
Nanoparticle-Based Rifampicin Delivery System Development


 ,
,  ,
,  , ,
, ,
Abstract

1. Introduction
2. Results and Discussion
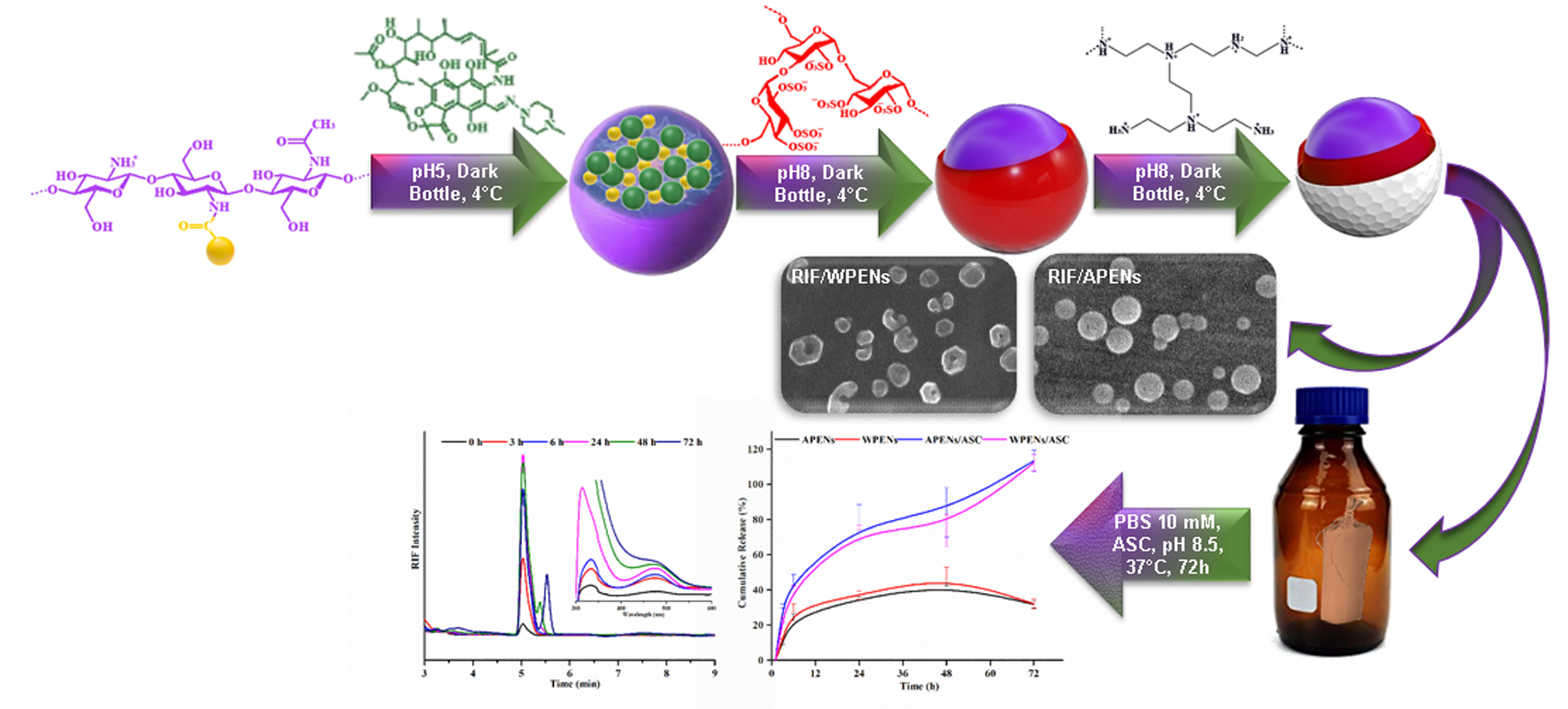
2.1. Evaluation of Size, Morphology, and ζ-Potential of PENs
2.2. Evaluation of Colloidal Stability of PENs
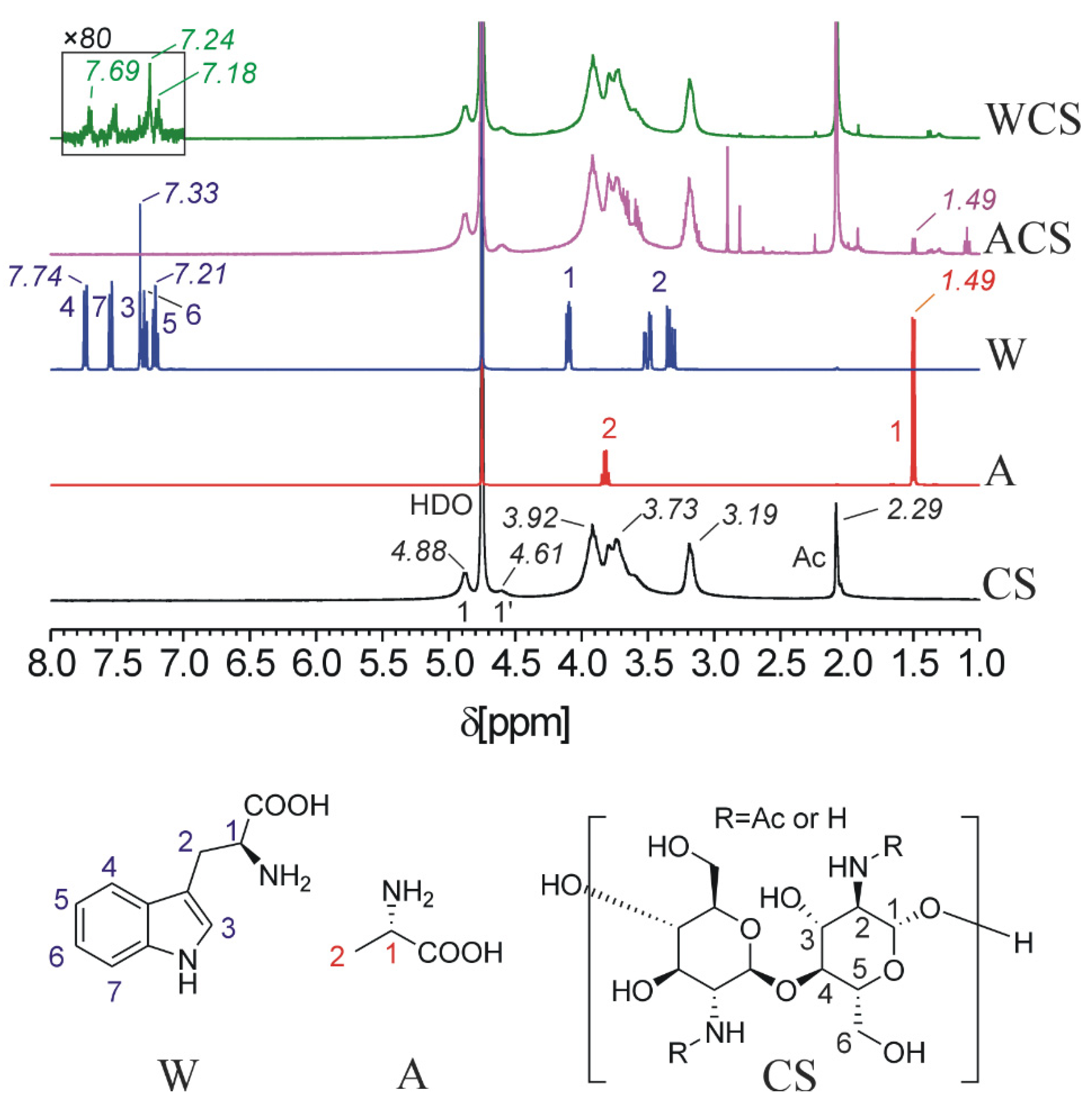
2.3. HNMR, Elemental Analysis and Infrared Spectrophotometry Analysis
2.4. XRD Analysis
2.5. Calorimetric Analysis
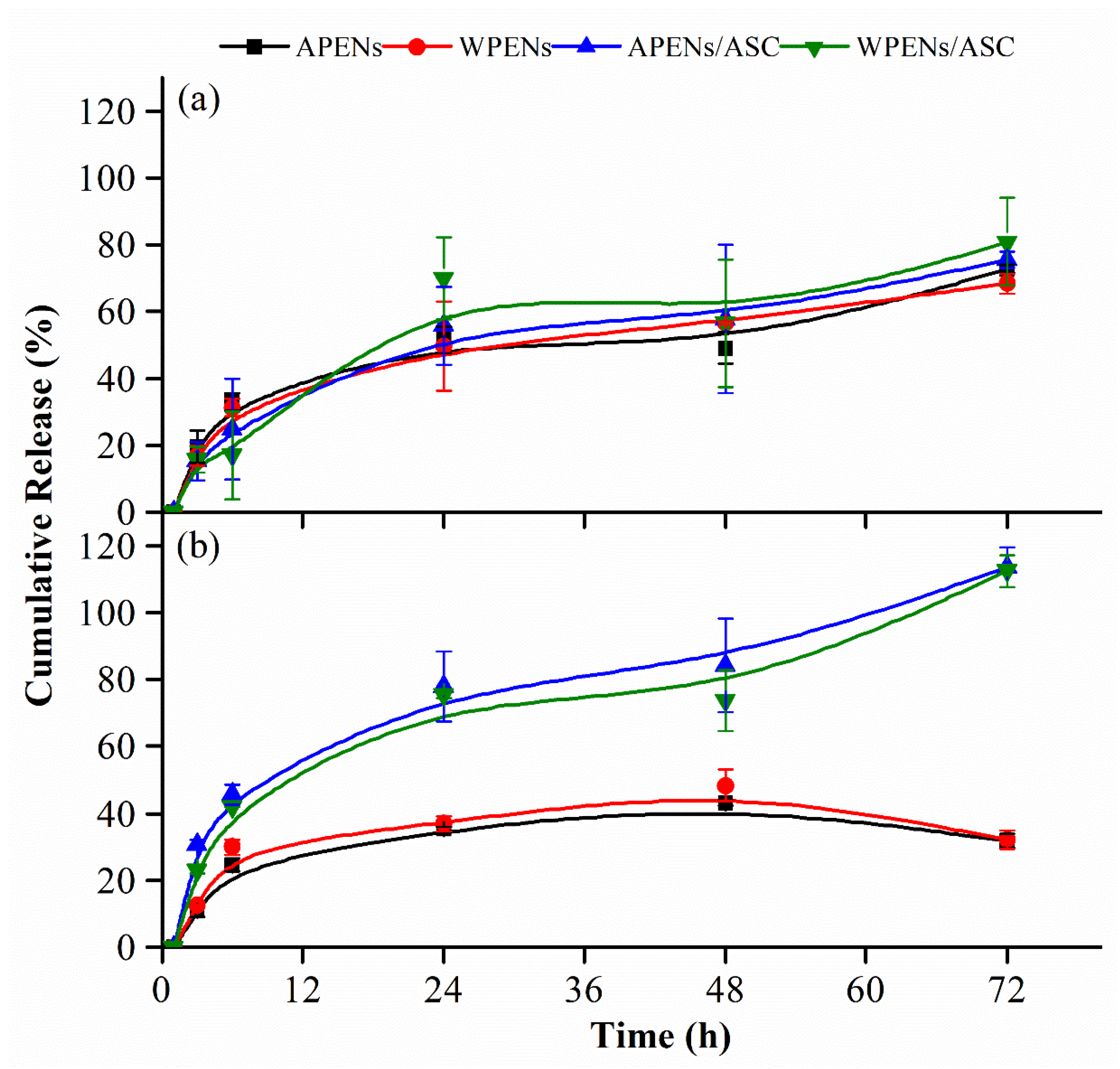
2.6. In Vitro Release Studies
2.7. Degradation Studies
3. Materials and Methods
3.1. Materials
3.2. Synthesizing and Characterization of CS Derivatives
3.3. Synthesizing and Characterization of PENs
3.4. Drug Loading and in Vitro Release Assays of PENs
3.5. RIF Degradation Assays
3.6. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Arca, H.Ç.; Mosquera-Giraldo, L.I.; Pereira, J.M.; Sriranganathan, N.; Taylor, L.S.; Edgar, K.J. Rifampin stability and solution concentration enhancement through amorphous solid dispersion in cellulose ω-carboxyalkanoate matrices. J. Pharm. Sci. 2018, 107, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Reinbold, J.; Hierlemann, T.; Urich, L.; Uhde, A.-K.; Müller, I.; Weindl, T.; Vogel, U.; Schlensak, C.; Wendel, H.P.; Krajewski, S. Biodegradable rifampicin-releasing coating of surgical meshes for the prevention of bacterial infections. Drug Des. Dev. Ther. 2017, 11, 2753. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-Y.; Huang, C.-H.; Lu, P.-L.; Ko, W.-C.; Chen, Y.-H.; Hsueh, P.-R. Role of rifampin for the treatment of bacterial infections other than mycobacteriosis. J. Infect. 2017, 75, 395–408. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Glackin, C.A.; Horwitz, M.A.; Zink, J.I. Nanomachines and other caps on mesoporous silica nanoparticles for drug delivery. Acc. Chem. Res. 2019, 52, 1531–1542. [Google Scholar] [CrossRef]
- Hakkimane, S.S.; Shenoy, V.P.; Gaonkar, S.L.; Bairy, I.; Guru, B.R. Antimycobacterial susceptibility evaluation of rifampicin and isoniazid benz-hydrazone in biodegradable polymeric nanoparticles against Mycobacterium tuberculosis H37Rv strain. Int. J. Nanomed. 2018, 13, 4303. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.; Pawar, R.-P.; Bose, D.; Durgbanshi, A.; Albiol-Chiva, J.; Peris-Vicente, J.; Esteve-Romero, J.; Jain, A. Stability studies of rifampicin in plasma and urine of tuberculosis patients according to the European Medicines Agency Guidelines. Bioanalysis 2019, 11, 713–726. [Google Scholar] [CrossRef] [PubMed]
- Peer, D.; Karp, J.M.; Hong, S.; Farokhzad, O.C.; Margalit, R.; Langer, R. Nanocarriers as an emerging platform for cancer therapy. Nat. Nanotechnol. 2007, 2, 751–760. [Google Scholar] [CrossRef] [PubMed]
- Rajaram, S.; Vemuri, V.D.; Natham, R. Ascorbic acid improves stability and pharmacokinetics of rifampicin in the presence of isoniazid. J. Pharm. Biomed. Anal. 2014, 100, 103–108. [Google Scholar] [CrossRef]
- Kruse, C.R.; Singh, M.; Targosinski, S.; Sinha, I.; Sørensen, J.A.; Eriksson, E.; Nuutila, K. The effect of pH on cell viability, cell migration, cell proliferation, wound closure, and wound reepithelialization: In vitro and in vivo study. Wound Repair Regen. 2017, 25, 260–269. [Google Scholar] [CrossRef]
- Vilchèze, C.; Kim, J.; Jacobs, W.R. Vitamin C potentiates the killing of Mycobacterium tuberculosis by the first-line tuberculosis drugs isoniazid and rifampin in mice. Antimicrob. Agents Chemother. 2018, 62, e02165-17. [Google Scholar] [CrossRef]
- Petkar, K.C.; Chavhan, S.; Kunda, N.; Saleem, I.; Somavarapu, S.; Taylor, K.M.; Sawant, K.K. Development of novel octanoyl chitosan nanoparticles for improved rifampicin pulmonary delivery: Optimization by factorial design. Aaps Pharm. Sci. Tech. 2018, 19, 1758–1772. [Google Scholar] [CrossRef]
- Mignani, S.; Tripathi, R.; Chen, L.; Caminade, A.-M.; Shi, X.; Majoral, J.-P. New ways to treat tuberculosis using dendrimers as nanocarriers. Pharmaceutics 2018, 10, 105. [Google Scholar] [CrossRef]
- Scolari, I.; Páez, P.; Musri, M.; Petiti, J.; Torres, A.; Granero, G. Rifampicin loaded in alginate/chitosan nanoparticles as a promising pulmonary carrier against Staphylococcus aureus. Drug Deliv. Trans. Res. 2020, 1–15. [Google Scholar] [CrossRef]
- Chokshi, N.V.; Khatri, H.N.; Patel, M.M. Formulation, optimization, and characterization of rifampicin-loaded solid lipid nanoparticles for the treatment of tuberculosis. Drug Dev. Ind. Pharm. 2018, 44, 1975–1989. [Google Scholar] [CrossRef]
- Ali, H.R.; Ali, M.R.; Wu, Y.; Selim, S.A.; Abdelaal, H.F.; Nasr, E.A.; El-Sayed, M.A. Gold nanorods as drug delivery vehicles for rifampicin greatly improve the efficacy of combating Mycobacterium tuberculosis with good biocompatibility with the host cells. Bioconjugate Chem. 2016, 27, 2486–2492. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, S.; Thomas, N.; Gustafsson, H.; Jambhrunkar, M.; Kidd, S.P.; Prestidge, C.A. Rifampicin-Loaded Mesoporous Silica Nanoparticles for the Treatment of Intracellular Infections. Antibiotics 2019, 8, 39. [Google Scholar] [CrossRef] [PubMed]
- Tarricone, A.; De La Mata, K.; Chen, S.; Krishnan, P.; Landau, S.; Soave, R. Relationship Between pH Shifts and Rate of Healing in Chronic Nonhealing Venous Stasis Lower-Extremity Wounds. J. Foot. Ankle Surg. 2020, 59, 748–752. [Google Scholar] [CrossRef] [PubMed]
- Jones, E.M.; Cochrane, C.A.; Percival, S.L. The effect of pH on the extracellular matrix and biofilms. Adv. Would Care 2015, 4, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Mihai, M.M.; Dima, M.B.; Dima, B.; Holban, A.M. Nanomaterials for Wound Healing and Infection Control. Materials 2019, 12, 2176. [Google Scholar] [CrossRef]
- Hu, S.; Bi, S.; Yan, D.; Zhou, Z.; Sun, G.; Cheng, X.; Chen, X. Preparation of composite hydroxybutyl chitosan sponge and its role in promoting wound healing. Carbohydr. Polym. 2018, 184, 154–163. [Google Scholar] [CrossRef]
- Xun, M.-M.; Huang, Z.; Xiao, Y.-P.; Liu, Y.-H.; Zhang, J.; Zhang, J.-H.; Yu, X.-Q. Synthesis and Properties of Low-Molecular-Weight PEI-Based Lipopolymers for Delivery of DNA. Polymers 2018, 10, 1060. [Google Scholar] [CrossRef] [PubMed]
- Gatti, T.H.H.; Eloy, J.O.; Ferreira, L.M.B.; Silva, I.C.; Pavan, F.R.; Gremião, M.P.D.; Chorilli, M. Insulin-loaded polymeric mucoadhesive nanoparticles: Development, characterization and cytotoxicity evaluation. Braz. J. Pharm. Sci 2018, 54, 1–10. [Google Scholar] [CrossRef]
- Motiei, M.; Kashanian, S.; Lucia, L.A.; Khazaei, M. Intrinsic parameters for the synthesis and tuned properties of amphiphilic chitosan drug delivery nanocarriers. J. Control. Release 2017, 260, 213–225. [Google Scholar] [CrossRef]
- Motiei, M.; Kashanian, S.; Taherpour, A. Hydrophobic amino acids grafted onto chitosan: A novel amphiphilic chitosan nanocarrier for hydrophobic drugs. Drug Dev. Ind. Pharm. 2017, 43, 1–11. [Google Scholar] [CrossRef]
- Motiei, M.; Kashanian, S. Novel amphiphilic chitosan nanocarriers for sustained oral delivery of hydrophobic drugs. Eur. J. Pharm. Sci 2017, 99, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Motiei, M.; Sedlařík, V.; Lucia, L.A.; Fei, H.; Münster, L. Stabilization of chitosan-based polyelectrolyte nanoparticle cargo delivery biomaterials by a multiple ionic cross-linking strategy. Carbohydr. Polym. 2020, 231, 115709. [Google Scholar] [CrossRef]
- Motiei, M.; Kashanian, S. Preparation of amphiphilic chitosan nanoparticles for controlled release of hydrophobic drugs. J. Nanosci. Nanotech. 2017, 17, 5226–5232. [Google Scholar] [CrossRef]
- Eaton, P.; Quaresma, P.; Soares, C.; Neves, C.; de Almeida, M.; Pereira, E.; West, P. A direct comparison of experimental methods to measure dimensions of synthetic nanoparticles. Ultramicroscopy 2017, 182, 179–190. [Google Scholar] [CrossRef]
- Fan, W.; Yan, W.; Xu, Z.; Ni, H. Formation mechanism of monodisperse, low molecular weight chitosan nanoparticles by ionic gelation technique. Colloids Surf. B Biointerfaces 2012, 90, 21–27. [Google Scholar] [CrossRef]
- Zielińska, A.; Carreiró, F.; Oliveira, A.M.; Neves, A.; Pires, B.; Venkatesh, D.N.; Durazzo, A.; Lucarini, M.; Eder, P.; Silva, A.M. Polymeric nanoparticles: Production, characterization, toxicology and ecotoxicology. Molecules 2020, 25, 3731. [Google Scholar] [CrossRef]
- Layek, B.; Singh, J. Amino acid grafted chitosan for high performance gene delivery: Comparison of amino acid hydrophobicity on vector and polyplex characteristics. Biomacromolecules 2013, 14, 485–494. [Google Scholar] [CrossRef] [PubMed]
- Selvakannan, P.; Mandal, S.; Phadtare, S.; Gole, A.; Pasricha, R.; Adyanthaya, S.; Sastry, M. Water-dispersible tryptophan-protected gold nanoparticles prepared by the spontaneous reduction of aqueous chloroaurate ions by the amino acid. J. Colloid Interface Sci. 2004, 269, 97–102. [Google Scholar] [CrossRef]
- Vaca-Garcia, C.; Borredon, M.-E.; Gaseta, A. Determination of the degree of substitution (DS) of mixed cellulose esters by elemental analysis. Cellulose 2001, 8, 225–231. [Google Scholar] [CrossRef]
- Chavan, C.; Bala, P.; Pal, K.; Kale, S. Cross-linked chitosan-dextran sulphate vehicle system for controlled release of ciprofloxaxin drug: An ophthalmic application. OpenNano 2017, 2, 28–36. [Google Scholar] [CrossRef]
- Nikolić, G.S.; Cakić, M.D.; Glišić, S.; Cvetković, D.J.; Mitić, Ž.J.; Marković, D.Z. Study of Green Nanoparticles and Biocomplexes Based on Exopolysaccharide by Modern Fourier Transform Spectroscopy. In Fourier Transforms-High-Tech Application and Current Trends; InTech: London, UK, 2017; pp. 149–174. [Google Scholar] [CrossRef]
- Sumisha, A.; Arthanareeswaran, G.; Ismail, A.F.; Kumar, D.P.; Shankar, M.V. Functionalized titanate nanotube–polyetherimide nanocomposite membrane for improved salt rejection under low pressure nanofiltration. RSC Adv. 2015, 5, 39464–39473. [Google Scholar] [CrossRef]
- Kassem, M.; Ali, A.; El-Assal, M.; El-badrawy, A. Formulation, characterization and in vivo application of oral insulin nanotechnology using different biodegradable polymers: Advanced drug delivery system. Int. J. Pharm. Sci. Res. 2018, 9, 3664–3677. [Google Scholar] [CrossRef]
- Li, H.; Huo, M.; Zhou, J.; Dai, Y.; Deng, Y.; Shi, X.; Masoud, J. Enhanced oral absorption of paclitaxel in N-deoxycholic acid-N, O-hydroxyethyl chitosan micellar system. J. Pharm. Sci 2010, 99, 4543–4553. [Google Scholar] [CrossRef]
- Hefni, H.H.; Nagy, M.; Azab, M.M.; Hussein, M.H. Esterification of chitosan with L-alanine and a study on their effect in removing the heavy metals and total organic carbon (TOC) from wastewater. Pure Appl. Chem. 2016, 88, 595–604. [Google Scholar] [CrossRef]
- Guo, J.; Ren, L.; Wang, R.; Zhang, C.; Yang, Y.; Liu, T. Water dispersible graphene noncovalently functionalized with tryptophan and its poly (vinyl alcohol) nanocomposite. Compos. Part B Eng. 2011, 42, 2130–2135. [Google Scholar] [CrossRef]
- Ge, W.; Li, D.; Chen, M.; Wang, X.; Liu, S.; Sun, R. Characterization and antioxidant activity of β-carotene loaded chitosan-graft-poly (lactide) nanomicelles. Carbohydr. Polym. 2015, 117, 169–176. [Google Scholar] [CrossRef]
- Antoniraj, M.G.; Kumar, C.S.; Kandasamy, R. Synthesis and characterization of poly (N-isopropylacrylamide)-g-carboxymethyl chitosan copolymer-based doxorubicin-loaded polymeric nanoparticles for thermoresponsive drug release. Colloid Polym. Sci. 2016, 294, 527–535. [Google Scholar] [CrossRef]
- Steinmann, W.; Walter, S.; Beckers, M.; Seide, G.; Gries, T. Thermal analysis of phase transitions and crystallization in polymeric fibers. Appl. Calor. Wide Cont. Differ. Scan. Calor. Isoth. Titra. Calor. Microcalor. 2013, 12, 279–305. [Google Scholar]
- Dudhani, A.R.; Kosaraju, S.L. Bioadhesive chitosan nanoparticles: Preparation and characterization. Carbohydr. Polym. 2010, 81, 243–251. [Google Scholar] [CrossRef]
- Righetti, M.C. Crystallization of polymers investigated by temperature-modulated DSC. Materials 2017, 10, 442. [Google Scholar] [CrossRef]
- Vu, H.; Nair, A.; Tran, L.; Pal, S.; Senkowsky, J.; Hu, W.; Tang, L. A Device to Predict Short-Term Healing Outcome of Chronic Wounds. Adv. Wound Care 2020, 9, 312–324. [Google Scholar] [CrossRef]
- Becker, C.; Dressman, J.; Junginger, H.; Kopp, S.; Midha, K.; Shah, V.; Stavchansky, S.; Barends, D. Biowaiver monographs for immediate release solid oral dosage forms: Rifampicin. J. Pharm. Sci. 2009, 98, 2252–2267. [Google Scholar] [CrossRef] [PubMed]
- Maniyar, S.A.; Jargar, J.G.; Das, S.N.; Dhundasi, S.A.; Das, K.K. Alteration of chemical behavior of L–ascorbic acid in combination with nickel sulfate at different pH solutions in vitro. Asian Pac. J. Trop. Biomed 2012, 2, 220–222. [Google Scholar] [CrossRef]
- Acuña, L.; Hamadat, S.; Corbalán, N.S.; González-Lizárraga, F.; dos-Santos-Pereira, M.; Rocca, J.; Sepúlveda Díaz, J.; Del-Bel, E.; Papy-García, D.; Chehín, R.N. Rifampicin and Its Derivative Rifampicin Quinone Reduce Microglial Inflammatory Responses and Neurodegeneration Induced In Vitro by α-Synuclein Fibrillary Aggregates. Cells 2019, 8, 776. [Google Scholar] [CrossRef] [PubMed]
- Alves, R.; Reis, T.V.S.; Silva, L.C.C.; Storpírtis, S.; Mercuri, L.P.; Matos, J.R. Thermal behavior and decomposition kinetics of rifampicin polymorphs under isothermal and non-isothermal conditions. Braz. J. Pharm. Sci. 2010, 46, 343–351. [Google Scholar] [CrossRef]
- Levy, R.; Okun, Z.; Shpigelman, A. The influence of chemical structure and the presence of ascorbic acid on anthocyanins stability and spectral properties in purified model systems. Foods 2019, 8, 207. [Google Scholar] [CrossRef] [PubMed]
- Amini Moghaddam, M.; Stloukal, P.; Kucharczyk, P.; Tow-Swiatek, A.; Garbacz, T.; Pummerova, M.; Klepka, T.; Sedlařík, V. Microcellular antibacterial polylactide-based systems prepared by additive extrusion with ALUM. Polym. Adv. Tech. 2019, 30, 2100–2108. [Google Scholar] [CrossRef]
- Osorio-Madrazo, A.; David, L.; Trombotto, S.; Lucas, J.-M.; Peniche-Covas, C.; Domard, A. Kinetics study of the solid-state acid hydrolysis of chitosan: Evolution of the crystallinity and macromolecular structure. Biomacromolecules 2010, 11, 1376–1386. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Cheng, C.-A.; Lee, B.-Y.; Clemens, D.L.; Huang, W.-Y.; Horwitz, M.A.; Zink, J.I. Facile strategy enabling both high loading and high release amounts of the water-insoluble drug clofazimine using mesoporous silica nanoparticles. ACS Appl. Mater. Interfaces. 2018, 10, 31870–31881. [Google Scholar] [CrossRef]
- Tang, F.; Li, L.; Chen, D. Mesoporous silica nanoparticles: Synthesis, biocompatibility and drug delivery. Adv. Mater. 2012, 24, 1504–1534. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| APENs | RIF/APENs | WPENs | RIF/WPENs | ||
|---|---|---|---|---|---|
| SEM | Size | 296.7 ± 28.2 a | 224.1 ± 50.9 b | 235.0 ± 46.5 b | 226.4 ± 50.7 b |
| TEM | Size | 271.9 ± 38.8 a | 215.0 ± 30.6 b | 220.0 ± 40.4 b | 184.9 ± 34.4 b |
| DLS | size | 313.8 ± 5.2 a | 241.9 ± 4.0 b | 249.1 ± 0.8 b | 224.7 ± 1.8 c |
| PDI | 0.31 ± 0.01 a | 0.31 ± 0.00 a | 0.28 ± 0.01 a | 0.29 ± 0.01 a | |
| ζ-potential | 29.0 ± 0.7 a | 42.0 ± 4.2 b | 26.3 ± 0.8 ac | 38.5 ± 2.8 b |
| Time (d) | Z-Average Size (nm) | PDI | Z-Average Size (nm) | PDI | |
|---|---|---|---|---|---|
| 1 | 302.1 ± 5.2 | 0.37 ± 0.03 | 233.5 ± 9.4 | 0.19 ± 0.02 | |
| H2O | 7 | 321.8 ± 13.3 | 0.27 ± 0.00 | 243.9 ± 2.0 | 0.25 ± 0.00 |
| 14 | 304.2 ± 7.4 | 0.25 ± 0.00 | 249.5 ± 5.2 | 0.18 ± 0.01 | |
| PBS 10mM | 7 | 311.4 ± 19.4 | 0.22 ± 0.01 | 240.0 ± 5.7 | 0.25 ± 0.00 |
| 14 | 294.1 ± 10.4 | 0.18 ± 0.01 | 239.1 ± 3.5 | 0.20 ± 0.01 |
| A-PENs | W-PENs | ||
|---|---|---|---|
| pH 7.4 | No ASC | 72.8 ± 1.1 a | 68.3 ± 3.0 a |
| ASC | 75.5 ± 2.4 a | 80.8 ± 13.0 a | |
| pH 8.5 | No ASC | 31.8 ± 2.1 b | 32.1 ± 2.8 b |
| ASC | 113.5 ± 6.0 c | 112.3 ± 4.7 c |
| 0 | 3 | 6 | 24 | 48 | 72 | |||
|---|---|---|---|---|---|---|---|---|
| ASC | pH 7.4 | 0.0 ± 0.0 | 1.1 ± 0.0 a | 2.4 ± 0.2 a | 8.9 ± 0.2 a | 28.8 ± 0.1 a | 58.3 ± 0.0 a | |
| pH 8.5 | 0.1 ± 0.0 | 0.1 ± 0.2 b | 1.7 ± 0.1 b | 14.1 ± 0.1 b | 46.8 ± 0.1 b | 72.2 ± 0.1 b | ||
| RIF | pH 7.4 | 0.0 ± 0.0 | 0.0 ± 0.0 | 5.1 ± 0.1 a | 20.7 ± 0.1 a | 37.1 ± 0.2 a | 50.6 ± 0.4 a | |
| pH 8.5 | 0.0 ± 0.0 | 0.1 ± 0.0 | 7.0 ± 0.2 b | 23.7 ± 0.2 b | 50.3 ± 0.4 b | 70.4 ± 0.1 b | ||
| ASC, pH 7.4 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 c | 0.0 ± 0.0 c | 3.1 ± 0.3 c | 9.2 ± 0.4 c | ||
| ASC, pH 8.5 | 0.0 ± 0.0 | 0.1 ± 0.0 | 0.9 ± 0.1 d | 5.2 ± 0.1 d | 8.8 ± 0.1 d | 12.6 ± 0.1 d | ||
| RIF/PENs | pH 7.4 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 | 4.1 ± 0.2 e | 4.1 ± 1.2 c | 3.7 ± 0.3 e | |
| pH 8.5 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 | 5.4 ± 0.5 f | 20.4 ± 0.2 e | 42.4 ± 1.7 f | ||
| ASC, pH 7.4 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 | 12.5 ± 1.1 b | 24.1 ± 2.7 e | 44.6 ± 4.6 a | ||
| ASC, pH 8.5 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 | 1.0 ± 0.0 g | 3.2 ± 0.1 c | 21.6 ± 3.0 g | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Motiei, M.; Pleno de Gouveia, L.; Šopík, T.; Vícha, R.; Škoda, D.; Císař, J.; Khalili, R.; Domincová Bergerová, E.; Münster, L.; Fei, H.; et al. Nanoparticle-Based Rifampicin Delivery System Development. Molecules 2021, 26, 2067. https://doi.org/10.3390/molecules26072067
Motiei M, Pleno de Gouveia L, Šopík T, Vícha R, Škoda D, Císař J, Khalili R, Domincová Bergerová E, Münster L, Fei H, et al. Nanoparticle-Based Rifampicin Delivery System Development. Molecules. 2021; 26(7):2067. https://doi.org/10.3390/molecules26072067
Chicago/Turabian StyleMotiei, Marjan, Luis Pleno de Gouveia, Tomáš Šopík, Robert Vícha, David Škoda, Jaroslav Císař, Reza Khalili, Eva Domincová Bergerová, Lukáš Münster, Haojie Fei, and et al. 2021. "Nanoparticle-Based Rifampicin Delivery System Development" Molecules 26, no. 7: 2067. https://doi.org/10.3390/molecules26072067
APA StyleMotiei, M., Pleno de Gouveia, L., Šopík, T., Vícha, R., Škoda, D., Císař, J., Khalili, R., Domincová Bergerová, E., Münster, L., Fei, H., Sedlařík, V., & Sáha, P. (2021). Nanoparticle-Based Rifampicin Delivery System Development. Molecules, 26(7), 2067. https://doi.org/10.3390/molecules26072067