
C2-Symmetric P-Stereogenic Ferrocene Ligands with Heavier Chalcogenophosphinous Acid Ester Donor Sites †


Abstract
1. Introduction
2. Results and Discussion
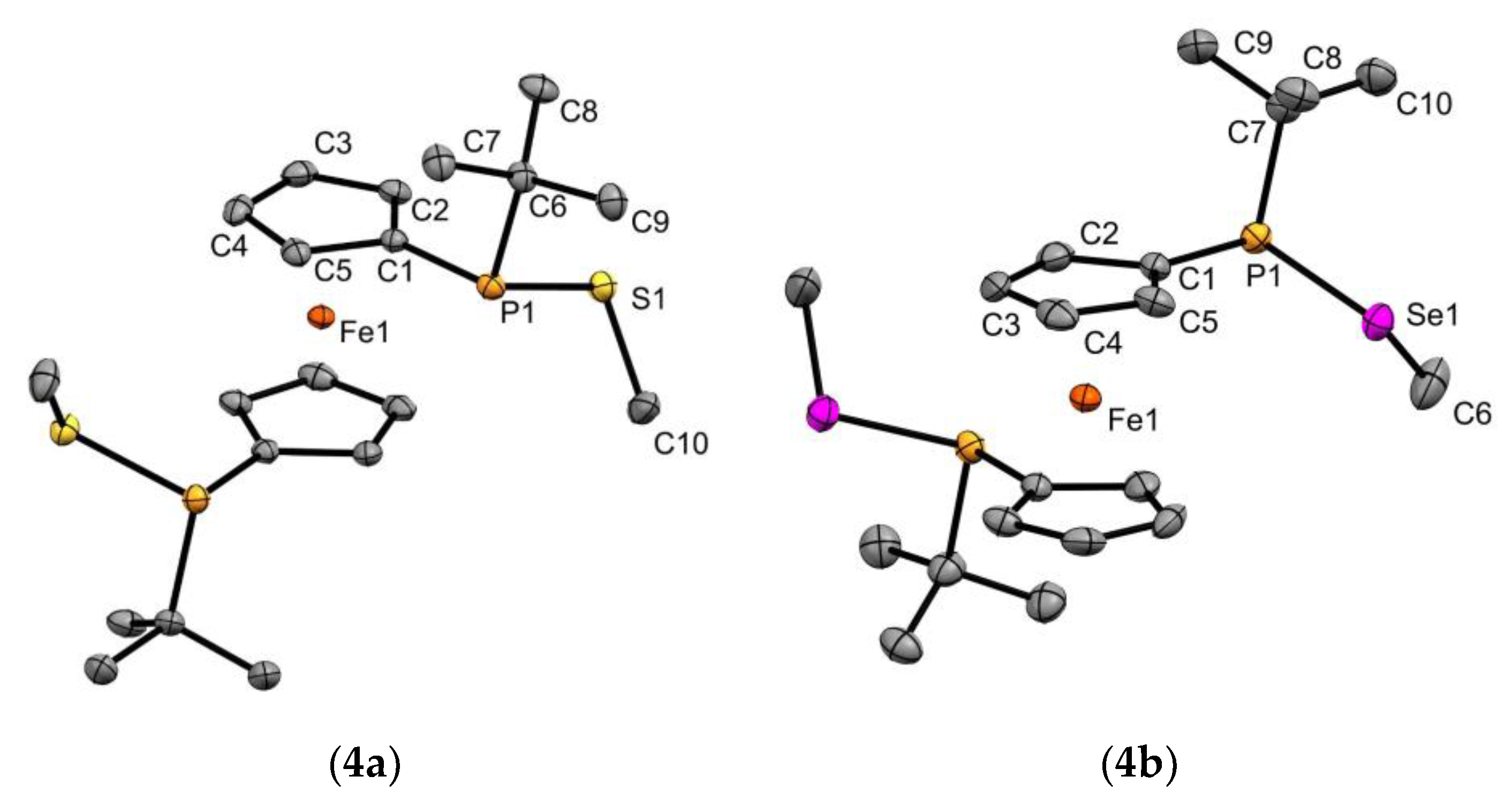
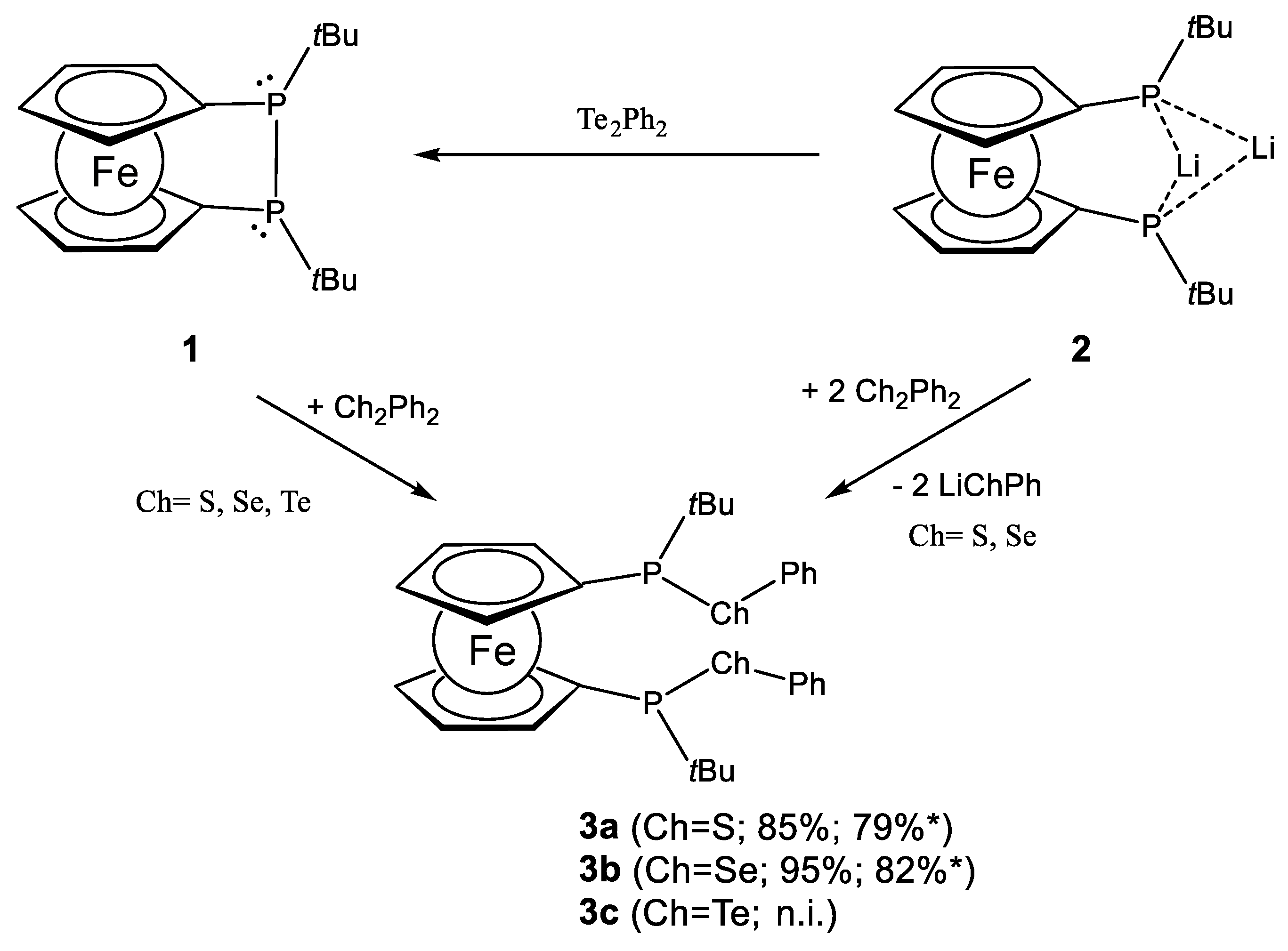
2.1. Ligand Preparation and Properties
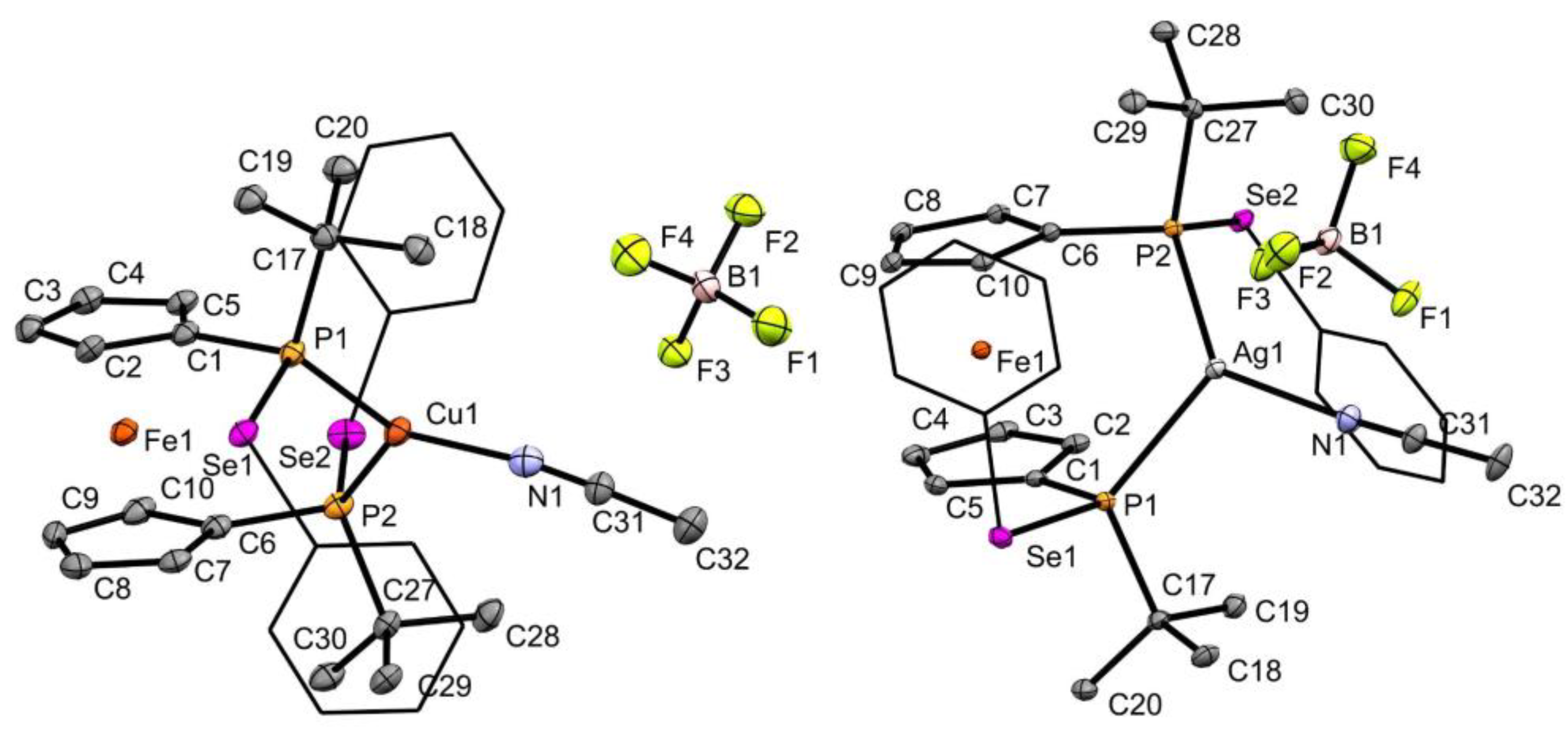
2.2. Coinage Metal Complexes
3. Materials and Methods
3.1. Experimental
3.2. X-ray Diffraction Measurements
3.3. Synthetic Protocols and Characterization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Rajanbabu, T.V. Phosphinite and Phosphonite Ligands. In Phosphorus(III) Ligands in Homogeneous Catalysis: Design and Synthesis; Kamer, P.C.J., van Leeuwen, P.W.N.M., Eds.; John Wiley & Sons, Ltd.: West Sussex, UK, 2012; pp. 159–232. [Google Scholar]
- DuMont, W.-W.; Kubiniok, S.; Severengiz, T. Eigenschaften von Tellur-Tellur-Bindungen. IV. Dismutationsreaktionen von Di-p-tolylditellurid mit Tetra-tert-butyldiphosphan und Tetra-isopropyldiphosphan. Z. Anorg. Allg. Chem. 1985, 531, 21–25. [Google Scholar] [CrossRef]
- Ashe, A.J., III; Ludwig, E.G., Jr. The exchange reaction of tetramethyldipnictogens with dimethyldichalcogenides. J. Organomet. Chem. 1986, 308, 289–296. [Google Scholar] [CrossRef]
- Medena, C.; Calogero, F.; Lemoine, Q.; Aubert, C.; Derat, E.; Fensterbank, L.; Gontard, G.; Khaled, O.; Vanthuyne, N.; Moussa, J.; et al. A HELIXOL-Derived Bisphosphinite Ligand: Synthesis and Application in Gold-Catalyzed Enynes Cycloisomerization. Eur. J. Org. Chem. 2019, 2019, 2129–2137. [Google Scholar] [CrossRef]
- Coburger, P.; Schulz, J.; Klose, J.; Schwarze, B.; Sárosi, M.B.; Hey-Hawkins, E. C2-Symmetric P,N Ligands Derived from Carborane-Based Diphosphetanes: Synthesis and Coordination Chemistry. Inorg. Chem. 2017, 56, 292–304. [Google Scholar] [CrossRef]
- Franz, R.; Nasemann, S.; Bruhn, C.; Kelemen, Z.; Pietschnig, R. Chalcogen transfer rearrangement: Exploring inter- vs. intramolecular P-P bond activation. Chem. Eur. J. 2021, 27, 641–648. [Google Scholar]
- Isenberg, S.; Weller, S.; Kargin, D.; Valić, S.; Schwederski, B.; Kelemen, Z.; Bruhn, C.; Krekić, K.; Maurer, M.; Feil, C.; et al. Bis-[3]ferrocenophanes with central >E-E’< bonds (E, E’ = P, SiH): Preparation, properties, and thermal activation. Chem. Open 2019, 8, 1235–1243. [Google Scholar] [CrossRef]
- Togni, A.; Hayashi, T. Ferrocenes—Homogeneous Catalysis, Organic Synthesis, Materials Science; VCH Verlagsgesellschaft: Weinheim, Germany, 1995. [Google Scholar]
- Štěpnička, P. Ferrocenes: Ligands, Materials and Biomolecules; John Wiley and Sons, Ltd.: West Sussex, UK, 2008. [Google Scholar]
- Dey, S.; Pietschnig, R. Chemistry of sterically demanding dppf-analogs. Coord. Chem. Rev. 2021, accepted. [Google Scholar]
- Kargin, D.; Kelemen, Z.; Krekić, K.; Maurer, M.; Bruhn, C.; Nyulászi, L.; Pietschnig, R. [3]Ferrocenophanes with bisphosphanotetryl bridge: Inorganic rings on the way to tetrylenes. Dalton Trans. 2016, 45, 2180–2189. [Google Scholar] [CrossRef] [PubMed]
- Isenberg, S.; Frenzel, L.-M.; Bruhn, C.; Pietschnig, R. Metallated [3]ferrocenophanes containing P3M bridges (M = Li, Na, K). Inorganics 2018, 6, 67. [Google Scholar] [CrossRef]
- Lik, A.; Kargin, D.; Isenberg, S.; Kelemen, Z.; Pietschnig, R.; Helten, H. PBP bridged [3]ferrocenophane: A bisphosphanylborane with a redox trigger Chem. Commun. 2018, 54, 2471–2474. [Google Scholar] [CrossRef]
- Koner, A.; Kelemen, Z.; Schnakenburg, G.; Nyulászi, L.; Streubel, R. 1,4-Additions of tricyclic 1,4-diphosphinines—A novel system to study σ-bond activation and π–π dispersion interactions. Chem. Commun. 2018, 54, 1182–1184. [Google Scholar] [CrossRef] [PubMed]
- Coburger, P.; Aures, R.; Schulz, P.; Hey-Hawkins, E. Exploiting the Ring Strain of Diphosphetanes: A Synthetic and Computational Approach towards 1,2,5-Selenadiphospholanes. ChemPlusChem 2018, 83, 1057–1064. [Google Scholar] [CrossRef] [PubMed]
- Ebels, J.; Pietschnig, R.; Kotila, S.; Dombrowski, A.; Niecke, E.; Nieger, M.; Schiffner, H.M. Bis(pentamethylcyclopentadienyl)-Substituted Phosphanes: Synthesis and Structure. Eur. J. Inorg. Chem. 1998, 1998, 331–337. [Google Scholar] [CrossRef]
- Graham, C.M.E.; Macdonald, C.L.B.; Boyle, P.D.; Wisner, J.A.; Ragogna, P.J. Addressing the Nature of Phosphinidene Sulfides via the Synthesis of P−S Heterocycles. Chem. Eur. J. 2018, 24, 743–749. [Google Scholar] [CrossRef]
- Tanimoto, Y.; Ishizu, Y.; Kubo, K.; Miyoshi, K.; Mizuta, T. Synthesis of [2]ferrocenophanes containing trivalent diphosphine units. J. Organomet. Chem. 2012, 713, 80–88. [Google Scholar] [CrossRef]
- Glodde, T.; Vishnevskiy, Y.V.; Zimmermann, L.; Stammler, H.-G.; Neumann, B.; Mitzel, N.W. The Nature of Chalcogen-Bonding-Type Tellurium–Nitrogen Interactions: A First Experimental Structure from the Gas Phase. Angew. Chem. Int. Ed. 2021, 60, 1519–1523. [Google Scholar] [CrossRef] [PubMed]
- Dey, S.; Buzsáki, D.; Bruhn, C.; Kelemen, Z.; Pietschnig, R. Bulky 1,1′-bisphosphanoferrocenes and their coordination behaviour towards Cu(i). Dalton Trans. 2020, 49, 6668–6681. [Google Scholar] [CrossRef]
- Trivedi, M.; Ujjain, S.K.; Singh, G.; Kumar, A.; Dubey, S.K.; Rath, N.P. Syntheses, characterization, and electrochemistry of compounds containing 1-diphenylphosphino-1′-(di-tert-butylphosphino)ferrocene (dppdtbpf). J. Organomet. Chem. 2014, 772-773, 202–209. [Google Scholar] [CrossRef]
- Trivedi, M.; Nagarajan, R.; Kumar, A.; Rath, N.P.; Valerga, P. Synthesis, characterization, crystal structures and photophysical properties of copper(I) complexes containing 1,1′-bis(diphenylphosphino)ferrocene (B-dppf) in doubly-bridged mode. Inorg. Chim. Acta 2011, 376, 549–556. [Google Scholar] [CrossRef]
- Borucki, S.; Kelemen, Z.; Maurer, M.; Bruhn, C.; Nyulaszi, L.; Pietschnig, R. Stereochemical Alignment in Triphospha[3]ferrocenophanes. Chem. Eur. J. 2017, 23, 10438–10450. [Google Scholar] [CrossRef]
- Laube, J.; Jäger, S.; Thöne, C. Synthesis and Structural Studies of Pyridine-2-selenolates − Reactions with Electrophilic Phosphorus(III) Compounds and Related Complex Chemistry. Eur. J. Inorg. Chem. 2001, 2001, 1983–1992. [Google Scholar] [CrossRef]
- Bowmaker, G.A.; Dyason, J.C.; Healy, P.C.; Engelhardt, L.M.; Pakawatchai, C.; White, A.H. Lewis-base adducts of Group 11 metal(I) compounds. Part 27. Solid-state phosphorus-31 cross-polarization magic-angle spinning nuclear magnetic resonance, far-infrared, and structural studies on the mononuclear 2: 1 adducts of triphenylphosphine with copper(I) and gold(I) halides. J. Chem. Soc. Dalton Trans. 1987. [Google Scholar] [CrossRef]
- Orthaber, A.; Borucki, S.; Shen, W.; Réau, R.; Lescop, C.; Pietschnig, R. Coordination behavior of a hexadentate 1,1’-ferrocenylene bridged bisphosphole towards coinage metal centers. Eur. J. Inorg. Chem 2014, 1751–1759. [Google Scholar] [CrossRef]
- Bachmann, S.; Gernert, B.; Stalke, D. Solution structures of alkali metal cyclopentadienides in THF estimated by ECC-DOSY NMR-spectroscopy (incl. software). Chem. Commun. 2016, 52, 12861–12864. [Google Scholar] [CrossRef]
- Kreyenschmidt, A.-K.; Bachmann, S.; Niklas, T.; Stalke, D. Molecular Weight Estimation of Molecules Incorporating Heavier Elements from van-der-Waals Corrected ECC-DOSY. ChemistrySelect 2017, 2, 6957–6960. [Google Scholar] [CrossRef]
- Sheldrick, G. Crystal structure refinement with SHELXL. Acta. Crystallogr. C. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Macrae, C.F.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Shields, G.P.; Taylor, R.; Towler, M.; van de Streek, J. Mercury: Visualization and analysis of crystal structures. J. Appl. Crystallogr. 2006, 39, 453–457. [Google Scholar] [CrossRef]
- Orthaber, A.; Belaj, F.; Pietschnig, R. Synthesis, Structure and π-Delocalization of a Phosphaalkenyl Based Neutral PNP-Pincer. Inorg. Chim. Acta 2011, 374, 211–215. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Orthaber, A.; Fuchs, M.; Belaj, F.; Rechberger, G.N.; Kappe, O.C.; Pietschnig, R. Bis(diethylamino)(pentafluorophenyl)phosphane—A Push–Pull Phosphane Available for Coordination. Eur. J. Inorg. Chem 2011, 2011, 2588–2596. [Google Scholar] [CrossRef][Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| δ(31P) [ppm] | JPCh [Hz] | |
|---|---|---|
| 3a | 51.2 (s); 50.2 (s) | --- |
| 3b | 57.8 (s); 57.1 (s) | 1J = 225; 2J = 5 |
| 3c | 45.8 (s); 45.3 (s) | 1J = 497; 2J = 5 |
| 4a | 56.8 (s); 56.5 (s) | --- |
| 4b | 58.7 (s); 58.4 (s) | 1J = 218; 2J = 6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Franz, R.; Bruhn, C.; Pietschnig, R. C2-Symmetric P-Stereogenic Ferrocene Ligands with Heavier Chalcogenophosphinous Acid Ester Donor Sites. Molecules 2021, 26, 1899. https://doi.org/10.3390/molecules26071899
Franz R, Bruhn C, Pietschnig R. C2-Symmetric P-Stereogenic Ferrocene Ligands with Heavier Chalcogenophosphinous Acid Ester Donor Sites. Molecules. 2021; 26(7):1899. https://doi.org/10.3390/molecules26071899
Chicago/Turabian StyleFranz, Roman, Clemens Bruhn, and Rudolf Pietschnig. 2021. "C2-Symmetric P-Stereogenic Ferrocene Ligands with Heavier Chalcogenophosphinous Acid Ester Donor Sites" Molecules 26, no. 7: 1899. https://doi.org/10.3390/molecules26071899
APA StyleFranz, R., Bruhn, C., & Pietschnig, R. (2021). C2-Symmetric P-Stereogenic Ferrocene Ligands with Heavier Chalcogenophosphinous Acid Ester Donor Sites. Molecules, 26(7), 1899. https://doi.org/10.3390/molecules26071899