
Charge Transfer Complexes of Ketotifen with 2,3-Dichloro-5,6-dicyano-p-benzoquinone and 7,7,8,8-Tetracyanoquodimethane: Spectroscopic Characterization Studies



Abstract
1. Introduction
2. Results and Discussion
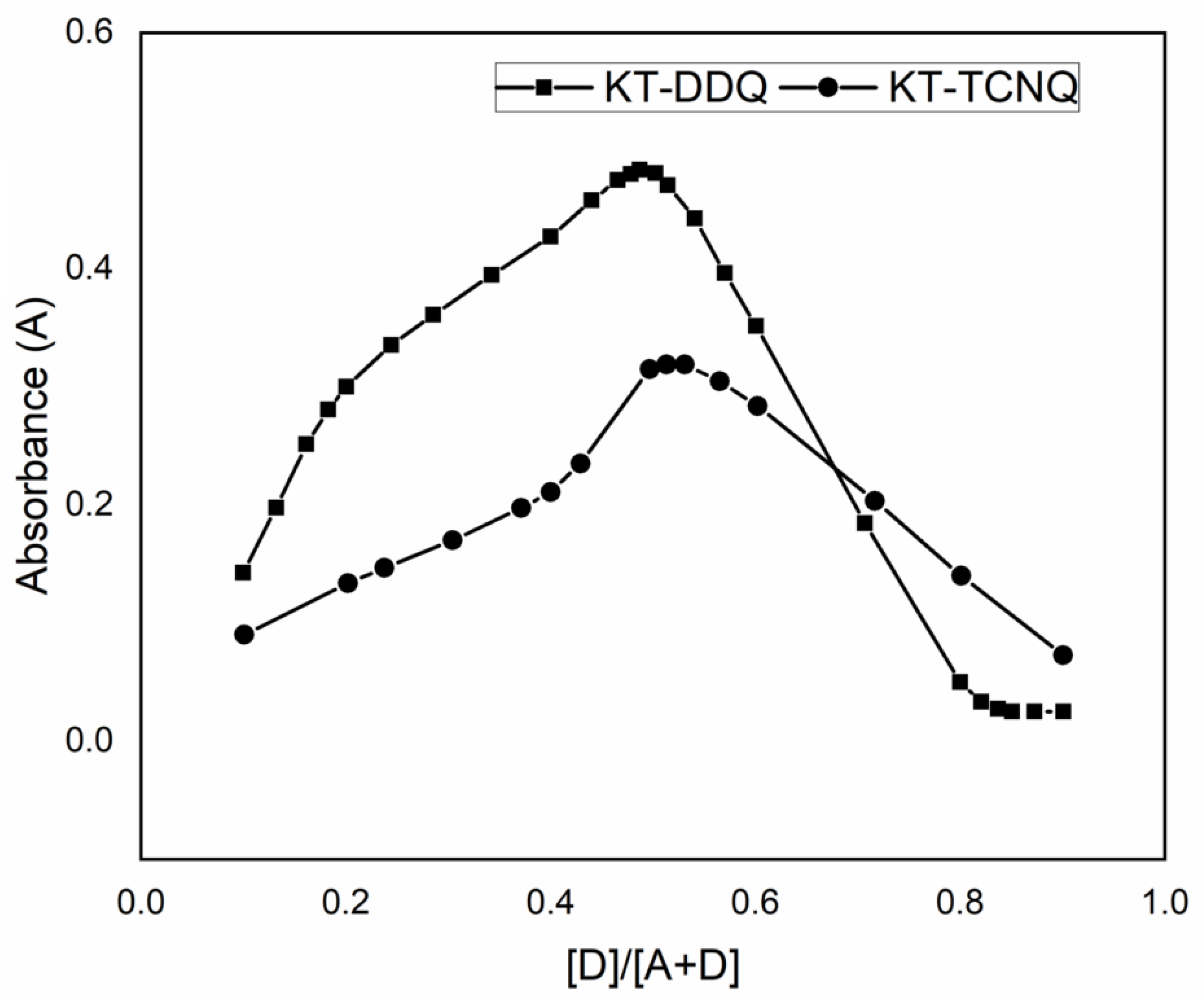
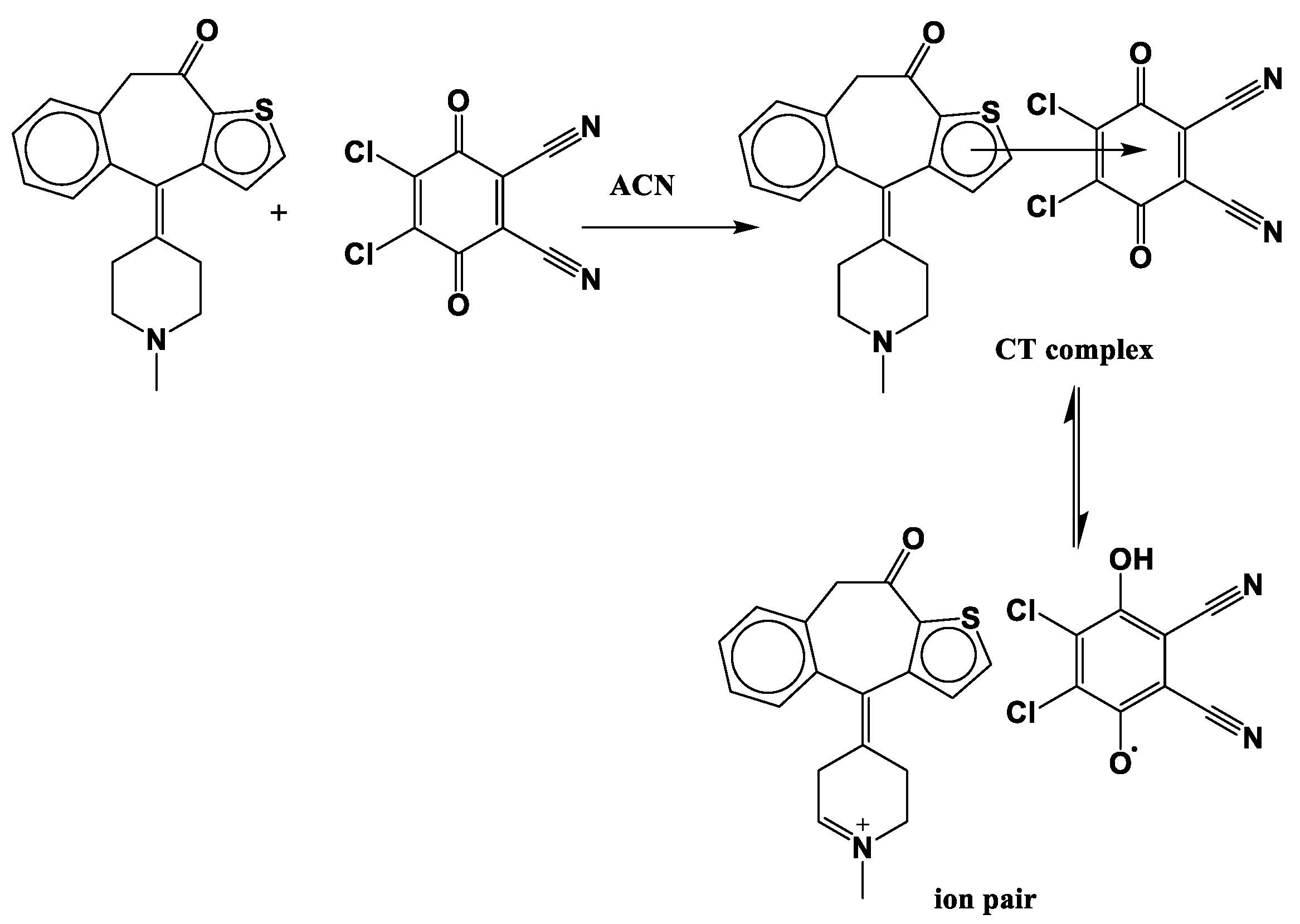
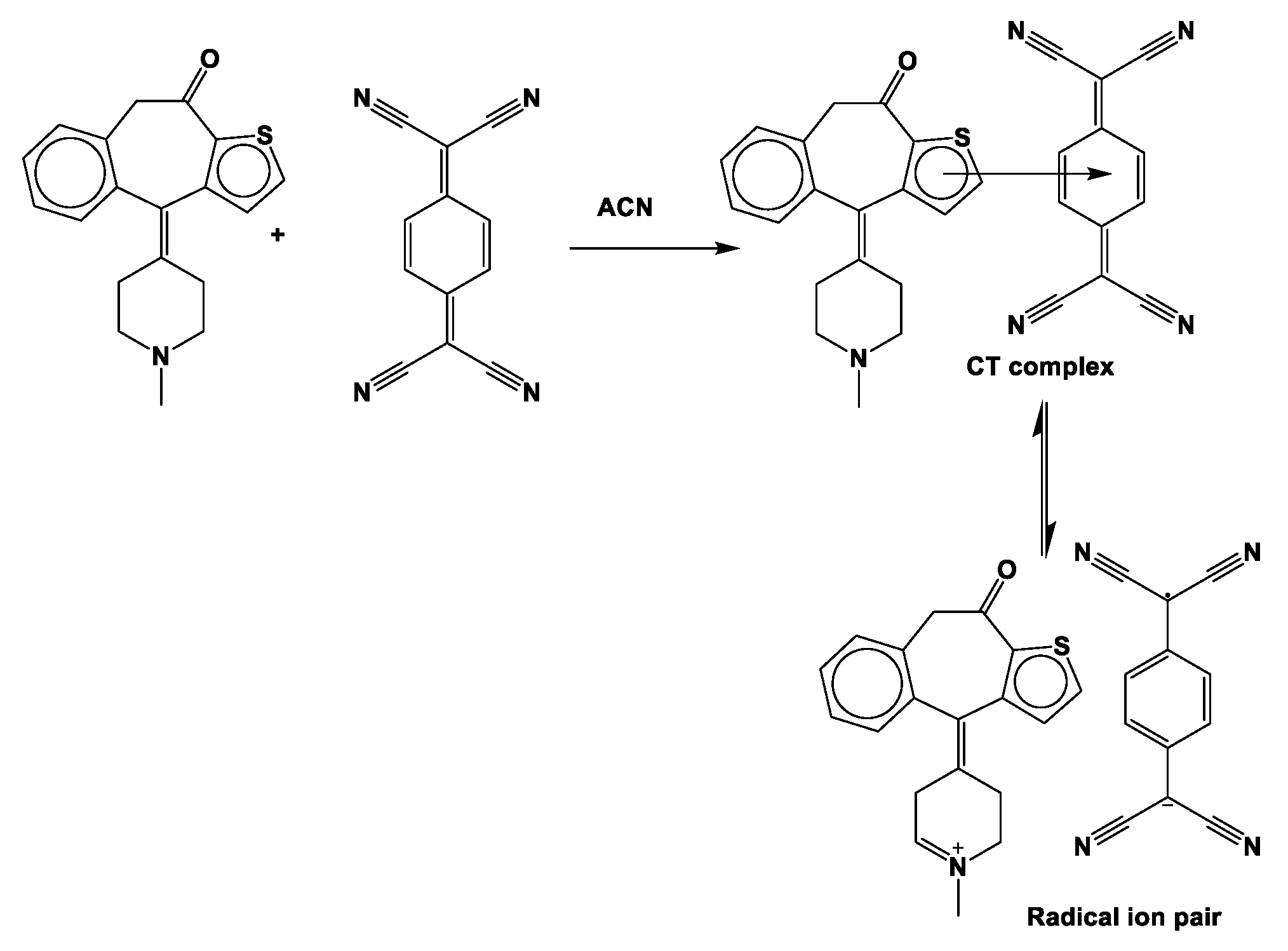
2.1. Molar Composition of KT Complexes
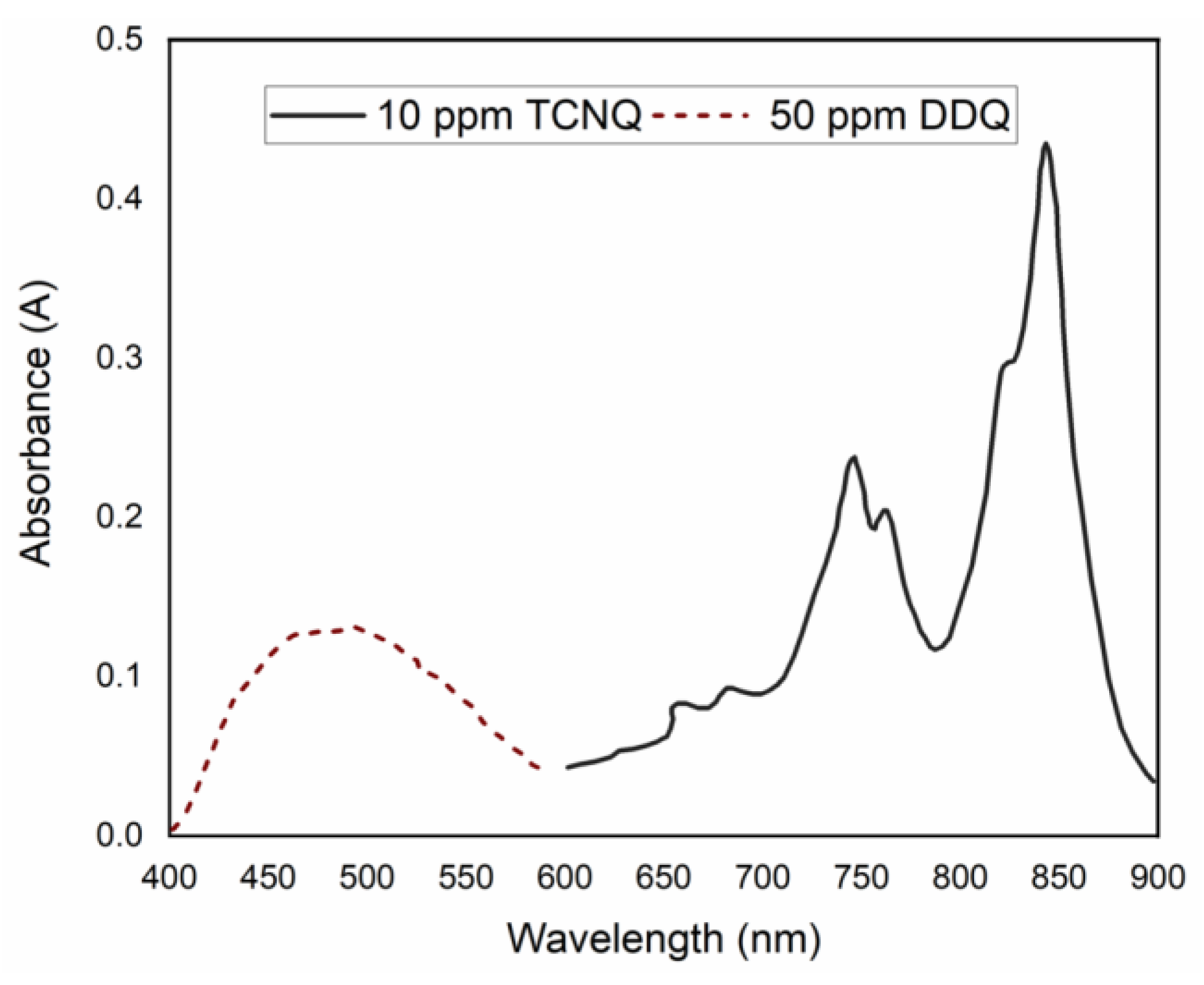
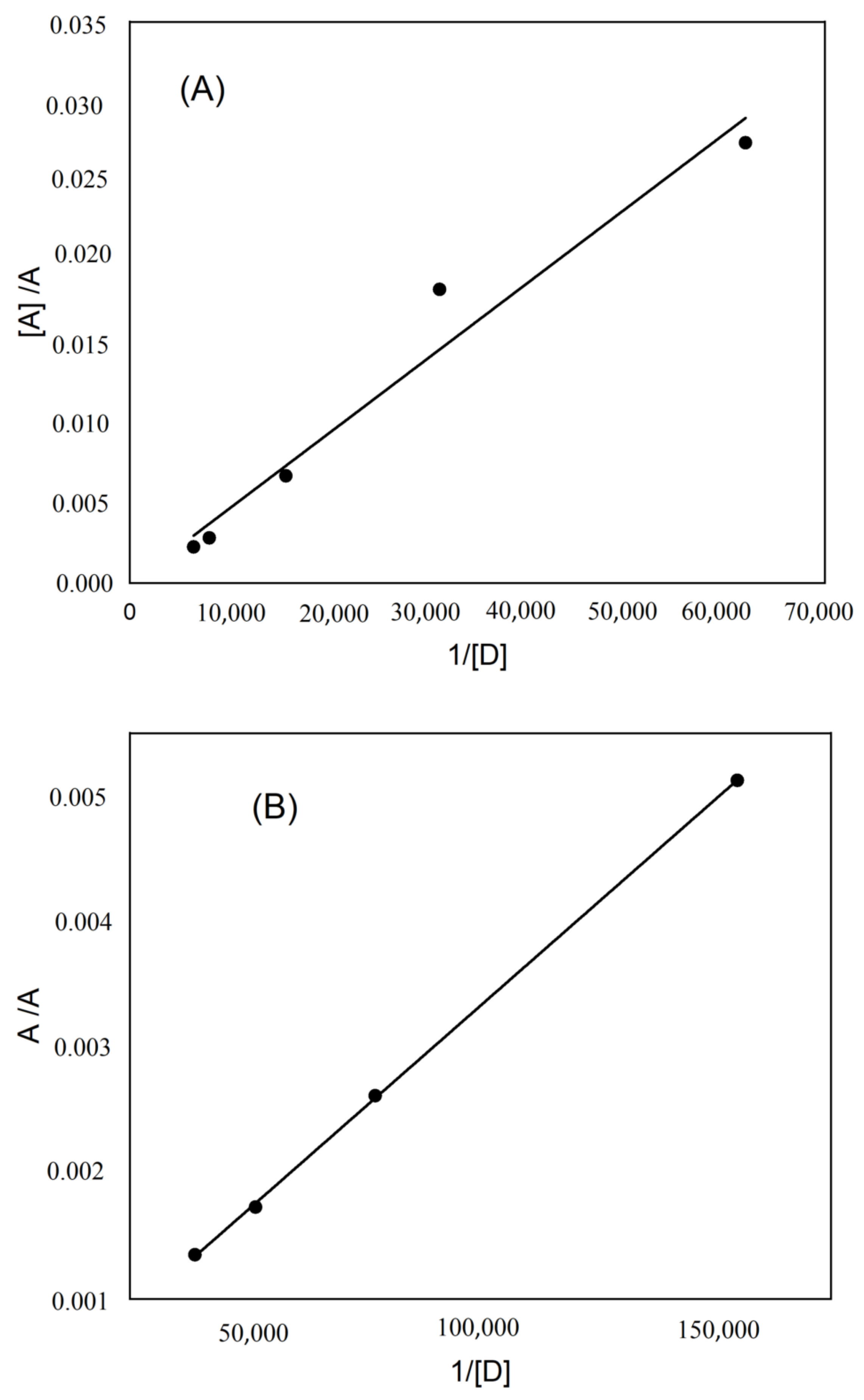
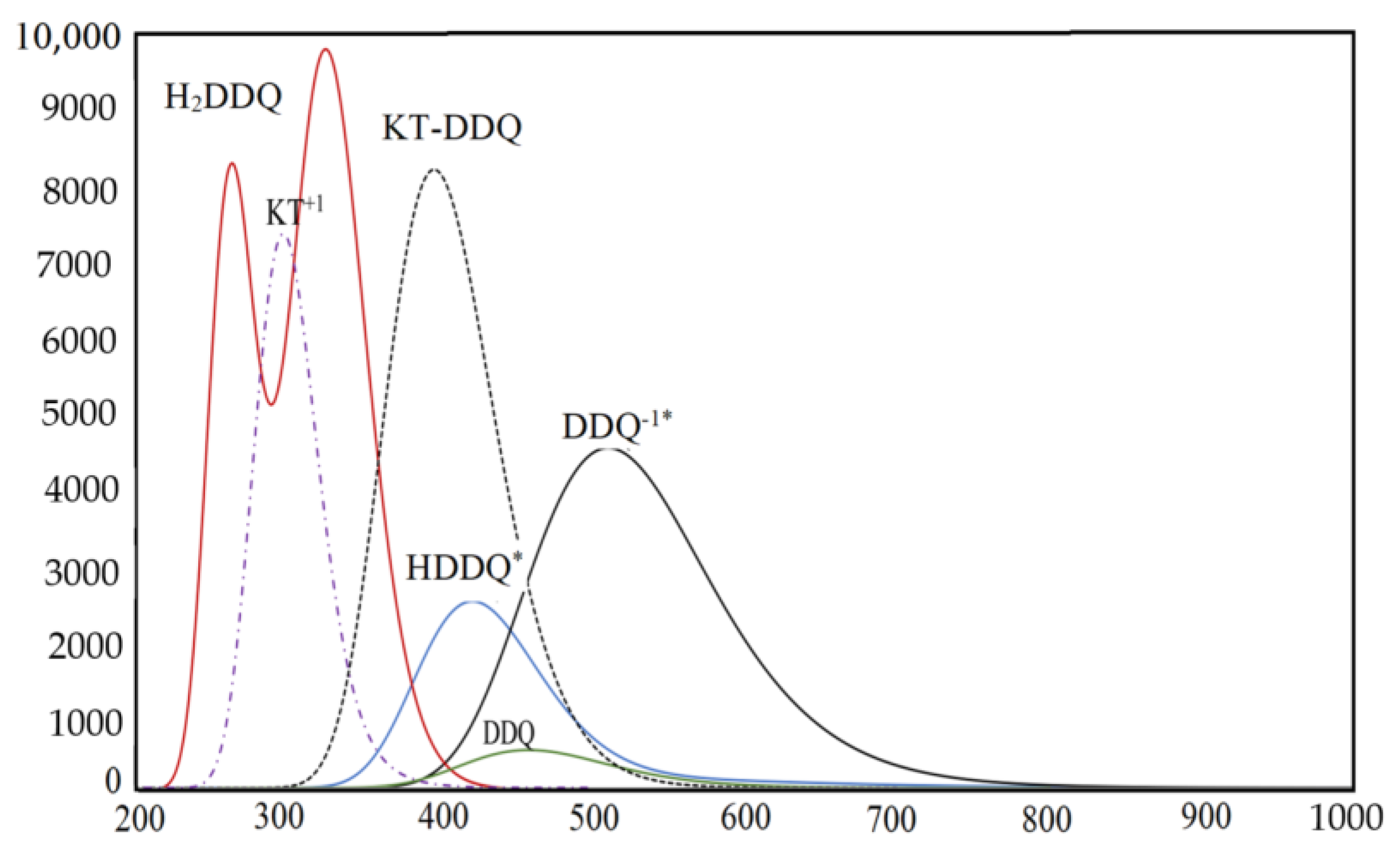
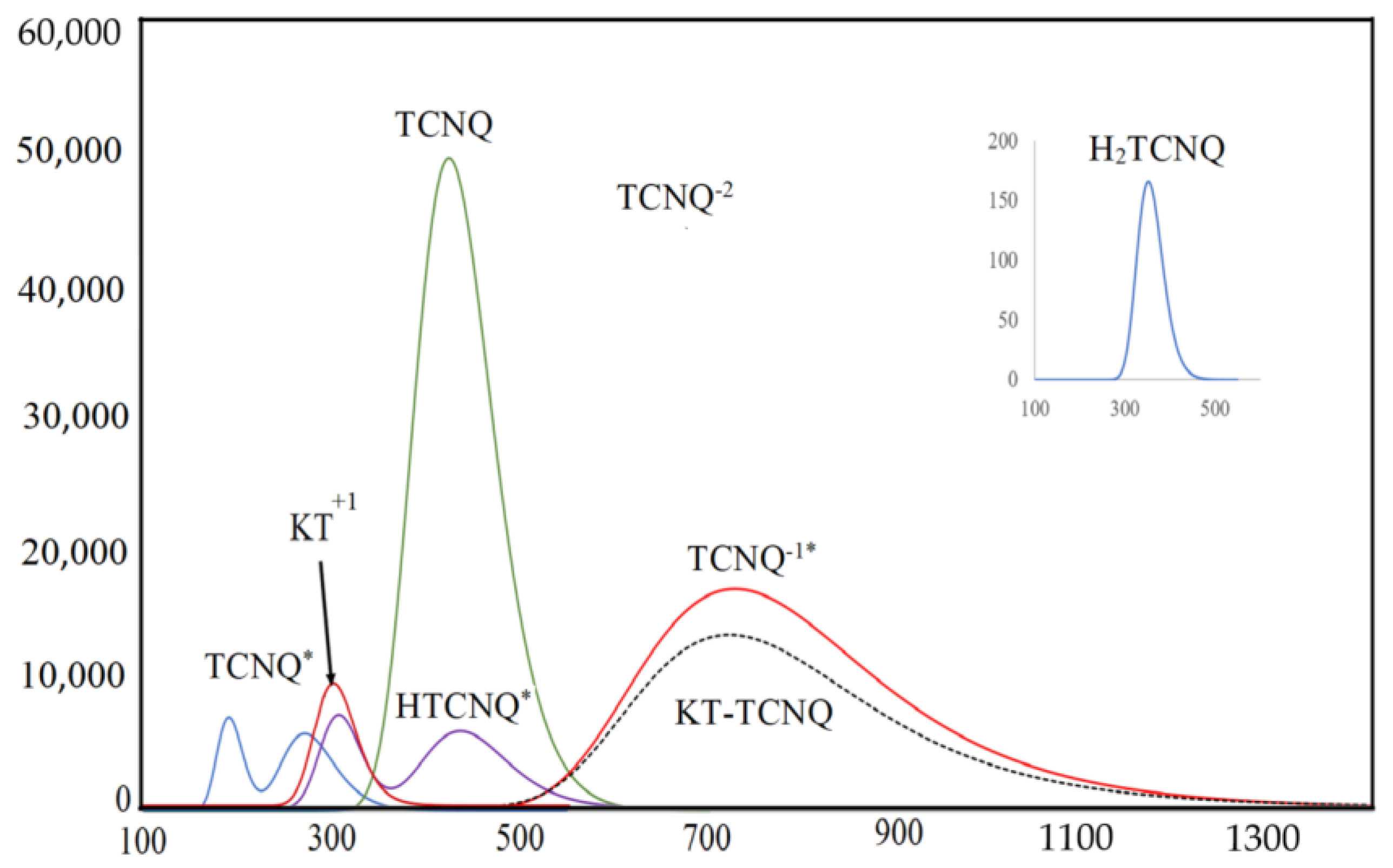
2.2. The Determination of Spectroscopic Physical Parameters
2.3. Validation
2.3.1. The Lower Limit of Detection and Quantification
2.3.2. Precision and Accuracy
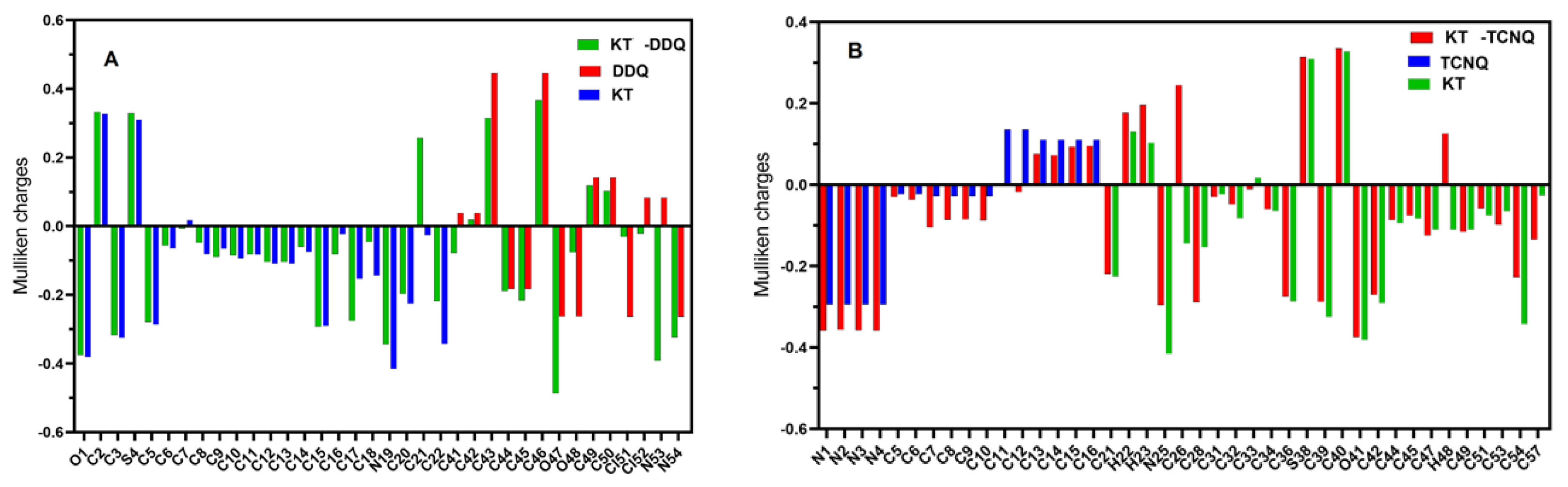
2.4. DFT/TD-DFT Calculations
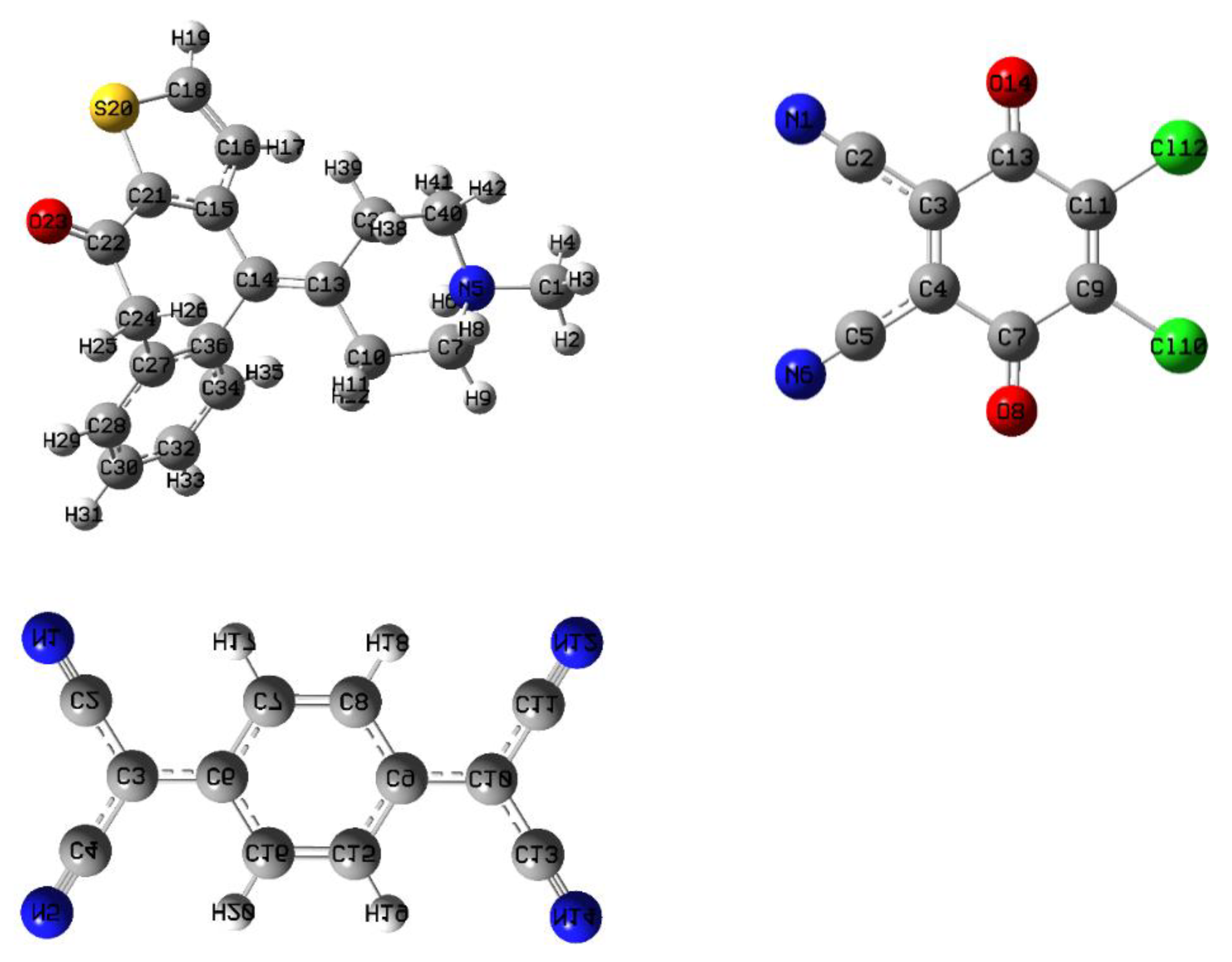
2.4.1. Optimized Geometrical Structures
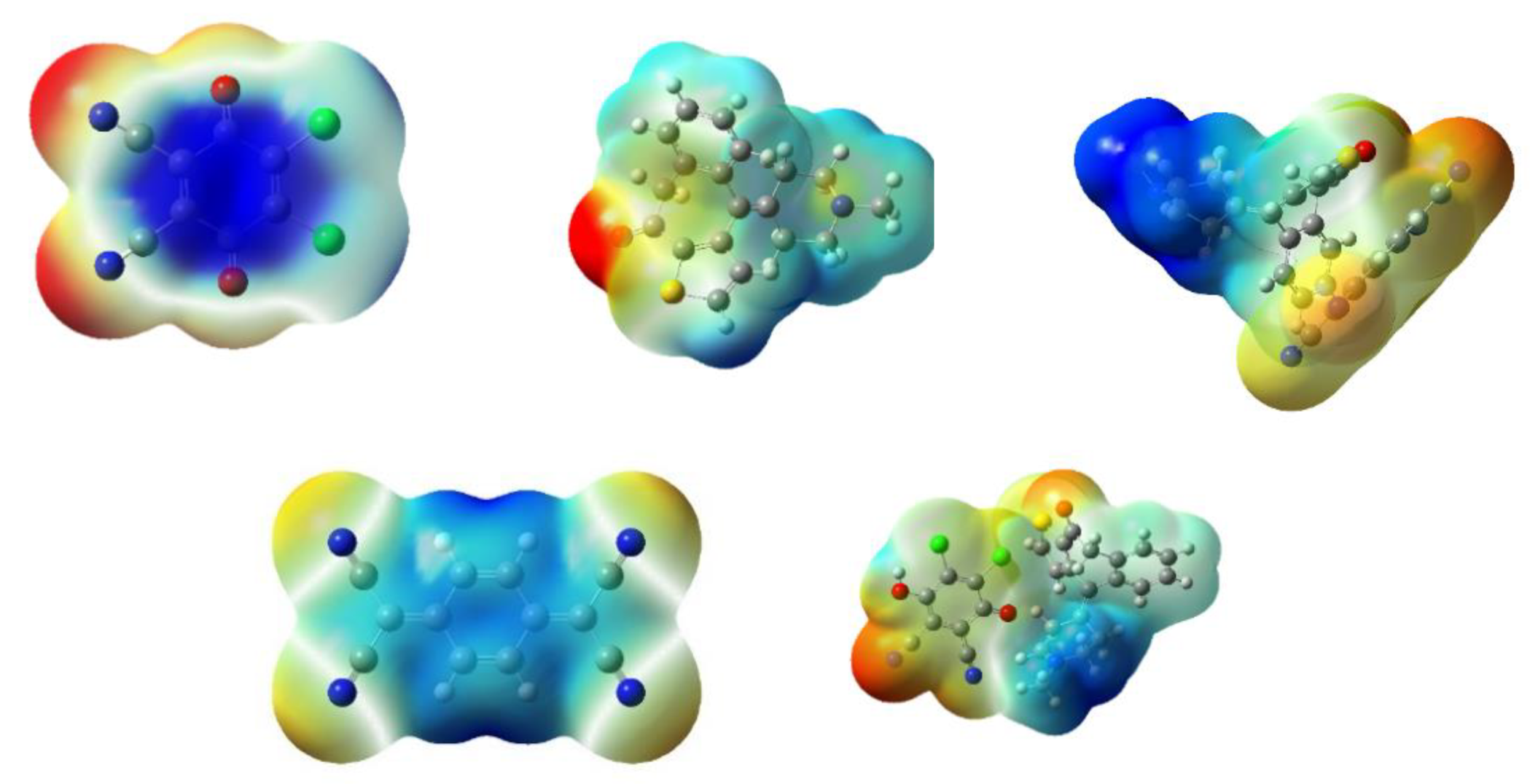
2.4.2. Molecular Electrostatic Potential (MEP) Surface Analysis
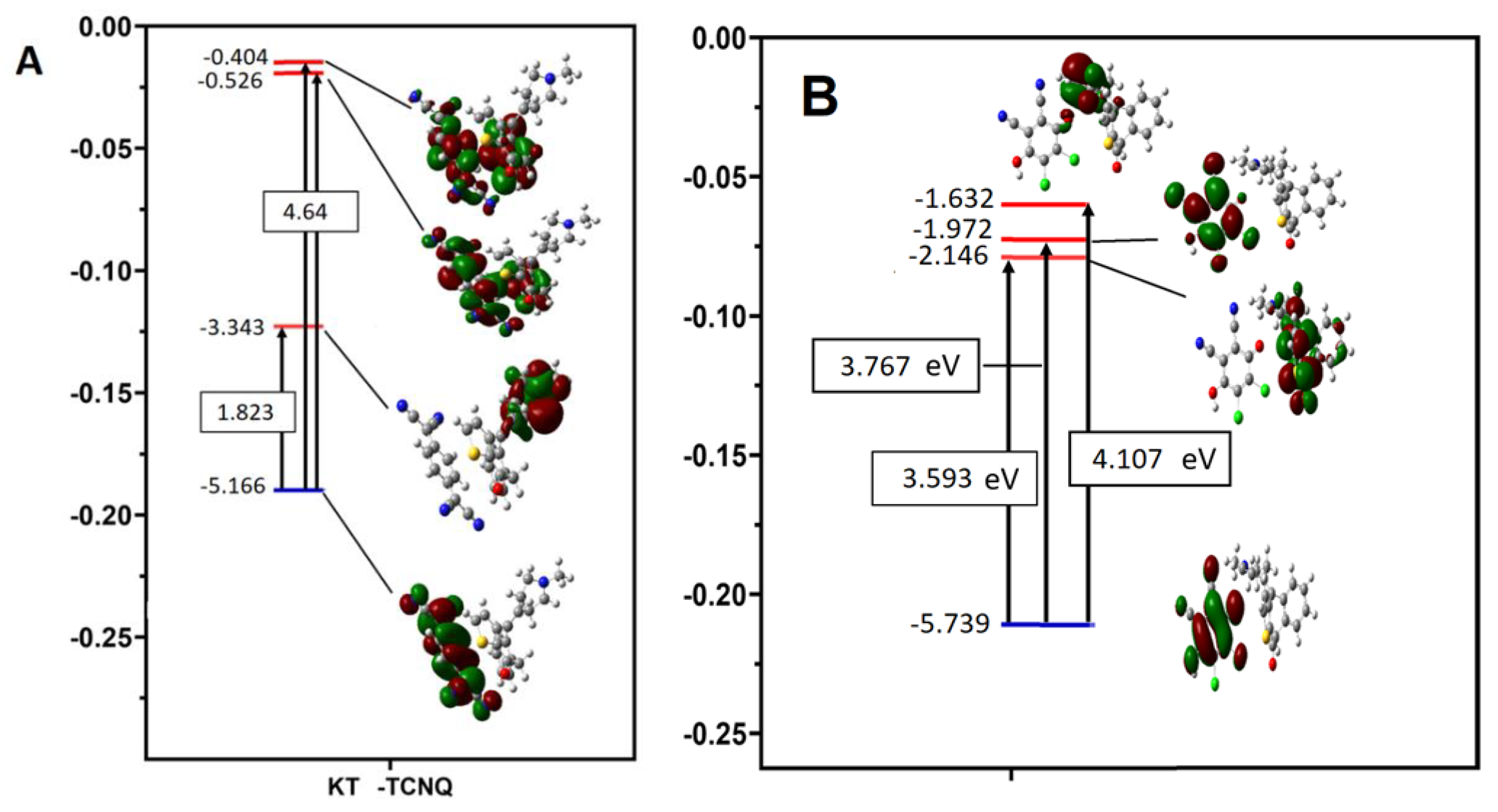
2.4.3. Thermodynamics of the KT-DDQ and KT-TCNQ Complexes
2.5. Applications
3. Experimental
3.1. Apparatus
3.2. Reagents and Materials
3.3. Procedure
3.4. Determination of the Molar Ratio in TK-DDQ and TK-TCNQ Complexes
3.5. Conductance Measurements
3.6. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CT | Charge transfer |
| KT | Ketotifen Fumarate |
| DDQ | 2,3-dichloro-5,6-dicyano-p-benzoquinone |
| TCNQ | 7,7,8, 8-tetracyanoquinodimethane |
| DFT | density functional theory |
| f | oscillator strength |
| μ | transition dipole moment |
| Ip | ionization potentials |
| energy of charge transfer complex | |
| W | dissociation energy |
| ΔG | Gibbs free energy |
| RN | Resonance energy |
| ε | extinction coefficient |
| MEP | molecular electrostatic potential |
| HOMO | highest occupied molecular orbital |
| LUMO | lowest unoccupied molecular orbital |
References
- Khan, I.M.; Alam, K.; Alam, M.J. Exploring charge transfer dynamics and photocatalytic behavior of designed donor-acceptor complex: Characterization, spectrophotometric and theoretical studies (DFT/TD-DFT). J. Mol. Liq. 2020, 310, 113213. [Google Scholar] [CrossRef]
- Kato, Y.; Matsumoto, H.; Mori, T. Absence of HOMO/LUMO Transition in Charge-Transfer Complexes of Thienoacenes. J. Phys. Chem. A 2021, 125, 146–153. [Google Scholar] [CrossRef]
- Mulliken, R.S. Molecular compounds and their spectra. J. Am. Chem. Soc. 1952, 74, 811. [Google Scholar] [CrossRef]
- Kosower, E.M.; Foster, R. Organic Charge-Transfer Complexes; Academic Press: New York, NY, USA, 1970. [Google Scholar]
- Adam, A.M.A. Synthesis, spectroscopic, thermal and antimicrobial investigations of charge-transfer complexes formed from the drug procaine hydrochloride with quinol, picric acid and TCNQ. J. Mol. Struct. 2012, 1030, 26–39. [Google Scholar] [CrossRef]
- Mohamed, M.E.; Frag, E.Y.; Hathoot, A.A.; Shalaby, E.A. Spectrophotometric determination of fenoprofen calcium drug in pure and pharmaceutical preparations. Spectroscopic characterization of the charge transfer solid complexes. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2018, 189, 357–365. [Google Scholar] [CrossRef]
- Xu, H.; Zhou, Y.; Zhang, J.; Jin, J.; Liu, G.; Li, Y.; Ganguly, R.; Huang, L.; Xu, W.; Zhu, D.; et al. Polymer-Assisted Single Crystal Engineering of Organic Semiconductors to Alter Electron Transport. ACS Appl. Mater. Interfaces 2018, 10, 11837–11842. [Google Scholar] [CrossRef]
- Wuest, J.D. Co-crystals give light a tune-up. Nat. Chem. 2012, 4, 74–75. [Google Scholar] [CrossRef]
- Wang, J.; Xu, S.; Li, A.; Chen, L.; Xu, W.; Zhang, H. Polymorphism-based luminescence and morphology-dependent optical waveguide properties in 1: 1 charge transfer cocrystals. Mater. Chem. Front. 2021, 5, 1477–1485. [Google Scholar] [CrossRef]
- Khan, I.M.; Islam, M.; Shakya, S.; Alam, K.; Alam, N.; Shahid, M. Synthesis, characterization, antimicrobial and DNA binding properties of an organic charge transfer complex obtained from pyrazole and chloranilic acid. Bioorg. Chem. 2020, 99, 103779. [Google Scholar] [CrossRef]
- Mohamed, G.G.; Hamed, M.M.; Zaki, N.G.; Abdou, M.M.; Mohamed, M.E.-B.; Abdallah, A.M. Melatonin charge transfer complex with 2,3-dichloro-5,6-dicyano-1,4-benzoquinone: Molecular structure, DFT studies, thermal analyses, evaluation of biological activity and utility for determination of melatonin in pure and dosage forms. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2017, 182, 143–159. [Google Scholar] [CrossRef]
- Khan, I.M.; Ahmad, A. Synthesis, characterization, structural, spectrophotometric and antimicrobial activity of charge transfer complex of p-phenylenediamine with 3,5-dinitrosalicylic acid. J. Mol. Struct. 2010, 975, 381–388. [Google Scholar] [CrossRef]
- Yousef, T.A.; Ezzeldin, E.; Abdel-Aziz, H.A.; Al-Agamy, M.H.; Mostafa, G.A. Charge Transfer Complex of Neostigmine with 2,3-Dichloro-5,6-Dicyano-1,4-Benzoquinone: Synthesis, Spectroscopic Characterization, Antimicrobial Activity, and Theoretical Study. Drug Des. Dev. Ther. 2020, 14, 4115–4129. [Google Scholar] [CrossRef]
- Shehab, O.R.; AlRabiah, H.; Abdel-Aziz, H.A.; Mostafa, G.A. Charge-transfer complexes of cefpodoxime proxetil with chloranilic acid and 2,3-dichloro-5,6-dicyano-1,4-benzoquinone: Experimental and theoretical studies. J. Mol. Liq. 2018, 257, 42–51. [Google Scholar] [CrossRef]
- Sweetman, S.C.M. The Complete Drug Reference, 34th ed.; Pharmaceutical Press: London, UK, 2005. [Google Scholar]
- Mokhtari, A.; Ghazaeian, M.; Maghsoudi, M.; Keyvanfard, M.; Emami, I. Simple chemiluminescence determination of ketotifen using tris (1, 10 phenanthroline) ruthenium (II)-Ce (IV) system. Luminescence 2015, 30, 1094–1100. [Google Scholar] [CrossRef]
- Daneshgar, P.; Norouzi, P.; Ganjali, M.R. Application of a Continuous Square-Wave Potential Program for Sub Nano Molar Determination of Ketotifen. Chem. Pharm. Bull. 2009, 57, 117–121. [Google Scholar] [CrossRef][Green Version]
- Elsayed, M.M.A. Development and Validation of a Rapid HPLC Method for the Determination of Ketotifen in Pharmaceuticals. Drug Dev. Ind. Pharm. 2006, 32, 457–461. [Google Scholar] [CrossRef]
- Chen, X.; Zhong, D.; Liu, D.; Wang, Y.; Han, Y.; Gu, J. Determination of ketotifen and its conjugated metabolite in human plasma by liquid chromatography/tandem mass spectrometry: Application to a pharmacokinetic study. Rapid Commun. Mass Spectrom. 2003, 17, 2459–2463. [Google Scholar] [CrossRef]
- Tzvetanov, S.; Vatsova, M.; Drenska, A.; Gorantcheva, J.; Tyutyulkova, N. Gas chromatographic-mass spectrometric method for quantitative determination of ketotifen in human plasma after enzyme hydrolysis of conjugated ketotifen. J. Chromatogr. B Biomed. Sci. Appl. 1999, 732, 251–256. [Google Scholar] [CrossRef]
- Zhou, M.; Li, Y.-J.; Ma, Y.-J.; Wang, W.-F.; Mi, J.; Chen, H. Determination of ketotifen fumarate by capillary electrophoresis with tris(2,2′-bipyridyl) ruthenium(II) electrochemiluminescence detection. Luminescence 2010, 26, 319–323. [Google Scholar] [CrossRef]
- Job, P. Advanced Physicochemical Experiments; Oliner and Boyd: Edinburgh, UK, 1964. [Google Scholar]
- Benesi, H.A.; Hildebrand, J.H. A Spectrophotometric Investigation of the Interaction of Iodine with Aromatic Hydrocarbons. J. Am. Chem. Soc. 1949, 71, 2703–2707. [Google Scholar] [CrossRef]
- Aloisi, G.G.; Pignataro, S. Molecular complexes of substituted thiophens with σ and π acceptors. Charge transfer spectra and ionization potentials of the donors. J. Chem. Soc. Faraday Trans. Phys. Chem. Condens. Phases 1973, 69, 534–539. [Google Scholar] [CrossRef]
- Briegleb, G.; Czekalla, J. Electron transfer by light absorption and emission in electron donor-acceptor complexes. Angew. Chem. 1960, 72, 401–413. [Google Scholar] [CrossRef]
- Miller, J.; Miller, J.C. Statistics and Chemometrics for Analytical Chemistry, 5th ed.; Pearson Education: Tottenham, UK, 2005. [Google Scholar]
- Mulliken, R.S. Electronic Population Analysis on LCAO–MO Molecular Wave Functions. I. J. Chem. Phys. 1955, 23, 1833–1840. [Google Scholar] [CrossRef]
- Ibrahim, O.B.; Al-Majthoub, M.M.; Mohamed, M.A.; Adam, A.M.; Refat, M.S. Quick and simple formation of charge transfer complexes of brain and nerves phenytoin drug with different π–acceptors: Chemical and biological studies. Int. J. Electrochem. Sci. 2015, 10, 1065–1080. [Google Scholar]
- Parr, R.G.; Donnelly, R.A.; Levy, M.; Palke, W.E. Electronegativity: The density functional viewpoint. J. Chem. Phys. 1978, 68, 3801–3807. [Google Scholar] [CrossRef]
- Chattaraj, P.K.; Maiti, B.; Sarkar, U. Philicity: A Unified Treatment of Chemical Reactivity and Selectivity. J. Phys. Chem. A 2003, 107, 4973–4975. [Google Scholar] [CrossRef]
- Parr, R.G.; Szentpály, L.V.; Liu, S. Electrophilicity Index. J. Am. Chem. Soc. 1999, 121, 1922–1924. [Google Scholar] [CrossRef]
- Palnati, M.K.; Baindla, N.; Tigulla, P. Spectrophotometric, Thermodynamic and Density Functional Studies of Charge Transfer Complex Between Benzhydryl Piperazine and 2,3-Dichloro-5,6-dicyano-1,4-benzoquinone. J. Solut. Chem. 2018, 47, 975–992. [Google Scholar] [CrossRef]
- Shehab, O.R.; Mansour, A.M. Sparfloxacin charge transfer complexes with 2,3-dichloro-5,6-dicyano-1,4-benzoquinone and tetracyanoquinodimethane: Molecular structures, spectral, and DFT studies. J. Mol. Struct. 2015, 1093, 186–194. [Google Scholar] [CrossRef]
- Scrocco, E.; Tomasi, J. The electrostatic molecular potential as a tool for the interpretation of molecular properties. In Astrophysics and Space Science Library; Metzler, J.B., Ed.; Springer Science+Business: Berlin/Heidelberg, Germany, 2007; Volume 42, pp. 95–170. [Google Scholar]
- Miertuš, S.; Scrocco, E.; Tomasi, J. Electrostatic interaction of a solute with a continuum. A direct utilizaion of AB initio molecular potentials for the prevision of solvent effects. Chem. Phys. 1981, 55, 117–129. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision, D. 01; Gaussian Inc.: Wallingford, CT, USA, 2009; Volume 121, pp. 150–166. [Google Scholar]
- Zhao, Y.; Tishchenko, O.; Truhlar, D.G. How Well Can Density Functional Methods Describe Hydrogen Bonds to π Acceptors? J. Phys. Chem. B 2005, 109, 19046–19051. [Google Scholar] [CrossRef]
- Lynch, B.J.; Fast, P.L.; Harris, A.M.; Truhlar, D.G. Adiabatic Connection for Kinetics. J. Phys. Chem. A 2000, 104, 4811–4815. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| [A], M | [D], M | Absorbance (A) | 1/[D] | [A]/A |
|---|---|---|---|---|
| KT–DDQ | ||||
| 0.00044 | 1.62 × 10−5 | 0.016 | 61,885.2 | 0.0275 |
| 0.00044 | 3.23 × 10−5 | 0.024 | 30,942.6 | 0.018333 |
| 0.00044 | 6.46 × 10−5 | 0.065 | 15,471.3 | 0.006769 |
| 0.00044 | 0.00012927 | 0.157 | 7735.65 | 0.002803 |
| 0.00044 | 0.00016159 | 0.196 | 6188.52 | 0.002245 |
| KT–TCNQ | ||||
| 0.000489 | 6.46 × 10−5 | 0.095 | 154,712.5 | 0.005147 |
| 0.000489 | 1.293 × 10−5 | 0.186 | 77,356.25 | 0.002629 |
| 0.000489 | 1.94 × 10−5 | 0.28 | 51,570.83 | 0.001746 |
| 0.000489 | 2.585 × 10−5 | 0.36 | 38,678.13 | 0.001358 |
| Parameters | KT-DDQ | KT-TCNQ |
|---|---|---|
| Wavelength, nm | 485 | 843 |
| Extinction coefficient: ε = L mol−1 Cm−1 | 1.1 × 103 | 1.3 × 104 |
| Association constant: K = L mol−1 | 0.2 × 103 | 2.56 × 103 |
| Energy: hv (eV) | 2.56 | 1.47 |
| Ionization Potential: Ip (eV) | 8.91 | 7.57 |
| Resonance energy: RN (eV) | 3.5 | 3.5 |
| Dissociation energy: W (eV) | 3.28 | 3.32 |
| Gibbs free energy: ΔG (kJ mol−1) | −4.5 × 103 | −4.6 × 103 |
| F | 0.34 | 5.91 |
| µ | 5.91 | 12.9 |
| Parameters | KT-DDQ | KT-TCNQ |
|---|---|---|
| Wavelength, nm | 485 | 843 |
| Calibration range | 5–100 | 2.5–40 |
| Extinction coefficient: ε (L mol−1 cm−1) | 1.1 × 103 | 1.3 × 104 |
| Lower limit of quantification | 5 | 2.5 |
| Lower limit of detection | 1.5 | 0.75 |
| Slope | 0.0042 | 0.0321 |
| Intercept | 0.0126 | 0.0741 |
| Correlation coefficient, r2 | 0.998 | 0.998 |
| Conc, μg/mL | Intra-Day Recovery % ± RSD | Inter-Day Recovery % ± RSD | |||
|---|---|---|---|---|---|
| KT-DDQ | KT-TCNQ | KT-DDQ | KT-TCNQ | KT-DDQ | KT-TCNQ |
| 15 | 10 | 97.5 ± 2.7 | 97.0 ± 2.7 | 97.0 ± 2.9 | 97.5 ± 2.9 |
| 35 | 20 | 97.5 ± 2.6 | 97.5 ± 2.5 | 97.5 ± 2.9 | 98.0 ± 2.8 |
| 75 | 30 | 98.0 ± 2.5 | 98.0 ± 2.5 | 98.0 ± 2.8 | 98.0 ± 3.0 |
| Parameters (eV) | DDQ | TCNQ | KTN | KTN-TCNQ | KTN-DDQ |
|---|---|---|---|---|---|
| ELUMO (eV) | −4.804 | −4.529 | −1.975 | −3.343 | −2.147 |
| EHOMO (eV) | −8.409 | −7.419 | −6.205 | −5.167 | −5.739 |
| Energy band gap(eV) [∆E = ELUMO − EHOMO] | 3.605 | 2.89 | 4.23 | 1.824 | 3.592 |
| Chemical hardness (η) | 1.802 | 1.445 | 2.115 | 0.912 | 1.796 |
| Softness (ς) | 0.555 | 0.692 | 0.473 | 1.097 | 0.557 |
| Dipole moment (µ) (Debye) | 1.802 | 1.445 | 2.115 | 0.912 | 1.796 |
| Electrophilicity index (ω) | 0.901 | 0.723 | 1.058 | 0.456 | 0.898 |
| Ip(eV) | 8.409 | 7.419 | 6.205 | 5.167 | 5.739 |
| Electron affinity (EA) | 4.804 | 4.529 | 1.975 | 3.343 | 2.147 |
| Electronegativity (χ) | 6.607 | 5.974 | 4.09 | 4.255 | 3.943 |
| CT Complex | G0 (kcal/mol) | H0 (kcal/mol) | S0 (cal/K mol) |
|---|---|---|---|
| KT-DDQ | −18.1827 | −30.1029 | −0.04 |
| KT-TCNQ | 389.4368 | 359.9338 | −0.099 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mostafa, G.A.E.; Bakheit, A.; AlMasoud, N.; AlRabiah, H. Charge Transfer Complexes of Ketotifen with 2,3-Dichloro-5,6-dicyano-p-benzoquinone and 7,7,8,8-Tetracyanoquodimethane: Spectroscopic Characterization Studies. Molecules 2021, 26, 2039. https://doi.org/10.3390/molecules26072039
Mostafa GAE, Bakheit A, AlMasoud N, AlRabiah H. Charge Transfer Complexes of Ketotifen with 2,3-Dichloro-5,6-dicyano-p-benzoquinone and 7,7,8,8-Tetracyanoquodimethane: Spectroscopic Characterization Studies. Molecules. 2021; 26(7):2039. https://doi.org/10.3390/molecules26072039
Chicago/Turabian StyleMostafa, Gamal A. E., Ahmed Bakheit, Najla AlMasoud, and Haitham AlRabiah. 2021. "Charge Transfer Complexes of Ketotifen with 2,3-Dichloro-5,6-dicyano-p-benzoquinone and 7,7,8,8-Tetracyanoquodimethane: Spectroscopic Characterization Studies" Molecules 26, no. 7: 2039. https://doi.org/10.3390/molecules26072039
APA StyleMostafa, G. A. E., Bakheit, A., AlMasoud, N., & AlRabiah, H. (2021). Charge Transfer Complexes of Ketotifen with 2,3-Dichloro-5,6-dicyano-p-benzoquinone and 7,7,8,8-Tetracyanoquodimethane: Spectroscopic Characterization Studies. Molecules, 26(7), 2039. https://doi.org/10.3390/molecules26072039