2.1. Structure of the Studied Catalysts
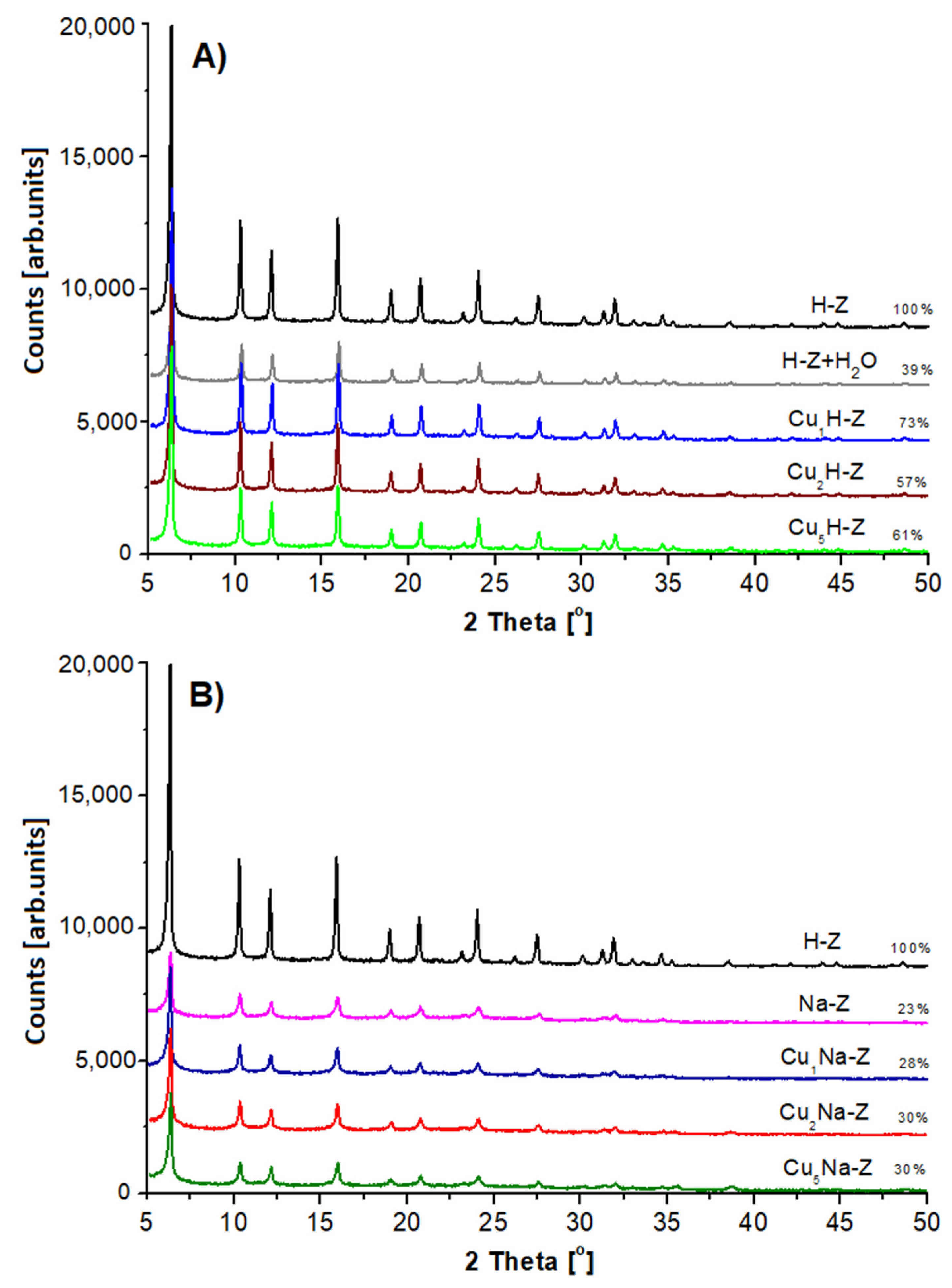
The X-ray diffraction (XRD) patterns collected for the studied catalysts are presented in
Figure 2. For all samples the presence of FAU type phase was found [
45]. The way of the catalyst modification plays an important role in the crystallinity of the final form of the samples. FAU31-based catalysts impregnated with Cu solution (Cu
xH-Z) are characterized by lower crystallinity (57–73%) in respect to the parent sample (H-Z-100%). The crystallinity loss depended weakly on the amount of the introduced copper.
Another effect was observed in case of the samples pretreated with sodium nitrate series (Na-Z). The application of the procedure referring to the removal of Brønsted acid sites with NaNO3 (pH = 7) led to a significant deterioration of zeolite crystallinity (from 100 to 23%). Further treatment with copper (CuxNa-Z) leads to no apparent changes in the crystallinity of the samples (28–30%).
The explanation of the observed effect seems to be complex. At first sight, observed phenomenon could be explained as a result of hydrolysis of Si-O-Al groups occurring in zeolites [
46]. H-Z treated with a pure distilled water (at the same pH and temperature) subjected to a distinct decrease of crystallinity (from 100 to 39%). On the other hand, it is worth emphasizing that the partially dealuminated H-Z (used here as a parent sample) has a fractured structure, which is more sensitive to any modification in relation to the virgin FAU, as shown by Gackowski et al. [
17,
47]. That leads to the amorphization of the “nibbled” FAU-type zeolite.
Observed differences in the crystallinity loss of the final form of prepared samples depending on the chemical composition of used mixture for H-Z modification might be associated with an ionic strength of the employed aqueous solutions. Further research concerning this matter will be continued.
For all Cu-zeolites (both CuxH-Z and CuxNa-Z), no XRD patterns of copper phase were found. This indicates that the incorporated copper forms crystals, which can be too difficult for identification by this technique.
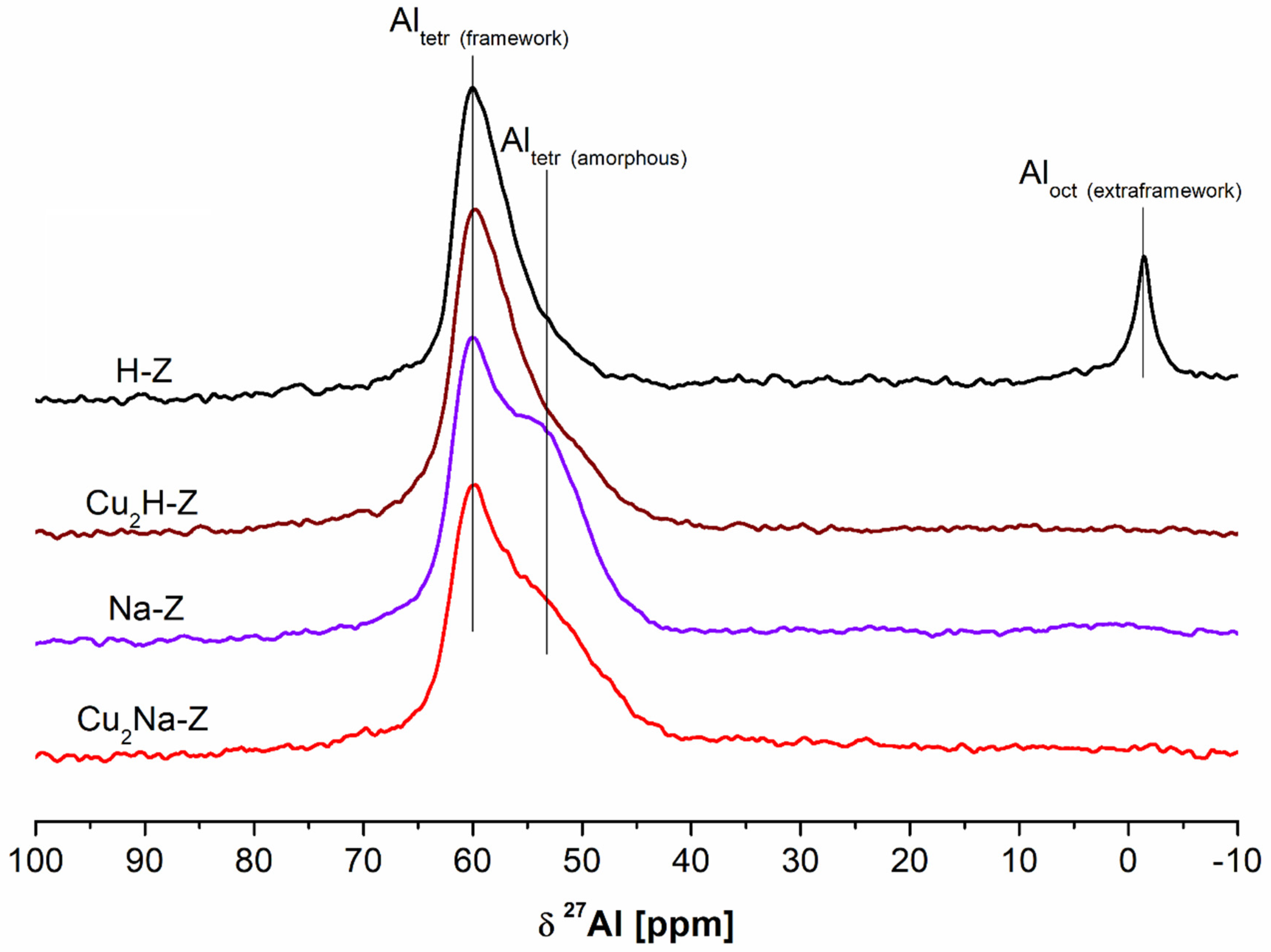
27Al MAS NMR spectra of the parent and modified samples are visualized in
Figure 3. The employed modification procedures have an influence on the status of aluminum.
Spectrum of the parent H-Z shows two signals: at 61.4 ppm related to “zeolitic” tetrahedral Al and at 0 ppm associated with octahedrally coordinated extraframework Al.
Treatment of H-Z either with NaNO
3 or Cu(NO
3)
2·3H
2O leads to the disappearance of the signal at 0 ppm coming from extraframewok Al. Simultaneously, a new signal at ca. −53 ppm appeared. The signal is typical of tetrahedral (non-zeolitic) Al present in amorphous aluminosilicates, which was also reported by Gackowski et al. [
17] and Kuterasiński et al. [
48]. This signal is particularly strong in the spectra of the sample modified with NaNO
3 alone (Na-Z), for which the most noticeable decrease of crystallinity was found. Hence, it may be concluded that the contribution of this type of aluminum species increases with the amorphization of parent H-Z zeolite [
49]. Another possibility is the change of the status of aluminum as a result of the interaction between Al species coming from zeolitic structure and extraframework cations originating from modifiers, such as pyridine, ammonia and salts (Na
+ or K
+), which was reported by Bourgeat et al. [
50]. Based on the data summarized in [
50], we may conclude that similar situation in the case of the treatment of our FAU-type zeolite with aqueous Cu(NO
3)
2 solution takes place.
2.5. Character of the Active Sites as Evidenced by IR Spectroscopy with CO and NH3 as Probe Molecules
Figure 6 shows the differential IR spectra recorded for the samples activated at 400 °C and after adsorption of ammonia at 130 °C. In the case of H-Z, Cu
1H-Z and Cu
2H-Z, the presence of Brønsted acid sites was evidenced. Raising Cu content causes a decrease of the intensity of the bands at 3620, 3550 and 1450 cm
−1 assigned to the acidic OH groups from the zeolite structure. That implies from ionic-exchange of protons by copper cations. Simultaneously, the increase of 1620 cm
−1 band intensity related to Lewis acid sites was found. The occurrence of Lewis acid sites originated both from aluminum in FAU31-type zeolite as well as from the copper species [
56,
57,
58]. In this case, the introduction of copper species into FAU31-zeolite structure is responsible for the increase of Lewis acidity. For H-Z, Cu
1H-Z, Cu
2H-Z and Cu
5H-Z, concentrations of Brønsted and Lewis acid sites formed the following sequences: (160 vs. 90 μmol/g), (80 vs. 211 μmol/g), (20 vs. 314 μmol/g) and (0 vs. 441 μmol/g), respectively (
Table 1). (Cu
xH-Z). In all cases the presence of terminal OH group was detected (see bands at 3740 cm
−1). The sample treated with NaNO
3 (Na-Z) was deprived of protonic acidity—no bands at 3620, 3550 and 1450 cm
−1 were found.
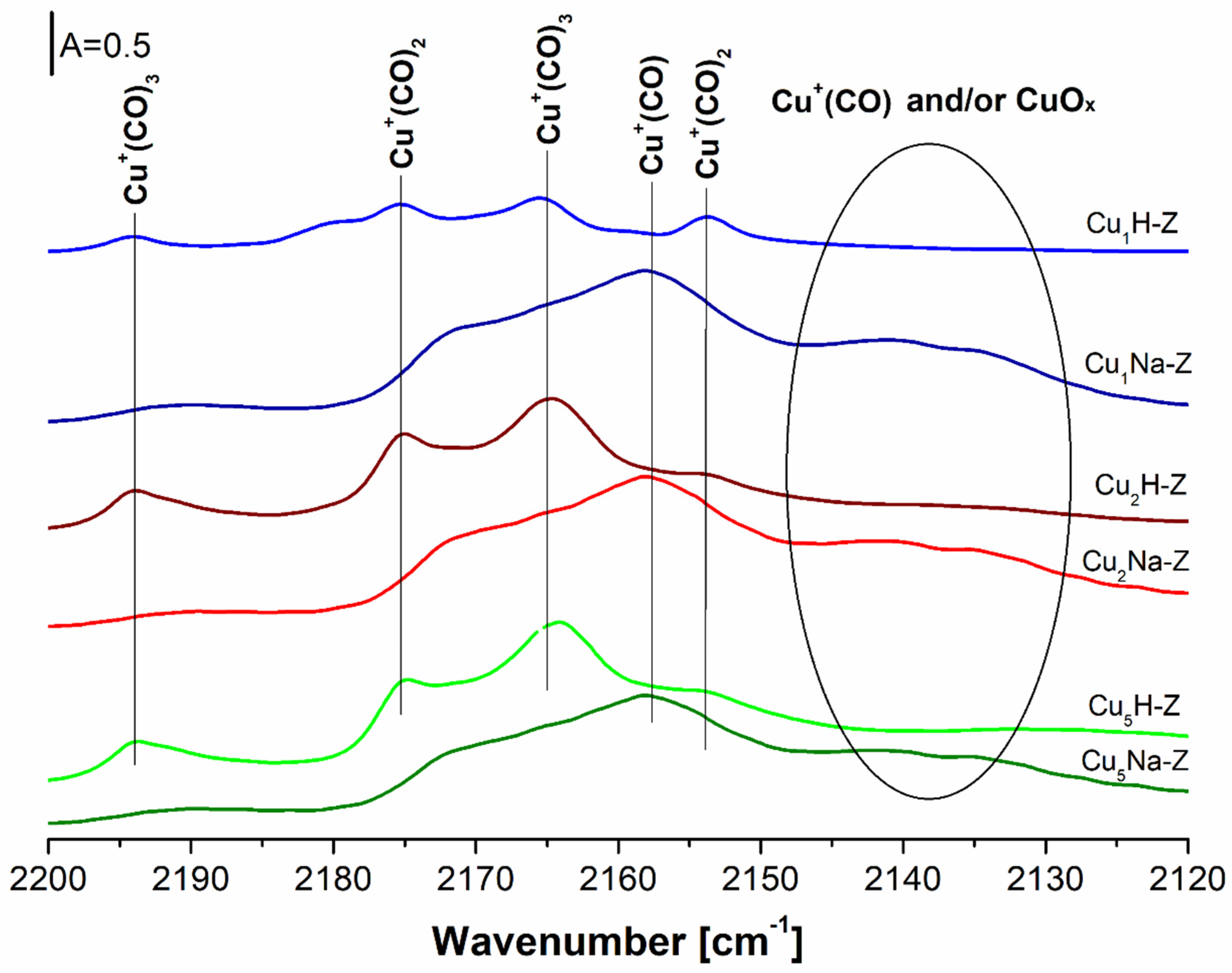
The results from the differential IR spectra and the quantitative analysis obtained from the experiments with the CO adsorption on the Cu-containing samples at room temperature are reported in
Figure 7 and
Table 1. For the samples directly impregnated with copper (Cu
xH-Z), the increasing Cu content from 1%wt. to 2%wt. led to much higher concentration of Cu
+ (from 75 to 160 μmol/g). Further rising of the amount of copper up to 5%wt. had no additional effect (160 vs. 170 μmol/g). It may be concluded that the cationic exchange positions were filled fully by Cu
+ present in high amount in the FAU-based samples.
The data obtained for the zeolites pretreated with NaNO3 and then modified with Cu (CuxNa-Z) indicated low concentration of Cu+ (ca 30 μmol/g), independent on the copper content in the studied samples. The distinct amorphization of the zeolitic structures led to the drastic deterioration of exchange capacity of such modified catalysts.
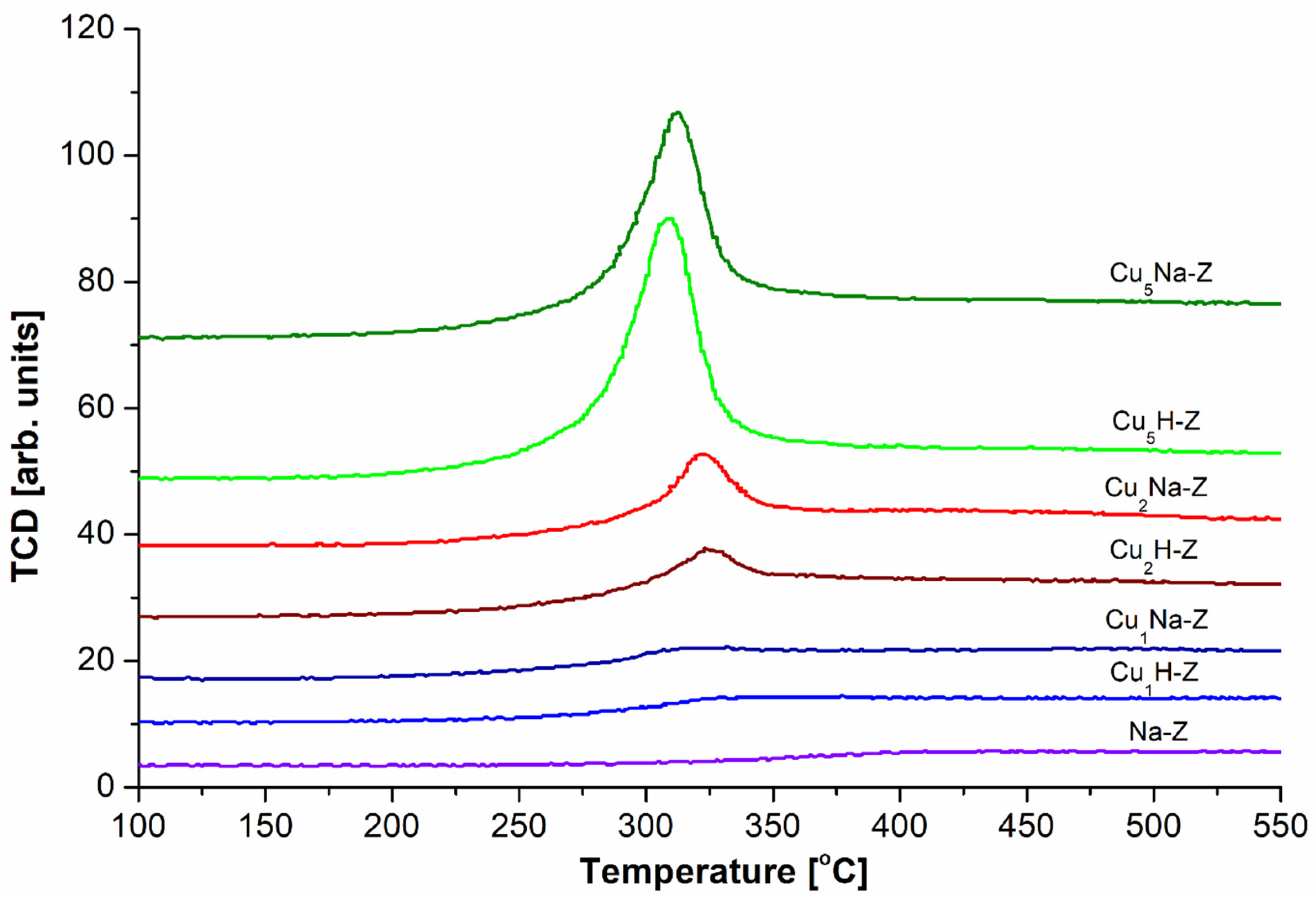
The presence of Cu
+ in our samples subjected to FT-IR studies originates from the so-called “auto-reduction” of Cu
2+ species in vacuum at high temperature (400 °C and higher) in the absence of a reducing agent. This phenomenon takes place during the activation procedure prior to the FT-IR measurements of our Cu-samples with adsorbed probe molecules. The auto-reduction mechanism of divalent copper species was previously described for Cu-loaded both zeolites [
59] and alumina [
60]. For comparison, in the H
2-TPR results (
Figure 5), we observed direct reduction of Cu
2+ species to Cu
0 (without the formation of Cu
+ which is generally difficult to keep stable). This ostensible contradiction between TPR and FT-IR data is due to different pretreatment conditions. In the former case, an activation prior to the H
2-TPR experiment was performed in the absence of vacuum (in a helium flow) at much lower temperature compared to FT-IR measurements (100 °C vs. 400 °C in vacuum). Hence, during the activation preceding H
2-TPR experiment, the auto-reduction of divalent Cu species did not take place.
In our previous work [
61], we reported that for the sample in protonic form and containing 1%wt. of Cu (Cu
1H-Z), the concentration of Cu
+ was twice smaller compared to the Cu content originating from its introduction by the impregnation method (160 μmol/g). This phenomenon may be explained by the fact that in our zeolite (FAU31), Cu
+ may be located both in supercages and inside hexagonal prisms and cuboctahedra. The latter location of cations makes them inaccessible to adsorbed particles, such as CO. The “hidden” sites are occupied by cations in the first place, due to the highest effectiveness of the stabilization of cations by framework oxygens in these positions.
In the case of samples containing higher Cu contents (2 and 5%wt. both for protonic and sodium series), lower amount of Cu interacting with CO in relation to the total copper content introduced during impregnation may be also explained by the status of Cu species found in our FAU-31-based samples. It was indicated that the copper was in the form both of cations in exchange positions (Cu
+exch, Cu
2+exch.) and in the form of oxides (Cu
+oxide, CuO). Zeolites in sodium form (Cu
xNa-Z) were characterized by much lower Cu
+exch. contents and higher amounts of Cu
+ and Cu
2+ in the oxide forms [
61].
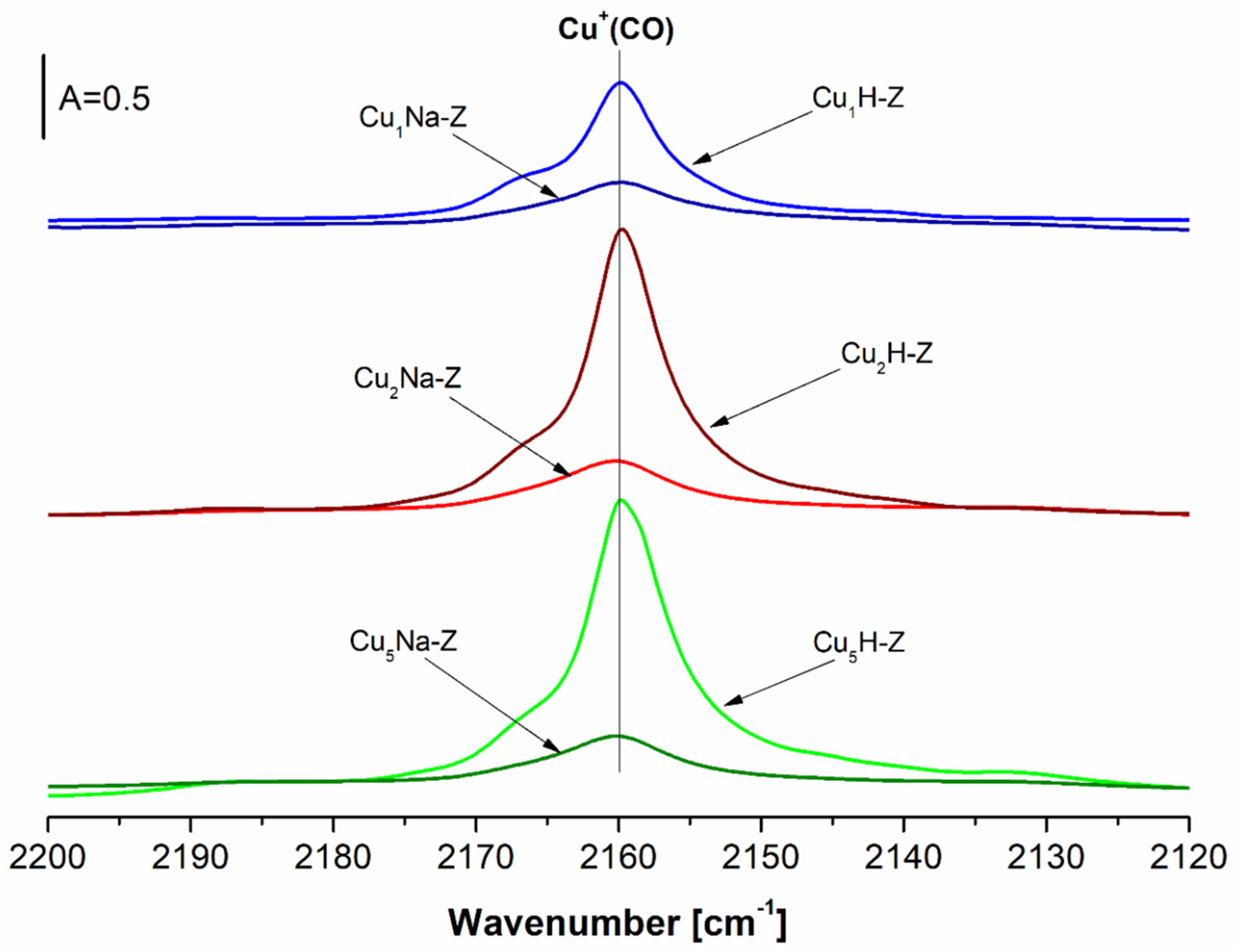
The differential IR spectra of the CO adsorbed at −100 °C on Cu-zeolite samples (Cu
xH-Z) are presented in
Figure 8. The adsorption of CO resulted in the appearance of the following bands: ν
asym at 2153 cm
−1 and ν
sym at 2175 cm
−1, corresponding to dicarbonyls Cu
+(CO)
2 formed at higher CO loadings [
62,
63]. The occurrence of new bands at 2165 and 2193 cm
−1 assigned to tricarbonyls Cu
+(CO)
3 was also found [
64,
65]. The intensity of the mentioned bands increases along with the increased Cu content in a similar way as in the case of bands at 2160 cm
−1 corresponding to Cu
+(CO), which were recorded at room temperature (
Figure 7).
For the samples additionally pretreated with NaNO
3 (Cu
xNa-Z), the bands at 2158, 2135 and 2140 cm
−1 were registered (
Figure 8). While the band at 2158 cm
−1 was assigned to Cu
+(CO), the interpretation of the bands at 2135 and at 2140 cm
−1 was not straightforward. They may be assigned to another kind of copper site [
66] or to the associated Cu ions [
67]. Datka et al. [
62] evidenced simultaneous occurrence of species by the presence of the bands at 2158 and 2135 cm
−1 which allowed us to suppose that the adsorption of CO was not an equilibrium process at low temperature (−100 °C). CO interacted with copper by adsorbing on all accessible sites. Subsequently, the redistribution took place: CO desorbed from sites of lower adsorption heat and re-adsorbed on sites having higher binding energy. Due to the low rate of desorption at −100 °C (requiring CO molecules to overcome the activation energy), the redistribution was also slow. Góra-Marek et al. [
68] suggested that the bands at ca 2130 cm
−1 may correspond to CuO
x species.
In all cases (CuxH-Z, CuxNa-Z), the absence of the bands at 2206 and 2124 cm−1 proves that neither Cu0 nor Cu2+ species were present in the studied systems.
In our previous work [
69], we reported the FT-IR analysis of Cu containing FAU31 zeolites in order to investigate the process of reduction of Cu sites with hydrogen and the properties of reduced zeolites. IR spectra of OH groups for two types of Cu-zeolites: in protonic (Cu
xH-Z) and sodium form (Cu
xNa-Z) were recorded. In all cases, the reduction of Cu sites with hydrogen resulted in the formation of Brønsted acid sites—the growth of IR bands of acidic hydroxyls was observed. Furthermore, quantitative analysis (NH
3 sorption) indicated the production of protonic acidity, which agreed with the fact that OH bands increased. The formation mechanism of protonic acidity may be explained by the reduction of copper species (Cu
+exch, Cu
+oxide, Cu
2+) with hydrogen according to the reactions, as follows: 2 Cu
+ + H
2 = 2 Cu
0 + 2H
+ and Cu
2+ + H
2 = Cu
0 + 2H
+. Furthermore, based on FT-IR studies of CO and NO sorption over Cu-FAU31 zeolites, it was indicated that the susceptibility of Cu sites for the reduction with hydrogen depended strictly on the type of Cu species. Order of reduction Cu sites formed the following sequence: Cu
+oxide > Cu
2+ > Cu
+exch [
69].
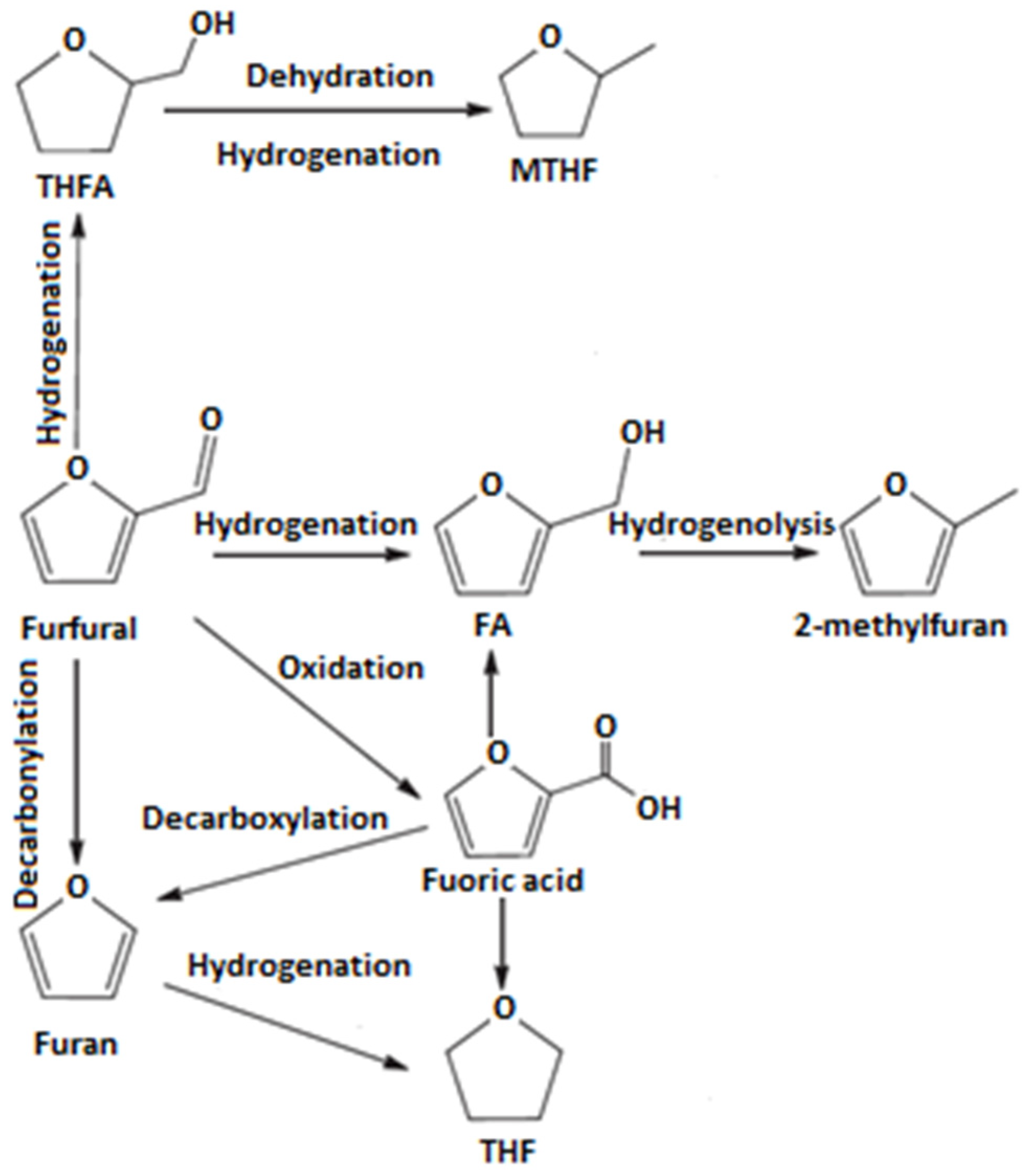
2.6. Catalytic Properties
Table 2 gathers the results of the catalytic tests for all four types of the samples: H-Z containing only Brønsted and Lewis acid sites coming from zeolite, Cu
2H-Z containing both protonic and Lewis acid sites including also copper redox active centers, Na-Z samples possessing only Lewis acid sites originating from zeolite structure and Cu
2Na-Z containing Lewis acid sites from both zeolite and copper species.
The data obtained at 300 °C (
Table 2) indicated that Brønsted acid sites and copper active sites were similarly responsible for the catalytic activity of the studied materials. The best catalytic properties were found for the system containing both protonic and copper active sites (Cu
2H-Z), for which the conversion of furfural was 73%, while furan yield was 22%. Somewhat worse catalytic properties were observed for the sample containing only acid sites attributed to zeolitic structure (H-Z). Conversion of furfural and yield of furan were 33% and 16%, respectively. In the case of the “Brønsted -free” catalyst (Na-Z) no catalytic activity was found, which evidenced no catalytic participation of Lewis acid sites assigned to Al species in this reaction. In turn, the presence of the copper redox sites in sodium form of FAU31-type zeolite (Cu
2Na-Z) resulted in an apparent furfural conversion (53%), but very low furan yield (10%) in comparison with protonic counterpart (Cu
2H-Z).
The increase of temperature up to 400 °C resulted in a significant improvement of catalytic properties of the parent sample (H-Z), for which furfural conversion and furan yield rose from 33 to 77% and from 16 to 60%, respectively. For the catalyst containing both protonic acidity and copper active sites (Cu2H-Z), the conversion of furfural decreased slightly from 73 to 64% and furan yield rose from slightly from 22 to 28%, respectively. When the reaction was carried out over Cu2Na-Z catalyst, higher temperature led to a very slight increase of both the conversion of furfural and furan yield (from 53 to 56% and from 10 to 14%, respectively). At 400 °C, no catalytic activity was found for the sodium sample (Na-Z).
Next, the influence of the amount of copper on the catalytic activity was checked—See
Table 3 and
Table 4. For the catalysts containing both protonic acid sites and copper active centers (Cu
xH-Z—
Table 3), the appearance of copper in FAU31 zeolite led to increase of both conversion of furfural (from 33 to 73–78%) and yield of furan (from 16 to 18–36%) at 300 °C. Nevertheless, the increase of the copper content led to significant deterioration in furan yield from 36% (Cu
1H-Z) to 18% (Cu
5H-Z).
Elevation of reaction temperature up to 400 °C resulted in a slight decrease of furfural conversion from 73–78% to 64% and some changes in furan yield: from 36 to 23%, from 22 to 28%, and from 18 to 19% for Cu1H-Z Cu2H-Z and Cu5H-Z, respectively. For the samples with higher copper content (Cu2H-Z and Cu5H-Z), trace of 2-methylfuran yield was found (1%).
In the case of catalysts deprived of Brønsted acid sites (Cu
xNa-Z), studied at 300 °C, the increase of the copper content from 1 to 5%wt. resulted in a significant improvement of furfural conversion (47 vs. 71%) without relevant changes of furan yield (7–10%—see
Table 4). Furthermore, rising copper loading caused increasing (but still very low) of the 2-methylfuran yield (from 2 to 4%).
At 400 °C, the increase of copper loading from 1%wt. to 5%wt. led to higher furfural conversion from 21 to 60%, but with very poor production of furan (5–14%). Another effect of higher copper content was increasing yield of 2-methylfuran (from 1 to 3%).
The application of the Cu-containing catalysts implies from their lower hydrogenation ability compared with noble metals, which allows it to avoid the hydrogenation of furan ring [
70,
71]. The nature of the active species has been a subject of discussion for many years [
72]. It was found that the Cu
+/Cu
0 ratio in the catalyst decides about catalytic activity. Rao et al. [
73] indicated that the turnover frequency (TOF) decreased with lowering Cu
+/Cu
0 ratio from 0.3 to 0 over carbon-supported Cu catalysts. This phenomenon may be explained by the activation of H
2 by metallic copper species, while Cu
+ sites plays a role of electrophilic or Lewis acid sites able to polarize C=O bond via electron lone pair of oxygen [
74,
75]. The Cu
+/Cu
0 ratio depends strongly on many factors, such as the kind of support, the way of preparation and the conditions of catalytic tests (including reduction meant as a pretreatment procedure).
In our previous work [
61], we reported the status and properties of Cu species in HFAU and NaFAU of Si/Al = 31 by IR spectroscopy with CO and NO as probe molecules. Both Cu
+ and Cu
2+ were found in the zeolites in the form both of exchange cations (Cu
+exch, Cu
2+exch.
) and in oxide form (Cu
+oxide, CuO). The proportion between various forms of Cu depended significantly on both the amount of Cu introduced into the materials and the way of treatment of FAU-type zeolite (HFAU or NaFAU). In CuHFAU containing 1wt.% of Cu, all of Cu species were in the form of Cu
+exch. neutralizing AlO
4-. Higher copper content resulted in the co-existence of Cu
+oxide as well as the presence of Cu
2+ as Cu
2+exch. and CuO species. Zeolites Cu
xNaFAU contained much lower amount of Cu
+exch. and higher contribution of Cu
+ and Cu
2+ in the oxide forms. Direct comparison of the data given in
Table 4 led to the conclusion that Cu
+oxide and CuO are the most active Cu species in this reaction. Furthermore, copper oxide species favor slightly the formation of 2-methylfuran.
The results of the catalytic experiments described above indicate that two types of active sites present in the tested samples contribute to their catalytic activity. Decarbonylation of furfural is realized mainly on the protonic acid sites.
High activity of the H-Z sample in furfural decarbonylation should be attributed to the existence of strong Brønsted acid sites. The similar reactivity of samples of acidic character was reported earlier for catalysts based on zeolites and heteropolyacids—see, e.g., references [
76,
77,
78] for reports on transformation of lactic acid and its derivatives to acetaldehyde. The activity in decarbonylation was ascribed to high density of acid sites which directly interact with the carbonyl group which is to be removed from the substrate.
Furfural decarbonylation to furan also proceeds on Cu sites.
Our findings are in line with the previous reports on the application of the copper-based systems to furfural hydrogenation and decarbonylation [
79]. A similar effect was also found for γ-valerolactone valorization [
80]. It is claimed that furfural can be activated on copper phase via C-H bond rupture from the aldehyde group. Thus, formed furfuryl residue is stabilized on the metal center due to its interactions with un-filled orbitals of copper. The reactivity of furfural is also determined by the geometry of the substrate adsorption on the catalytic active sites [
72].
Further, it is believed that the copper centers are also responsible for hydrogen activation during hydrogenation of biomass-derived products [
80,
81].
The contribution of copper redox active centers in the decarbonylation of furfural may be also explained by the production of Brønsted acid sites as a result the reduction of copper species with hydrogen according to the following reactions: 2 Cu
+ + H
2 = 2 Cu
0 + 2H
+ and Cu
2+ + H
2 = Cu
0 + 2H
+, which was reported in our previous work [
69]. Protonic acid sites generated in this way could participate in the formation of furan.
We also investigated an effect of the application of the pretreatment in hydrogen (prior to catalytic experiment) on the catalytic behavior of the samples containing various copper content in protonic form—Cu
xH-Z (
Table 5). It was indicated that for Cu-containing catalysts, the use of hydrogen did not change furfural conversion (64%), but caused a slight increase of furan yield from 19 to 23% or from 17 to 28% or from 11 to 19% for Cu
1H-Z or Cu
2H-Z or Cu
5H-Z, respectively. Analysis of these results suggests that the pretreatment of our catalysts in hydrogen (directly before catalytic measurements) led to the formation of new protonic acid sites, which could enhance the production of furan.
All studied catalysts revealed very high susceptibility to coking (
Table 2,
Table 3,
Table 4 and
Table 5). This effect was particularly strong for Cu-containing catalysts and generally increased with the amount of copper in the prepared samples. Deactivation is the main problem for Cu-based hydrogenation catalysts, which hampers their industrial application. This phenomenon also occurs over the catalysts containing acid sites like zeolites [
81]. Hence, it is possible that newly created protonic acid sites take part in the production of coke. It may be also related with the loss of the Cu
+ species by the reduction and poisoning of active Cu sites by the adsorption of reaction intermediates.
In order to determine how the studied catalysts changed after the reaction, we performed XRD, SEM and FT-IR experiments for the spent samples. The results concerning the crystallinity, morphology, acidic properties and an optical appearance of the catalysts after catalytic experiments are summarized in
Appendix A (
Figure A1,
Figure A2,
Figure A3 and
Figure A4).
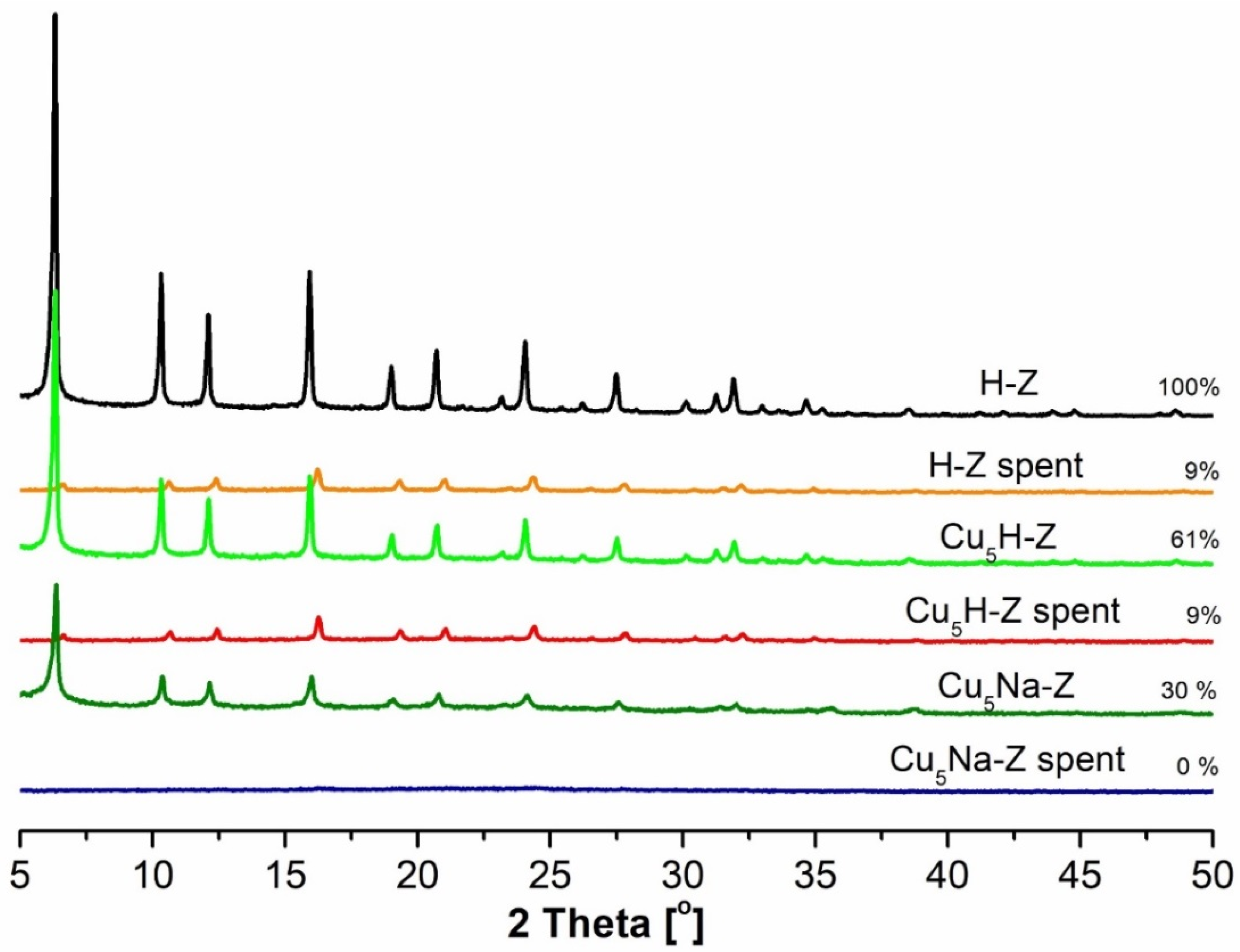
In all cases, the analysis of XRD data (
Figure A1) indicated that, during catalytic tests, the crystallinity of the studied samples underwent a drastic deterioration probably due to the interaction between vaporized furfural and very a sensitive structure of FAU31-type zeolite. Final crystallinity was ca 9% for the samples in protonic series (H-Z and Cu
5H-Z), while Cu
5Na-Z was characterized by zero crystallinity.
For the parent sample (H-Z), the contact with the reaction mixture led to apparent changes in its morphology (
Figure A2). Well defined edges of irregular-shaped crystals were subjected to blurring, which could be an effect of the FAU31 amorphization.
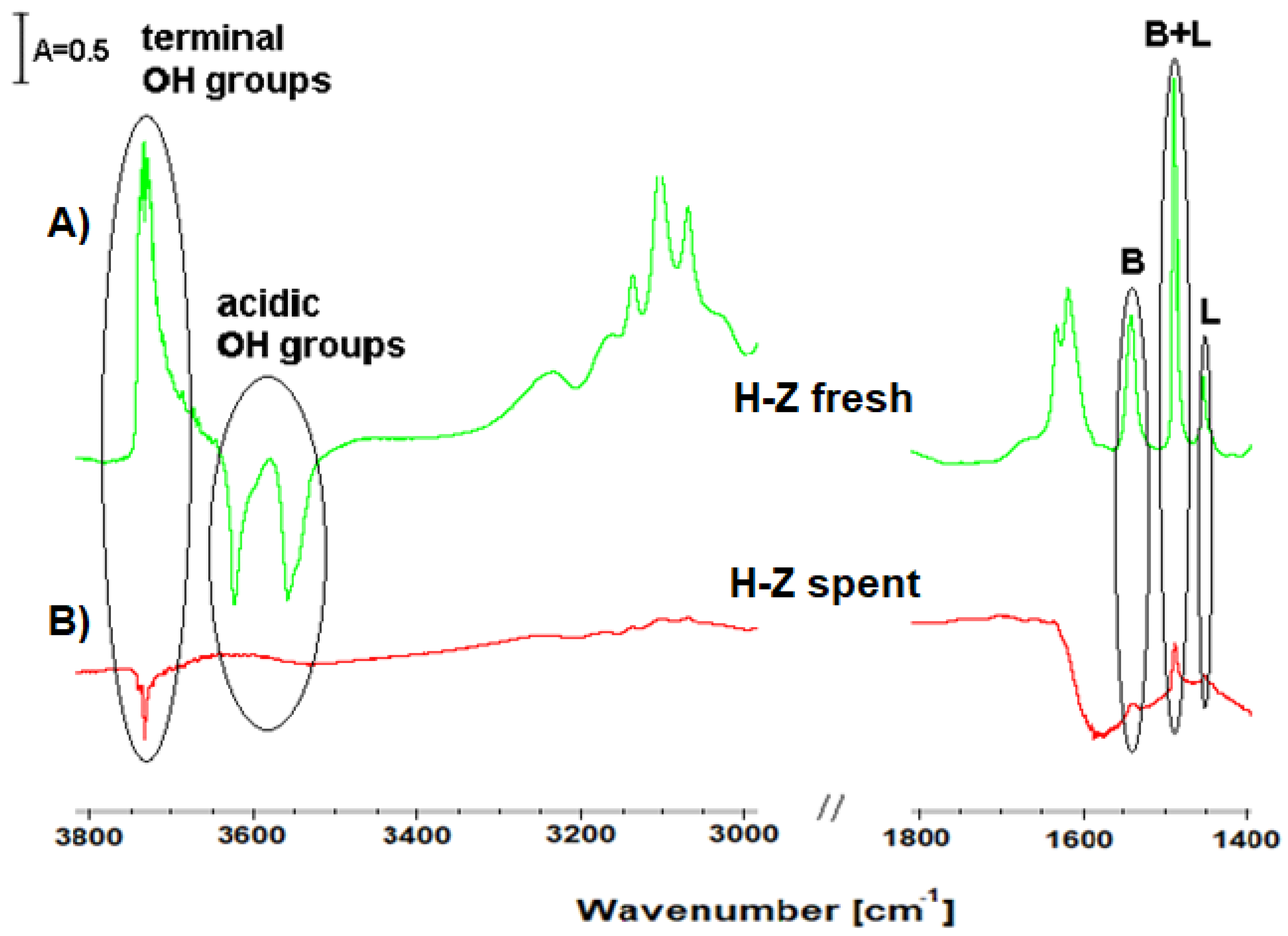
Comparison of FT-IR spectra of pyridine adsorbed on fresh and spent H-Z catalyst led to the conclusion that studies catalyst underwent coking. The decrease of the bands assigned to the acid sites was probably due to blocking of acid sites by coke depositions.
Last evidence for the coking of the studied catalysts is a direct comparison of the optical appearance of samples before and after catalytic experiment (
Figure A4). Both Cu-free and samples containing Cu became black during catalytic tests.
For the H-Z catalyst, a stability test was performed at 350 °C (
Table A1). From the presented data, it may be concluded that the investigated catalyst was not highly stable due to coking. Both furfural conversion and yield of furan decreased from 74 to 35% and from 61 to 26%, respectively. The deterioration of catalytic properties of the studied sample during the time of experiment was in line with the loss of Brønsted acid sites accessibility (
Figure A3), as well as corresponded to the changes in the crystallinity (
Figure A1) and morphology (
Figure A2) of the studied catalyst.

 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}








