
Recent Developments in New Therapeutic Agents against Alzheimer and Parkinson Diseases: In-Silico Approaches

 ,
,  ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Alzheimer’s Disease
2.1. Acetylcholinesterase
2.2. N-Methyl-D-aspartate Receptor
2.3. Secretases
2.3.1. β-Secretase
2.3.2. γ-Secretase
2.4. Sirtuins
2.5. Caspases
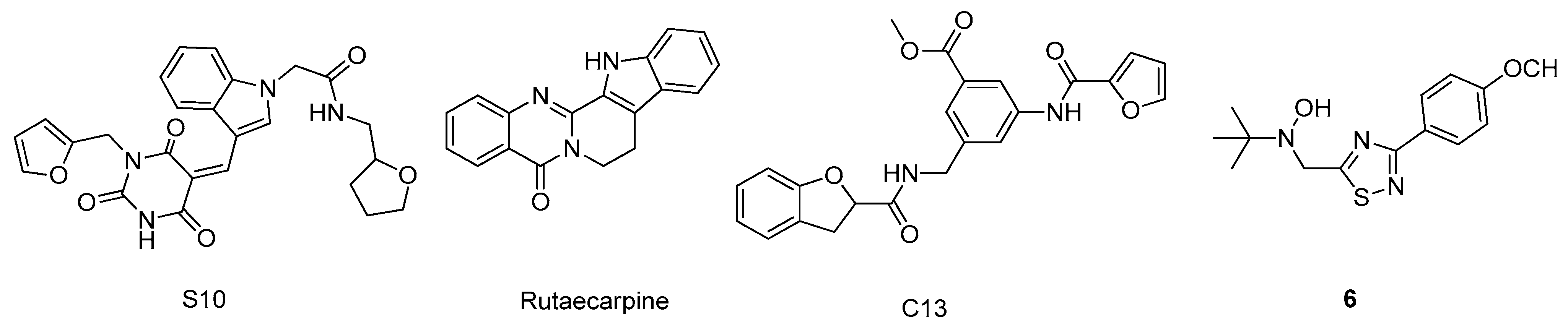
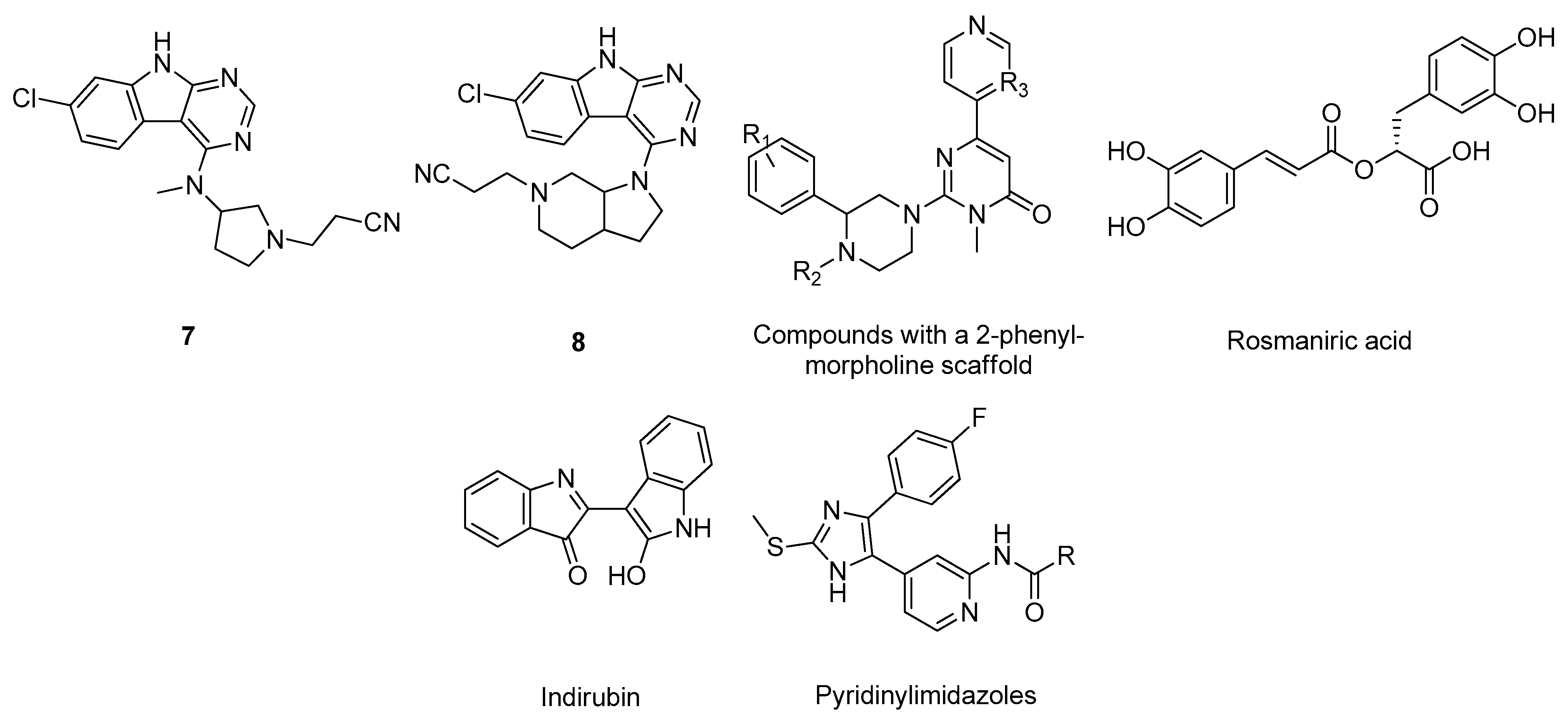
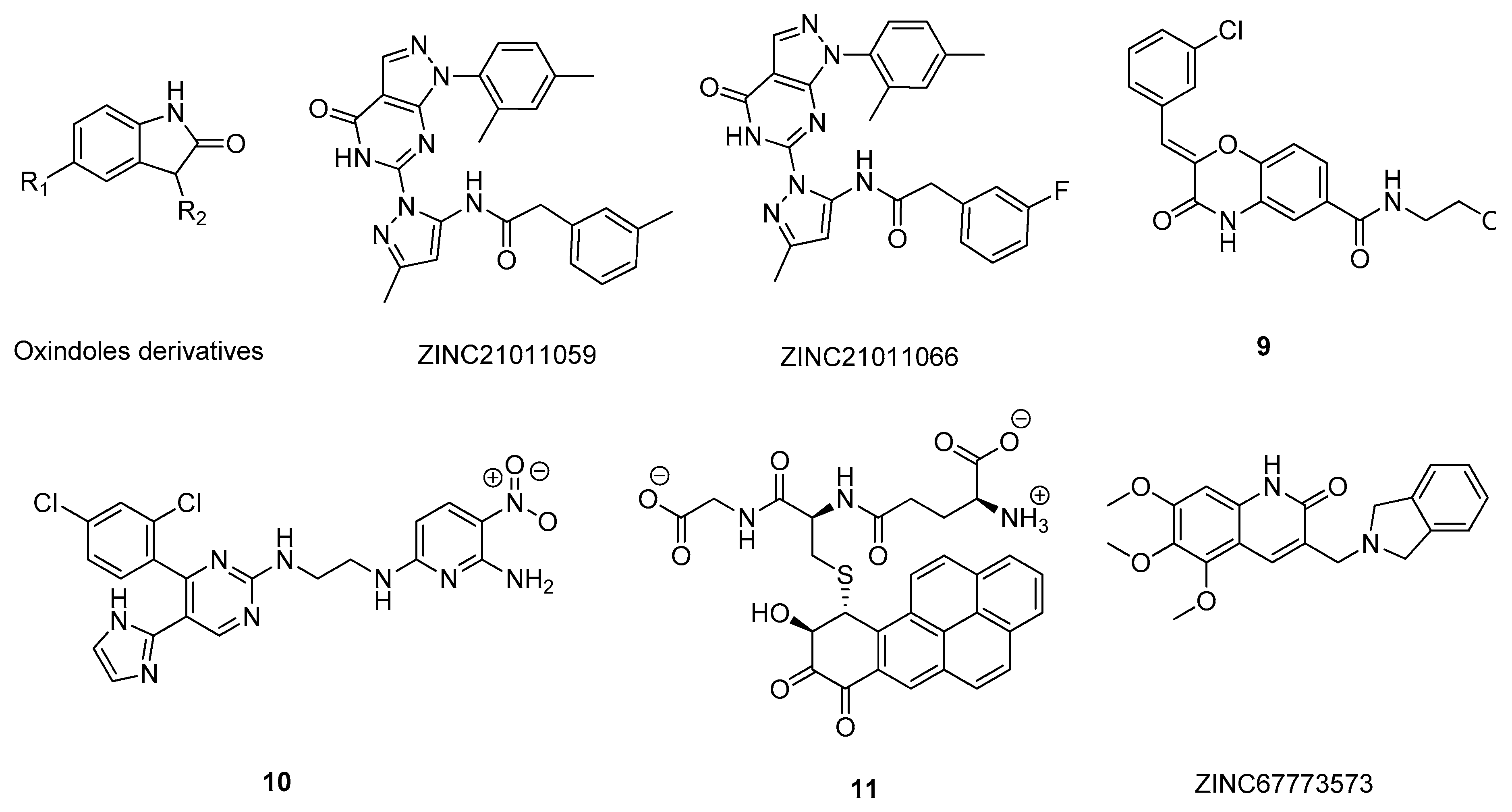
2.6. Glycogen Synthase Kinase-3
3. Parkinson’s Disease
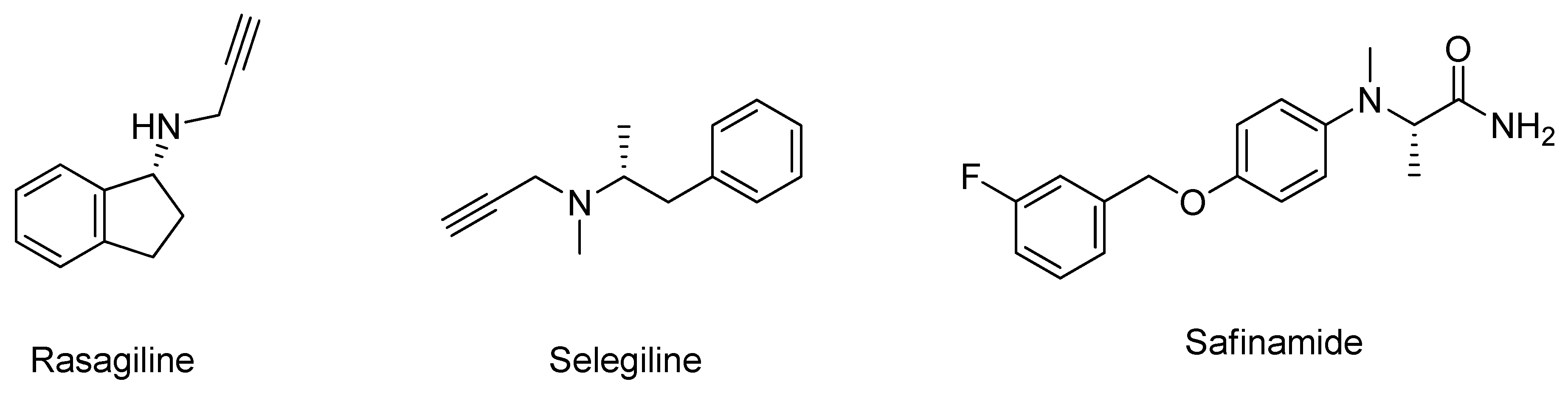
3.1. Monoamine Oxidase Type B Inhibitors
3.2. Dopamine Agonists
3.3. Adenosine Receptors Antagonists
3.4. Catechol-O-methyltransferase Inhibitors
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- World Population Ageing [highlights], (n.d.). Available online: https://www.un.org/en/development/desa/population/publications/pdf/ageing/WPA2017_Highlights.pdf (accessed on 2 March 2021).
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef]
- Van Bulck, M.; Sierra-Magro, A.; Alarcon-Gil, J.; Perez-Castillo, A.; Morales-Garcia, J. Novel Approaches for the Treatment of Alzheimer’s and Parkinson’s Disease. Int. J. Mol. Sci. 2019, 20, 719. [Google Scholar] [CrossRef]
- Feigin, V.L.; Nichols, E.; Alam, T.; Bannick, M.S.; Beghi, E.; Blake, N.; Culpepper, W.J.; Dorsey, E.R.; Elbaz, A.; Ellenbogen, R.G.; et al. Global, regional, and national burden of neurological disorders, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 459–480. [Google Scholar] [CrossRef]
- Gale, S.A.; Acar, D.; Daffner, K.R. Dementia. Am. J. Med. 2018, 131, 1161–1169. [Google Scholar] [CrossRef] [PubMed]
- McFarthing, K.; Buff, S.; Rafaloff, G.; Dominey, T.; Wyse, R.K.; Stott, S.R.W. Parkinson’s Disease Drug Therapies in the Clinical Trial Pipeline: 2020. J. Parkinsons. Dis. 2020, 10, 757–774. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.; Lee, G.; Ritter, A.; Sabbagh, M.; Zhong, K. Alzheimer’s disease drug development pipeline: 2020. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2020, 6. [Google Scholar] [CrossRef]
- Goodsell, D.S.; Zardecki, C.; Di Costanzo, L.; Duarte, J.M.; Hudson, B.P.; Persikova, I.; Segura, J.; Shao, C.; Voigt, M.; Westbrook, J.D.; et al. RCSB Protein Data Bank: Enabling biomedical research and drug discovery. Protein Sci. 2020, 29, 52–65. [Google Scholar] [CrossRef]
- Sehgal, S.A.; Hammad, M.A.; Tahir, R.A.; Akram, H.N.; Ahmad, F. Current Therapeutic Molecules and Targets in Neurodegenerative Diseases Based on in silico Drug Design. Curr. Neuropharm. 2018, 16, 649–663. [Google Scholar] [CrossRef]
- Soria Lopez, J.A.; González, H.M.; Léger, G.C. Alzheimer’s disease. In Handbook of Clinical Neurology; Elsevier B.V.: Amsterdam, The Netherlands, 2019; Volume 167, pp. 231–255. [Google Scholar]
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s disease. Eur. J. Neurol. 2018, 25, 59–70. [Google Scholar] [CrossRef]
- Vaz, M.; Silvestre, S. Alzheimer’s disease: Recent treatment strategies. Eur. J. Pharmacol. 2020, 887, 173554. [Google Scholar] [CrossRef]
- Silva, T.; Reis, J.; Teixeira, J.; Borges, F. Alzheimer’s disease, enzyme targets and drug discovery struggles: From natural products to drug prototypes. Ageing Res. Rev. 2014, 15, 116–145. [Google Scholar] [CrossRef]
- Schliebs, R.; Arendt, T. The cholinergic system in aging and neuronal degeneration. Behav. Brain Res. 2011, 221, 555–563. [Google Scholar] [CrossRef]
- Colovic, M.B.; Krstic, D.Z.; Lazarevic-Pasti, T.D.; Bondzic, A.M.; Vasic, V.M. Acetylcholinesterase Inhibitors: Pharmacology and Toxicology. Curr. Neuropharmacol. 2013, 11, 315–335. [Google Scholar] [CrossRef] [PubMed]
- Dvir, H.; Silman, I.; Harel, M.; Rosenberry, T.L.; Sussman, J.L. Acetylcholinesterase: From 3D structure to function. Chem. Biol. Interact. 2010, 187, 10–22. [Google Scholar] [CrossRef]
- Raves, M.L.; Harel, M.; Pang, Y.-P.; Silman, I.; Kozikowski, A.P.; Sussman, J.L. Structure of acetylcholinesterase complexed with the nootropic alkaloid, (–)-huperzine A. Nat. Struct. Mol. Biol. 1997, 4, 57–63. [Google Scholar] [CrossRef] [PubMed]
- da Silva Mesquita, R.; Kyrylchuk, A.; Costa de Oliveira, R.; Costa Sá, I.S.; Coutinho Borges Camargo, G.; Soares Pontes, G.; Moura Araújo da Silva, F.; de Saraiva Nunomura, R.C.; Grafov, A. Alkaloids of Abuta panurensis Eichler: In silico and in vitro study of acetylcholinesterase inhibition, cytotoxic and immunomodulatory activities. PLoS ONE 2020, 15, e0239364. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, J.E.; Garro, A.; Pigni, N.B.; Agüero, M.B.; Roitman, G.; Slanis, A.; Enriz, R.D.; Feresin, G.E.; Bastida, J.; Tapia, A. Cholinesterase-inhibitory effect and in silico analysis of alkaloids from bulbs of Hieronymiella species. Phytomedicine 2018, 39, 66–74. [Google Scholar] [CrossRef] [PubMed]
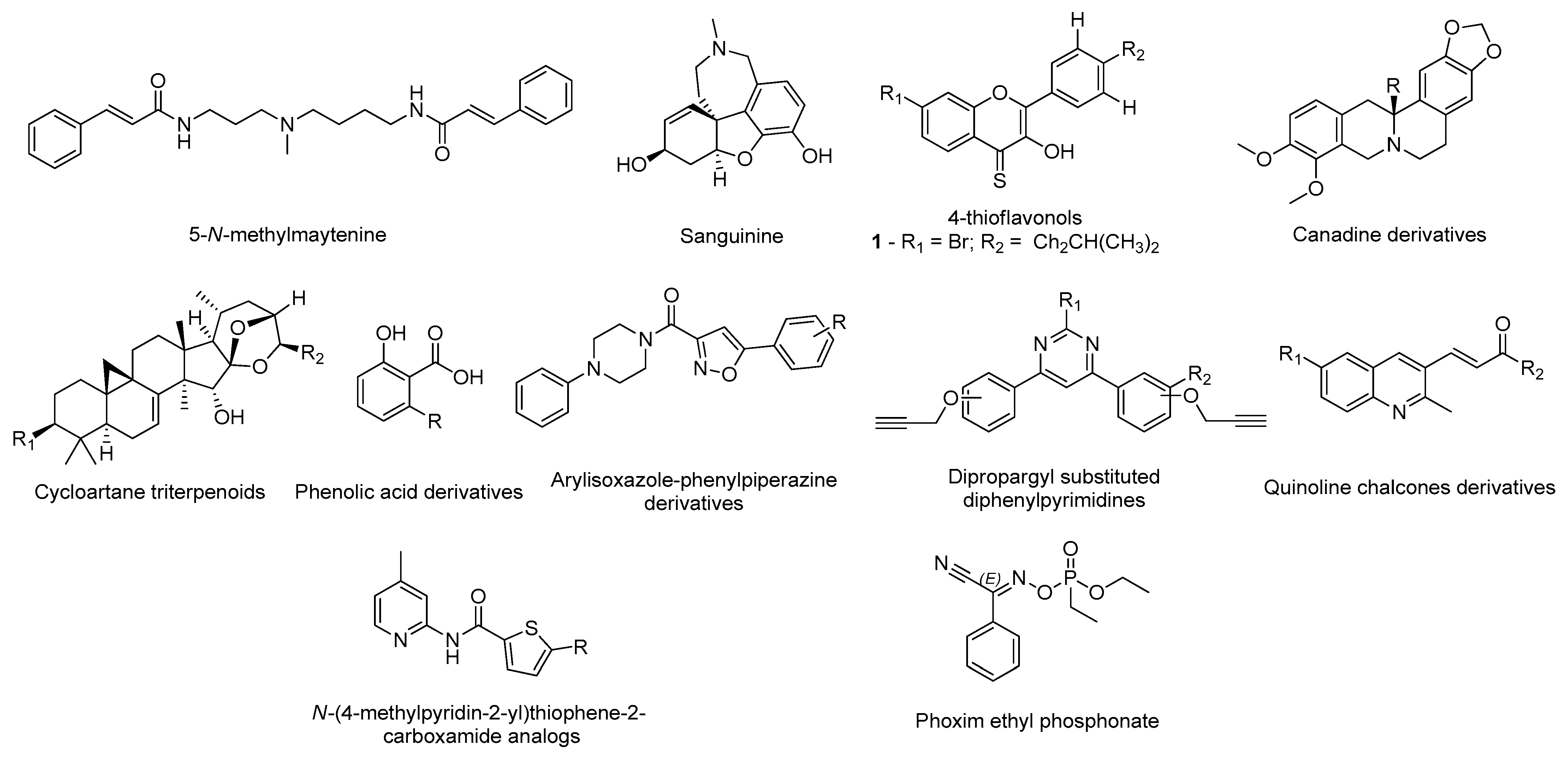
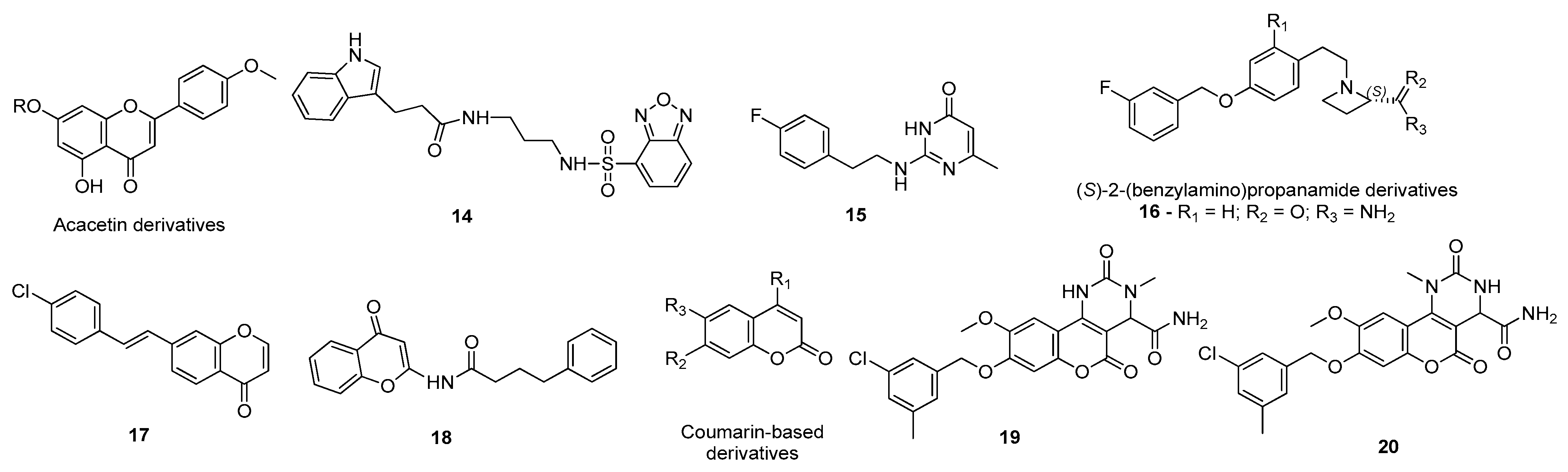
- Mughal, E.U.; Sadiq, A.; Ashraf, J.; Zafar, M.N.; Sumrra, S.H.; Tariq, R.; Mumtaz, A.; Javid, A.; Khan, B.A.; Ali, A.; et al. Flavonols and 4-thioflavonols as potential acetylcholinesterase and butyrylcholinesterase inhibitors: Synthesis, structure-activity relationship and molecular docking studies. Bioorg. Chem. 2019, 91, 103124. [Google Scholar] [CrossRef]
- Chlebek, J.; Korábečný, J.; Doležal, R.; Štěpánková, Š.; Pérez, D.; Hošťálková, A.; Opletal, L.; Cahlíková, L.; Macáková, K.; Kučera, T.; et al. In Vitro and In Silico Acetylcholinesterase Inhibitory Activity of Thalictricavine and Canadine and Their Predicted Penetration across the Blood-Brain Barrier. Molecules 2019, 24, 1340. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Thao, N.P.; Han, Y.K.; Lee, Y.S.; Luyen, B.T.T.; Van Oanh, H.; Kim, Y.H.; Yang, S.Y. The insight of in vitro and in silico studies on cholinesterase inhibitors from the roots of Cimicifuga dahurica (Turcz.) Maxim. J. Enzym. Inhib. Med. Chem. 2018, 33, 1174–1180. [Google Scholar] [CrossRef] [PubMed]
- Kiametis, A.S.; Silva, M.A.; Romeiro, L.A.S.; Martins, J.B.L.; Gargano, R. Potential acetylcholinesterase inhibitors: Molecular docking, molecular dynamics, and in silico prediction. J. Mol. Model. 2017, 23, 67. [Google Scholar] [CrossRef]
- Saeedi, M.; Mohtadi-Haghighi, D.; Mirfazli, S.S.; Mahdavi, M.; Hariri, R.; Lotfian, H.; Edraki, N.; Iraji, A.; Firuzi, O.; Akbarzadeh, T. Design and Synthesis of Selective Acetylcholinesterase Inhibitors: Arylisoxazole-Phenylpiperazine Derivatives. Chem. Biodivers. 2019, 16, e1800433. [Google Scholar] [CrossRef]
- Kumar, B.; Kumar, V.; Prashar, V.; Saini, S.; Dwivedi, A.R.; Bajaj, B.; Mehta, D.; Parkash, J.; Kumar, V. Dipropargyl substituted diphenylpyrimidines as dual inhibitors of monoamine oxidase and acetylcholinesterase. Eur. J. Med. Chem. 2019, 177, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.S.; Najam-ul-Haq, M.; Shah, H.S.; Farooq Rizvi, S.U.; Iqbal, J. Quinoline containing chalcone derivatives as cholinesterase inhibitors and their in silico modeling studies. Comput. Biol. Chem. 2018, 76, 310–317. [Google Scholar] [CrossRef]
- Ahmad, G.; Rasool, N.; Rizwan, K.; Imran, I.; Zahoor, A.F.; Zubair, M.; Sadiq, A.; Rashid, U. Synthesis, in-vitro cholinesterase inhibition, in-vivo anticonvulsant activity and in-silico exploration of N-(4-methylpyridin-2-yl)thiophene-2-carboxamide analogs. Bioorg. Chem. 2019, 92, 103216. [Google Scholar] [CrossRef]
- Ranjan, A.; Chauhan, A.; Jindal, T. In-silico and in-vitro evaluation of human acetylcholinesterase inhibition by organophosphates. Environ. Toxicol. Pharmacol. 2018, 57, 131–140. [Google Scholar] [CrossRef]
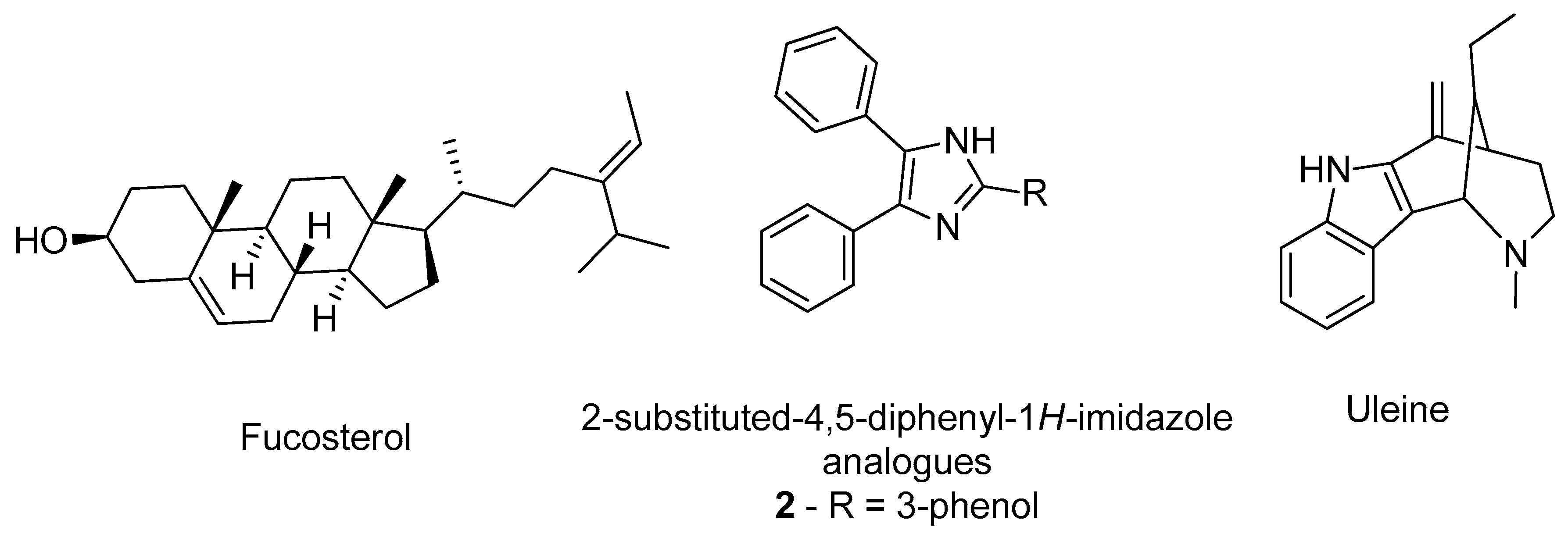
- Castro-Silva, E.S.; Bello, M.; Hernández-Rodríguez, M.; Correa-Basurto, J.; Murillo-Álvarez, J.I.; Rosales-Hernández, M.C.; Muñoz-Ochoa, M. In vitro and in silico evaluation of fucosterol from Sargassum horridum as potential human acetylcholinesterase inhibitor. J. Biomol. Struct. Dyn. 2019, 37, 3259–3268. [Google Scholar] [CrossRef]
- Gurjar, A.S.; Darekar, M.N.; Yeong, K.Y.; Ooi, L. In silico studies, synthesis and pharmacological evaluation to explore multi-targeted approach for imidazole analogues as potential cholinesterase inhibitors with neuroprotective role for Alzheimer’s disease. Bioorg. Med. Chem. 2018, 26, 1511–1522. [Google Scholar] [CrossRef] [PubMed]
- Pereira Rocha, M.; Rodrigues Valadares Campana, P.; de Oliveira Scoaris, D.; de Almeida, V.L.; Dias Lopes, J.C.; Fonseca Silva, A.; Pieters, L.; Gontijo Silva, C. Biological activities of extracts from Aspidosperma subincanum Mart. and in silico prediction for inhibition of acetylcholinesterase. Phyther. Res. 2018, 32, 2021–2033. [Google Scholar] [CrossRef] [PubMed]
- Niu, B.; Zhao, M.; Su, Q.; Zhang, M.; Lv, W.; Chen, Q.; Chen, F.; Chu, D.; Du, D.; Zhang, Y. 2D-SAR and 3D-QSAR analyses for acetylcholinesterase inhibitors. Mol. Divers. 2017, 21, 413–426. [Google Scholar] [CrossRef]
- Wang, R.; Reddy, P.H. Role of Glutamate and NMDA Receptors in Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 57, 1041–1048. [Google Scholar] [CrossRef] [PubMed]
- Yiannopoulou, K.G.; Papageorgiou, S.G. Current and future treatments for Alzheimer’s disease. Ther. Adv. Neurol. Disord. 2013, 6, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Shearman, E.; Rossi, S.; Szasz, B.; Juranyi, Z.; Fallon, S.; Pomara, N.; Sershen, H.; Lajtha, A. Changes in cerebral neurotransmitters and metabolites induced by acute donepezil and memantine administrations: A microdialysis study. Brain Res. Bull. 2006, 69, 204–213. [Google Scholar] [CrossRef]
- Ivanova, L.; Karelson, M.; Dobchev, D. Identification of Natural Compounds against Neurodegenerative Diseases Using In Silico Techniques. Molecules 2018, 23, 1847. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Mittal, A.; Singh, A.; Jainarayanan, A.K.; Sharma, S.; Paliwal, S. Pharmacophore-driven identification of N-methyl-D-receptor antagonists as potent neuroprotective agents validated using in vivo studies. Biol. Methods Protoc. 2020, 5. [Google Scholar] [CrossRef]
- Waqar, M.; Batool, S. In silico analysis of binding interaction of conantokins with NMDA receptors for potential therapeutic use in Alzheimer’s disease. J. Venom. Anim. Toxins Incl. Trop. Dis. 2017, 23, 42. [Google Scholar] [CrossRef]
- Hu, S.; Hu, H.; Mak, S.; Cui, G.; Lee, M.; Shan, L.; Wang, Y.; Lin, H.; Zhang, Z.; Han, Y. A Novel Tetramethylpyrazine Derivative Prophylactically Protects against Glutamate-Induced Excitotoxicity in Primary Neurons through the Blockage of N-Methyl-D-aspartate Receptor. Front. Pharmacol. 2018, 9. [Google Scholar] [CrossRef]
- Kumar, S.; Chowdhury, S.; Kumar, S. In silico repurposing of antipsychotic drugs for Alzheimer’s disease. BMC Neurosci. 2017, 18, 76. [Google Scholar] [CrossRef]
- Singh, R.; Ganeshpurkar, A.; Kumar, D.; Kumar, D.; Kumar, A.; Singh, S.K. Identifying potential GluN2B subunit containing N-Methyl-D-aspartate receptor inhibitors: An integrative in silico and molecular modeling approach. J. Biomol. Struct. Dyn. 2020, 38, 2533–2545. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P. Pathways towards and away from Alzheimer’s disease. Nature 2004, 430, 631–639. [Google Scholar] [CrossRef]
- Imbimbo, B.P. Alzheimer’s disease: γ-secretase inhibitors. Drug Discov. Today Ther. Strateg. 2008, 5, 169–175. [Google Scholar] [CrossRef]
- Boddapati, S.; Levites, Y.; Sierks, M.R. Inhibiting β-Secretase Activity in Alzheimer’s Disease Cell Models with Single-Chain Antibodies Specifically Targeting APP. J. Mol. Biol. 2011, 405, 436–447. [Google Scholar] [CrossRef]
- De Strooper, B.; Vassar, R.; Golde, T. The secretases: Enzymes with therapeutic potential in Alzheimer disease. Nat. Rev. Neurol. 2010, 6, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Cole, S.L.; Vassar, R. The Alzheimer’s disease Beta-secretase enzyme, BACE1. Mol. Neurodegener. 2007, 2, 22. [Google Scholar] [CrossRef]
- Citron, M. Emerging Alzheimer’s disease therapies: Inhibition of β-secretase. Neurobiol. Aging 2002, 23, 1017–1022. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Brindisi, M.; Tang, J. Developing β-secretase inhibitors for treatment of Alzheimer’s disease. J. Neurochem. 2012, 120, 71–83. [Google Scholar] [CrossRef] [PubMed]
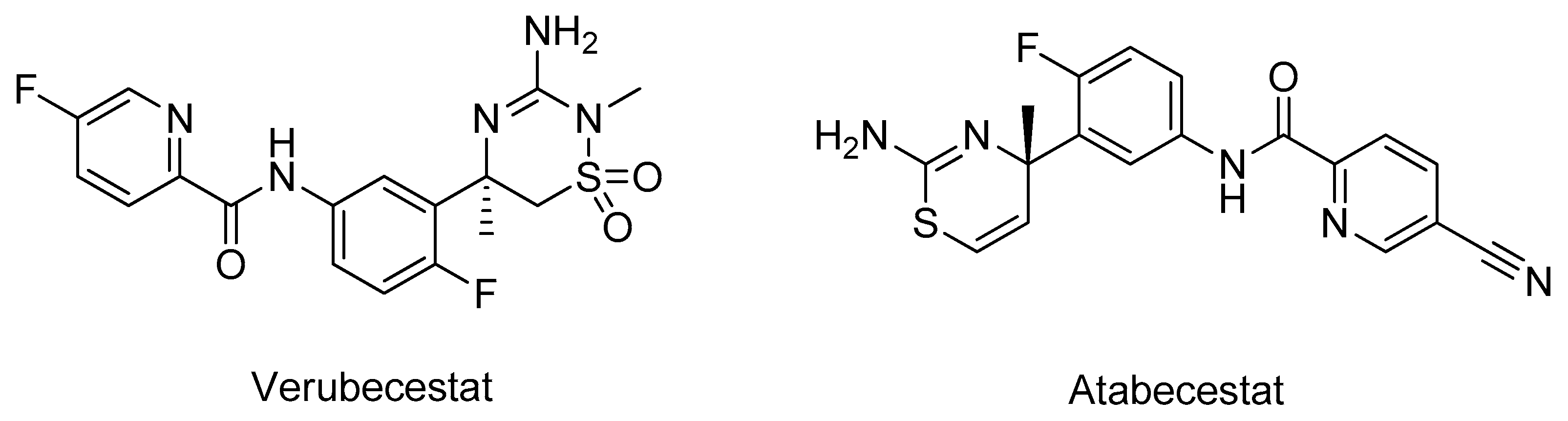
- Moussa-Pacha, N.M.; Abdin, S.M.; Omar, H.A.; Alniss, H.; Al-Tel, T.H. BACE1 inhibitors: Current status and future directions in treating Alzheimer’s disease. Med. Res. Rev. 2020, 40, 339–384. [Google Scholar] [CrossRef] [PubMed]
- Gueto-Tettay, C.; Martinez-Consuegra, A.; Zuchniarz, J.; Gueto-Tettay, L.R.; Drosos-Ramírez, J.C. A PM7 dynamic residue-ligand interactions energy landscape of the BACE1 inhibitory pathway by hydroxyethylamine compounds. Part I: The flap closure process. J. Mol. Graph. Model. 2017, 76, 274–288. [Google Scholar] [CrossRef]
- Vitale, R.M.; Rispoli, V.; Desiderio, D.; Sgammato, R.; Thellung, S.; Canale, C.; Vassalli, M.; Carbone, M.; Ciavatta, M.L.; Mollo, E.; et al. In Silico Identification and Experimental Validation of Novel Anti-Alzheimer’s Multitargeted Ligands from a Marine Source Featuring a “2-Aminoimidazole plus Aromatic Group” Scaffold. ACS Chem. Neurosci. 2018, 9, 1290–1303. [Google Scholar] [CrossRef]
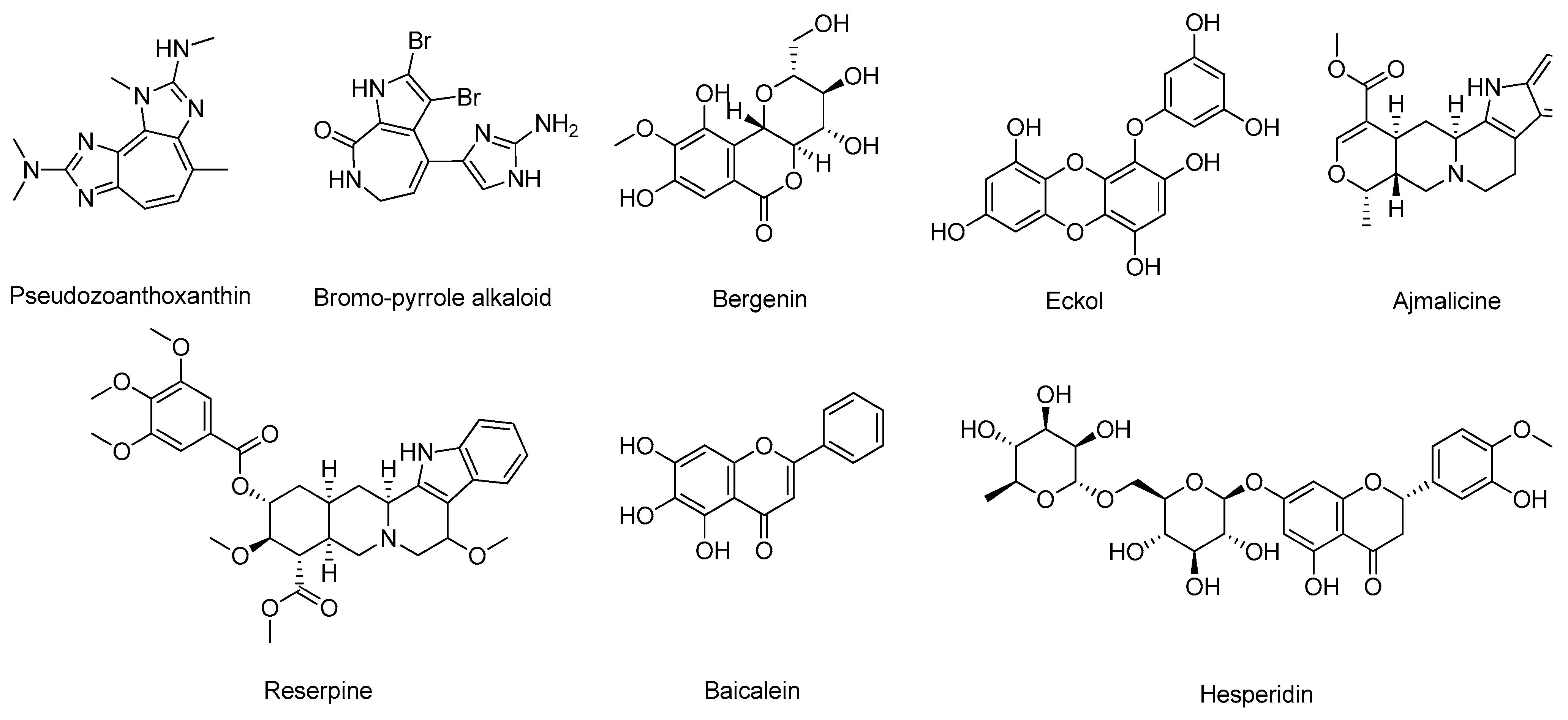
- Barai, P.; Raval, N.; Acharya, S.; Borisa, A.; Bhatt, H.; Acharya, N. Neuroprotective effects of bergenin in Alzheimer’s disease: Investigation through molecular docking, in vitro and in vivo studies. Behav. Brain Res. 2019, 356, 18–40. [Google Scholar] [CrossRef]
- Lee, J.; Jun, M. Dual BACE1 and Cholinesterase Inhibitory Effects of Phlorotannins from Ecklonia cava—An In Vitro and in Silico Study. Mar. Drugs 2019, 17, 91. [Google Scholar] [CrossRef]
- Kashyap, P.; Kalaiselvan, V.; Kumar, R.; Kumar, S. Ajmalicine and Reserpine: Indole Alkaloids as Multi-Target Directed Ligands Towards Factors Implicated in Alzheimer’s Disease. Molecules 2020, 25, 1609. [Google Scholar] [CrossRef]
- Han, J.; Ji, Y.; Youn, K.; Lim, G.; Lee, J.; Kim, D.H.; Jun, M. Baicalein as a Potential Inhibitor against BACE1 and AChE: Mechanistic Comprehension through In Vitro and Computational Approaches. Nutrients 2019, 11, 2694. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Youn, K.; Lim, G.; Lee, J.; Jun, M. In Silico Docking and In Vitro Approaches towards BACE1 and Cholinesterases Inhibitory Effect of Citrus Flavanones. Molecules 2018, 23, 1509. [Google Scholar] [CrossRef]
- Tran, T.-S.; Tran, T.-D.; Tran, T.-H.; Mai, T.-T.; Nguyen, N.-L.; Thai, K.-M.; Le, M.-T. Synthesis, In Silico and In Vitro Evaluation of Some Flavone Derivatives for Acetylcholinesterase and BACE-1 Inhibitory Activity. Molecules 2020, 25, 4064. [Google Scholar] [CrossRef] [PubMed]
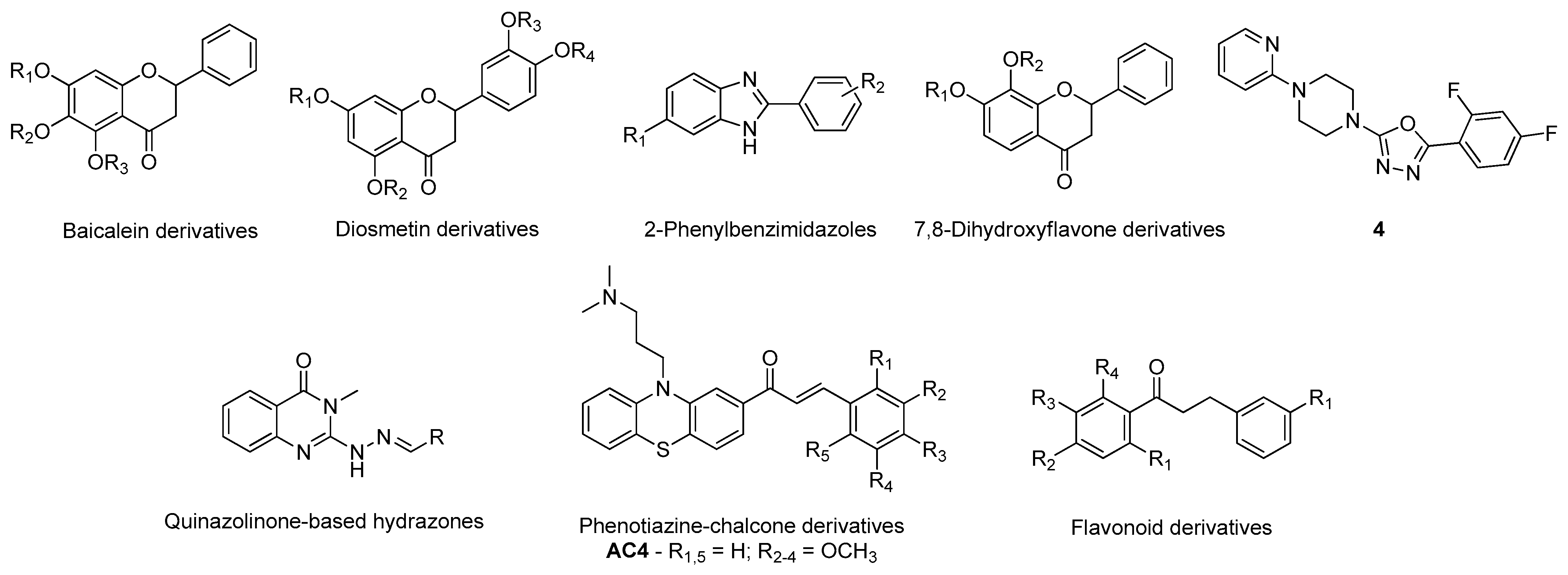
- Ali, S.; Asad, M.H.H.B.; Maity, S.; Zada, W.; Rizvanov, A.A.; Iqbal, J.; Babak, B.; Hussain, I. Fluoro-benzimidazole derivatives to cure Alzheimer’s disease: In-silico studies, synthesis, structure-activity relationship and in vivo evaluation for β secretase enzyme inhibition. Bioorg. Chem. 2019, 88, 102936. [Google Scholar] [CrossRef] [PubMed]
- Pandey, R.P.; Parajuli, P.; Pokhrel, A.R.; Sohng, J.K. Biosynthesis of novel 7,8-dihydroxyflavone glycoside derivatives and in silico study of their effects on BACE1 inhibition. Biotechnol. Appl. Biochem. 2018, 65, 128–137. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, A.; Choubey, P.K.; Sharma, P.; Seth, A.; Tripathi, P.N.; Tripathi, M.K.; Prajapati, S.K.; Krishnamurthy, S.; Shrivastava, S.K. Design and development of molecular hybrids of 2-pyridylpiperazine and 5-phenyl-1,3,4-oxadiazoles as potential multifunctional agents to treat Alzheimer’s disease. Eur. J. Med. Chem. 2019, 183, 111707. [Google Scholar] [CrossRef]
- Haghighijoo, Z.; Firuzi, O.; Hemmateenejad, B.; Emami, S.; Edraki, N.; Miri, R. Synthesis and biological evaluation of quinazolinone-based hydrazones with potential use in Alzheimer’s disease. Bioorg. Chem. 2017, 74, 126–133. [Google Scholar] [CrossRef]
- Tran, T.-S.; Le, M.-T.; Nguyen, T.-C.-V.; Tran, T.-H.; Tran, T.-D.; Thai, K.-M. Synthesis, In Silico and In Vitro Evaluation for Acetylcholinesterase and BACE-1 Inhibitory Activity of Some N-Substituted-4-Phenothiazine-Chalcones. Molecules 2020, 25, 3916. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, G.; Poda, G. In silico ligand-based modeling of h BACE-1 inhibitors. Chem. Biol. Drug Des. 2018, 91, 817–827. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.-S.; Le, M.-T.; Tran, T.-D.; Tran, T.-H.; Thai, K.-M. Design of Curcumin and Flavonoid Derivatives with Acetylcholinesterase and Beta-Secretase Inhibitory Activities Using in Silico Approaches. Molecules 2020, 25, 3644. [Google Scholar] [CrossRef]
- Coimbra, J.R.M.; Baptista, S.J.; Dinis, T.C.P.; Silva, M.M.C.; Moreira, P.I.; Santos, A.E.; Salvador, J.A.R. Combining Virtual Screening Protocol and In Vitro Evaluation towards the Discovery of BACE1 Inhibitors. Biomolecules 2020, 10, 535. [Google Scholar] [CrossRef] [PubMed]
- Turner, A.J. Targeting Amyloid-Degrading Enzymes as Therapeutic Strategies in Neurodegeneration. Ann. N. Y. Acad. Sci. 2004, 1035, 1–20. [Google Scholar] [CrossRef]
- Gupta, M.K.; Vadde, R. In silico identification of natural product inhibitors for γ-secretase activating protein, a therapeutic target for Alzheimer’s disease. J. Cell. Biochem. 2019, 120, 10323–10336. [Google Scholar] [CrossRef]
- Hitzenberger, M.; Zacharias, M. Uncovering the Binding Mode of γ -Secretase Inhibitors. ACS Chem. Neurosci. 2019, 10, 3398–3403. [Google Scholar] [CrossRef] [PubMed]
- Haigis, M.C.; Sinclair, D.A. Mammalian Sirtuins: Biological Insights and Disease Relevance. Annu. Rev. Pathol. Mech. Dis. 2010, 5, 253–295. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, M.M.; Tomkinson, E.M.; Nobles, J.; Wizeman, J.W.; Amore, A.M.; Quinti, L.; Chopra, V.; Hersch, S.M.; Kazantsev, A.G. The Sirtuin 2 microtubule deacetylase is an abundant neuronal protein that accumulates in the aging CNS. Hum. Mol. Genet. 2011, 20, 3986–3996. [Google Scholar] [CrossRef]
- Suzuki, T.; Khan, M.N.A.; Sawada, H.; Imai, E.; Itoh, Y.; Yamatsuta, K.; Tokuda, N.; Takeuchi, J.; Seko, T.; Nakagawa, H.; et al. Design, Synthesis, and Biological Activity of a Novel Series of Human Sirtuin-2-Selective Inhibitors. J. Med. Chem. 2012, 55, 5760–5773. [Google Scholar] [CrossRef]
- Huhtiniemi, T.; Salo, H.S.; Suuronen, T.; Poso, A.; Salminen, A.; Leppänen, J.; Jarho, E.; Lahtela-Kakkonen, M. Structure-Based Design of Pseudopeptidic Inhibitors for SIRT1 and SIRT2. J. Med. Chem. 2011, 54, 6456–6468. [Google Scholar] [CrossRef]
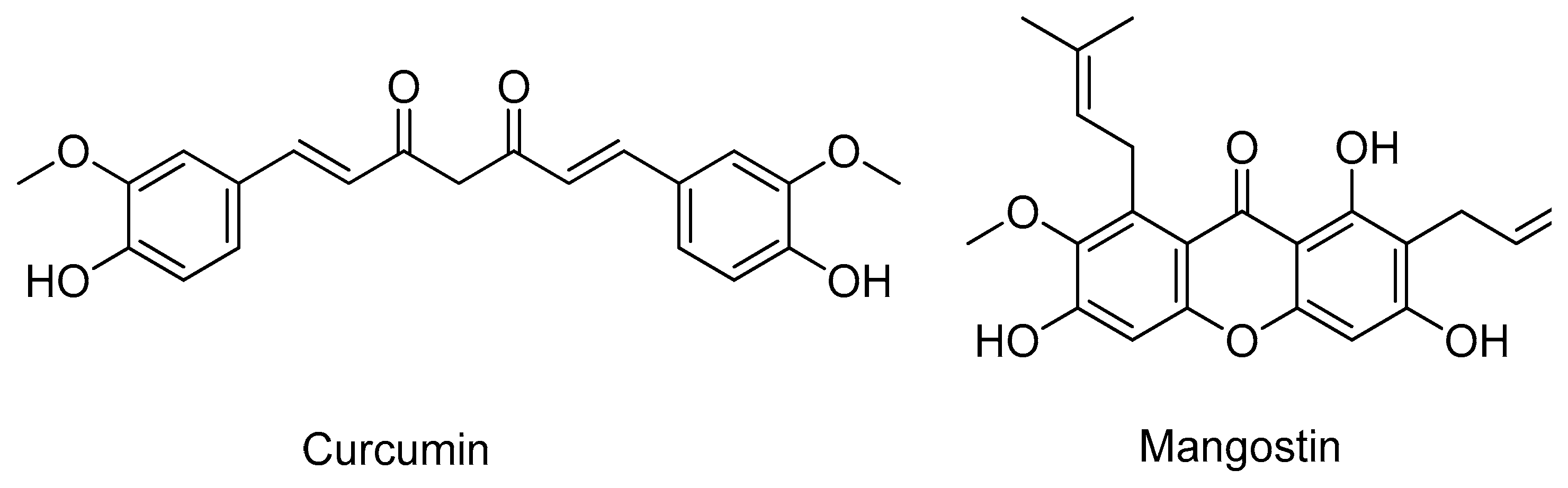
- Furlan, V.; Konc, J.; Bren, U. Inverse Molecular Docking as a Novel Approach to Study Anticarcinogenic and Anti-Neuroinflammatory Effects of Curcumin. Molecules 2018, 23, 3351. [Google Scholar] [CrossRef]
- Yeong, K.Y.; Khaw, K.Y.; Takahashi, Y.; Itoh, Y.; Murugaiyah, V.; Suzuki, T. Discovery of gamma-mangostin from Garcinia mangostana as a potent and selective natural SIRT2 inhibitor. Bioorg. Chem. 2020, 94, 103403. [Google Scholar] [CrossRef]
- Shalini, S.; Dorstyn, L.; Dawar, S.; Kumar, S. Old, new and emerging functions of caspases. Cell Death Differ. 2015, 22, 526–539. [Google Scholar] [CrossRef]
- Cavallucci, V.; D’Amelio, M. Matter of Life and Death: The Pharmacological Approaches Targeting Apoptosis in Brain Diseases. Curr. Pharm. Des. 2011, 17, 215–229. [Google Scholar] [CrossRef] [PubMed]
- Rohn, T.T.; Head, E. Caspases as therapeutic targets in Alzheimer’s disease: Is it time to “cut” to the chase? Int. J. Clin. Exp. Pathol. 2009, 2, 108–118. [Google Scholar] [PubMed]
- Tubeleviciute-Aydin, A.; Beautrait, A.; Lynham, J.; Sharma, G.; Gorelik, A.; Deny, L.J.; Soya, N.; Lukacs, G.L.; Nagar, B.; Marinier, A.; et al. Identification of Allosteric Inhibitors against Active Caspase-6. Sci. Rep. 2019, 9, 5504. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.S.; Sinha, M.; Ahmad, K.; Khalid, M.; Choi, I. Study of Caspase 8 Inhibition for the Management of Alzheimer’s Disease: A Molecular Docking and Dynamics Simulation. Molecules 2020, 25, 2071. [Google Scholar] [CrossRef]
- Kumi, R.O.; Agoni, C.; Issahaku, A.R.; Olotu, F.A.; Soliman, M.E.S. ‘Polymorphism-aided’ Selective Targeting and Inhibition of Caspase-6 by a Novel Allosteric Inhibitor Towards Efficient Alzheimer’s Disease Treatment. Cell Biochem. Biophys. 2020, 78, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Cancela, S.; Canclini, L.; Mourglia-Ettlin, G.; Hernández, P.; Merlino, A. Neuroprotective effects of novel nitrones: In vitro and in silico studies. Eur. J. Pharmacol. 2020, 871, 172926. [Google Scholar] [CrossRef]
- Kim, W.-Y.; Snider, W.D. Functions of GSK-3 Signaling in Development of the Nervous System. Front. Mol. Neurosci. 2011, 4. [Google Scholar] [CrossRef]
- Medina, M.; Wandosell, F. Deconstructing GSK-3: The Fine Regulation of Its Activity. Int. J. Alzheimers. Dis. 2011, 2011, 1–12. [Google Scholar] [CrossRef]
- Hooper, C.; Killick, R.; Lovestone, S. The GSK3 hypothesis of Alzheimer’s disease. J. Neurochem. 2008, 104, 1433–1439. [Google Scholar] [CrossRef]
- Andreev, S.; Pantsar, T.; Ansideri, F.; Kudolo, M.; Forster, M.; Schollmeyer, D.; Laufer, S.A.; Koch, P. Design, Synthesis and Biological Evaluation of 7-Chloro-9H-pyrimido[4,5-b]indole-based Glycogen Synthase Kinase-3β Inhibitors. Molecules 2019, 24, 2331. [Google Scholar] [CrossRef] [PubMed]
- Usui, Y.; Uehara, F.; Hiki, S.; Watanabe, K.; Tanaka, H.; Shouda, A.; Yokoshima, S.; Aritomo, K.; Adachi, T.; Fukunaga, K.; et al. Discovery of novel 2-(3-phenylpiperazin-1-yl)-pyrimidin-4-ones as glycogen synthase kinase-3β inhibitors. Bioorg. Med. Chem. Lett. 2017, 27, 3726–3732. [Google Scholar] [CrossRef]
- Paudel, P.; Seong, S.; Zhou, Y.; Park, C.; Yokozawa, T.; Jung, H.; Choi, J. Rosmarinic Acid Derivatives’ Inhibition of Glycogen Synthase Kinase-3β Is the Pharmacological Basis of Kangen-Karyu in Alzheimer’s Disease. Molecules 2018, 23, 2919. [Google Scholar] [CrossRef]
- Nisha, C.M.; Kumar, A.; Vimal, A.; Bai, B.M.; Pal, D.; Kumar, A. Docking and ADMET prediction of few GSK-3 inhibitors divulges 6-bromoindirubin-3-oxime as a potential inhibitor. J. Mol. Graph. Model. 2016, 65, 100–107. [Google Scholar] [CrossRef]
- Heider, F.; Ansideri, F.; Tesch, R.; Pantsar, T.; Haun, U.; Döring, E.; Kudolo, M.; Poso, A.; Albrecht, W.; Laufer, S.A.; et al. Pyridinylimidazoles as dual glycogen synthase kinase 3β/p38α mitogen-activated protein kinase inhibitors. Eur. J. Med. Chem. 2019, 175, 309–329. [Google Scholar] [CrossRef]
- Lozinskaya, N.A.; Babkov, D.A.; Zaryanova, E.V.; Bezsonova, E.N.; Efremov, A.M.; Tsymlyakov, M.D.; Anikina, L.V.; Zakharyascheva, O.Y.; Borisov, A.V.; Perfilova, V.N.; et al. Synthesis and biological evaluation of 3-substituted 2-oxindole derivatives as new glycogen synthase kinase 3β inhibitors. Bioorg. Med. Chem. 2019, 27, 1804–1817. [Google Scholar] [CrossRef] [PubMed]
- Shukla, R.; Munjal, N.S.; Singh, T.R. Identification of novel small molecules against GSK3β for Alzheimer’s disease using chemoinformatics approach. J. Mol. Graph. Model. 2019, 91, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Joshi, P.; Gupta, M.; Vishwakarma, R.A.; Kumar, A.; Bharate, S.B. (Z)-2-(3-Chlorobenzylidene)-3,4-dihydro- N -(2-methoxyethyl)-3-oxo-2H-benzo[b][1,4]oxazine-6-carboxamide as GSK-3β inhibitor: Identification by virtual screening and its validation in enzyme- and cell-based assay. Chem. Biol. Drug Des. 2017, 89, 964–971. [Google Scholar] [CrossRef]
- He, Q.; Han, C.; Li, G.; Guo, H.; Wang, Y.; Hu, Y.; Lin, Z.; Wang, Y. In silico design novel (5-imidazol-2-yl-4-phenylpyrimidin-2-yl)[2-(2-pyridylamino)ethyl]amine derivatives as inhibitors for glycogen synthase kinase 3 based on 3D-QSAR, molecular docking and molecular dynamics simulation. Comput. Biol. Chem. 2020, 88, 107328. [Google Scholar] [CrossRef]
- Natarajan, P.; Priyadarshini, V.; Pradhan, D.; Manne, M.; Swargam, S.; Kanipakam, H.; Bhuma, V.; Amineni, U. E-pharmacophore-based virtual screening to identify GSK-3β inhibitors. J. Recept. Signal Transduct. 2016, 36, 445–458. [Google Scholar] [CrossRef]
- El Kerdawy, A.M.; Osman, A.A.; Zaater, M.A. Receptor-based pharmacophore modeling, virtual screening, and molecular docking studies for the discovery of novel GSK-3β inhibitors. J. Mol. Model. 2019, 25, 171. [Google Scholar] [CrossRef] [PubMed]
- Lotankar, S.; Prabhavalkar, K.S.; Bhatt, L.K. Biomarkers for Parkinson’s Disease: Recent Advancement. Neurosci. Bull. 2017, 33, 585–597. [Google Scholar] [CrossRef] [PubMed]
- Diaz, N.L.; Waters, C.H. Current strategies in the treatment of Parkinson’s disease and a personalized approach to management. Expert Rev. Neurother. 2009, 9, 1781–1789. [Google Scholar] [CrossRef]
- Cacabelos, R. Parkinson’s Disease: From Pathogenesis to Pharmacogenomics. Int. J. Mol. Sci. 2017, 18, 551. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.E.; Yun, J.Y.; Jeon, B.S. Effect of intravenous amantadine on dopaminergic-drug-resistant freezing of gait. Parkinsonism Relat. Disord. 2011, 17, 491–492. [Google Scholar] [CrossRef]
- Marjama-Lyons, J.; Koller, W. Tremor-Predominant Parkinson’s Disease. Drugs Aging 2000, 16, 273–278. [Google Scholar] [CrossRef]
- Bymaster, F.P.; Felder, C.C.; Tzavara, E.; Nomikos, G.G.; Calligaro, D.O.; Mckinzie, D.L. Muscarinic mechanisms of antipsychotic atypicality. Prog. Neuro Psychopharmacol. Biol. Psychiatry 2003, 27, 1125–1143. [Google Scholar] [CrossRef]
- Schneider, R.B.; Iourinets, J.; Richard, I.H. Parkinson’s disease psychosis: Presentation, diagnosis and management. Neurodegener. Dis. Manag. 2017, 7, 365–376. [Google Scholar] [CrossRef]
- Vaikath, N.N.; Hmila, I.; Gupta, V.; Erskine, D.; Ingelsson, M.; El-Agnaf, O.M.A. Antibodies against alpha-synuclein: Tools and therapies. J. Neurochem. 2019, 150, 612–625. [Google Scholar] [CrossRef] [PubMed]
- Henderson, M.X.; Covell, D.J.; Chung, C.H.-Y.; Pitkin, R.M.; Sandler, R.M.; Decker, S.C.; Riddle, D.M.; Zhang, B.; Gathagan, R.J.; James, M.J.; et al. Characterization of novel conformation-selective α-synuclein antibodies as potential immunotherapeutic agents for Parkinson’s disease. Neurobiol. Dis. 2020, 136, 104712. [Google Scholar] [CrossRef] [PubMed]
- Riederer, P.; Laux, G. MAO-inhibitors in Parkinson’s Disease. Exp. Neurobiol. 2011, 20, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Elmer, L.W.; Bertoni, J.M. The increasing role of monoamine oxidase type B inhibitors in Parkinson’s disease therapy. Expert Opin. Pharmacother. 2008, 9, 2759–2772. [Google Scholar] [CrossRef] [PubMed]
- Müller, T. Safinamide: An add-on treatment for managing Parkinson’s disease. Clin. Pharmacol. Adv. Appl. 2018, 10, 31–41. [Google Scholar] [CrossRef]
- Milczek, E.M.; Binda, C.; Rovida, S.; Mattevi, A.; Edmondson, D.E. The ‘gating’ residues Ile199 and Tyr326 in human monoamine oxidase B function in substrate and inhibitor recognition. FEBS J. 2011, 278, 4860–4869. [Google Scholar] [CrossRef]
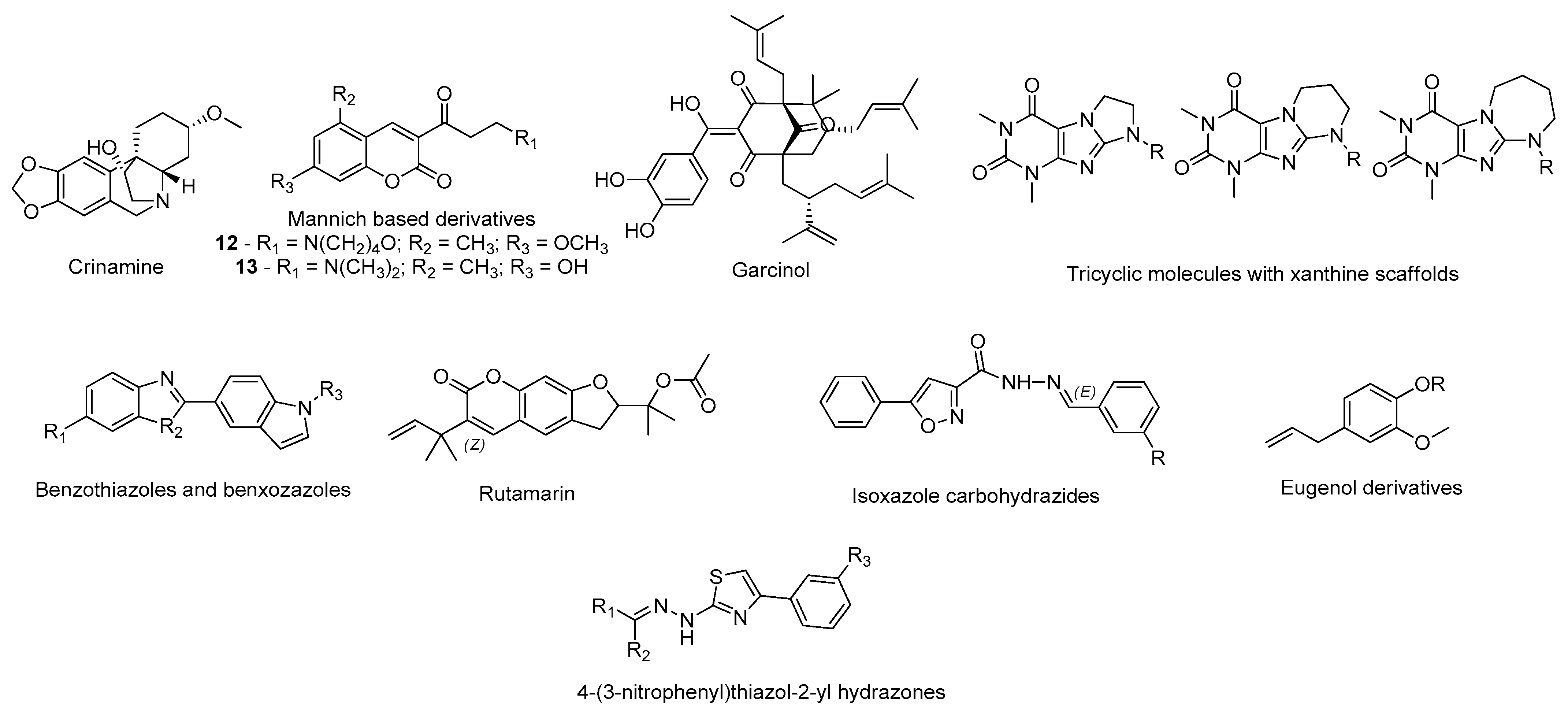
- Naidoo, D.; Roy, A.; Slavětínská, L.P.; Chukwujekwu, J.C.; Gupta, S.; Van Staden, J. New role for crinamine as a potent, safe and selective inhibitor of human monoamine oxidase B: In vitro and in silico pharmacology and modeling. J. Ethnopharmacol. 2020, 248, 112305. [Google Scholar] [CrossRef]
- Tao, D.; Wang, Y.; Bao, X.-Q.; Yang, B.-B.; Gao, F.; Wang, L.; Zhang, D.; Li, L. Discovery of coumarin Mannich base derivatives as multifunctional agents against monoamine oxidase B and neuroinflammation for the treatment of Parkinson’s disease. Eur. J. Med. Chem. 2019, 173, 203–212. [Google Scholar] [CrossRef]
- Mazumder, M.K.; Paul, R.; Phukan, B.C.; Dutta, A.; Chakrabarty, J.; Bhattacharya, P.; Borah, A. Garcinol, an effective monoamine oxidase-B inhibitor for the treatment of Parkinson’s disease. Med. Hypotheses 2018, 117, 54–58. [Google Scholar] [CrossRef] [PubMed]
- Załuski, M.; Schabikowski, J.; Schlenk, M.; Olejarz-Maciej, A.; Kubas, B.; Karcz, T.; Kuder, K.; Latacz, G.; Zygmunt, M.; Synak, D.; et al. Novel multi-target directed ligands based on annelated xanthine scaffold with aromatic substituents acting on adenosine receptor and monoamine oxidase B. Synthesis, in vitro and in silico studies. Bioorg. Med. Chem. 2019, 27, 1195–1210. [Google Scholar] [CrossRef]
- Nam, M.-H.; Park, M.; Park, H.; Kim, Y.; Yoon, S.; Sawant, V.S.; Choi, J.W.; Park, J.-H.; Park, K.D.; Min, S.-J.; et al. Indole-Substituted Benzothiazoles and Benzoxazoles as Selective and Reversible MAO-B Inhibitors for Treatment of Parkinson’s Disease. ACS Chem. Neurosci. 2017, 8, 1519–1529. [Google Scholar] [CrossRef] [PubMed]
- Kozioł, E.; Luca, S.V.; Ağalar, H.G.; Sağlık, B.N.; Demirci, F.; Marcourt, L.; Wolfender, J.-L.; Jóźwiak, K.; Skalicka-Woźniak, K. Rutamarin: Efficient Liquid–Liquid Chromatographic Isolation from Ruta graveolens L. and Evaluation of Its In Vitro and In Silico MAO-B Inhibitory Activity. Molecules 2020, 25, 2678. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, N.; Mishra, P. Synthesis, monoamine oxidase inhibitory activity and computational study of novel isoxazole derivatives as potential antiparkinson agents. Comput. Biol. Chem. 2019, 79, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Dhiman, P.; Malik, N.; Khatkar, A. Lead optimization for promising monoamine oxidase inhibitor from eugenol for the treatment of neurological disorder: Synthesis and in silico based study. BMC Chem. 2019, 13, 38. [Google Scholar] [CrossRef]
- Secci, D.; Carradori, S.; Petzer, A.; Guglielmi, P.; D’Ascenzio, M.; Chimenti, P.; Bagetta, D.; Alcaro, S.; Zengin, G.; Petzer, J.P.; et al. 4-(3-Nitrophenyl)thiazol-2-ylhydrazone derivatives as antioxidants and selective hMAO-B inhibitors: Synthesis, biological activity and computational analysis. J. Enzym. Inhib. Med. Chem. 2019, 34, 597–612. [Google Scholar] [CrossRef]
- Chaurasiya, N.; Zhao, J.; Pandey, P.; Doerksen, R.; Muhammad, I.; Tekwani, B. Selective Inhibition of Human Monoamine Oxidase B by Acacetin 7-Methyl Ether Isolated from Turnera diffusa (Damiana). Molecules 2019, 24, 810. [Google Scholar] [CrossRef]
- Is, Y.S.; Durdagi, S.; Aksoydan, B.; Yurtsever, M. Proposing Novel MAO-B Hit Inhibitors Using Multidimensional Molecular Modeling Approaches and Application of Binary QSAR Models for Prediction of Their Therapeutic Activity, Pharmacokinetic and Toxicity Properties. ACS Chem. Neurosci. 2018, 9, 1768–1782. [Google Scholar] [CrossRef]
- Jin, C.-F.; Wang, Z.-Z.; Chen, K.-Z.; Xu, T.-F.; Hao, G.-F. Computational Fragment-Based Design Facilitates Discovery of Potent and Selective Monoamine Oxidase-B (MAO-B) Inhibitor. J. Med. Chem. 2020, 63, 15021–15036. [Google Scholar] [CrossRef]
- Cruz-Monteagudo, M.; Borges, F.; Cordeiro, M.N.D.S.; Helguera, A.M.; Tejera, E.; Paz-y-Mino, C.; Sanchez-Rodriguez, A.; Perera-Sardina, Y.; Perez-Castillo, Y. Chemoinformatics Profiling of the Chromone Nucleus as a MAO-B/A2AAR Dual Binding Scaffold. Curr. Neuropharm. 2017, 15, 1117. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mladenović, M.; Patsilinakos, A.; Pirolli, A.; Sabatino, M.; Ragno, R. Understanding the Molecular Determinant of Reversible Human Monoamine Oxidase B Inhibitors Containing 2 H -Chromen-2-One Core: Structure-Based and Ligand-Based Derived Three-Dimensional Quantitative Structure–Activity Relationships Predictive Models. J. Chem. Inf. Model. 2017, 57, 787–814. [Google Scholar] [CrossRef] [PubMed]
- Hisahara, S.; Shimohama, S. Dopamine Receptors and Parkinson’s Disease. Int. J. Med. Chem. 2011, 2011, 1–16. [Google Scholar] [CrossRef] [PubMed]
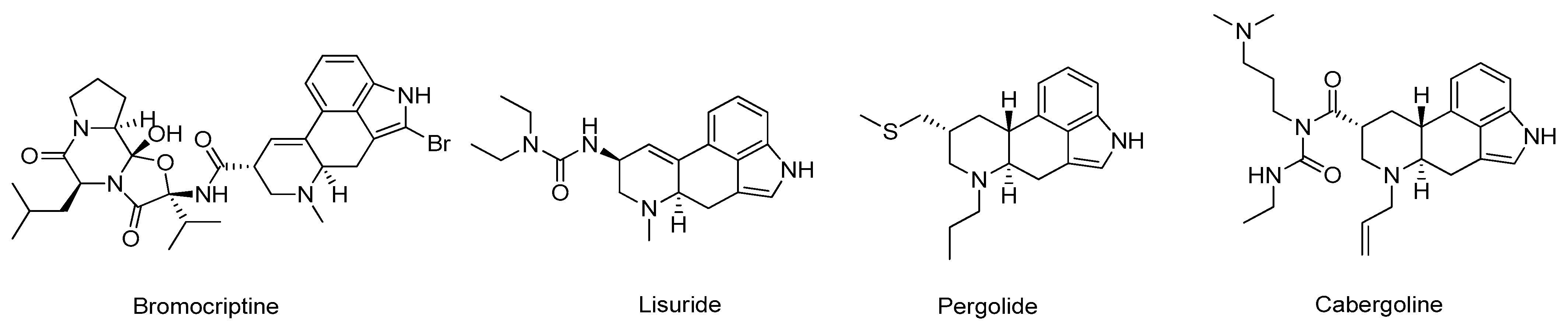
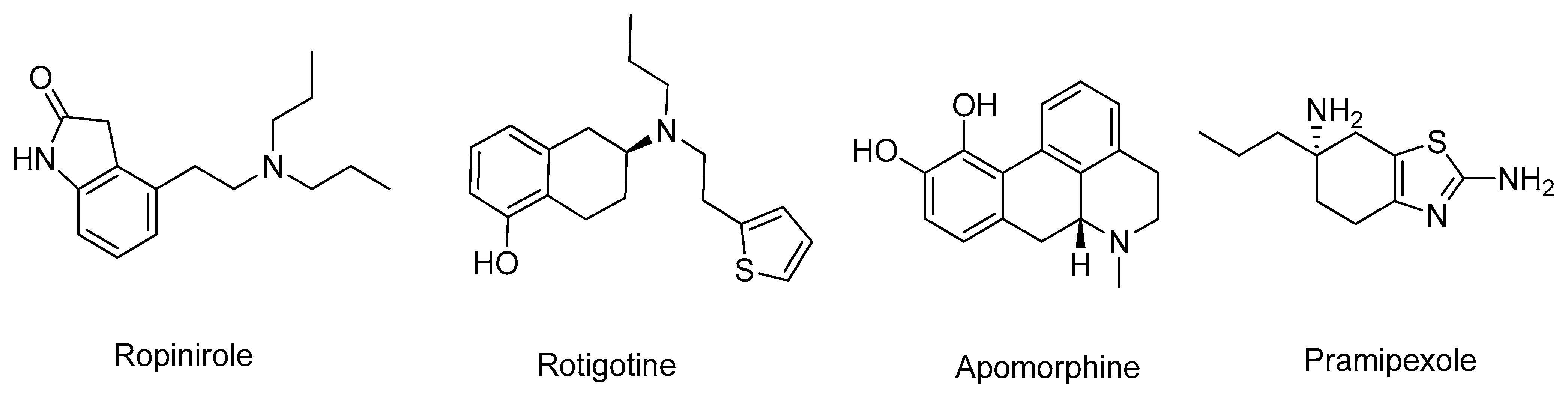
- Brooks, D.J. Dopamine agonists: Their role in the treatment of Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 2000, 68, 685–689. [Google Scholar] [CrossRef] [PubMed]
- Jankovic, J.; Tan, E.K. Parkinson’s disease: Etiopathogenesis and treatment. J. Neurol. Neurosurg. Psychiatry 2020, 91, 795–808. [Google Scholar] [CrossRef]
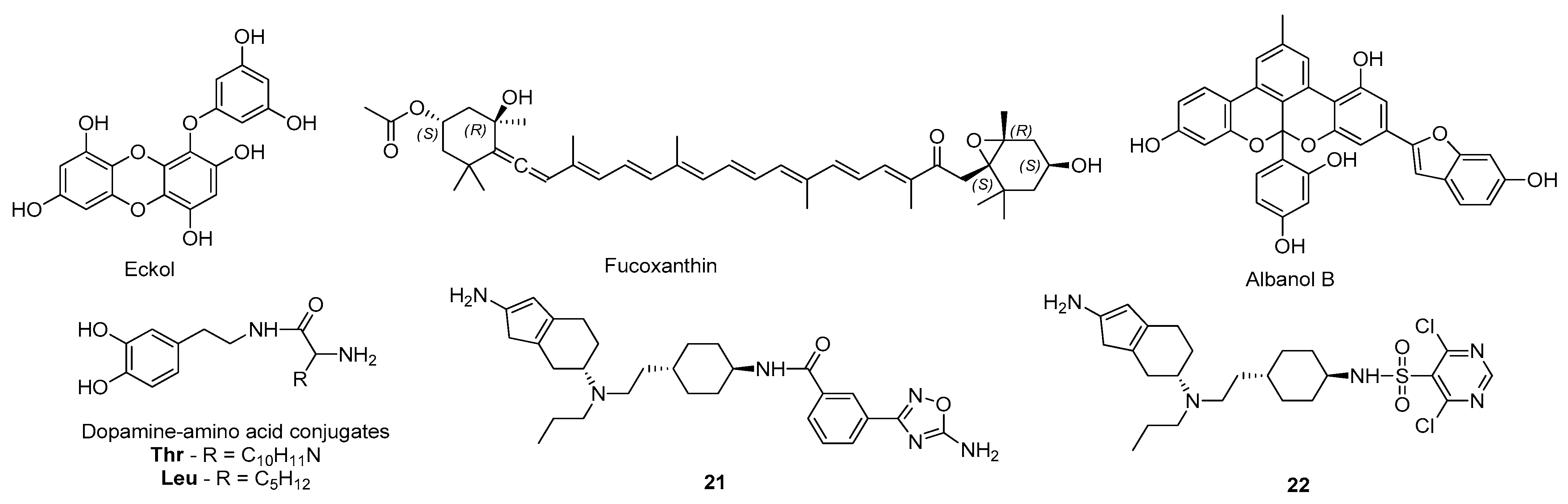
- Paudel, P.; Seong, S.H.; Wu, S.; Park, S.; Jung, H.A.; Choi, J.S. Eckol as a Potential Therapeutic against Neurodegenerative Diseases Targeting Dopamine D3/D4 Receptors. Mar. Drugs 2019, 17, 108. [Google Scholar] [CrossRef]
- Paudel, P.; Seong, S.H.; Jung, H.A.; Choi, J.S. Characterizing fucoxanthin as a selective dopamine D3/D4 receptor agonist: Relevance to Parkinson’s disease. Chem. Biol. Interact. 2019, 310, 108757. [Google Scholar] [CrossRef]
- Paudel, P.; Park, S.E.; Seong, S.H.; Jung, H.A.; Choi, J.S. Novel Diels–Alder Type Adducts from Morus alba Root Bark Targeting Human Monoamine Oxidase and Dopaminergic Receptors for the Management of Neurodegenerative Diseases. Int. J. Mol. Sci. 2019, 20, 6232. [Google Scholar] [CrossRef]
- Tutone, M.; Chinnici, A.; Almerico, A.M.; Perricone, U.; Sutera, F.M.; De Caro, V. Design, synthesis and preliminary evaluation of dopamine-amino acid conjugates as potential D1 dopaminergic modulators. Eur. J. Med. Chem. 2016, 124, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Duan, X.; Zhang, X.; Xu, B.; Wang, F.; Lei, M. Computational Study and Modified Design of Selective Dopamine D 3 Receptor Agonists. Chem. Biol. Drug Des. 2016, 88, 142–154. [Google Scholar] [CrossRef]
- Chen, J.; Levant, B.; Wang, S. High-affinity and selective dopamine D3 receptor full agonists. Bioorg. Med. Chem. Lett. 2012, 22, 5612–5617. [Google Scholar] [CrossRef]
- Zheng, J.; Zhang, X.; Zhen, X. Development of Adenosine A 2A Receptor Antagonists for the Treatment of Parkinson’s Disease: A Recent Update and Challenge. ACS Chem. Neurosci. 2019, 10, 783–791. [Google Scholar] [CrossRef]
- Jazayeri, A.; Andrews, S.P.; Marshall, F.H. Structurally Enabled Discovery of Adenosine A 2A Receptor Antagonists. Chem. Rev. 2017, 117, 21–37. [Google Scholar] [CrossRef] [PubMed]
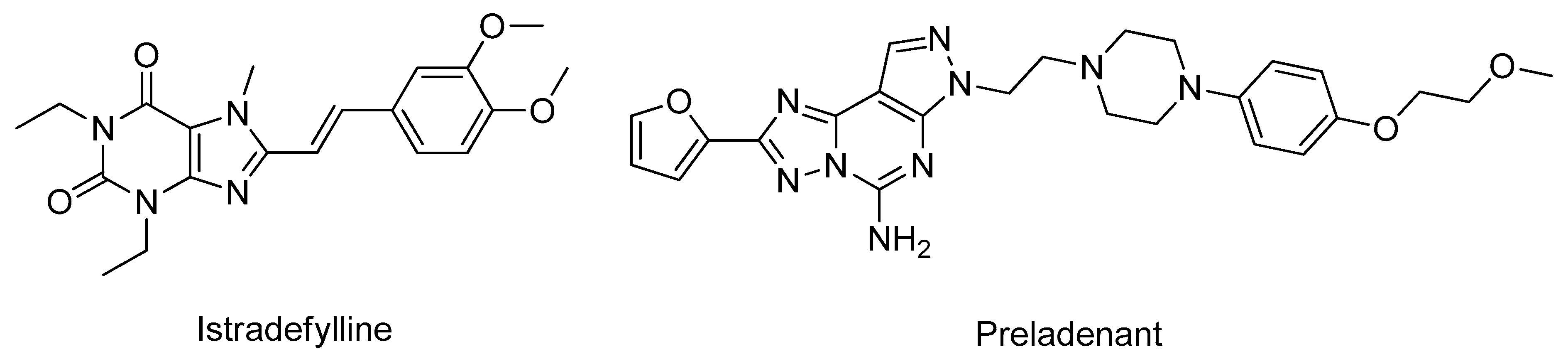
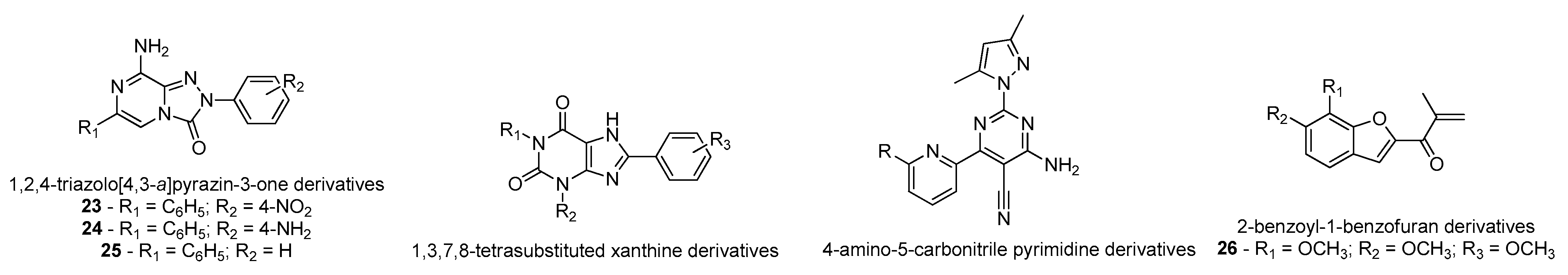
- Falsini, M.; Squarcialupi, L.; Catarzi, D.; Varano, F.; Betti, M.; Dal Ben, D.; Marucci, G.; Buccioni, M.; Volpini, R.; De Vita, T.; et al. The 1,2,4-Triazolo[4,3- a ]pyrazin-3-one as a Versatile Scaffold for the Design of Potent Adenosine Human Receptor Antagonists. Structural Investigations to Target the A 2A Receptor Subtype. J. Med. Chem. 2017, 60, 5772–5790. [Google Scholar] [CrossRef] [PubMed]
- Rohilla, S.; Bansal, R.; Kachler, S.; Klotz, K.-N. Synthesis, biological evaluation and molecular modelling studies of 1,3,7,8-tetrasubstituted xanthines as potent and selective A2A AR ligands with in vivo efficacy against animal model of Parkinson’s disease. Bioorg. Chem. 2019, 87, 601–612. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Li, L.; Zheng, J.; Ma, H.; Tian, S.; Li, J.; Zhang, H.; Zhen, X.; Zhang, X. Identification of a New Series of Potent Adenosine A 2A Receptor Antagonists Based on 4-Amino-5-carbonitrile Pyrimidine Template for the Treatment of Parkinson’s Disease. ACS Chem. Neurosci. 2016, 7, 1575–1584. [Google Scholar] [CrossRef]
- Janse van Rensburg, H.D.; Legoabe, L.J.; Terre’Blanche, G.; Aucamp, J. Synthesis and evaluation of methoxy substituted 2-benzoyl-1-benzofuran derivatives as lead compounds for the development adenosine A1 and/or A2A receptor antagonists. Bioorg. Chem. 2020, 94, 103459. [Google Scholar] [CrossRef]
- Bonifácio, M.J.; Palma, P.N.; Almeida, L.; Soares-da-Silva, P. Catechol-O-methyltransferase and Its Inhibitors in Parkinson’s Disease. CNS Drug Rev. 2007, 13, 352–379. [Google Scholar] [CrossRef]
- Kiss, L.E.; Soares-da-Silva, P. Medicinal Chemistry of Catechol O Methyltransferase (COMT) Inhibitors and Their Therapeutic Utility. J. Med. Chem. 2014, 57, 8692–8717. [Google Scholar] [CrossRef]
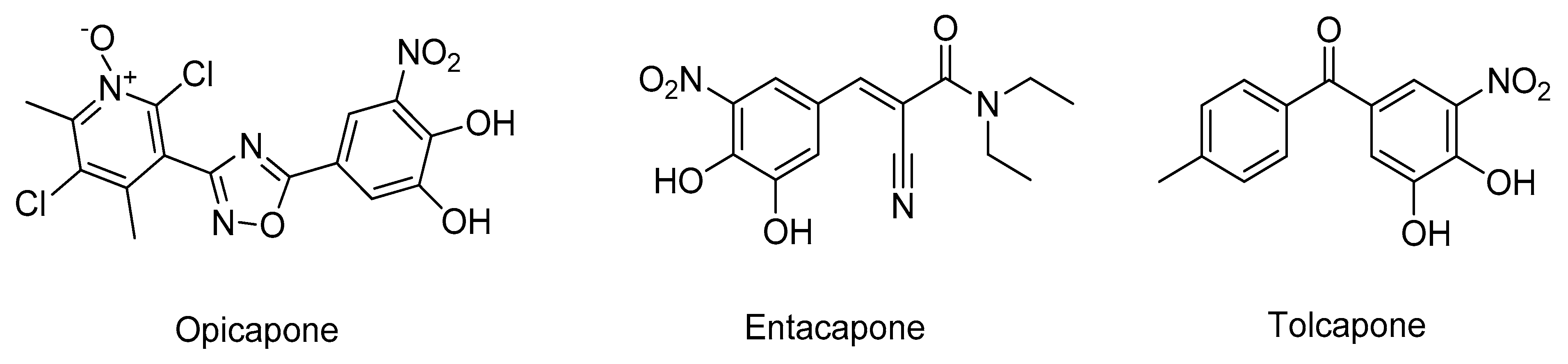
- Castro Caldas, A.; Teodoro, T.; Ferreira, J.J. The launch of opicapone for Parkinson’s disease: Negatives versus positives. Expert Opin. Drug Saf. 2018, 17, 331–337. [Google Scholar] [CrossRef]
- Ma, Z.; Liu, H.; Wu, B. Structure-based drug design of catechol-O-methyltransferase inhibitors for CNS disorders. Br. J. Clin. Pharmacol. 2014, 77, 410–420. [Google Scholar] [CrossRef]
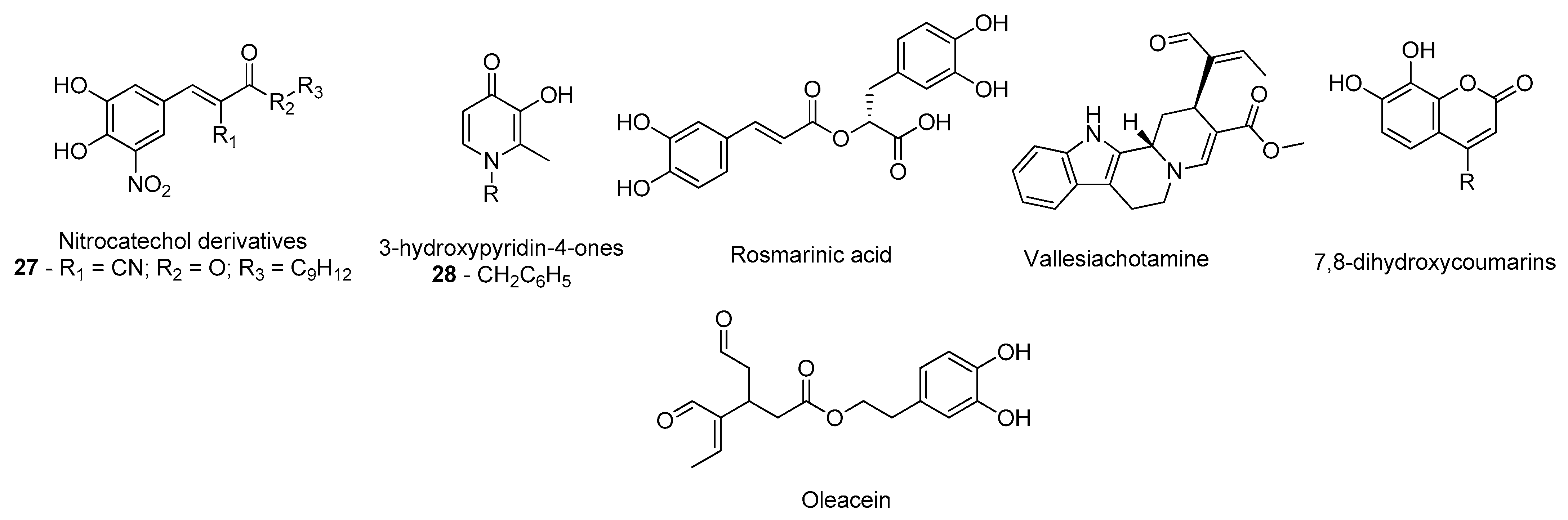
- Silva, T.; Mohamed, T.; Shakeri, A.; Rao, P.P.N.; Martínez-González, L.; Pérez, D.I.; Martínez, A.; Valente, M.J.; Garrido, J.; Uriarte, E.; et al. Development of Blood–Brain Barrier Permeable Nitrocatechol-Based Catechol O -Methyltransferase Inhibitors with Reduced Potential for Hepatotoxicity. J. Med. Chem. 2016, 59, 7584–7597. [Google Scholar] [CrossRef]
- de Beer, J.; Petzer, J.P.; Lourens, A.C.U.; Petzer, A. Design, synthesis and evaluation of 3-hydroxypyridin-4-ones as inhibitors of catechol-O-methyltransferase. Mol. Divers. 2020, 1–10. [Google Scholar] [CrossRef]
- Andrade, J.M.D.M.; dos Santos Passos, C.; Kieling Rubio, M.A.; Mendonça, J.N.; Lopes, N.P.; Henriques, A.T. Combining in vitro and in silico approaches to evaluate the multifunctional profile of rosmarinic acid from Blechnum brasiliense on targets related to neurodegeneration. Chem. Biol. Interact. 2016, 254, 135–145. [Google Scholar] [CrossRef]
- dos Santos Passos, C.; Klein-Júnior, L.C.; de Mello Andrade, J.M.; Matté, C.; Henriques, A.T. The catechol-O-methyltransferase inhibitory potential of Z-vallesiachotamine by in silico and in vitro approaches. Rev. Bras. Farmacogn. 2015, 25, 382–386. [Google Scholar] [CrossRef]
- Xia, Y.-L.; Dou, T.-Y.; Liu, Y.; Wang, P.; Ge, G.-B.; Yang, L. In vitro evaluation of the effect of C-4 substitution on methylation of 7,8-dihydroxycoumarin: Metabolic profile and catalytic kinetics. R. Soc. Open Sci. 2018, 5, 171271. [Google Scholar] [CrossRef] [PubMed]
- Cuyàs, E.; Verdura, S.; Lozano-Sánchez, J.; Viciano, I.; Llorach-Parés, L.; Nonell-Canals, A.; Bosch-Barrera, J.; Brunet, J.; Segura-Carretero, A.; Sanchez-Martinez, M.; et al. The extra virgin olive oil phenolic oleacein is a dual substrate-inhibitor of catechol-O-methyltransferase. Food Chem. Toxicol. 2019, 128, 35–45. [Google Scholar] [CrossRef] [PubMed]
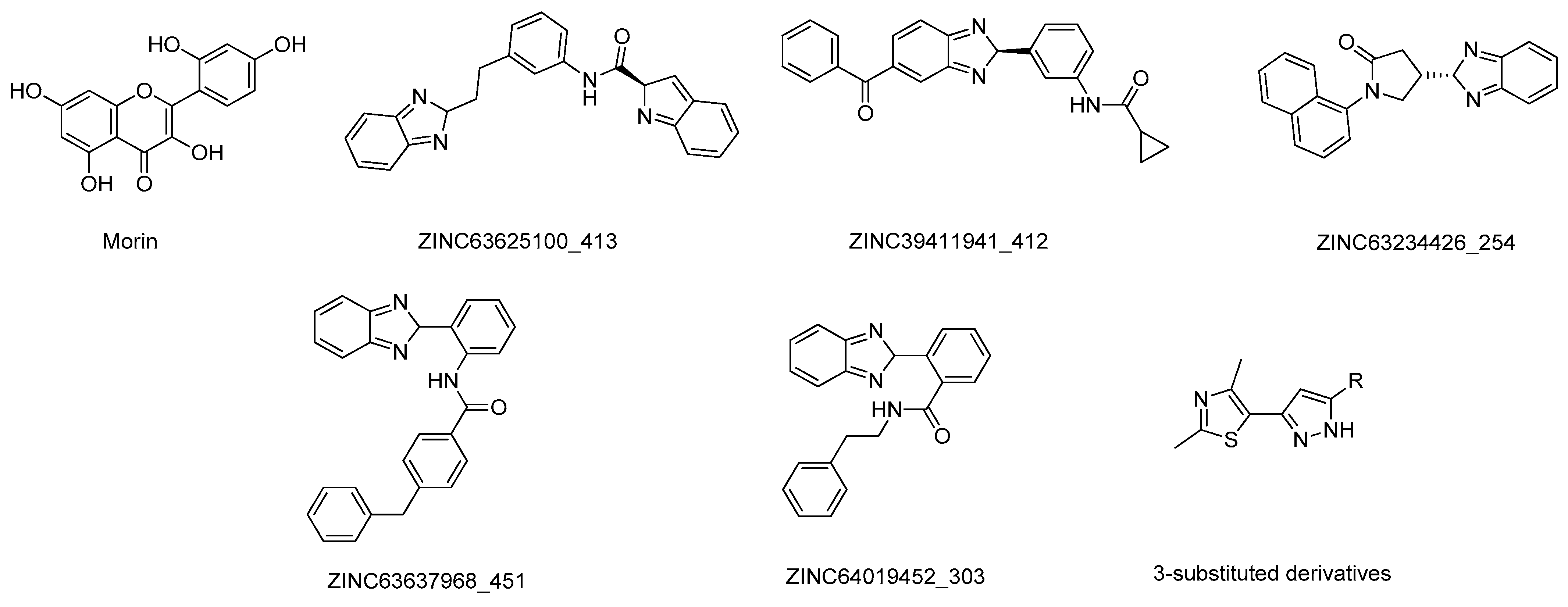
- Govindasamy, H.; Magudeeswaran, S.; Poomani, K. Identification of novel flavonoid inhibitor of Catechol-O-Methyltransferase enzyme by molecular screening, quantum mechanics/molecular mechanics and molecular dynamics simulations. J. Biomol. Struct. Dyn. 2019, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Patel, C.N.; Georrge, J.J.; Modi, K.M.; Narechania, M.B.; Patel, D.P.; Gonzalez, F.J.; Pandya, H.A. Pharmacophore-based virtual screening of catechol-o-methyltransferase (COMT) inhibitors to combat Alzheimer’s disease. J. Biomol. Struct. Dyn. 2018, 36, 3938–3957. [Google Scholar] [CrossRef] [PubMed]
- Lerner, C.; Jakob-Roetne, R.; Buettelmann, B.; Ehler, A.; Rudolph, M.; Rodríguez Sarmiento, R.M. Design of Potent and Druglike Nonphenolic Inhibitors for Catechol O -Methyltransferase Derived from a Fragment Screening Approach Targeting the S -Adenosyl- l -methionine Pocket. J. Med. Chem. 2016, 59, 10163–10175. [Google Scholar] [CrossRef]
























Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cruz-Vicente, P.; Passarinha, L.A.; Silvestre, S.; Gallardo, E. Recent Developments in New Therapeutic Agents against Alzheimer and Parkinson Diseases: In-Silico Approaches. Molecules 2021, 26, 2193. https://doi.org/10.3390/molecules26082193
Cruz-Vicente P, Passarinha LA, Silvestre S, Gallardo E. Recent Developments in New Therapeutic Agents against Alzheimer and Parkinson Diseases: In-Silico Approaches. Molecules. 2021; 26(8):2193. https://doi.org/10.3390/molecules26082193
Chicago/Turabian StyleCruz-Vicente, Pedro, Luís A. Passarinha, Samuel Silvestre, and Eugenia Gallardo. 2021. "Recent Developments in New Therapeutic Agents against Alzheimer and Parkinson Diseases: In-Silico Approaches" Molecules 26, no. 8: 2193. https://doi.org/10.3390/molecules26082193
APA StyleCruz-Vicente, P., Passarinha, L. A., Silvestre, S., & Gallardo, E. (2021). Recent Developments in New Therapeutic Agents against Alzheimer and Parkinson Diseases: In-Silico Approaches. Molecules, 26(8), 2193. https://doi.org/10.3390/molecules26082193