | 6 | 100 | 89 | 3 | |||||||
| Glucobrassicanapin | 50 | 99 | 9 | 2.95 | 8.85 | 5 | 90 | 14 | 0.34 | 1.03 | y = 9.8891x − 75.1189 | 0.997 |
| 250 | 99 | 7 | 25 | 94 | 2 | |||||||
| 1000 | 91 | 6 | 100 | 95 | 1 | |||||||
| Progoitrin | 50 | 95 | 9 | 3.47 | 10.42 | 5 | 92 | 4 | 0.37 | 1.11 | y = 73.4885x − 376.377 | 0.995 |
| 250 | 76 | 4 | 25 | 90 | 3 | |||||||
| 1000 | 74 | 6 | 100 | 86 | 8 | |||||||
| Glucoerucin | 50 | 112 | 4 | 2.11 | 6.34 | 5 | 82 | 15 | 0.29 | 0.86 | y = 123.675x − 670.066 | 0.997 |
| 250 | 100 | 2 | 25 | 85 | 4 | |||||||
| 1000 | 81 | 7 | 100 | 88 | 0 | |||||||
| Glucoraphenin | 50 | 100 | 8 | 5.51 | 16.53 | 5 | 100 | 6 | 0.45 | 1.35 | y = 12.0899x − 30.9665 | 1.000 |
| 250 | 91 | 4 | 25 | 95 | 6 | |||||||
| 1000 | 77 | 7 | 100 | 96 | 1 | |||||||
| Glucoraphanin | 50 | 88 | 6 | 1.91 | 5.72 | 5 | 103 | 6 | 0.27 | 0.80 | y = 41.6557x − 63.9675 | 0.997 |
| 250 | 99 | 3 | 25 | 98 | 6 | |||||||
| 1000 | 79 | 7 | 100 | 104 | 7 | |||||||
| Glucoalyssin | 50 | 86 | 7 | 3.32 | 9.95 | 5 | 94 | 5 | 0.33 | 0.99 | y = 37.7662x − 31.3535 | 0.991 |
| 250 | 87 | 3 | 25 | 102 | 4 | |||||||
| 1000 | 79 | 6 | 100 | 101 | 1 | |||||||
| Glucobrassicin | 50 | 119 | 4 | 5.38 | 16.14 | 5 | 86 | 2 | 0.44 | 1.32 | y = 4.62456x − 33.7449 | 0.997 |
| 250 | 92 | 9 | 25 | 79 | 13 | |||||||
| 1000 | 92 | 10 | 100 | 87 | 10 | |||||||
| 4-Methoxyglucobrassicin | 50 | 104 | 10 | 2.64 | 7.92 | 5 | 84 | 13 | 0.29 | 0.86 | y = 120.093x − 267.184 | 0.999 |
| 250 | 98 | 7 | 25 | 86 | 8 | |||||||
| 1000 | 79 | 9 | 100 | 83 | 2 | |||||||
| Neoglucobrassicin | 50 | 109 | 10 | 2.87 | 8.62 | 5 | 78 | 6 | 0.30 | 0.90 | y = 106.49x − 98.5261 | 0.999 |
| 250 | 108 | 6 | 25 | 77 | 4 | |||||||
| 1000 | 82 | 8 | 100 | 80 | 4 | |||||||
| Glucotropaeolin | 50 | 106 | 5 | 5.80 | 17.40 | 5 | 79 | 12 | 0.48 | 1.43 | y = 10.493x − 62.1323 | 0.998 |
| 250 | 101 | 7 | 25 | 79 | 4 | |||||||
| 1000 | 82 | 6 | 100 | 80 | 1 | |||||||
| Gluconasturtiin | 50 | 96 | 5 | 3.60 | 10.79 | 5 | 78 | 12 | 0.36 | 1.08 | y = 10.6905x − 53.0295 | 0.996 |
| 250 | 97 | 1 | 25 | 77 | 4 | |||||||
| 1000 | 77 | 6 | 100 | 79 | 1 | |||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, X.; He, H.; Zhao, X.; Liu, G.; Hu, L.; Cheng, B.; Wang, Y. Determination of 18 Intact Glucosinolates in Brassicaceae Vegetables by UHPLC-MS/MS: Comparing Tissue Disruption Methods for Sample Preparation. Molecules 2022, 27, 231. https://doi.org/10.3390/molecules27010231
Yu X, He H, Zhao X, Liu G, Hu L, Cheng B, Wang Y. Determination of 18 Intact Glucosinolates in Brassicaceae Vegetables by UHPLC-MS/MS: Comparing Tissue Disruption Methods for Sample Preparation. Molecules. 2022; 27(1):231. https://doi.org/10.3390/molecules27010231
Chicago/Turabian StyleYu, Xiaolu, Hongju He, Xuezhi Zhao, Guangmin Liu, Liping Hu, Bing Cheng, and Yaqin Wang. 2022. "Determination of 18 Intact Glucosinolates in Brassicaceae Vegetables by UHPLC-MS/MS: Comparing Tissue Disruption Methods for Sample Preparation" Molecules 27, no. 1: 231. https://doi.org/10.3390/molecules27010231
APA StyleYu, X., He, H., Zhao, X., Liu, G., Hu, L., Cheng, B., & Wang, Y. (2022). Determination of 18 Intact Glucosinolates in Brassicaceae Vegetables by UHPLC-MS/MS: Comparing Tissue Disruption Methods for Sample Preparation. Molecules, 27(1), 231. https://doi.org/10.3390/molecules27010231