
Tubulin Inhibitors: A Chemoinformatic Analysis Using Cell-Based Data




Abstract
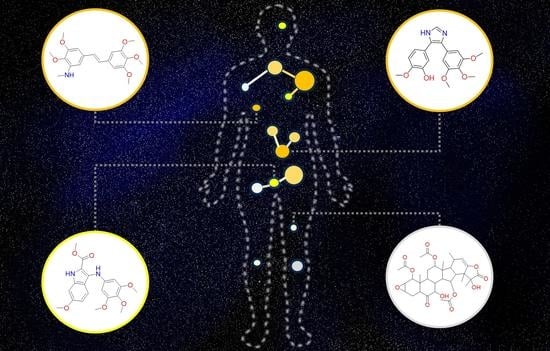
{kind=link}
{kind=link}
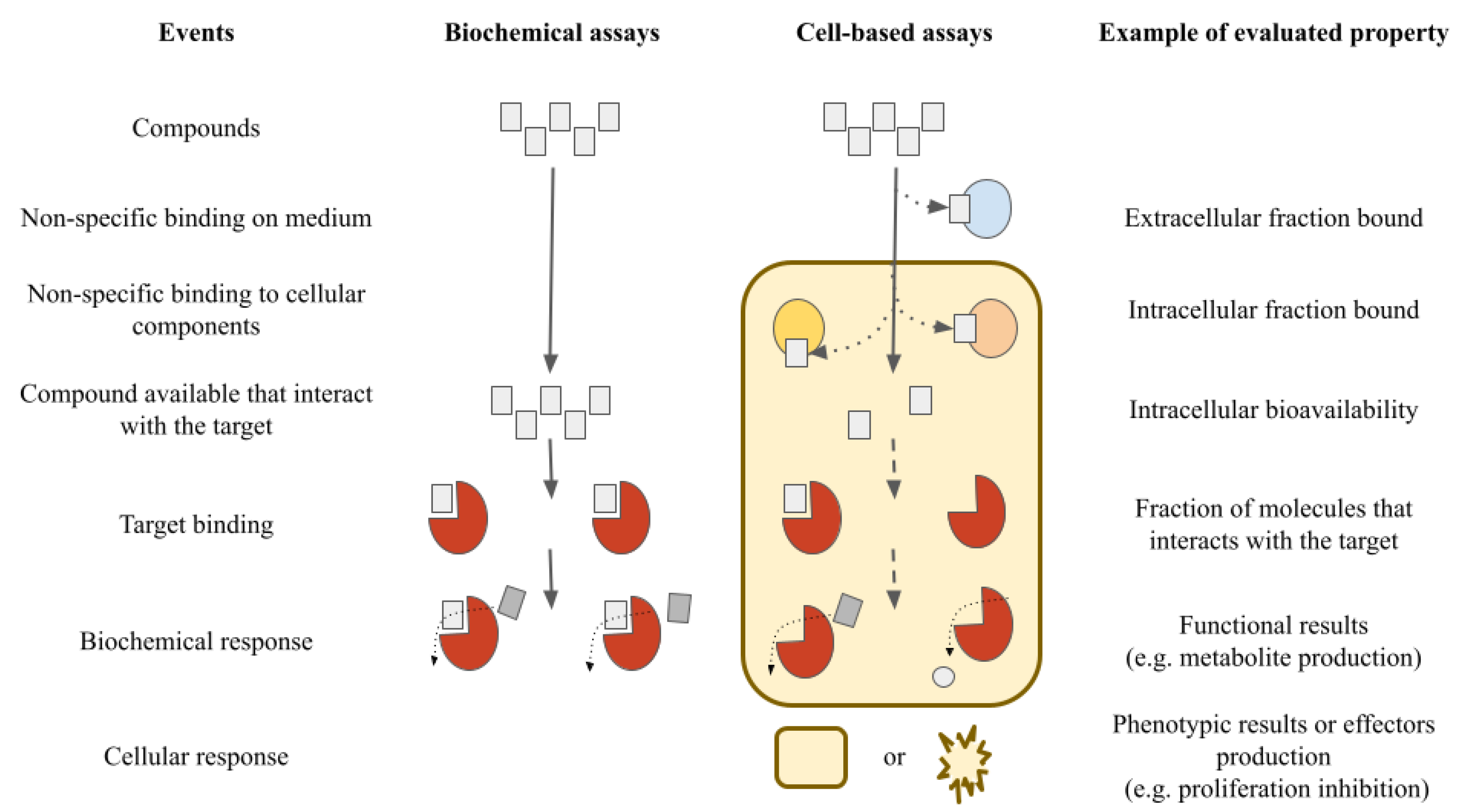{kind=link}
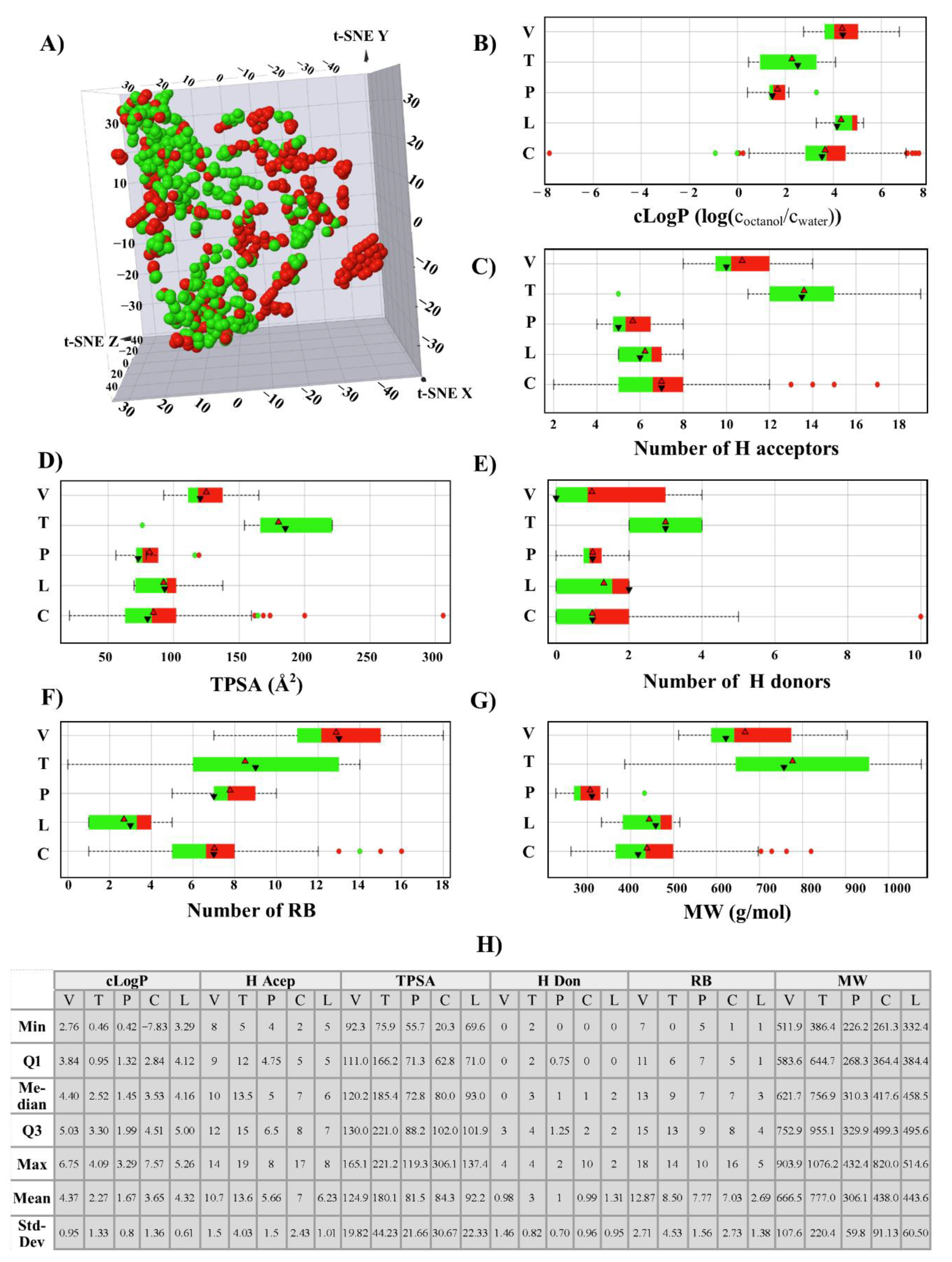{kind=link}
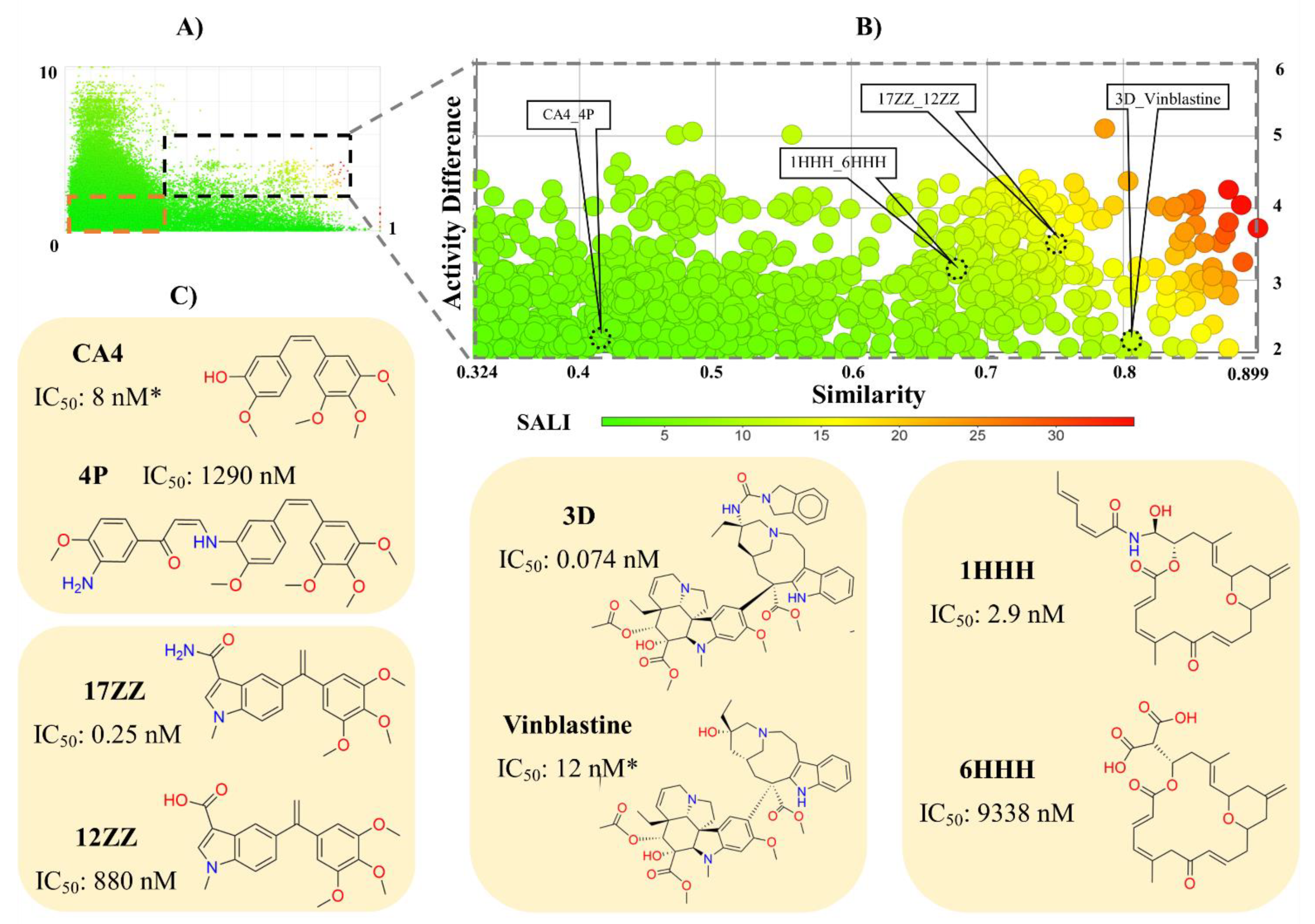{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Dataset
2.2. Chemical Space
2.3. Activity Landscape Modeling
2.4. Scaffold Content Analysis
2.5. Constellation Plot
3. Results and Discussion
3.1. Chemoinformatics Approaches
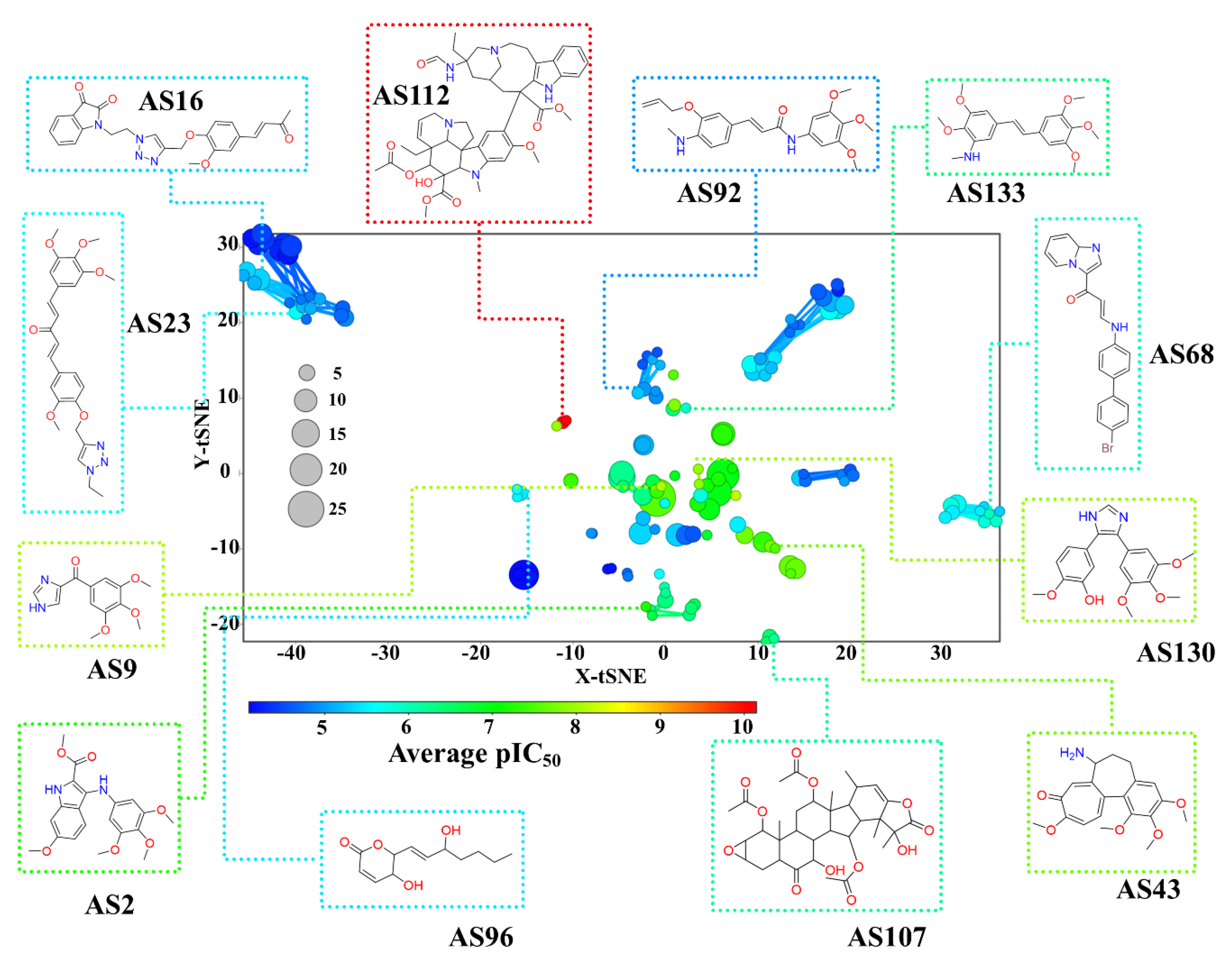
3.1.1. Chemical Space
3.1.2. Activity Landscape Modeling
3.1.3. Scaffold Content Analysis
3.1.4. Constellation Plot
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Arnst, K.E.; Banerjee, S.; Chen, H.; Deng, S.; Hwang, D.; Li, W.; Miller, D.D. Current Advances of Tubulin Inhibitors as Dual Acting Small Molecules for Cancer Therapy. Med. Res. Rev. 2019, 39, 1398–1426. [Google Scholar] [CrossRef]
- Lu, Y.; Chen, J.; Xiao, M.; Li, W.; Miller, D.D. An Overview of Tubulin Inhibitors That Interact with the Colchicine Binding Site. Pharm. Res. 2012, 29, 2943–2971. [Google Scholar] [CrossRef]
- Coulup, S.K.; Georg, G.I. Revisiting Microtubule Targeting Agents: α-Tubulin and the Pironetin Binding Site as Unexplored Targets for Cancer Therapeutics. Bioorg. Med. Chem. Lett. 2019, 29, 1865–1873. [Google Scholar] [CrossRef]
- Chen, H.; Deng, S.; Wang, Y.; Albadari, N.; Kumar, G.; Ma, D.; Li, W.; White, S.W.; Miller, D.D.; Li, W. Structure–Activity Relationship Study of Novel 6-Aryl-2-Benzoyl-Pyridines as Tubulin Polymerization Inhibitors with Potent Antiproliferative Properties. J. Med. Chem. 2020, 63, 827–846. [Google Scholar] [CrossRef]
- Xu, Q.; Bao, K.; Sun, M.; Xu, J.; Wang, Y.; Tian, H.; Zuo, D.; Guan, Q.; Wu, Y.; Zhang, W. Design, Synthesis and Structure-Activity Relationship of 3,6-Diaryl-7H-[1,2,4]Triazolo[3,4-b][1,3,4]Thiadiazines as Novel Tubulin Inhibitors. Sci. Rep. 2017, 7, 11997. [Google Scholar] [CrossRef] [PubMed]
- Ducki, S.; Mackenzie, G.; Lawrence, N.J.; Snyder, J.P. Quantitative Structure−Activity Relationship (5D-QSAR) Study of Combretastatin-like Analogues as Inhibitors of Tubulin Assembly. J. Med. Chem. 2005, 48, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Fu, D.-J.; Fu, L.; Liu, Y.-C.; Wang, J.-W.; Wang, Y.-Q.; Han, B.-K.; Li, X.-R.; Zhang, C.; Li, F.; Song, J.; et al. Structure-Activity Relationship Studies of β-Lactam-Azide Analogues as Orally Active Antitumor Agents Targeting the Tubulin Colchicine Site. Sci. Rep. 2017, 7, 12788. [Google Scholar] [CrossRef] [PubMed]
- Aziz, J.; Brachet, E.; Hamze, A.; Peyrat, J.-F.; Bernadat, G.; Morvan, E.; Bignon, J.; Wdzieczak-Bakala, J.; Desravines, D.; Dubois, J.; et al. Synthesis, Biological Evaluation, and Structure–Activity Relationships of Tri- and Tetrasubstituted Olefins Related to Isocombretastatin A-4 as New Tubulin Inhibitors. Org. Biomol. Chem. 2013, 11, 430–442. [Google Scholar] [CrossRef]
- Romagnoli, R.; Baraldi, P.G.; Carrion, M.D.; Cara, C.L.; Cruz-Lopez, O.; Tolomeo, M.; Grimaudo, S.; Cristina, A.D.; Pipitone, M.R.; Balzarini, J.; et al. Design, Synthesis and Structure–Activity Relationship of 2-(3′,4′,5′-Trimethoxybenzoyl)-Benzo[b]Furan Derivatives as a Novel Class of Inhibitors of Tubulin Polymerization. Bioorg. Med. Chem. 2009, 17, 6862–6871. [Google Scholar] [CrossRef]
- Mirzaei, S.; Hadizadeh, F.; Eisvand, F.; Mosaffa, F.; Ghodsi, R. Synthesis, Structure-Activity Relationship and Molecular Docking Studies of Novel Quinoline-Chalcone Hybrids as Potential Anticancer Agents and Tubulin Inhibitors. J. Mol. Struct. 2020, 1202, 127310. [Google Scholar] [CrossRef]
- Guo, Q.; Zhang, H.; Deng, Y.; Zhai, S.; Jiang, Z.; Zhu, D.; Wang, L. Ligand- and Structural-Based Discovery of Potential Small Molecules That Target the Colchicine Site of Tubulin for Cancer Treatment. Eur. J. Med. Chem. 2020, 196, 112328. [Google Scholar] [CrossRef] [PubMed]
- Bajorath, J.; Peltason, L.; Wawer, M.; Guha, R.; Lajiness, M.S.; Van Drie, J.H. Navigating Structure–Activity Landscapes. Drug Discov. Today 2009, 14, 698–705. [Google Scholar] [CrossRef]
- Medina-Franco, J.L.; Navarrete-Vázquez, G.; Méndez-Lucio, O. Activity and Property Landscape Modeling Is at the Interface of Chemoinformatics and Medicinal Chemistry. Future Med. Chem. 2015, 7, 1197–1211. [Google Scholar] [CrossRef]
- Maggiora, G.M. On Outliers and Activity CliffsWhy QSAR Often Disappoints. J. Chem. Inf. Model. 2006, 46, 1535. [Google Scholar] [CrossRef]
- Maggiora, G.; Medina-Franco, J.L.; Iqbal, J.; Vogt, M.; Bajorath, J. From Qualitative to Quantitative Analysis of Activity and Property Landscapes. J. Chem. Inf. Model. 2020, 60, 5873–5880. [Google Scholar] [CrossRef] [PubMed]
- Sink, R.; Gobec, S.; Pecar, S.; Zega, A. False Positives in the Early Stages of Drug Discovery. Curr. Med. Chem. 2010, 17, 4231–4255. [Google Scholar] [CrossRef] [PubMed]
- Mateus, A.; Gordon, L.J.; Wayne, G.J.; Almqvist, H.; Axelsson, H.; Seashore-Ludlow, B.; Treyer, A.; Matsson, P.; Lundbäck, T.; West, A.; et al. Prediction of Intracellular Exposure Bridges the Gap between Target- and Cell-Based Drug Discovery. Proc. Natl. Acad. Sci. USA 2017, 114, E6231–E6239. [Google Scholar] [CrossRef]
- Mendez, D.; Gaulton, A.; Bento, A.P.; Chambers, J.; De Veij, M.; Félix, E.; Magariños, M.P.; Mosquera, J.F.; Mutowo, P.; Nowotka, M.; et al. ChEMBL: Towards Direct Deposition of Bioassay Data. Nucleic Acids Res. 2019, 47, D930–D940. [Google Scholar] [CrossRef]
- López-López, E.; Rabal, O.; Oyarzabal, J.; Medina-Franco, J.L. Towards the Understanding of the Activity of G9a Inhibitors: An Activity Landscape and Molecular Modeling Approach. J. Comput. Aided Mol. Des. 2020, 34, 659–669. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.-N.; Lin, L.; Qiu, H.-Y.; Zou, W.-Y.; Qian, Y.; Zhu, H.-L. The Design, Synthesis, in Vitro Biological Evaluation and Molecular Modeling of Novel Benzenesulfonate Derivatives Bearing Chalcone Moieties as Potent Anti-Microtubulin Polymerization Agents. RSC Adv. 2015, 5, 23767–23777. [Google Scholar] [CrossRef]
- Ghawanmeh, A.A.; Al-Bajalan, H.M.; Mackeen, M.M.; Alali, F.Q.; Chong, K.F. Recent Developments on (−)-Colchicine Derivatives: Synthesis and Structure-Activity Relationship. Eur. J. Med. Chem. 2020, 185, 111788. [Google Scholar] [CrossRef]
- Li, W.; Xu, F.; Shuai, W.; Sun, H.; Yao, H.; Ma, C.; Xu, S.; Yao, H.; Zhu, Z.; Yang, D.-H.; et al. Discovery of Novel Quinoline–Chalcone Derivatives as Potent Antitumor Agents with Microtubule Polymerization Inhibitory Activity. J. Med. Chem. 2019, 62, 993–1013. [Google Scholar] [CrossRef]
- Kamal, A.; Kumar, G.B.; Polepalli, S.; Shaik, A.B.; Reddy, V.S.; Reddy, M.K.; Reddy, C.R.; Mahesh, R.; Kapure, J.S.; Jain, N. Design and Synthesis of Aminostilbene-Arylpropenones as Tubulin Polymerization Inhibitors. ChemMedChem 2014, 9, 2565–2579. [Google Scholar] [CrossRef] [PubMed]
- Miao, T.-T.; Tao, X.-B.; Li, D.-D.; Chen, H.; Jin, X.-Y.; Geng, Y.; Wang, S.-F.; Gu, W. Synthesis and Biological Evaluation of 2-Aryl-Benzimidazole Derivatives of Dehydroabietic Acid as Novel Tubulin Polymerization Inhibitors. RSC Adv. 2018, 8, 17511–17526. [Google Scholar] [CrossRef]
- Wang, G.; Liu, W.; Gong, Z.; Huang, Y.; Li, Y.; Peng, Z. Synthesis, Biological Evaluation, and Molecular Modelling of New Naphthalene-Chalcone Derivatives as Potential Anticancer Agents on MCF-7 Breast Cancer Cells by Targeting Tubulin Colchicine Binding Site. J. Enzym. Inhib. Med. Chem. 2020, 35, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Sankara Rao, N.; Lakshma Nayak, V.; Subba Rao, A.V.; Ali Hussaini, S.M.; Sunkari, S.; Alarifi, A.; Kamal, A. Arylcinnamido-Propionone Conjugates as Tubulin Polymerization Inhibitors and Apoptotic Inducers. Arab. J. Chem. 2019, 12, 4740–4755. [Google Scholar] [CrossRef]
- Wang, C.; Li, Y.; Liu, T.; Wang, Z.; Zhang, Y.; Bao, K.; Wu, Y.; Guan, Q.; Zuo, D.; Zhang, W. Design, Synthesis and Evaluation of Antiproliferative and Antitubulin Activities of 5-Methyl-4-Aryl-3-(4-Arylpiperazine-1-Carbonyl)-4H-1,2,4-Triazoles. Bioorg. Chem. 2020, 104, 103909. [Google Scholar] [CrossRef]
- Yang, F.; Jian, X.-E.; Diao, P.-C.; Huo, X.-S.; You, W.-W.; Zhao, P.-L. Synthesis, and Biological Evaluation of 3,6-Diaryl-[1,2,4]Triazolo[4,3-a]Pyridine Analogues as New Potent Tubulin Polymerization Inhibitors. Eur. J. Med. Chem. 2020, 204, 112625. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Jian, X.-E.; Chen, Z.-R.; Chen, L.; Huo, X.-S.; Li, Z.-H.; You, W.-W.; Rao, J.-J.; Zhao, P.-L. Synthesis and Biological Evaluation of Benzofuran-Based 3,4,5-Trimethoxybenzamide Derivatives as Novel Tubulin Polymerization Inhibitors. Bioorg. Chem. 2020, 102, 104076. [Google Scholar] [CrossRef]
- Huang, L.; Liu, M.; Man, S.; Ma, D.; Feng, D.; Sun, Z.; Guan, Q.; Zuo, D.; Wu, Y.; Zhang, W.; et al. Design, Synthesis and Bio-Evaluation of Novel 2-Aryl-4-(3,4,5-Trimethoxy-Benzoyl)-5-Substituted-1,2,3-Triazoles as the Tubulin Polymerization Inhibitors. Eur. J. Med. Chem. 2020, 186, 111846. [Google Scholar] [CrossRef]
- Diao, P.-C.; Jian, X.-E.; Chen, P.; Huang, C.; Yin, J.; Huang, J.C.; Li, J.-S.; Zhao, P.-L. Design, Synthesis and Biological Evaluation of Novel Indole-Based Oxalamide and Aminoacetamide Derivatives as Tubulin Polymerization Inhibitors. Bioorg. Med. Chem. Lett. 2020, 30, 126816. [Google Scholar] [CrossRef] [PubMed]
- Oliva, P.; Romagnoli, R.; Manfredini, S.; Brancale, A.; Ferla, S.; Hamel, E.; Ronca, R.; Maccarinelli, F.; Giacomini, A.; Rruga, F.; et al. Design, Synthesis, in Vitro and in Vivo Biological Evaluation of 2-Amino-3-Aroylbenzo[b]Furan Derivatives as Highly Potent Tubulin Polymerization Inhibitors. Eur. J. Med. Chem. 2020, 200, 112448. [Google Scholar] [CrossRef]
- Shao, Y.-Y.; Yin, Y.; Lian, B.-P.; Leng, J.-F.; Xia, Y.-Z.; Kong, L.-Y. Synthesis and Biological Evaluation of Novel Shikonin-Benzo[b]Furan Derivatives as Tubulin Polymerization Inhibitors Targeting the Colchicine Binding Site. Eur. J. Med. Chem. 2020, 190, 112105. [Google Scholar] [CrossRef] [PubMed]
- Romagnoli, R.; Prencipe, F.; Oliva, P.; Kimatrai Salvador, M.; Brancale, A.; Ferla, S.; Hamel, E.; Viola, G.; Bortolozzi, R.; Persoons, L.; et al. Design, Synthesis and Biological Evaluation of 2-Alkoxycarbonyl-3-Anilinoindoles as a New Class of Potent Inhibitors of Tubulin Polymerization. Bioorg. Chem. 2020, 97, 103665. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Cheng, J.; Liang, Y.; Wang, Y. Discovery of a Chiral Fluorinated Azetidin-2-One as a Tubulin Polymerisation Inhibitor with Potent Antitumour Efficacy. Eur. J. Med. Chem. 2020, 197, 112323. [Google Scholar] [CrossRef] [PubMed]
- Mirzaei, S.; Eisvand, F.; Hadizadeh, F.; Mosaffa, F.; Ghasemi, A.; Ghodsi, R. Design, Synthesis and Biological Evaluation of Novel 5,6,7-Trimethoxy-N-Aryl-2-Styrylquinolin-4-Amines as Potential Anticancer Agents and Tubulin Polymerization Inhibitors. Bioorg. Chem. 2020, 98, 103711. [Google Scholar] [CrossRef]
- Li, G.; Wang, Y.; Li, L.; Ren, Y.; Deng, X.; Liu, J.; Wang, W.; Luo, M.; Liu, S.; Chen, J. Design, Synthesis, and Bioevaluation of Pyrazolo[1,5-a]Pyrimidine Derivatives as Tubulin Polymerization Inhibitors Targeting the Colchicine Binding Site with Potent Anticancer Activities. Eur. J. Med. Chem. 2020, 202, 112519. [Google Scholar] [CrossRef]
- Poornima, B.; Siva, B.; Venkanna, A.; Shankaraiah, G.; Jain, N.; Yadav, D.K.; Misra, S.; Babu, K.S. Novel Gomisin B Analogues as Potential Cytotoxic Agents: Design, Synthesis, Biological Evaluation and Docking Studies. Eur. J. Med. Chem. 2017, 139, 441–453. [Google Scholar] [CrossRef]
- Martino, E.; Casamassima, G.; Castiglione, S.; Cellupica, E.; Pantalone, S.; Papagni, F.; Rui, M.; Siciliano, A.M.; Collina, S. Vinca Alkaloids and Analogues as Anti-Cancer Agents: Looking Back, Peering Ahead. Bioorg. Med. Chem. Lett. 2018, 28, 2816–2826. [Google Scholar] [CrossRef]
- Maklad, R.M.; AbdelHafez, E.-S.M.N.; Abdelhamid, D.; Aly, O.M. Tubulin Inhibitors: Discovery of a New Scaffold Targeting Extra-Binding Residues within the Colchicine Site through Anchoring Substituents Properly Adapted to Their Pocket by a Semi-Flexible Linker. Bioorg. Chem. 2020, 99, 103767. [Google Scholar] [CrossRef]
- Weininger, D. SMILES, a Chemical Language and Information System. 1. Introduction to Methodology and Encoding Rules. J. Chem. Inf. Model. 1988, 28, 31–36. [Google Scholar] [CrossRef]
- Chemical Computing Group Inc. Molecular Operating Environment (MOE); Chemical Computing Group Inc.: Montreal, QC, Canada, 2020; Available online: http://www.chemcomp.com (accessed on 8 January 2021).
- Van der Maaten, L.; Hinton, G. Visualizing Data Using T-SNE. J. Mach. Learn. Res. 2008, 9, 2579–2605. [Google Scholar]
- López-López, E.; Naveja, J.J.; Medina-Franco, J.L. DataWarrior: An Evaluation of the Open-Source Drug Discovery Tool. Expert Opin. Drug Dis. 2019, 14, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Medina-Franco, J.L. Scanning Structure–Activity Relationships with Structure–Activity Similarity and Related Maps: From Consensus Activity Cliffs to Selectivity Switches. J. Chem. Inf. Model. 2012, 52, 2485–2493. [Google Scholar] [CrossRef]
- Rogers, D.; Hahn, M. Extended-Connectivity Fingerprints. J. Chem. Inf. Model. 2010, 50, 742–754. [Google Scholar] [CrossRef] [PubMed]
- Guha, R.; Van Drie, J.H. Structure−Activity Landscape Index: Identifying and Quantifying Activity Cliffs. J. Chem. Inf. Model. 2008, 48, 646–658. [Google Scholar] [CrossRef] [PubMed]
- González-Medina, M.; Méndez-Lucio, O.; Medina-Franco, J.L. Activity Landscape Plotter: A Web-Based Application for the Analysis of Structure–Activity Relationships. J. Chem. Inf. Model. 2017, 57, 397–402. [Google Scholar] [CrossRef] [PubMed]
- Bemis, G.W.; Murcko, M.A. The Properties of Known Drugs. 1. Molecular Frameworks. J. Med. Chem. 1996, 39, 2887–2893. [Google Scholar] [CrossRef] [PubMed]
- Berthold, M.R.; Cebron, N.; Dill, F.; Gabriel, T.R.; Kötter, T.; Meinl, T.; Ohl, P.; Sieb, C.; Thiel, K.; Wiswedel, B. KNIME: The Konstanz Information Miner. In Data Analysis, Machine Learning and Applications; Studies in Classification, Data Analysis; Preisach, C., Burkhardt, H., Schmidt-Thieme, L., Decker, R., Eds.; Springer: Berlin/Heidelberg, Germany, 2008; pp. 319–326. ISBN 978-3-540-78239-1. [Google Scholar]
- Medina-Franco, J.L.; Naveja, J.J.; López-López, E. Reaching for the Bright StARs in Chemical Space. Drug Discov. Today 2019, 24, 2162–2169. [Google Scholar] [CrossRef]
- Naveja, J.J.; Medina-Franco, J.L. Finding Constellation in Chemical Space Trough Core Analysis. Front. Chem. 2019, 7, 510. [Google Scholar] [CrossRef]
- Naveja, J.J.; Vogt, M.; Stumpfe, D.; Medina-Franco, J.L.; Bajorath, J. Systematic Extraction of Analogue Series from Large Compound Collections Using a New Computational Compound–Core Relationship Method. ACS Omega 2019, 4, 1027–1032. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
López-López, E.; Cerda-García-Rojas, C.M.; Medina-Franco, J.L. Tubulin Inhibitors: A Chemoinformatic Analysis Using Cell-Based Data. Molecules 2021, 26, 2483. https://doi.org/10.3390/molecules26092483
López-López E, Cerda-García-Rojas CM, Medina-Franco JL. Tubulin Inhibitors: A Chemoinformatic Analysis Using Cell-Based Data. Molecules. 2021; 26(9):2483. https://doi.org/10.3390/molecules26092483
Chicago/Turabian StyleLópez-López, Edgar, Carlos M. Cerda-García-Rojas, and José L. Medina-Franco. 2021. "Tubulin Inhibitors: A Chemoinformatic Analysis Using Cell-Based Data" Molecules 26, no. 9: 2483. https://doi.org/10.3390/molecules26092483
APA StyleLópez-López, E., Cerda-García-Rojas, C. M., & Medina-Franco, J. L. (2021). Tubulin Inhibitors: A Chemoinformatic Analysis Using Cell-Based Data. Molecules, 26(9), 2483. https://doi.org/10.3390/molecules26092483