
Liposomes as Antibiotic Delivery Systems: A Promising Nanotechnological Strategy against Antimicrobial Resistance


 ,
,  , ,
, ,  and
and 
Abstract
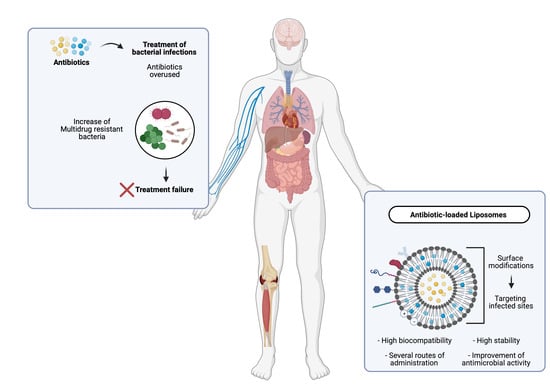
1. Introduction
2. Nanotechnological Approaches for Treatment of Bacterial Infections
3. Structure and Properties of Liposomes
4. Advantages of Liposomes as Antibiotic Carriers
4.1. Stability
4.1.1. Controlled and Sustained Release of Antibiotics
4.1.2. Prolonged Plasma Circulation Time
4.2. Infection Targeting
4.3. Improved Bactericidal Potency and Efficacy
4.4. Overcoming Bacterial Resistance Mechanisms

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathogen | Emerging Resistance Patterns | Formulations Developed | Effect | Ref. | |
|---|---|---|---|---|---|
| Active Compound | Lipid Composition | ||||
| Acinetobacter baumannii | Carbapenem Polymyxin | Polymyxin B | Chitosan–DPPC:DSPE:Chol Chitosan–DPPC:DSPE:Chol with USMB (DPPC:DSPE:Chol) | The combination of the two systems revealed an antibacterial synergetic effect that could almost eliminate the biofilm-producing bacterium. | [106] |
| Fusidic acid | DOPE:DPPC:CHEMS DPPC:Chol | An increased antibacterial effect of fusogenic liposomes (DOPE:DPPC:CHEMS) against clinical isolates in comparison to non-fusogenic formulation (DPPC:Chol) was observed (MICs of 37.5–300.0 µg/mL versus >833.0 µg/mL). Free fusidic acid did not present antibacterial effect against Gram-negative bacteria. | [107] | ||
| Vancomycin | DOPE:DPPC:CHEMS DPPC:Chol | Fusogenic liposomes (DOPE:DPPC:CHEMS) displayed MICs of 6–12.5 µg/mL against clinical isolates, while free vancomycin and non-fusogenic formulation (DPPC:Chol) showed no antibacterial activity. | [17] | ||
| Polymyxin B | DPPC:Chol POPC:Chol | Higher incorporation parameters for DPPC:Chol were achieved. MIC was 16-fold lower for liposomal formulation than for free antibiotic. | [100] | ||
| Pseudomonas aeruginosa | Carbapenem | Amikacin Gentamicin Tobramycin | DPPC:Chol | With liposomal formulations, MICs have been maintained or reduced against all tested clinical isolates, for all antibiotics incorporated in relation to respective free antibiotics (MICs reductions were antibiotic- and strain-dependent: amikacin, 2–64-fold; gentamicin, 2–64-fold; tobramycin, 1–128-fold). | [92] |
| Polymyxin B | DPPC:Chol POPC:Chol | Higher incorporation parameters for DPPC:Chol were achieved. MICs against clinical isolates were 4–32-fold lower for liposomal formulation in relation to free antibiotic. Liposomal formulation promoted the antibiotic penetration into a resistant strain in higher extent than free form. | [100] | ||
| Gentamicin | DMPC:Chol | MICs against clinical isolates and a laboratory strain were 2–16- and 4-fold lower, respectively, for liposomal gentamicin in comparison with free form. Time–kill values of liposomal formulation were equivalent to the free antibiotic, for the laboratory strain and one clinical isolate, while for the other clinical isolate the bactericidal effect was achieved at 4× MIC for liposomal formulation and free gentamicin, after 6 and 24 h, respectively. | [108] | ||
| Norfloxacin | PCT1–EPC:Chol:α tocopherol PCT2–EPC:Chol:α tocopherol | An increased antibacterial effect against a multi-resistant strain for both formulations in comparison with free antibiotic was achieved (MIC of 3.2 µg/mL versus >30.0 µg/mL). No toxic effects were observed for any of the formulations, evaluated through an in vivo embryo chicken model. | [109] | ||
| Ofloxacin | DMPC:Chol:DP DMPC:Chol:DPPS DMPC:Chol:DPPE DMPC:Chol:DPPA | After a susceptibility screening against reference strains of all developed formulations, DMPC:Chol:DP and DMPC:Chol:DPPS were chosen for further studies. An increased antibacterial effect against clinical isolates resistant to quinolones, mainly with DMPC:Chol:DP formulations was observed, resulting in MICs of 2–4-fold lower than free antibiotic. Higher intracellular antibiotic concentrations were obtained for both strains tested, when antibiotic was loaded in DMPC:Chol:DP. | [110] | ||
| Enterobacteriacea | Carbapenem ESBL+ Fluoroquinolones | Cefepime | EPC:Chol EPC:Chol:12NBr DOPE:12NBr | The formulation EPC:Chol:12NBr demonstrated higher incorporation parameters and, thus, was used for antibacterial study. Cefepime-loaded liposomes presented similar antibacterial activity to its free form, against an E. coli strain. | [111] |
| Azithromycin | EPC:EPG: EPC:HSPC-3 EPC:EPG:HSPC-3 EPC:Pg EPC:EPG:Pg EPC:SLPC:-80:Pg EPC:EPG:SLPC-80:Pg | Liposomes incorporation parameters and stability assays promoted the selection of EPC:HSPC-3, EPC:Pg and EPC:SLPC:-80:Pg formulations for further experiments. MIC50 for all strains tested, were similar for liposomal formulations and for free antibiotic, while against bacteria in biofilm form the activity was lipid composition-dependent. Antibiotic-loaded EPC:EPG:HSPC-3 demonstrated the lower MBIC50 against the E. coli k-12 strain (8-fold lower in relation to free antibiotic). | [112] | ||
| Ofloxacin | DMPC:Chol:DP DMPC:Chol:DPPS DMPC:Chol:DPPE DMPC:Chol:DPPA | After a susceptibility screening against reference strains of all developed formulations, DMPC:Chol:DP and DMPC:Chol:DPPS were chosen for further studies. MICs against E. coli clinical isolates were 4-fold lower for both formulations in relation to free antibiotic. Higher intracellular antibiotic concentrations were achieved when antibiotic was loaded in DMPC:Chol:DP. | [110] | ||
| Norfloxacin | PCT1–EPC:Chol:α tocopherol PCT2–EPC:Chol:α tocopherol | An increased antibacterial effect against an E. coli strain, mainly with PCT1–EPC:Chol:α tocopherol formulation was observed, resulting in a MIC 9-fold lower than free antibiotic. In case of Salmonella strains, PCT2–EPC:Chol:α tocopherol presented the highest antibacterial effect with MICs of 2–17- and 16–42-fold lower than the other formulation and free antibiotic, respectively. No toxic effects were observed for any of the formulations, evaluated though an in vivo embryo chicken model. | [109] | ||
| Polymyxin B | DPPC:Chol POPC:Chol | Higher incorporation parameters for DPPC:Chol were achieved, thus further studies were conducted with this formulation. MICs against E. coli and K. pneumoniae were 8–16- and 16-fold, respectively, for the liposomal formulation in comparison with free polymyxin B. | [100] | ||
| Ciprofloxacin | DPPC:Chol DSPC:Chol SM:Chol | The SM:Chol formulation presented higher circulation lifetime than the remaining formulations. In this way, the efficacy of antibiotic-loaded SM:Chol was evaluated in a Salmonella typhimurium infection model, resulting in viable bacteria 103–104-fold lower in the livers and spleens of infected mice than the free antibiotic. | [113] | ||
| Staphylococcus aureus | Methicillin Vancomycin | Ofloxacin | DMPC:Chol:DP DMPC:Chol:DPPS DMPC:Chol:DPPE DMPC:Chol:DPPA | After a susceptibility screening against reference strains of all developed formulations, DMPC:Chol:DP and DMPC:Chol:DPPS were chosen for further studies. An increased antibacterial effect against S. aureus clinical isolates, mainly for DMPC:Chol:DPPS, was observed, with values 3- and 4-fold lower than free antibiotic. | [110] |
| Piperacillin | PC:Chol | Antibiotic incorporated in liposomes inhibited 3-fold higher a S. aureus clinical isolate growth, than its free form. Experiments using exogenous staphylococcal β-lactamase demonstrated that the liposomal formulation promoted the highest degree of protection against hydrolysis by staphylococcal β-lactamase. | [101] | ||
| Vancomycin | DSPC:DcP:Chol DSPC:DMPG:Chol | MICs and MBCs against MRSA strains were 2–4- and 4-fold lower, respectively, for both formulations in relation to free antibiotic. The DSPC:DcP:Chol formulation showed the highest efficacy. In a systemic MRSA murine model, the liposomal formulation displayed a higher therapeutic effect, improving kidney clearance by 1-log in comparison with free antibiotic. | [69] | ||
| Vancomycin | DSPC:Chol DSPC:Chol:DSPE-PEG | At the highest antibiotic concentration tested, DSPC:Chol formulation (non-pegylated liposomes) reduced the intracellular MRSA growth inside macrophages in approximately 2- and 3-fold higher in relation to pegylated formulation (DSPC:Chol:DSPE-PEG) and free antibiotic, respectively. | [103] | ||
| Azithromycin | Lipoid S75 Lipoid S75:SDCh Lipoid S75:Pg DPPC:DODAB | MIC and MBIC were maintained or reduced for all formulations in relation to free antibiotic. The DPPC:DODAB formulation presented the highest antibacterial activity against both planktonic and biofilm forms of all clinical isolates tested. The MICs and MBICs were 8–32- and 16–32-fold lower than free azithromycin. Liposomal formulations demonstrated biocompatibility with keratinocytes and fibroblasts. | [114] | ||
| Methicillin | DOPE:DPPC:CHEMS: DSPE-PEG-MAL DOPE:DPPC:CHEMS:DSPE-PEG-Tat | Antibacterial activity reductions were observed for both formulations, especially for DOPE:DPPC:CHEMS:DSPE-PEG-Tat formulation. MICs against a MRSA strain were 3.3, 5.0 and >5.0 µg/mL for DOPE:DPPC:CHEMS:DSPE-PEG-Tat, DOPE:DPPC:CHEMS:DSPE-PEG-MAL and free methicillin, respectively. | [115] | ||
| Helicobacter pylori | Clarithromycin | Ampicillin Metronidazole | DPPC:Chol:NBD-PC DPPC:Fuc-E4-Chol:Chol:NBD-PC Epikuron 170:Chol:NBD-PC Epikuron 170:Fuc-E4-Chol:Chol:NBD-PC | Liposome–bacteria interaction results obtained by epifluorescence microscopy demonstrated to be strain- and lipid composition-dependent. Formulations without Epikuron 170 displayed superior interaction levels in both strains tested. However, DPPC:Fuc-E4-Chol:Chol:NBD-PC showed the highest interaction levels in the strain that express the babA2 gene (H. pylori 17875), due to the specifically link between the BabA2 protein and the fucose at the surface of liposomes. | [116] |
| Amoxicillin | LC:Chol:DDAB PCT-LC:Chol:DDAB | Although both formulations presented similar antibacterial effect, the experimental assays developed in this study evidenced a specific interaction of PCT-coating liposomes with mucins and surface structures of bacteria. | [117] | ||
| Campylobacter | Fluoroquinolones | Norfloxacin | PCT1–EPC:Chol:α tocoferol PCT2–EPC:Chol:α tocoferol | An increased antibacterial activity against a Campylobacter jejuni strain, mainly with PCT–EPC:Chol:α tocoferol formulation was observed. MIC was 10-fold lower than free antibiotic. No toxic effects were observed for any of the formulations, evaluated in an in vivo embryo chicken model. | [109] |
| Streptococcus pneumoniae | Penicillin | Vancomycin | DOPE:DPPC:CHEMS: DSPE-PEG-MAL DOPE:DPPC:CHEMS:DSPE-PEG-Tat | MICs were approximately 2-fold lower for both formulations than respective free antibiotic. For the lowest concentrations tested (0.6 µg/mL) the formulation. DOPE:DPPC:CHEMS:DSPE-PEG-Tat displayed more favorable results, with a reduction of viable bacteria of approximately 1- and 2-fold in relation to the other formulation and to free vancomycin, respectively. | [115] |
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Rodríguez-Rojas, A.; Rodríguez-Beltrán, J.; Couce, A.; Blázquez, J. Antibiotics and Antibiotic Resistance: A Bitter Fight against Evolution. Int. J. Med. Microbiol. 2013, 303, 293–297. [Google Scholar] [CrossRef]
- Luo, W.; Chen, D.; Wu, M.; Li, Z.; Tao, Y.; Liu, Q.; Pan, Y.; Qu, W.; Yuan, Z.; Xie, S. Pharmacokinetics/Pharmacodynamics Models of Veterinary Antimicrobial Agents. J. Vet. Sci. 2019, 20, e40. [Google Scholar] [CrossRef]
- World Health Organization. Antibiotic Resistance. Available online: https://www.who.int/news-room/fact-sheets/detail/antibiotic-resistance (accessed on 29 January 2020).
- WHO. Antibacterial Agents In Clinical Development. Available online: http://www.who.int/medicines/areas/rational_use/antibacterial_agents_clinical_development/en/ (accessed on 22 April 2020).
- Metz, M.; Shlaes, D.M. Eight More Ways To Deal with Antibiotic Resistance. Antimicrob. Agents Chemother. 2014, 4253–4256. [Google Scholar] [CrossRef]
- Wang, C.H.; Hsieh, Y.H.; Powers, Z.M.; Kao, C.Y. Defeating Antibiotic-Resistant Bacteria: Exploring Alternative Therapies for a Post-Antibiotic Era. Int. J. Mol. Sci. 2020, 21, 1061. [Google Scholar] [CrossRef]
- Aslam, B.; Wang, W.; Arshad, M.I.; Khurshid, M.; Muzammil, S.; Rasool, M.H.; Nisar, M.A.; Alvi, R.F.; Aslam, M.A.; Qamar, M.U.; et al. Antibiotic Resistance: A Rundown of a Global Crisis. Infect. Drug Resist. 2018, 11, 1645–1658. [Google Scholar] [CrossRef]
- Zhang, L.; Pornpattananangku, D.; Hu, C.M.J.; Huang, C.M. Development of Nanoparticles for Antimicrobial Drug Delivery. Curr. Med. Chem. 2010, 17, 585–594. [Google Scholar] [CrossRef] [PubMed]
- Huh, A.J.; Kwon, Y.J. “Nanoantibiotics”: A New Paradigm for Treating Infectious Diseases Using Nanomaterials in the Antibiotics Resistant Era. J. Control. Release 2011, 156, 128–145. [Google Scholar] [CrossRef]
- Liu, D.; Yang, F.; Xiong, F.; Gu, N. The Smart Drug Delivery System and Its Clinical Potential. Theranostics 2016, 6, 1306–1323. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Rai, D.B.; Jangid, A.K.; Kulhari, H. Chapter 7—Use of nanotechnology in antimicrobial therapy. In Methods in Microbiology; Gurtler, V., Ball, A.S., Soni, S., Eds.; Nanotechnology; Academic Press: Cambridge, MA, USA, 2019; Volume 46, pp. 143–172. [Google Scholar]
- Pinto-Alphandary, H.; Andremont, A.; Couvreur, P. Targeted Delivery of Antibiotics Using Liposomes and Nanoparticles: Research and Applications. Int. J. Antimicrob. Agents 2000, 13, 155–168. [Google Scholar] [CrossRef]
- Drulis-Kawa, Z.; Dorotkiewicz-Jach, A. Liposomes as Delivery Systems for Antibiotics. Int. J. Pharm. 2010, 387, 187–198. [Google Scholar] [CrossRef]
- Gomez, A.G.; Hosseinidoust, Z. Liposomes for Antibiotic Encapsulation and Delivery. ACS Infect. Dis. 2020, 6, 896–908. [Google Scholar] [CrossRef] [PubMed]
- Lima, R.; Del Fiol, F.S.; Balcão, V.M. Prospects for the Use of New Technologies to Combat Multidrug-Resistant Bacteria. Front. Pharmacol. 2019, 10, 692. [Google Scholar] [CrossRef] [PubMed]
- Brooks, B.D.; Brooks, A.E. Therapeutic Strategies to Combat Antibiotic Resistance. Adv. Drug Deliv. Rev. 2014, 78, 14–27. [Google Scholar] [CrossRef] [PubMed]
- Nicolosi, D.; Scalia, M.; Nicolosi, V.M.; Pignatello, R. Encapsulation in Fusogenic Liposomes Broadens the Spectrum of Action of Vancomycin against Gram-Negative Bacteria. Int. J. Antimicrob. Agents 2010, 35, 553–558. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Chen, Y.; Zhang, Y.; Zhang, Q.; Zhang, L. Nanoparticle-Based Local Antimicrobial Drug Delivery. Adv. Drug Deliv. Rev. 2018, 127, 46–57. [Google Scholar] [CrossRef]
- dos Santos Ramos, M.A.; dos Santos, K.C.; da Silva, P.B.; de Toledo, L.G.; Marena, G.D.; Rodero, C.F.; de Camargo, B.A.F.; Fortunato, G.C.; Bauab, T.M.; Chorilli, M. Nanotechnological Strategies for Systemic Microbial Infections Treatment: A Review. Int. J. Pharm. 2020, 589, 119780. [Google Scholar] [CrossRef]
- Li, M.; Du, C.; Guo, N.; Teng, Y.; Meng, X.; Sun, H.; Li, S.; Yu, P.; Galons, H. Composition Design and Medical Application of Liposomes. Eur. J. Med. Chem. 2019, 164, 640–653. [Google Scholar] [CrossRef]
- Abu Lila, A.S.; Ishida, T. Liposomal Delivery Systems: Design Optimization and Current Applications. Biol. Pharm. Bull. 2017, 40, 1–10. [Google Scholar] [CrossRef]
- Ferreira, M.; Aguiar, S.; Bettencourt, A.; Gaspar, M.M. Lipid-Based Nanosystems for Targeting Bone Implant-Associated Infections: Current Approaches and Future Endeavors. Drug Deliv. Transl. Res. 2020. [Google Scholar] [CrossRef]
- Crommelin, D.J.A.; van Hoogevest, P.; Storm, G. The Role of Liposomes in Clinical Nanomedicine Development. What Now? Now What? J. Control. Release 2019, 318, 256–263. [Google Scholar] [CrossRef]
- Santos, R.S.; Figueiredo, C.; Azevedo, N.F.; Braeckmans, K.; De Smedt, S.C. Nanomaterials and Molecular Transporters to Overcome the Bacterial Envelope Barrier: Towards Advanced Delivery of Antibiotics. Adv. Drug Deliv. Rev. 2018, 136–137, 28–48. [Google Scholar] [CrossRef]
- Sercombe, L.; Veerati, T.; Moheimani, F.; Wu, S.Y.; Sood, A.K.; Hua, S. Advances and Challenges of Liposome Assisted Drug Delivery. Front. Pharmacol. 2015, 6, 286. [Google Scholar] [CrossRef] [PubMed]
- Akbarzadeh, A.; Rezaei-Sadabady, R.; Davaran, S.; Joo, S.W.; Zarghami, N.; Hanifehpour, Y.; Samiei, M.; Kouhi, M.; Nejati-Koshki, K. Liposome: Classification, Preparation and Applications. Nanoscale Res. Lett. 2013, 8, 102. [Google Scholar] [CrossRef] [PubMed]
- Cruz, M.E.M.; Manuela Gaspar, M.; Bárbara, M.; Martins, F.; Luísa Corvo, M. Liposomal Superoxide Dismutases and Their Use in the Treatment of Experimental Arthritis. Meth. Enzymol. 2005, 391, 395–413. [Google Scholar] [CrossRef]
- Cruz, M.E.M.; Simoes, S.I.; Corvo, M.L.; Martins, M.B.F.; Gaspar, M.M. Formulation of NPDDS for Macromolecules. In Drug Delivery Nanoparticles Formulation and Characterization; CRC Press: Boca Raton, FL, USA, 2016; Volume 191, pp. 35–49. [Google Scholar]
- Lian, T.; Ho, R.J. Trends and Developments in Liposome Drug Delivery Systems. J. Pharm. Sci. 2001, 90, 667–680. [Google Scholar] [CrossRef] [PubMed]
- Eleraky, N.E.; Allam, A.; Hassan, S.B.; Omar, M.M. Nanomedicine Fight against Antibacterial Resistance: An Overview of the Recent Pharmaceutical Innovations. Pharmaceutics 2020, 12, 142. [Google Scholar] [CrossRef]
- Boidin, C.; Moshiri, P.; Dahyot-Fizelier, C.; Goutelle, S.; Lefeuvre, S. Pharmacokinetic Variability of Beta-Lactams in Critically Ill Patients: A Narrative Review. Anaesth. Crit. Care Pain Med. 2019, 39, 87–109. [Google Scholar] [CrossRef] [PubMed]
- Levison, M.E.; Levison, J.H. Pharmacokinetics and Pharmacodynamics of Antibacterial Agents. Infect. Dis. Clin. North Am. 2009, 23, 791-vii. [Google Scholar] [CrossRef] [PubMed]
- Olofsson, S.K.; Cars, O. Optimizing Drug Exposure to Minimize Selection of Antibiotic Resistance. Clin. Infect. Dis. 2007, 45, S129–S136. [Google Scholar] [CrossRef]
- Buijk, S.E.; Mouton, J.W.; Gyssens, I.C.; Verbrugh, H.A.; Bruining, H.A. Experience with a Once-Daily Dosing Program of Aminoglycosides in Critically Ill Patients. Intensive Care Med. 2002, 28, 936–942. [Google Scholar] [CrossRef]
- Leekha, S.; Terrell, C.L.; Edson, R.S. General Principles of Antimicrobial Therapy. Mayo Clin. Proc. 2011, 86, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Zazo, H.; Colino, C.I.; Lanao, J.M. Current Applications of Nanoparticles in Infectious Diseases. J. Control. Release 2016, 224, 86–102. [Google Scholar] [CrossRef]
- Lim, S.B.; Banerjee, A.; Önyüksel, H. Improvement of Drug Safety by the Use of Lipid-Based Nanocarriers. J. Control. Release 2012, 163, 34–45. [Google Scholar] [CrossRef]
- Allen, T.M.; Cullis, P.R. Liposomal Drug Delivery Systems: From Concept to Clinical Applications. Adv. Drug Deliv. Rev. 2013, 65, 36–48. [Google Scholar] [CrossRef] [PubMed]
- Zeitlinger, M.A.; Derendorf, H.; Mouton, J.W.; Cars, O.; Craig, W.A.; Andes, D.; Theuretzbacher, U. Protein Binding: Do We Ever Learn? Antimicrob. Agents Chemother. 2011, 55, 3067–3074. [Google Scholar] [CrossRef]
- Swenson, C.E.; Popescu, M.C.; Ginsberg, R.S. Preparation and Use of Liposomes in the Treatment of Microbial Infections. Crit. Rev. Microbiol. 1988, 15 Suppl. 1, S1–S31. [Google Scholar] [CrossRef]
- Samuelsson, E.; Shen, H.; Blanco, E.; Ferrari, M.; Wolfram, J. Contribution of Kupffer Cells to Liposome Accumulation in the Liver. Colloids Surf. B Biointerfaces 2017, 158, 356–362. [Google Scholar] [CrossRef]
- Gaspar, M.M.; Calado, S.; Pereira, J.; Ferronha, H.; Correia, I.; Castro, H.; Tomás, A.M.; Cruz, M.E.M. Targeted Delivery of Paromomycin in Murine Infectious Diseases through Association to Nano Lipid Systems. Nanomedicine 2015, 11, 1851–1860. [Google Scholar] [CrossRef]
- van Etten, E.W.; ten Kate, M.T.; Snijders, S.V.; Bakker-Woudenberg, I.A. Administration of Liposomal Agents and Blood Clearance Capacity of the Mononuclear Phagocyte System. Antimicrob. Agents Chemother. 1998, 42, 1677–1681. [Google Scholar] [CrossRef] [PubMed]
- Hamidi, M.; Azadi, A.; Rafiei, P. Pharmacokinetic Consequences of Pegylation. Drug Deliv. 2006, 13, 399–409. [Google Scholar] [CrossRef] [PubMed]
- Zylberberg, C.; Matosevic, S. Pharmaceutical Liposomal Drug Delivery: A Review of New Delivery Systems and a Look at the Regulatory Landscape. Drug Deliv. 2016, 23, 3319–3329. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, M.M.; Boerman, O.C.; Laverman, P.; Corvo, M.L.; Storm, G.; Cruz, M.E.M. Enzymosomes with Surface-Exposed Superoxide Dismutase: In Vivo Behaviour and Therapeutic Activity in a Model of Adjuvant Arthritis. J. Control. Release 2007, 117, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Ellbogen, M.H.; Olsen, K.M.; Gentry-Nielsen, M.J.; Preheim, L.C. Efficacy of Liposome-Encapsulated Ciprofloxacin Compared with Ciprofloxacin and Ceftriaxone in a Rat Model of Pneumococcal Pneumonia. J. Antimicrob. Chemother. 2003, 51, 83–91. [Google Scholar] [CrossRef][Green Version]
- Muppidi, K.; Wang, J.; Betageri, G.; Pumerantz, A.S. PEGylated Liposome Encapsulation Increases the Lung Tissue Concentration of Vancomycin. Antimicrob. Agents Chemother. 2011, 55, 4537–4542. [Google Scholar] [CrossRef]
- Abed, N.; Couvreur, P. Nanocarriers for Antibiotics: A Promising Solution to Treat Intracellular Bacterial Infections. Int. J. Antimicrob. Agents 2014, 43, 485–496. [Google Scholar] [CrossRef] [PubMed]
- Zarogoulidis, P.; Kioumis, I.; Porpodis, K.; Spyratos, D.; Tsakiridis, K.; Huang, H.; Li, Q.; Turner, J.F.; Browning, R.; Hohenforst-Schmidt, W.; et al. Clinical Experimentation with Aerosol Antibiotics: Current and Future Methods of Administration. Drug Des. Devel. Ther. 2013, 7, 1115–1134. [Google Scholar] [CrossRef]
- Khatib, I.; Khanal, D.; Ruan, J.; Cipolla, D.; Dayton, F.; Blanchard, J.D.; Chan, H.-K. Ciprofloxacin Nanocrystals Liposomal Powders for Controlled Drug Release via Inhalation. Int. J. Pharm. 2019, 566, 641–651. [Google Scholar] [CrossRef]
- Bassetti, M.; Vena, A.; Russo, A.; Peghin, M. Inhaled Liposomal Antimicrobial Delivery in Lung Infections. Drugs 2020, 80, 1309–1318. [Google Scholar] [CrossRef]
- Liu, C.; Shi, J.; Dai, Q.; Yin, X.; Zhang, X.; Zheng, A. In-Vitro and in-Vivo Evaluation of Ciprofloxacin Liposomes for Pulmonary Administration. Drug Dev. Ind. Pharm. 2015, 41, 272–278. [Google Scholar] [CrossRef]
- Serisier, D.J. Inhaled Antibiotics for Lower Respiratory Tract Infections: Focus on Ciprofloxacin. Drugs Today (Barc) 2012, 48, 339–351. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Yu, S.; Lin, Y.; Zou, P.; Chai, G.; Yu, H.H.; Wickremasinghe, H.; Shetty, N.; Ling, J.; Li, J.; et al. Co-Delivery of Ciprofloxacin and Colistin in Liposomal Formulations with Enhanced In Vitro Antimicrobial Activities against Multidrug Resistant Pseudomonas Aeruginosa. Pharm. Res. 2018, 35, 187. [Google Scholar] [CrossRef] [PubMed]
- Lipsky, B.A.; Hoey, C. Topical Antimicrobial Therapy for Treating Chronic Wounds. Clin. Infect. Dis. 2009, 49, 1541–1549. [Google Scholar] [CrossRef] [PubMed]
- Ki, V.; Rotstein, C. Bacterial Skin and Soft Tissue Infections in Adults: A Review of Their Epidemiology, Pathogenesis, Diagnosis, Treatment and Site of Care. Can. J. Infect. Dis. Med. Microbiol. 2008, 19, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Lu, K.; Yu, C.; Huang, Q.; Du, Y.-Z. Nano-Drug Delivery Systems in Wound Treatment and Skin Regeneration. J. Nanobiotechnol. 2019, 17, 82. [Google Scholar] [CrossRef]
- Price, C.I.; Horton, J.W.; Baxter, C.R. Topical Liposomal Delivery of Antibiotics in Soft Tissue Infection. J. Surg. Res. 1990, 49, 174–178. [Google Scholar] [CrossRef]
- Augustin, M.; Goepel, L.; Jacobi, A.; Bosse, B.; Mueller, S.; Hopp, M. Efficacy and Tolerability of Liposomal Polyvinylpyrrolidone-Iodine Hydrogel for the Localized Treatment of Chronic Infective, Inflammatory, Dermatoses: An Uncontrolled Pilot Study. Clin. Cosmet. Investig. Dermatol. 2017, 10, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Sun, D.; Fan, Q.; Ma, Q.; Dong, Z.; Tao, W.; Tao, H.; Liu, Z.; Wang, C. The Enhanced Permeability and Retention Effect Based Nanomedicine at the Site of Injury. Nano Res. 2020, 13, 564–569. [Google Scholar] [CrossRef]
- Drulis-Kawa, Z.; Dorotkiewicz-Jach, A.; Gubernator, J.; Gula, G.; Bocer, T.; Doroszkiewicz, W. The Interaction between Pseudomonas Aeruginosa Cells and Cationic PC:Chol:DOTAP Liposomal Vesicles versus Outer-Membrane Structure and Envelope Properties of Bacterial Cell. Int. J. Pharm. 2009, 367, 211–219. [Google Scholar] [CrossRef]
- Wang, D.-Y.; van der Mei, H.C.; Ren, Y.; Busscher, H.J.; Shi, L. Lipid-Based Antimicrobial Delivery-Systems for the Treatment of Bacterial Infections. Front. Chem. 2019, 7, 872. [Google Scholar] [CrossRef]
- Nisini, R.; Poerio, N.; Mariotti, S.; De Santis, F.; Fraziano, M. The Multirole of Liposomes in Therapy and Prevention of Infectious Diseases. Front. Immunol. 2018, 9, 155. [Google Scholar] [CrossRef]
- Kneidl, B.; Peller, M.; Winter, G.; Lindner, L.H.; Hossann, M. Thermosensitive Liposomal Drug Delivery Systems: State of the Art Review. Int. J. Nanomedicine 2014, 9, 4387–4398. [Google Scholar] [CrossRef]
- Gaspar, M.M.; Radomska, A.; Gobbo, O.L.; Bakowsky, U.; Radomski, M.W.; Ehrhardt, C. Targeted Delivery of Transferrin-Conjugated Liposomes to an Orthotopic Model of Lung Cancer in Nude Rats. J. Aerosol Med. Pulm. Drug Deliv. 2012, 25, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, E.; Gomes, A.C.; Preto, A.; Cavaco-Paulo, A. Design of Liposomal Formulations for Cell Targeting. Colloids Surf. B Biointerfaces 2015, 136, 514–526. [Google Scholar] [CrossRef]
- Alhariri, M.; Azghani, A.; Omri, A. Liposomal Antibiotics for the Treatment of Infectious Diseases. Expert Opin. Drug Deliv. 2013, 10, 1515–1532. [Google Scholar] [CrossRef] [PubMed]
- Sande, L.; Sanchez, M.; Montes, J.; Wolf, A.J.; Morgan, M.A.; Omri, A.; Liu, G.Y. Liposomal Encapsulation of Vancomycin Improves Killing of Methicillin-Resistant Staphylococcus Aureus in a Murine Infection Model. J. Antimicrob. Chemother. 2012, 67, 2191–2194. [Google Scholar] [CrossRef] [PubMed]
- Hajiahmadi, F.; Alikhani, M.Y.; Shariatifar, H.; Arabestani, M.R.; Ahmadvand, D. The Bactericidal Effect of Lysostaphin Coupled with Liposomal Vancomycin as a Dual Combating System Applied Directly on Methicillin-Resistant Staphylococcus Aureus Infected Skin Wounds in Mice. Int. J. Nanomedicine 2019, 14, 5943–5955. [Google Scholar] [CrossRef]
- Santos-Ferreira, I.; Bettencourt, A.; Almeida, A.J. Nanoparticulate Platforms for Targeting Bone Infections: Meeting a Major Therapeutic Challenge. Nanomedicine (London) 2015, 10, 3131–3145. [Google Scholar] [CrossRef] [PubMed]
- Soares, D.; Leite, P.; Barreira, P.; Aido, R.; Sousa, R. Antibiotic-Loaded Bone Cement in Total Joint Arthroplasty. Acta Orthop. Belg. 2015, 81, 184–190. [Google Scholar]
- Jiranek, W.A.; Hanssen, A.D.; Greenwald, A.S. Antibiotic-Loaded Bone Cement for Infection Prophylaxis in Total Joint Replacement. J. Bone Joint Surg. Am. 2006, 88, 2487–2500. [Google Scholar] [CrossRef]
- Bistolfi, A.; Massazza, G.; Verné, E.; Massè, A.; Deledda, D.; Ferraris, S.; Miola, M.; Galetto, F.; Crova, M. Antibiotic-Loaded Cement in Orthopedic Surgery: A Review. ISRN Orthop. 2011, 2011. [Google Scholar] [CrossRef]
- Ferreira, M.; Rzhepishevska, O.; Grenho, L.; Malheiros, D.; Gonçalves, L.; Almeida, A.J.; Jordão, L.; Ribeiro, I.A.; Ramstedt, M.; Gomes, P.; et al. Levofloxacin-Loaded Bone Cement Delivery System: Highly Effective against Intracellular Bacteria and Staphylococcus Aureus Biofilms. Int. J. Pharm. 2017, 532, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Snoddy, B.; Jayasuriya, A.C. The Use of Nanomaterials to Treat Bone Infections. Mater. Sci. Eng. C Mater. Biol. Appl. 2016, 67, 822–833. [Google Scholar] [CrossRef]
- Bastari, K.; Arshath, M.; Ng, Z.H.M.; Chia, J.H.; Yow, Z.X.D.; Sana, B.; Tan, M.F.C.; Lim, S.; Loo, S.C.J. A Controlled Release of Antibiotics from Calcium Phosphate-Coated Poly(Lactic-Co-Glycolic Acid) Particles and Their in Vitro Efficacy against Staphylococcus Aureus Biofilm. J. Mater. Sci. Mater. Med. 2014, 25, 747–757. [Google Scholar] [CrossRef]
- Hui, T.; Yongqing, X.; Tiane, Z.; Gang, L.; Yonggang, Y.; Muyao, J.; Jun, L.; Jing, D. Treatment of Osteomyelitis by Liposomal Gentamicin-Impregnated Calcium Sulfate. Arch. Orthop. Trauma Surg. 2009, 129, 1301–1308. [Google Scholar] [CrossRef] [PubMed]
- Koedel, U.; Scheld, W.M.; Pfister, H.-W. Pathogenesis and Pathophysiology of Pneumococcal Meningitis. Lancet Infect. Dis. 2002, 2, 721–736. [Google Scholar] [CrossRef]
- Viladrich, P.F. Management of Meningitis caused by resistant Streptococcus pneumoniae. In Management of Multiple Drug-resistant Infections; Humana Press Inc.: Totowa, NJ, USA, 2004; pp. 31–48. [Google Scholar]
- Mook-Kanamori, B.B.; Geldhoff, M.; van der Poll, T.; van de Beek, D. Pathogenesis and Pathophysiology of Pneumococcal Meningitis. Clin. Microbiol. Rev. 2011, 24, 557–591. [Google Scholar] [CrossRef]
- Brouwer, M.C.; Tunkel, A.R.; van de Beek, D. Epidemiology, Diagnosis and Antimicrobial Treatment of Acute Bacterial Meningitis. Clin. Microbiol. Rev. 2010, 23, 467–492. [Google Scholar] [CrossRef]
- Pardridge, W.M. Drug Transport across the Blood–Brain Barrier. J. Cereb. Blood Flow Metab. 2012, 32, 1959–1972. [Google Scholar] [CrossRef]
- Nau, R.; Sörgel, F.; Eiffert, H. Penetration of Drugs through the Blood-Cerebrospinal Fluid/Blood-Brain Barrier for Treatment of Central Nervous System Infections. Clin. Microbiol. Rev. 2010, 23, 858–883. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, S.H. Management of Multiple Drug-Resistant Infections; Humana Press Inc.: Totowa, NJ, USA, 2004. [Google Scholar]
- Vieira, D.B.; Gamarra, L.F. Getting into the Brain: Liposome-Based Strategies for Effective Drug Delivery across the Blood–Brain Barrier. Int. J. Nanomedicine 2016, 11, 5381–5414. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.; Singh-Moon, R.P.; Ellis, J.A.; Chaudhuri, D.B.; Wang, M.; Reif, R.; Bruce, J.N.; Bigio, I.J.; Straubinger, R.M. Cerebral Hypoperfusion-Assisted Intra-Arterial Deposition of Liposomes in Normal and Glioma-Bearing Rats. Neurosurgery 2015, 76, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Schnyder, A.; Huwyler, J. Drug Transport to Brain with Targeted Liposomes. NeuroRx. 2005, 2, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Neves, V.; Aires-da-Silva, F.; Corte-Real, S.; Castanho, M.A.R.B. Antibody Approaches To Treat Brain Diseases. Trends Biotechnol. 2016, 34, 36–48. [Google Scholar] [CrossRef]
- Loureiro, J.A.; Gomes, B.; Fricker, G.; Cardoso, I.; Ribeiro, C.A.; Gaiteiro, C.; Coelho, M.A.N.; Pereira, M.d.C.; Rocha, S. Dual Ligand Immunoliposomes for Drug Delivery to the Brain. Colloids Surf. B Biointerfaces 2015, 134, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Tsibouklis, J.; Weng, T.; Zhang, B.; Yin, G.; Feng, G.; Cui, Y.; Savina, I.N.; Mikhalovska, L.I.; Sandeman, S.R.; et al. Nano Carriers for Drug Transport across the Blood-Brain Barrier. J. Drug Target. 2017, 25, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Mugabe, C.; Halwani, M.; Azghani, A.O.; Lafrenie, R.M.; Omri, A. Mechanism of Enhanced Activity of Liposome-Entrapped Aminoglycosides against Resistant Strains of Pseudomonas Aeruginosa. Antimicrob. Agents Chemother. 2006, 50, 2016–2022. [Google Scholar] [CrossRef] [PubMed]
- Gubernator, J.; Drulis-Kawa, Z.; Dorotkiewicz-Jach, A.; Doroszkiewicz, W.; Kozubek, A. In Vitro Antimicrobial Activity of Liposomes Containing Ciprofloxacin, Meropenem and Gentamicin Against Gram-Negative Clinical Bacterial Strains. Int. J. Pharm. 2007, 315, 59–66. [Google Scholar] [CrossRef]
- Bakker-Woudenberg, I.A.J.M.; ten Kate, M.T.; Guo, L.; Working, P.; Mouton, J.W. Improved Efficacy of Ciprofloxacin Administered in Polyethylene Glycol-Coated Liposomes for Treatment of Klebsiella Pneumoniae Pneumonia in Rats. Antimicrob. Agents Chemother. 2001, 45, 1487–1492. [Google Scholar] [CrossRef]
- Wang, Z.; Ma, Y.; Khalil, H.; Wang, R.; Lu, T.; Zhao, W.; Zhang, Y.; Chen, J.; Chen, T. Fusion between Fluid Liposomes and Intact Bacteria: Study of Driving Parameters and in Vitro Bactericidal Efficacy. Int. J. Nanomedicine 2016, 11, 4025–4036. [Google Scholar] [CrossRef]
- Beaulac, C.; Clément-Major, S.; Hawari, J.; Lagacé, J. Eradication of Mucoid Pseudomonas Aeruginosa with Fluid Liposome-Encapsulated Tobramycin in an Animal Model of Chronic Pulmonary Infection. Antimicrob. Agents Chemother. 1996, 40, 665–669. [Google Scholar] [CrossRef]
- Sachetelli, S.; Khalil, H.; Chen, T.; Beaulac, C.; Sénéchal, S.; Lagacé, J. Demonstration of a Fusion Mechanism between a Fluid Bactericidal Liposomal Formulation and Bacterial Cells. Biochim. Biophys. Acta 2000, 1463, 254–266. [Google Scholar] [CrossRef]
- Ma, Y.; Wang, Z.; Zhao, W.; Lu, T.; Wang, R.; Mei, Q.; Chen, T. Enhanced Bactericidal Potency of Nanoliposomes by Modification of the Fusion Activity between Liposomes and Bacterium. Int. J. Nanomedicine 2013, 8, 2351–2360. [Google Scholar] [CrossRef]
- Beaulac, C.; Sachetelli, S.; Lagace, J. In-Vitro Bactericidal Efficacy of Sub-MIC Concentrations of Liposome-Encapsulated Antibiotic against Gram-Negative and Gram-Positive Bacteria. J. Antimicrob. Chemother. 1998, 41, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Alipour, M.; Halwani, M.; Omri, A.; Suntres, Z.E. Antimicrobial Effectiveness of Liposomal Polymyxin B against Resistant Gram-Negative Bacterial Strains. Int. J. Pharm. 2008, 355, 293–298. [Google Scholar] [CrossRef]
- Nacucchio, M.C.; Bellora, M.J.; Sordelli, D.O.; D’Aquino, M. Enhanced Liposome-Mediated Activity of Piperacillin against Staphylococci. Antimicrob. Agents Chemother. 1985, 27, 137–139. [Google Scholar] [CrossRef]
- Gaspar, M.M.; Cruz, A.; Penha, A.F.; Reymão, J.; Sousa, A.C.; Eleutério, C.V.; Domingues, S.A.; Fraga, A.G.; Filho, A.L.; Cruz, M.E.M.; et al. Rifabutin Encapsulated in Liposomes Exhibits Increased Therapeutic Activity in a Model of Disseminated Tuberculosis. Int. J. Antimicrob. Agents 2008, 31, 37–45. [Google Scholar] [CrossRef]
- Pumerantz, A.; Muppidi, K.; Agnihotri, S.; Guerra, C.; Venketaraman, V.; Wang, J.; Betageri, G. Preparation of Liposomal Vancomycin and Intracellular Killing of Meticillin-Resistant Staphylococcus Aureus (MRSA). Int. J. Antimicrob. Agents 2011, 37, 140–144. [Google Scholar] [CrossRef]
- Ferreira, M.; Pinto, S.N.; Aires-da-Silva, F.; Bettencourt, A.; Aguiar, S.I.; Gaspar, M.M. Liposomes as a Nanoplatform to Improve the Delivery of Antibiotics into Staphylococcus Aureus Biofilms. Pharmaceutics 2021, 13, 321. [Google Scholar] [CrossRef]
- Kadry, A.A.; Al-Suwayeh, S.A.; Abd-Allah, A.R.A.; Bayomi, M.A. Treatment of Experimental Osteomyelitis by Liposomal Antibiotics. J. Antimicrob. Chemother. 2004, 54, 1103–1108. [Google Scholar] [CrossRef]
- Fu, Y.-Y.; Zhang, L.; Yang, Y.; Liu, C.-W.; He, Y.-N.; Li, P.; Yu, X. Synergistic Antibacterial Effect of Ultrasound Microbubbles Combined with Chitosan-Modified Polymyxin B-Loaded Liposomes on Biofilm-Producing Acinetobacter Baumannii. Int. J. Nanomed. 2019, 14, 1805–1815. [Google Scholar] [CrossRef] [PubMed]
- Nicolosi, D.; Cupri, S.; Genovese, C.; Tempera, G.; Mattina, R.; Pignatello, R. Nanotechnology Approaches for Antibacterial Drug Delivery: Preparation and Microbiological Evaluation of Fusogenic Liposomes Carrying Fusidic Acid. Int. J. Antimicrob. Agents 2015, 45, 622–626. [Google Scholar] [CrossRef] [PubMed]
- Rukholm, G.; Mugabe, C.; Azghani, A.O.; Omri, A. Antibacterial Activity of Liposomal Gentamicin against Pseudomonas Aeruginosa: A Time-Kill Study. Int. J. Antimicrob. Agents 2006, 27, 247–252. [Google Scholar] [CrossRef]
- Ribeiro, L.N.D.M.; de Paula, E.; Rossi, D.A.; Monteiro, G.P.; Júnior, E.C.V.; Silva, R.R.; Franco, R.R.; Espíndola, F.S.; Goulart, L.R.; Fonseca, B.B. Hybrid Pectin-Liposome Formulation against Multi-Resistant Bacterial Strains. Pharmaceutics 2020, 12, 769. [Google Scholar] [CrossRef] [PubMed]
- Furneri, P.M.; Fresta, M.; Puglisi, G.; Tempera, G. Ofloxacin-Loaded Liposomes: In Vitro Activity and Drug Accumulation in Bacteria. Antimicrob. Agents Chemother. 2000, 44, 2458–2464. [Google Scholar] [CrossRef] [PubMed]
- Moyá, M.L.; López-López, M.; Lebrón, J.A.; Ostos, F.J.; Pérez, D.; Camacho, V.; Beck, I.; Merino-Bohórquez, V.; Camean, M.; Madinabeitia, N.; et al. Preparation and Characterization of New Liposomes. Bactericidal Activity of Cefepime Encapsulated into Cationic Liposomes. Pharmaceutics 2019, 11, 69. [Google Scholar] [CrossRef]
- Vanić, Ž.; Rukavina, Z.; Manner, S.; Fallarero, A.; Uzelac, L.; Kralj, M.; Amidžić Klarić, D.; Bogdanov, A.; Raffai, T.; Virok, D.P.; et al. Azithromycin-Liposomes as a Novel Approach for Localized Therapy of Cervicovaginal Bacterial Infections. Int. J. Nanomed. 2019, 14, 5957–5976. [Google Scholar] [CrossRef]
- Webb, M.S.; Boman, N.L.; Wiseman, D.J.; Saxon, D.; Sutton, K.; Wong, K.F.; Logan, P.; Hope, M.J. Antibacterial Efficacy against an in Vivo Salmonella Typhimurium Infection Model and Pharmacokinetics of a Liposomal Ciprofloxacin Formulation. Antimicrob. Agents Chemother. 1998, 42, 45–52. [Google Scholar] [CrossRef]
- Rukavina, Z.; Šegvić Klarić, M.; Filipović-Grčić, J.; Lovrić, J.; Vanić, Ž. Azithromycin-Loaded Liposomes for Enhanced Topical Treatment of Methicillin-Resistant Staphyloccocus Aureus (MRSA) Infections. Int. J. Pharm. 2018, 553, 109–119. [Google Scholar] [CrossRef]
- Bartomeu Garcia, C.; Shi, D.; Webster, T.J. Tat-Functionalized Liposomes for the Treatment of Meningitis: An in Vitro Study. Int. J. Nanomed. 2017, 12, 3009–3021. [Google Scholar] [CrossRef] [PubMed]
- Bardonnet, P.-L.; Faivre, V.; Boullanger, P.; Piffaretti, J.-C.; Falson, F. Pre-Formulation of Liposomes against Helicobacter Pylori: Characterization and Interaction with the Bacteria. Eur. J. Pharm. Biopharm. 2008, 69, 908–922. [Google Scholar] [CrossRef]
- Gottesmann, M.; Goycoolea, F.M.; Steinbacher, T.; Menogni, T.; Hensel, A. Smart Drug Delivery against Helicobacter Pylori: Pectin-Coated, Mucoadhesive Liposomes with Antiadhesive Activity and Antibiotic Cargo. Appl. Microbiol. Biotechnol. 2020, 104, 5943–5957. [Google Scholar] [CrossRef] [PubMed]
- Allen, T.M. Liposomal Drug Formulations. Rationale for Development and What We Can Expect for the Future. Drugs 1998, 56, 747–756. [Google Scholar] [CrossRef] [PubMed]
- Underwood, C.; van Eps, A.W. Nanomedicine and Veterinary Science: The Reality and the Practicality. Vet. J. 2012, 193, 12–23. [Google Scholar] [CrossRef] [PubMed]



| Commercial Name | Company | Active Compound | Lipid Composition | Indication |
|---|---|---|---|---|
| Ambisome® | Gilead Sciences/ Fujisawa Healthcare | Amphotericin B | HSPC:DSPG:Chol | Fungal infections |
| Amphotec®/Amphocil® | Ben Venue Laboratories | Amphotericin B | Cholesteryl sulfate | Fungal infections |
| Abelcet® | Sigma-Tau Pharmaceuticals | Amphotericin B | DMPC:DMPG | Fungal infections |
| Epaxal® | Crucell | Formalin-inactivated Hepatitis A virus | DOPC:DOPE | Hepatitis A |
| Inflexal® | Crucell | Inactivated hemaglutinine of Influenza virus | DOPC:DOPE | Influenza |
| Arikayce® | Insmed, Inc. | Amikacin | DPPC:Chol | Mycobacterium avium complex (MAC) lung disease |
| Arikace TM | Transave, Inc. | Amikacin | DPPC:Chol | Pseudomonas aeruginosa infections (cystic fibrosis) |
| RTS,S/AS01 | GlaxoSmithKline | Recombinant fusion of P. falciparum circumsporozoite protein and Hepatitis B surface antigen | MPL:DOPC:Chol | Malaria |
| ALIS | Insmed, Inc. | Amikacin | DPPC:Chol | Nontuberculous Mycobacterial lung infection |
| Vaxisome | NasVax | Inactivated Influenza virus | CCS | Influenza |
| JVRS-100 | Juvaris BioTherapeutics | Inactivated Influenza virus | CLDC:Chol | Influenza |
| Nyotran | Aronex Pharmaceuticals | Nystatin | DMPC:DMPG:Chol | Fungal infections |
| CAF01 | Statens Serum Institut | Subunit protein antigen Ag85B-ESAT, DDA, TDB | DODAB:TDB | Tuberculosis |
| Vaxfectin | Vical | Plasmid DNA-encoded influenza proteins | VC1052:DPyPE | Influenza |
| MPER-656 Liposome Vaccine | National Institute of Allergy and Infectious Diseases (NIAID) | Immunogenicity of an HIV-1 gp41 MPER-656 | DOPC:DOPG | HIV infections |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferreira, M.; Ogren, M.; Dias, J.N.R.; Silva, M.; Gil, S.; Tavares, L.; Aires-da-Silva, F.; Gaspar, M.M.; Aguiar, S.I. Liposomes as Antibiotic Delivery Systems: A Promising Nanotechnological Strategy against Antimicrobial Resistance. Molecules 2021, 26, 2047. https://doi.org/10.3390/molecules26072047
Ferreira M, Ogren M, Dias JNR, Silva M, Gil S, Tavares L, Aires-da-Silva F, Gaspar MM, Aguiar SI. Liposomes as Antibiotic Delivery Systems: A Promising Nanotechnological Strategy against Antimicrobial Resistance. Molecules. 2021; 26(7):2047. https://doi.org/10.3390/molecules26072047
Chicago/Turabian StyleFerreira, Magda, Maria Ogren, Joana N. R. Dias, Marta Silva, Solange Gil, Luís Tavares, Frederico Aires-da-Silva, Maria Manuela Gaspar, and Sandra Isabel Aguiar. 2021. "Liposomes as Antibiotic Delivery Systems: A Promising Nanotechnological Strategy against Antimicrobial Resistance" Molecules 26, no. 7: 2047. https://doi.org/10.3390/molecules26072047
APA StyleFerreira, M., Ogren, M., Dias, J. N. R., Silva, M., Gil, S., Tavares, L., Aires-da-Silva, F., Gaspar, M. M., & Aguiar, S. I. (2021). Liposomes as Antibiotic Delivery Systems: A Promising Nanotechnological Strategy against Antimicrobial Resistance. Molecules, 26(7), 2047. https://doi.org/10.3390/molecules26072047