
Nature of the Hydrogen Bond Enhanced Halogen Bond



Abstract
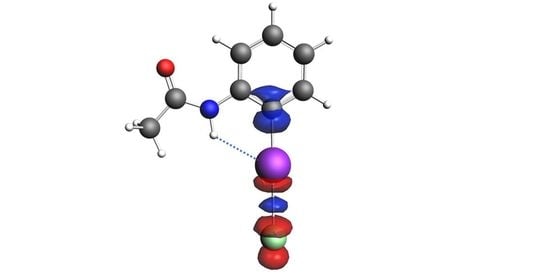
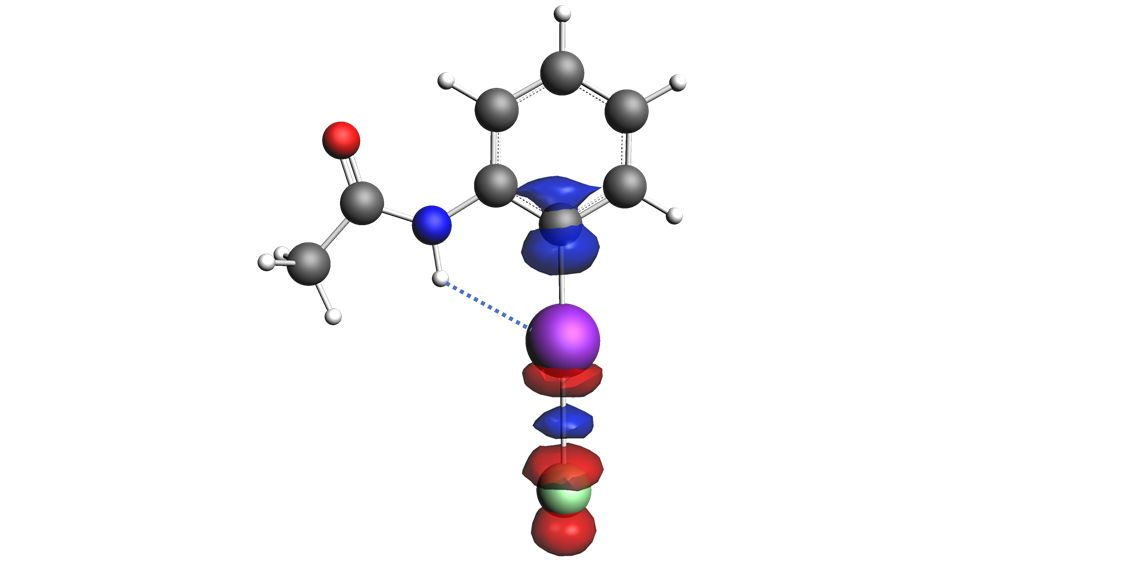
1. Introduction
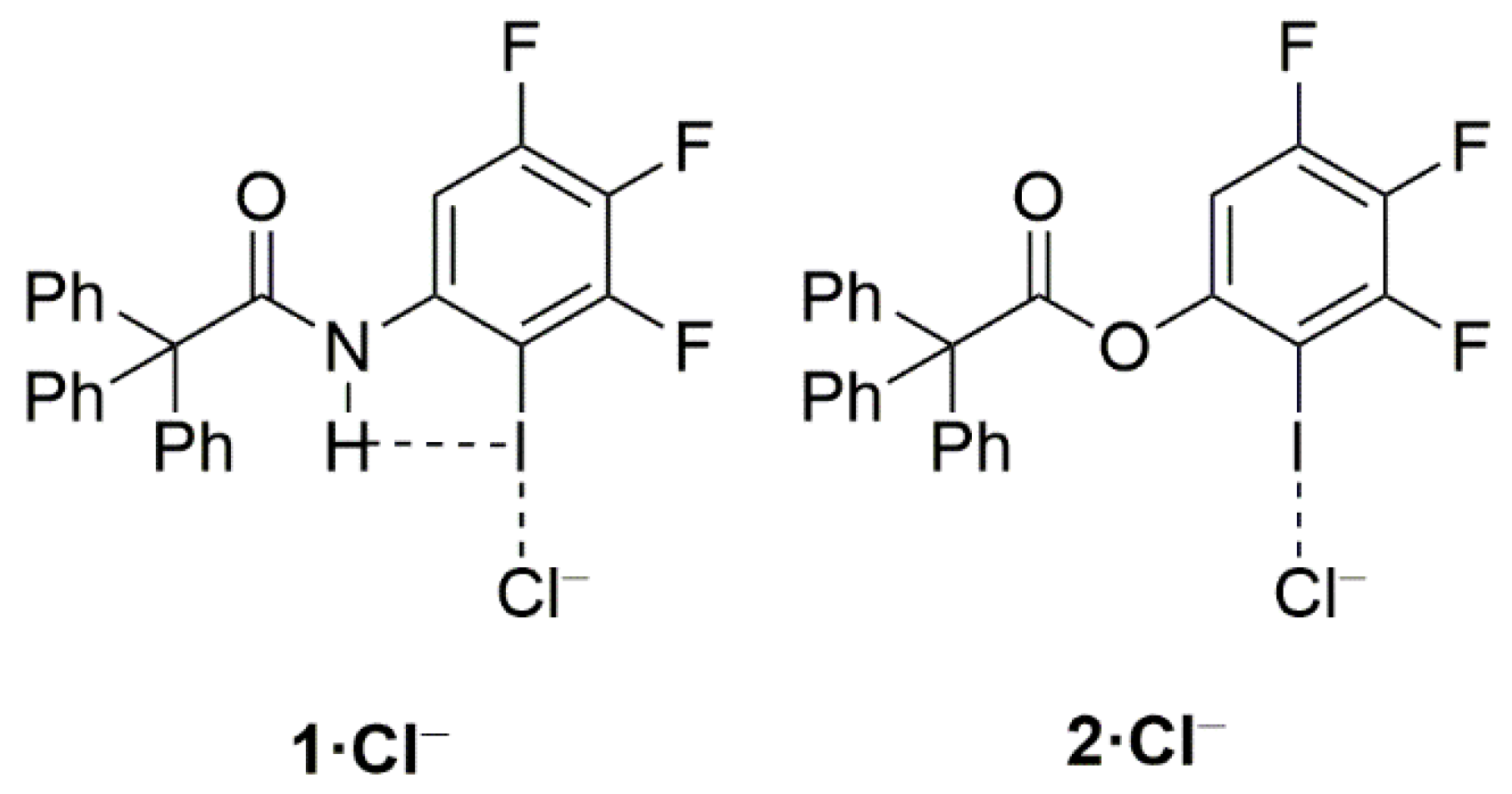
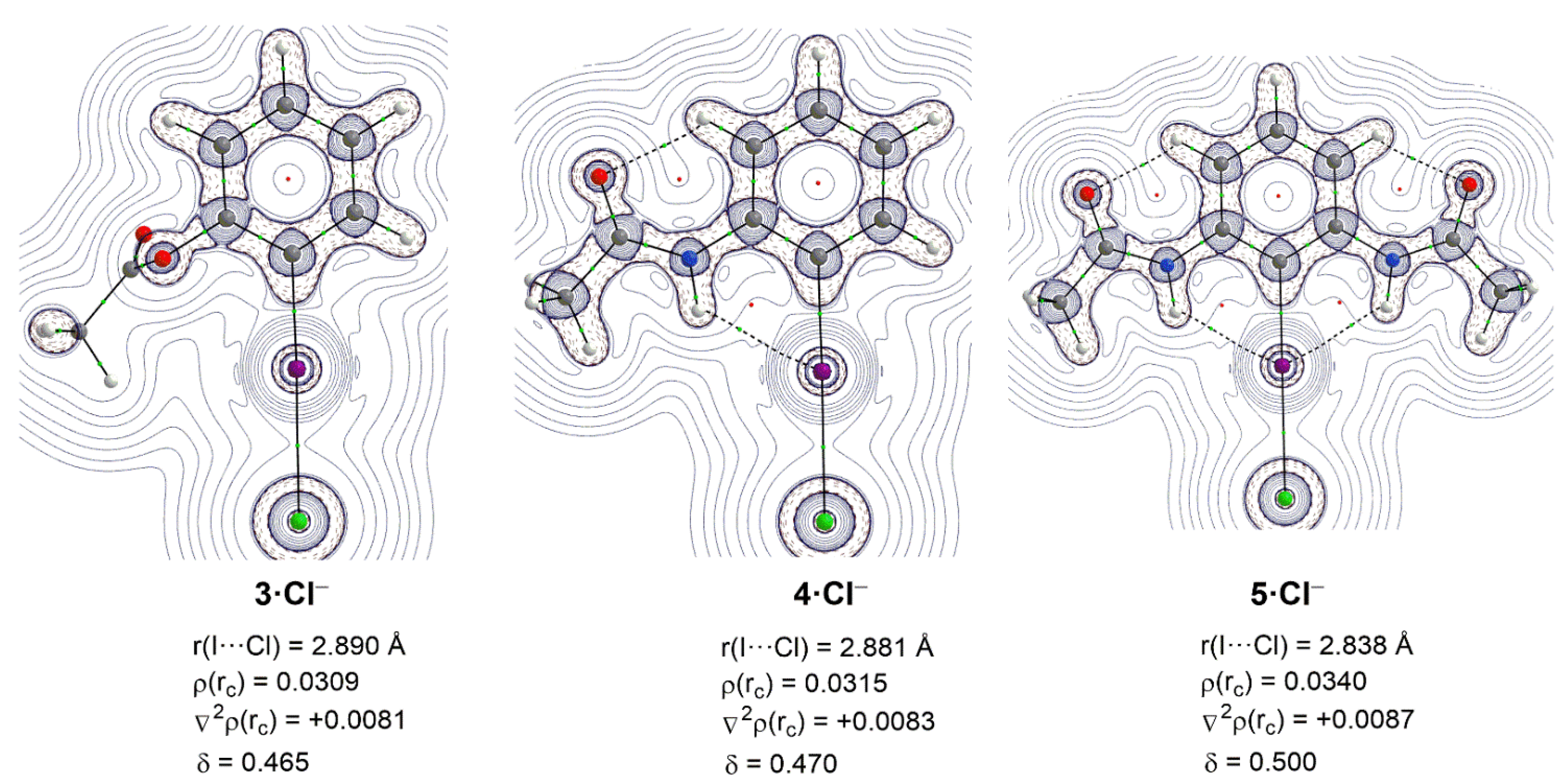
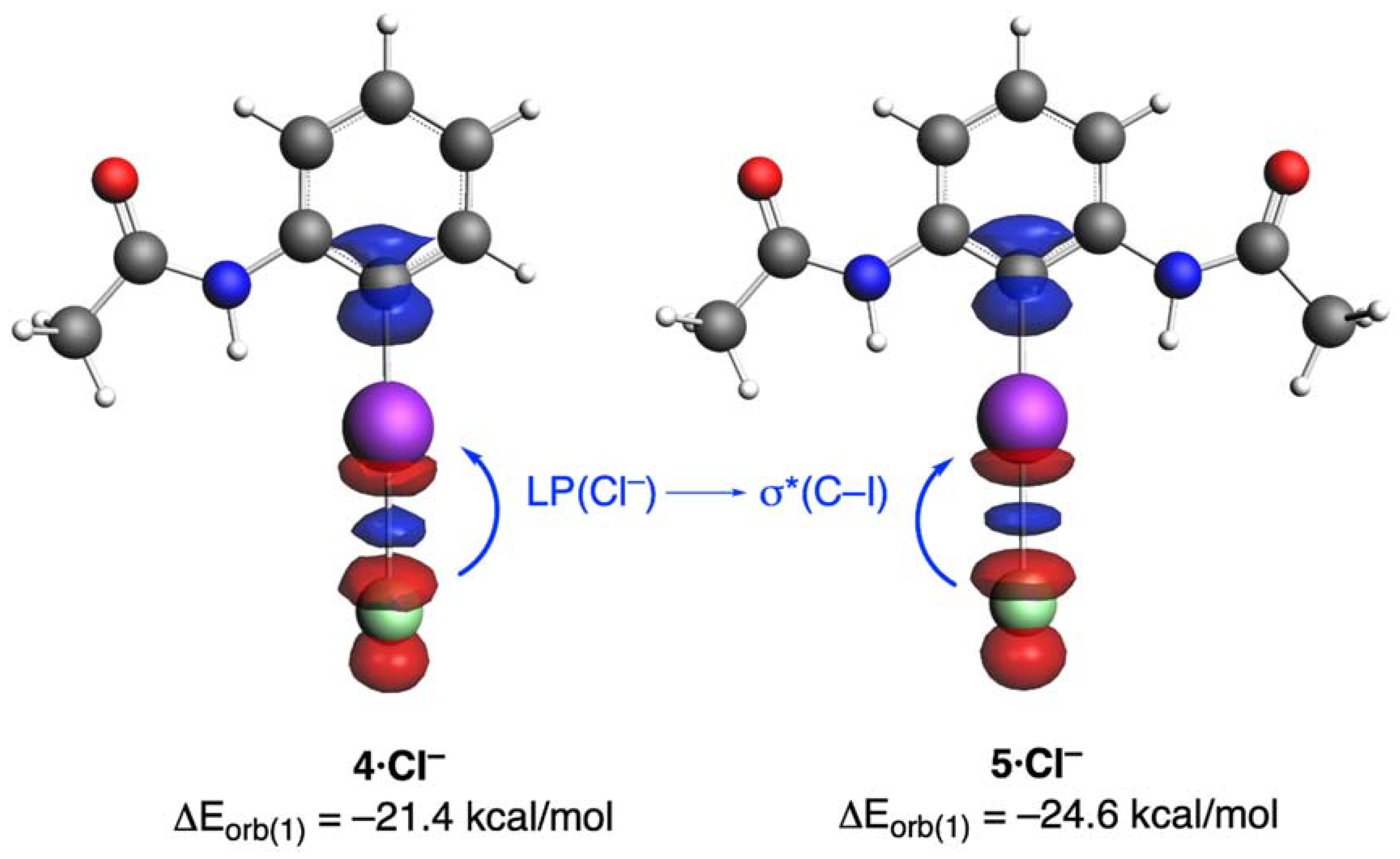
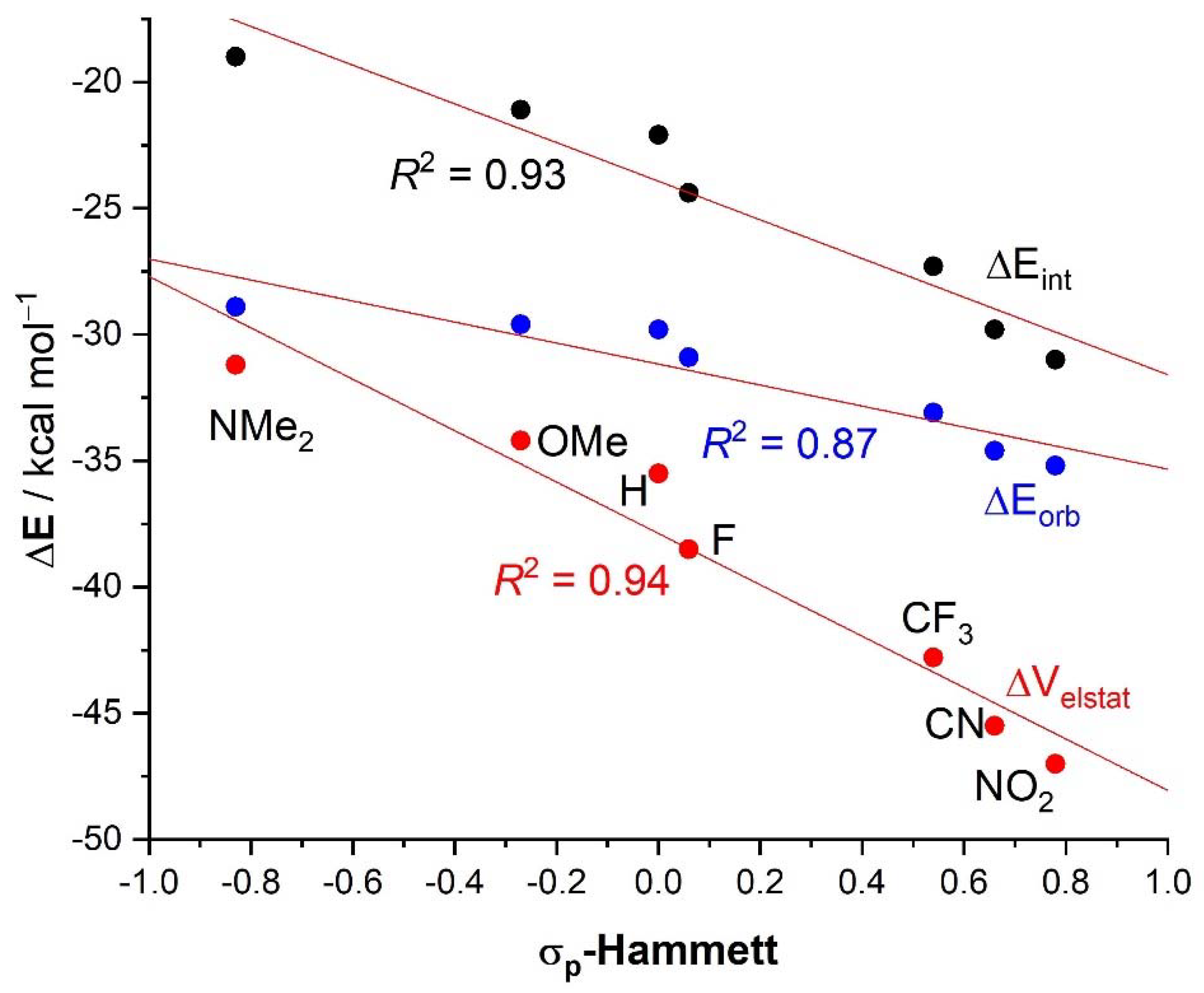
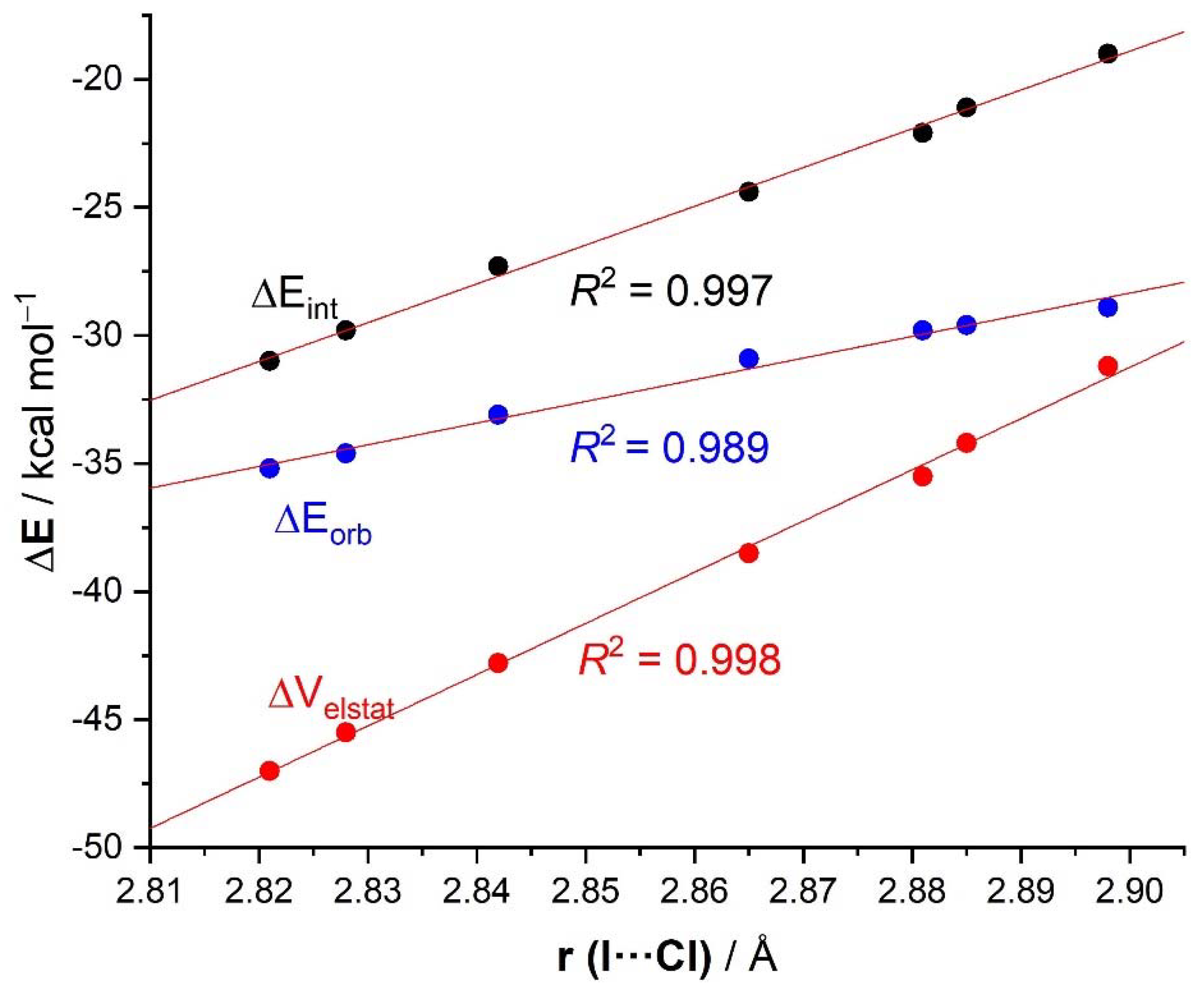
2. Results and Discussion
3. Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hobza, P.; Rezác, J. (Eds.) Noncovalent Interactions. Chem. Rev. 2016, 116, 4911–4912. [Google Scholar] [CrossRef] [PubMed]
- Halogen Bonding I; Metrangol, P., Resnati, G., Eds.; Springer: Berlin/Heidelberg, Germany, 2015. [Google Scholar]
- Halogen Bonding II; Metrangol, P., Resnati, G., Eds.; Springer: Berlin/Heidelberg, Germany, 2015. [Google Scholar]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The Halogen Bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef] [PubMed]
- Desiraju, G.R.; Ho, P.; Kloo, L.; Legon, A.C.; Marquardt, R.; Metrangolo, P.; Politzer, P.; Resnati, G.; Rissanen, K. Definition of the halogen bond (IUPAC Recommendations 2013). Pure Appl. Chem. 2013, 85, 1711–1713. [Google Scholar] [CrossRef]
- Mukherjee, A.; Tothadi, S.; Desiraju, G.R. Halogen Bonds in Crystal Engineering: Like Hydrogen Bonds yet Different. Acc. Chem. Res. 2014, 47, 2514–2524. [Google Scholar] [CrossRef] [PubMed]
- Pancholi, J.; Beer, P.D. Halogen bonding motifs for anion recognition. Coord. Chem. Rev. 2020, 416, 213281. [Google Scholar] [CrossRef]
- Danelius, E.; Andersson, H.; Jarvoll, P.; Lood, K.; Gräfenstein, J.; Erdélyi, M. Halogen Bonding: A Powerful Tool for Modulation of Peptide Conformation. Biochemistry 2017, 56, 3265–3272. [Google Scholar] [CrossRef]
- Sutar, R.L.; Huber, S.M. Catalysis of Organic Reactions through Halogen Bonding. ACS Catal. 2019, 9, 9622–9639. [Google Scholar] [CrossRef]
- Breugst, M.; Koenig, J.J. σ-Hole Interactions in Catalysis. Eur. J. Org. Chem. 2020, 5473–5487. [Google Scholar] [CrossRef]
- Clark, T.; Hennemann, M.; Murray, J.S.; Politzer, P. Halogen bonding: The σ-hole. J. Mol. Model. 2007, 13, 291–296. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S.; Clark, T.; Resnati, G. The σ-hole revisited. Phys. Chem. Chem. Phys. 2017, 19, 32166–32178. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen bonding and other σ-hole interactions: A perspective. Phys. Chem. Chem. Phys. 2013, 15, 11178–11189. [Google Scholar] [CrossRef] [PubMed]
- Decato, D.A.; Riel, A.M.S.; May, J.H.; Bryantsev, V.S.; Berryman, O.B. Theoretical, Solid-State and Solution Quantification of the Hydrogen Bond Enhanced Halogen Bond. Angew. Chem. Int. Ed. 2021, 60, 3685–3692. [Google Scholar] [CrossRef] [PubMed]
- Glendening, E.D.; Landis, C.R.; Weinhold, C.F. NBO 6.0: Natural bond orbital analysis program. J. Comput. Chem. 2013, 34, 1429–1437. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Clarendon Press: Oxford, UK, 1994. [Google Scholar]
- Outeiral, C.; Vincent, M.A.; Martín Pendás, A.; Popelier, P.L.A. Revitalizing the concept of bond order through delocalization measures in real space. Chem. Sci. 2018, 9, 5517–5529. [Google Scholar] [CrossRef]
- von Hopffgarten, M.; Frenking, G. Energy decomposition analysis. WIREs Comput. Mol. Sci. 2012, 2, 43–62. [Google Scholar] [CrossRef]
- Zhao, L.; von Hopffgarten, M.; Andrada, D.M.; Frenking, G. Energy decomposition analysis. WIREs Comput. Mol. Sci. 2018, 8, e1345. [Google Scholar] [CrossRef]
- Wolters, L.P.; Bickelhaupt, F.M. Halogen bonding versus hydrogen bonding: A molecular orbital perspective. Chem. Open 2012, 1, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Kurczab, R.; Mitoraj, M.P.; Michalak, A.; Ziegler, T. Theoretical analysis of the resonance assisted hydrogen bond based on the combined extended transition state method and natural orbitals for chemical valence scheme. J. Phys. Chem. A 2010, 114, 8581–8590. [Google Scholar] [CrossRef]
- Stasyuk, O.A.; Sedlak, R.; Fonseca Guerra, C.; Hobza, P. Comparison of the DFT-SAPT and Canonical EDA Schemes for the Energy Decomposition of Various Types of Noncovalent Interactions. J. Chem. Theory Comput. 2018, 14, 3440–3450. [Google Scholar] [CrossRef]
- Hamlin, T.A.; Fernández, I.; Bickelhaupt, F.M. How Dihalogens Catalyze Michael Addition Reactions. Angew. Chem. Int. Ed. 2019, 58, 8922–8926. [Google Scholar] [CrossRef]
- Kellett, C.W.; Kennepohl, P.; Berlinguette, C.P. π-Covalency in the halogen bond. Nat. Commun. 2020, 11, 3310. [Google Scholar]
- Politzer, P.; Murray, J.S. Electrostatics and Polarization in σ- and π-Hole Noncovalent Interactions: An Overview. ChemPhysChem 2020, 21, 579–588. [Google Scholar] [CrossRef]
- Lv, H.; Zhuo, H.-Y.; Li, Q.-Z.; Yang, X.; Li, W.-Z.; Cheng, J.-B. Halogen bonds with N-heterocyclic carbenes as halogen acceptors: A partially covalent character. Mol. Phys. 2014, 112, 3024–3032. [Google Scholar] [CrossRef]
- Sanyal, S.; Esterhuysen, C. Nature of Halogen Bond Adducts of Carbones with XCF3 (X = Cl, Br, I) species. Polyhedron 2021. [Google Scholar] [CrossRef]
- Mitoraj, M.P.; Michalak, A.; Ziegler, T. A Combined Charge and Energy Decomposition Scheme for Bond Analysis. J. Chem. Theory Comput. 2009, 5, 962–975. [Google Scholar] [CrossRef]
- Mayer, R.J.; Ofial, A.R.; Mayr, H.; Legault, C.Y. Lewis Acidity Scale of Diaryliodonium Ions toward Oxygen, Nitrogen, and Halogen Lewis Bases. J. Am. Chem. Soc. 2020, 142, 5221–5233. [Google Scholar] [CrossRef]
- Reinhard, D.L.; Heinen, F.; Stoesser, J.; Engelage, E.; Huber, S.M. Tuning the Halogen Bonding Strength of Cyclic Diaryliodonium Salts. Helv. Chem. Acta 2021, 104, e2000221. [Google Scholar] [CrossRef]
- Hansch, C.; Leo, A.; Taft, R.W. A survey of Hammett substituent constants and resonance and field parameters. Chem. Rev. 1991, 91, 165–195. [Google Scholar] [CrossRef]
- Wittkopp, A.; Schreiner, P.R. Metal-Free, Noncovalent Catalysis of Diels–Alder Reactions by Neutral Hydrogen Bond Donors in Organic Solvents and in Water. Chem. Eur. J. 2003, 9, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Schreiner, P.R. Metal-free organocatalysis through explicit hydrogen bonding interactions. Chem. Soc. Rev. 2003, 32, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Vermeeren, P.; Hamlin, T.A.; Bickelhaupt, F.M.; Fernández, I. Bifunctional Hydrogen Bond Donor-Catalyzed Diels–Alder Reactions: Origin of Stereoselectivity and Rate Enhancement. Chem. Eur. J. 2021, 27, 5180–5190. [Google Scholar] [CrossRef]
- Jakab, G.; Tancon, C.; Zhang, Z.; Lippert, K.M.; Schreiner, P.R. (Thio)urea Organocatalyst Equilibrium Acidities in DMSO. Org. Lett. 2012, 14, 1724–1727. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A. ; et. al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yan, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: A critical analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Huzinaga, S.; Klobukowski, M. Well-tempered Gaussian basis sets for the calculation of matrix Hartree-Fock wavefunctions. Chem. Phys. Lett. 1993, 212, 260–264. [Google Scholar] [CrossRef]
- Keith, T.A. AIMAll. 2010. Available online: http://tkgristmill.com (accessed on 9 March 2021).
- Shaik, S.S.; Hiberty, P.C. A Chemist’s Guide to Valence Bond. Theory; Wiley: New York, NY, USA, 2007. [Google Scholar]
- te Velde, G.; Bickelhaupt, F.M.; Baerends, E.J.; Fonseca Guerra, C.; van Gisbergen, S.J.A.; Snijders, J.G.; Ziegler, T. Chemistry with ADF. J. Comput. Chem. 2001, 22, 931–967. [Google Scholar] [CrossRef]
- Clark, T.; Murray, J.S.; Politzer, P. A perspective on quantum mechanics and chemical concepts in describing noncovalent interactions. Phys. Chem. Chem. Phys. 2018, 20, 30076–30082. [Google Scholar] [CrossRef] [PubMed]
- ADF, SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands. 2020. Available online: http://www.scm.com (accessed on 9 March 2021).
- Snijders, J.G.; Vernooijs, P.; Baerends, E.J. Roothaan-Hartree-Fock-Slater atomic wave functions: Single-zeta, double-zeta, and extended Slater-type basis sets for 87Fr-103Lr. At. Data Nucl. Data Tables 1981, 26, 483–509. [Google Scholar] [CrossRef]
- Krijn, J.; Baerends, E.J. Fit Functions in the HFS-Method, Internal Report; Vrije Universiteit: Amsterdam, The Netherlands, 1984. (In Dutch) [Google Scholar]
- van Lenthe, E.; Baerends, E.J.; Snijders, J.G. Relativistic regular two-component Hamiltonians. J. Chem. Phys. 1993, 99, 4597–4610. [Google Scholar] [CrossRef]
- van Lenthe, E.; Baerends, E.J.; Snijders, J.G. Relativistic total energy using regular approximations. J. Chem. Phys. 1994, 101, 9783–9792. [Google Scholar] [CrossRef]
- van Lenthe, E.; Ehlers, A.; Baerends, E.J. Geometry optimizations in the zero order regular approximation for relativistic effects. J. Chem. Phys. 1999, 110, 8943–8953. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | 3·Cl− | 4·Cl− | 5·Cl− |
|---|---|---|---|
| ΔEint | −19.0 | −22.1 | −28.8 |
| ΔEPauli | 42.0 | 43.6 | 49.7 |
| ΔVelstat a | −31.8 (52.2%) | −35.5 (54.0%) | −45.1 (57.4%) |
| ΔEorb a | −28.7 (47.0%) | −29.8 (45.4%) | −33.0 (42.0%) |
| ΔEorb(1) b | −20.6 (71.8%) | −21.4 (71.8%) | −24.6 (74.5%) |
| ΔEorb(2) b | −2.4 (8.4%) | −2.3 (7.7%) | −2.3 (7.0%) |
| ΔEorb(rest) b | −5.8 (20.2%) | −6.1 (20.5%) | −8.4 (25.5%) |
| ΔEdisp a | −0.5 (0.8%) | −0.4 (0.6%) | −0.5 (0.6%) |
| Complex |  |  |  | ||||||
|---|---|---|---|---|---|---|---|---|---|
| 4·Cl− | 4−NMe2·Cl− | 4−OMe·Cl− | 4−F·Cl− | 4−CF3·Cl− | 4−CN·Cl− | 4−NO2·Cl− | 1·Cl− | 6·Cl− | |
| ΔEint | −22.1 | −19.0 | −21.1 | −24.4 | −27.3 | −29.8 | −31.0 | −31.1 | −96.2 |
| ΔEPauli | 43.6 | 41.5 | 43.1 | 45.4 | 48.6 | 50.6 | 51.6 | 54.5 | 86.0 |
| ΔVelstat a | −35.5 (54.0%) | −31.2 (51.6%) | −34.2 (53.3%) | −38.5 (55.2%) | −42.8 (56.1%) | −45.5 (56.5%) | −47.0 (56.9%) | −48.1 (56.2%) | −121.0 (66.4%) |
| ΔEorb a | −29.8 (45.4%) | −28.9 (47.8%) | −29.6 (46.1%) | −30.9 (44.3%) | −33.1 (43.4%) | −34.6 (43.0%) | −35.2 (42.6%) | −37.1 (43.3%) | −60.8 (33.4%) |
| ΔEorb(1) b | −21.4 (71.8%) | −20.2 (69.9%) | −21.1 (71.3%) | −22.3 (72.2%) | −23.8 (71.9%) | −24.7 (71.4%) | −25.0 (71.0%) | −27.5 (74.1%) | −46.2 (76.0%) |
| ΔEorb(2) b | −2.3 (7.7%) | −2.5 (8.7%) | −2.3 (7.8%) | −2.3 (7.4%) | −2.6 (7.9%) | −3.0 (8.7%) | −3.4 (9.7%) | −2.4 (6.5%) | −3.6 (5.9%) |
| ΔEorb(rest) b | −6.1 (20.5%) | −6.1 (21.1%) | −6.2 (20.9%) | −6.3 (20.4%) | −6.6 (19.9%) | −6.8 (19.7%) | −6.8 (19.3%) | −7.2 (19.4%) | −11.0 (18.1%) |
| ΔEdisp | −0.4 (0.6%) | −0.4 (0.7%) | −0.4 (0.6%) | −0.4 (0.6%) | −0.4 (0.5%) | −0.4 (0.5%) | −0.4 (0.5%) | −0.4 (0.5%) | −0.4 (0.2%) |
| r(I···Cl)/Å | 2.881 | 2.898 | 2.885 | 2.865 | 2.842 | 2.828 | 2.821 | 2.796 | 2.632 |
 |  |  |  | |
|---|---|---|---|---|
| 4·Cl− | 7·Cl− | 8·Cl− | 9·Cl− | |
| ΔEint | −22.1 | −24.9 | −35.5 | −25.8 |
| ΔEPauli | 43.6 | 47.0 | 56.2 | 48.2 |
| ΔVelstat a | −35.5 (54.0%) | −39.2 (54.5%) | −52.0 (56.8%) | −40.4 (54.7%) |
| ΔEorb a | −29.8 (45.4%) | −32.3 (44.9%) | −39.0 (42.6%) | −33.1 (44.8%) |
| ΔEorb(1) b | −21.4 (71.8%) | −23.1 (71.5%) | −27.8 (71.3%) | −23.6 (71.2%) |
| ΔEorb(2) b | −2.3 (7.7%) | −2.4 (7.4%) | −2.5 (6.4%) | −2.4 (7.3%) |
| ΔEorb(rest) b | −6.1 (20.5%) | −6.8 (21.1%) | −8.7 (22.3%) | −7.1 (21.5%) |
| ΔEdisp | −0.4 (0.6%) | −0.4 (0.6%) | −0.5 (0.5%) | −0.4 (0.5%) |
| r(I···Cl)/Å | 2.881 | 2.855 | 2.796 | 2.847 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Portela, S.; Fernández, I. Nature of the Hydrogen Bond Enhanced Halogen Bond. Molecules 2021, 26, 1885. https://doi.org/10.3390/molecules26071885
Portela S, Fernández I. Nature of the Hydrogen Bond Enhanced Halogen Bond. Molecules. 2021; 26(7):1885. https://doi.org/10.3390/molecules26071885
Chicago/Turabian StylePortela, Susana, and Israel Fernández. 2021. "Nature of the Hydrogen Bond Enhanced Halogen Bond" Molecules 26, no. 7: 1885. https://doi.org/10.3390/molecules26071885
APA StylePortela, S., & Fernández, I. (2021). Nature of the Hydrogen Bond Enhanced Halogen Bond. Molecules, 26(7), 1885. https://doi.org/10.3390/molecules26071885