
Optimized Atomic Partial Charges and Radii Defined by Radical Voronoi Tessellation of Bulk Phase Simulations



Abstract
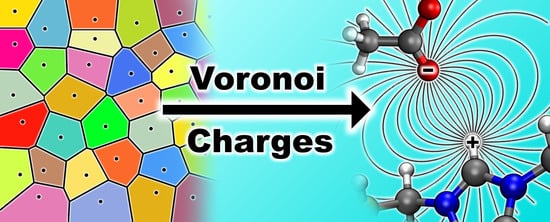
1. Introduction
2. Description of the Method
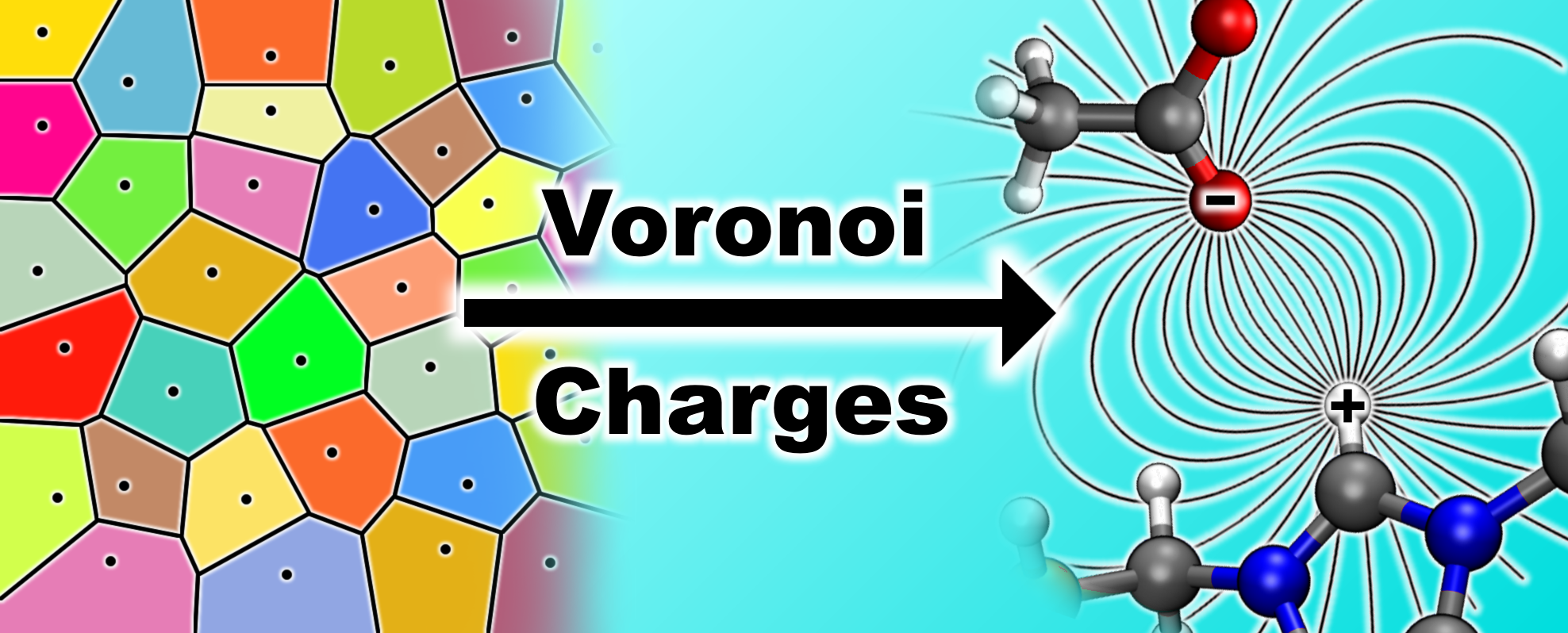
2.1. General Workflow
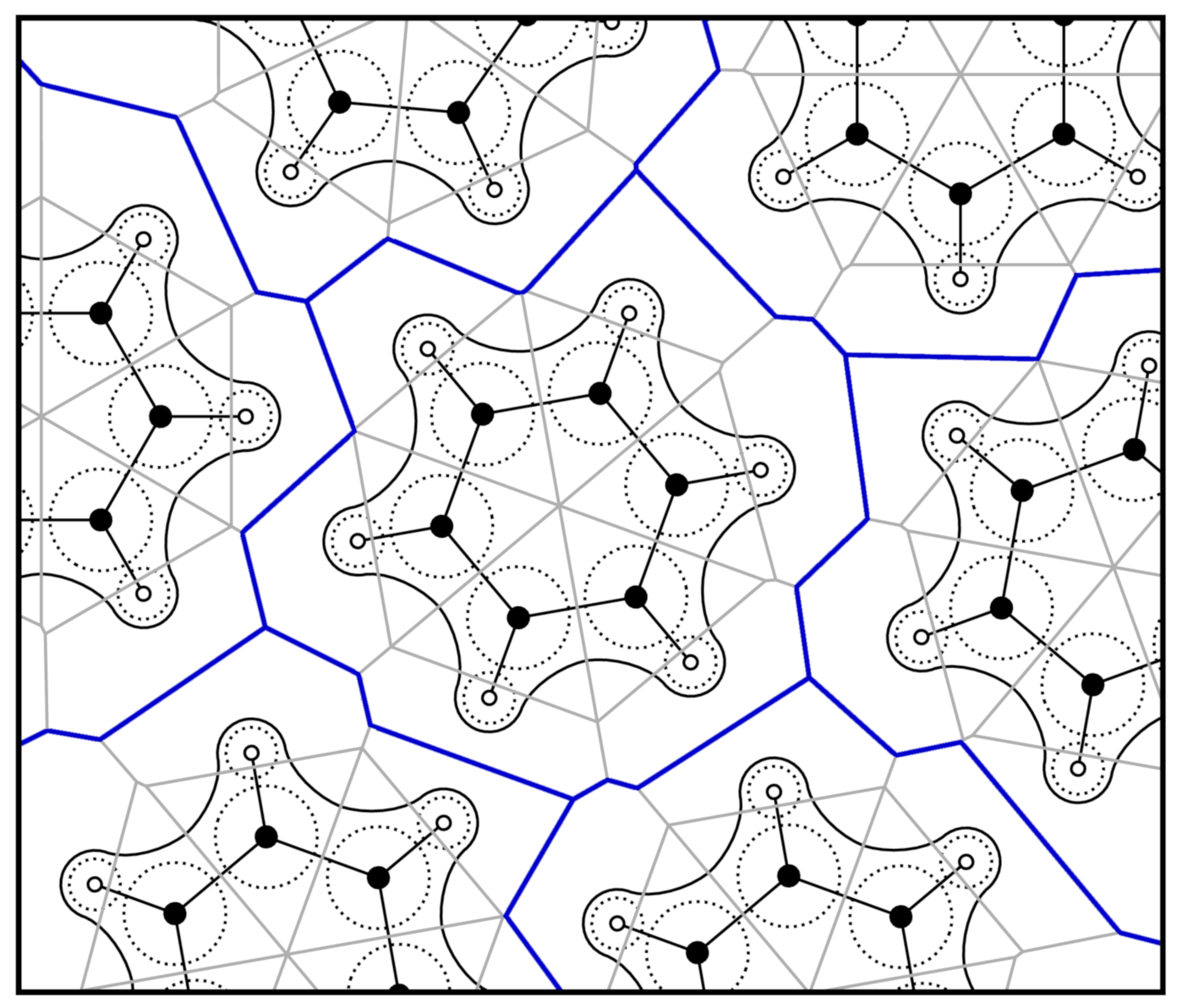
2.2. Voronoi Tessellation and Radical Voronoi Tessellation
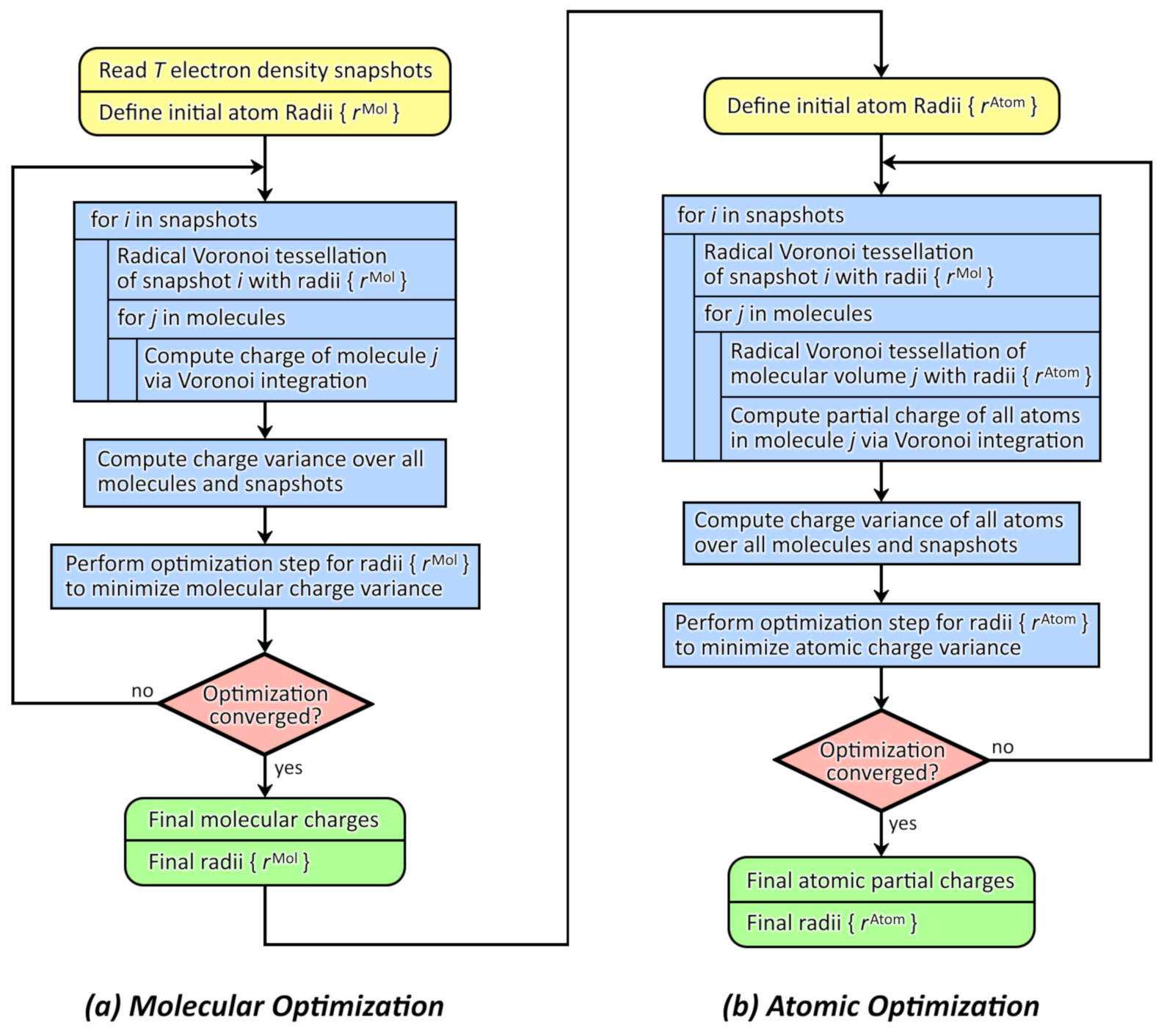
2.3. Integrating over Voronoi Cells
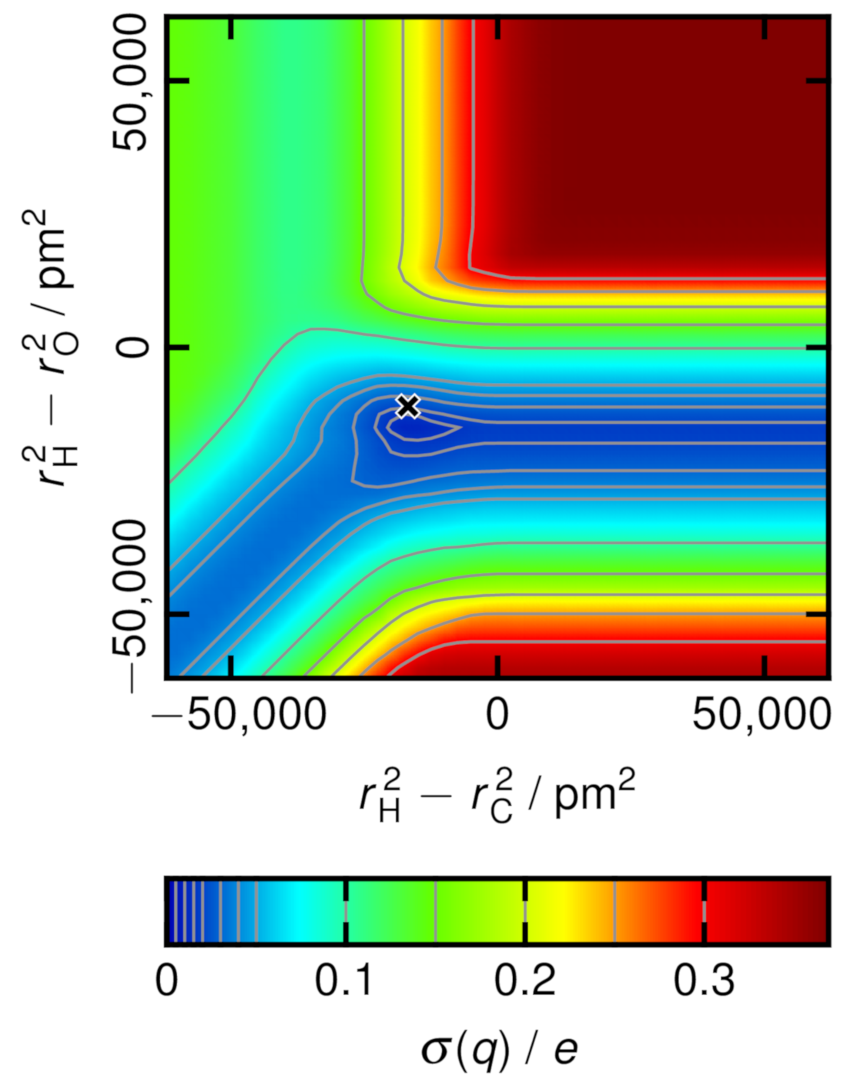
2.4. Charge Variance Minimization Algorithm
3. Results and Discussion
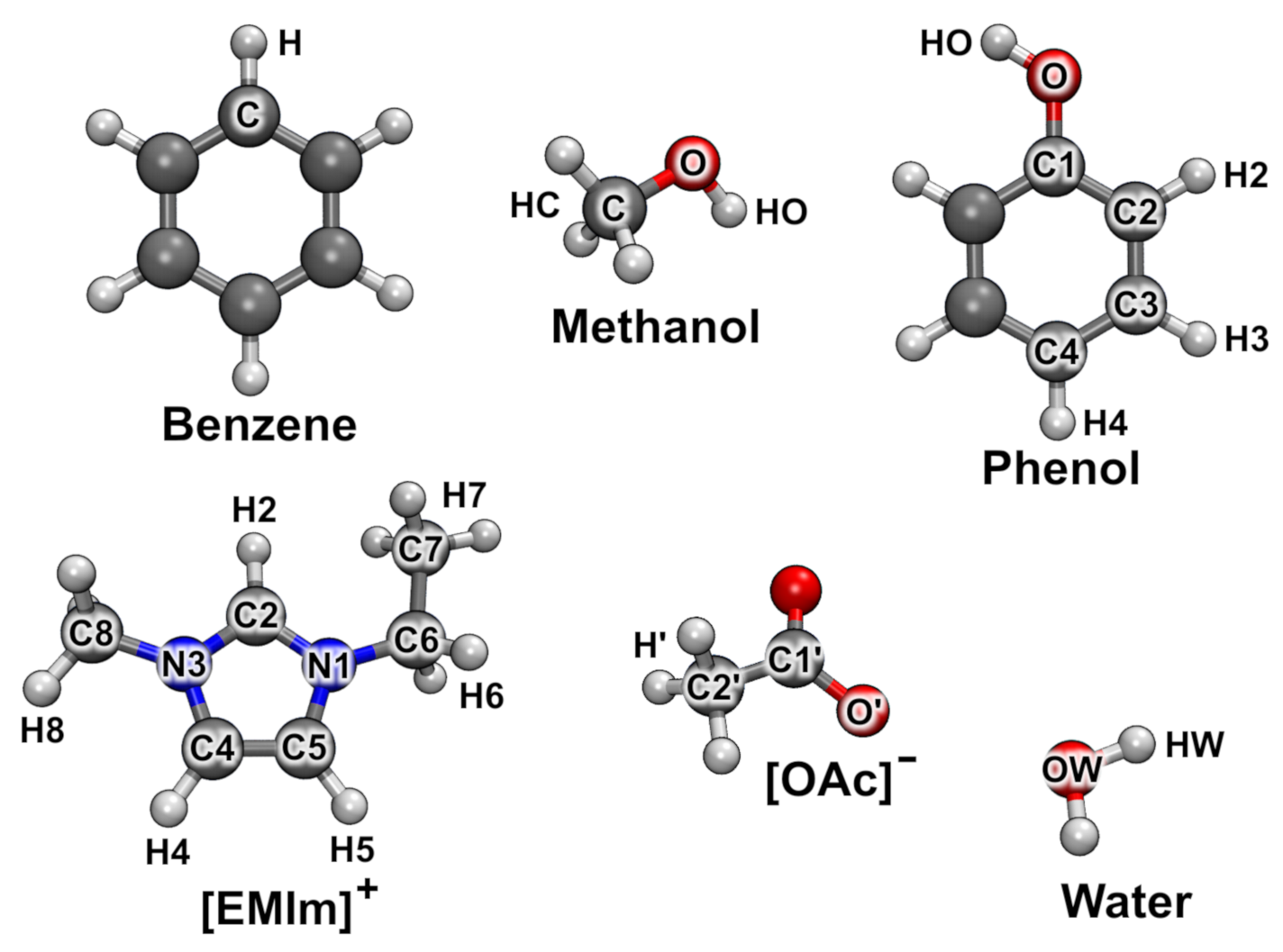
3.1. Molecular Charges
3.2. Atomic Partial Charges and Radii
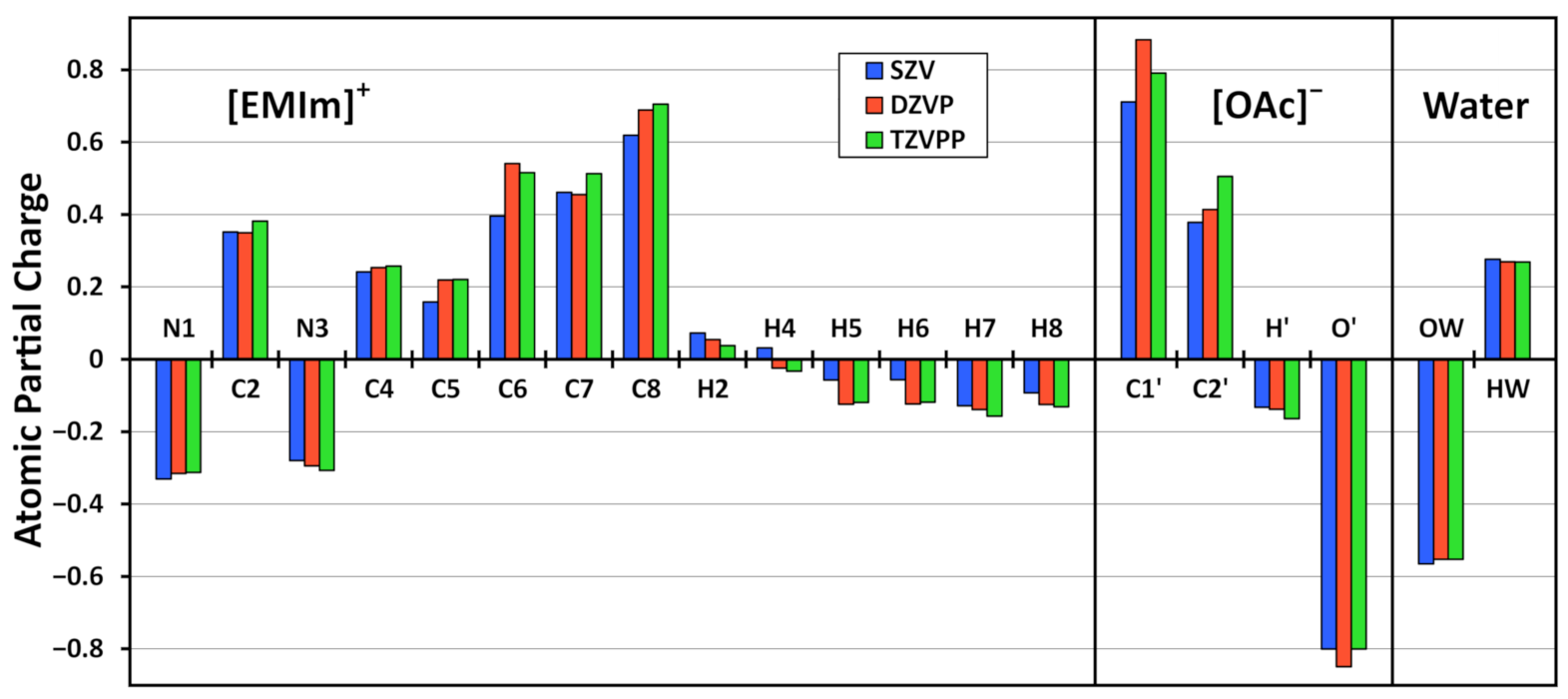
3.3. Basis Set Dependence
4. Computational Details
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| [EMIm] | 1-Ethyl-3-methylimidazolium cation |
| [OAc] | Acetate anion |
| BLYP | Becke–Lee–Yang–Parr density functional |
| bqb | The bqb file format for lossless compression |
| CG | Conjugate gradient optimization method |
| DDAPC | Density derived atomic point charges (Blöchl charges) |
| DFT | Density functional theory |
| DFTB | Density functional based tight binding |
| GTH | Goedecker–Teter–Hutter pseudopotentials |
| IL | Ionic liquid |
| MP2 | Møller–Plesset perturbation theory |
| RESP | Restrained electrostatic potential |
| ROA | Raman optical activity |
| VCD | Vibrational circular dichroism |
References
- Kohagen, M.; Brehm, M.; Thar, J.; Zhao, W.; Müller-Plathe, F.; Kirchner, B. Performance of Quantum Chemically Derived Charges and Persistence of Ion Cages in Ionic Liquids. A Molecular Dynamics Simulations Study of 1-n-Butyl-3-methylimidazolium Bromide. J. Phys. Chem. B 2011, 115, 693–702. [Google Scholar] [CrossRef] [PubMed]
- Kohagen, M.; Brehm, M.; Lingscheid, Y.; Giernoth, R.; Sangoro, J.; Kremer, F.; Naumov, S.; Iacob, C.; Kärger, J.; Valiullin, R.; et al. How Hydrogen Bonds Influence the Mobility of Imidazolium-Based Ionic Liquids. A Combined Theoretical and Experimental Study of 1-n-Butyl-3-methylimidazolium Bromide. J. Phys. Chem. B 2011, 115, 15280–15288. [Google Scholar] [CrossRef] [PubMed]
- Kirchner, B.; Hollóczki, O.; Canongia Lopes, J.N.; Pádua, A.A.H. Multiresolution calculation of ionic liquids. WIREs Comput. Mol. Sci. 2015, 5, 202–214. [Google Scholar] [CrossRef]
- Kristyán, S. Immediate estimation of correlation energy for molecular systems from the partial charges on atoms in the molecule. Chem. Phys. 1997, 224, 33–51. [Google Scholar] [CrossRef]
- Kristyán, S.; Ruzsinszky, A.; Csonka, G. The performance of the rapid estimation of basis set error and correlation energy from partial charges method on new molecules of the G3/99 test set. Theor. Chem. Acc. 2001, 106, 404–411. [Google Scholar] [CrossRef]
- Mulliken, R.S. Criteria for the Construction of Good Self-Consistent-Field Molecular Orbital Wave Functions, and the Significance of LCAO-MO Population Analysis. J. Chem. Phys. 1962, 36, 3428–3439. [Google Scholar] [CrossRef]
- Löwdin, P.O. On the Nonorthogonality Problem. Adv. Quant. Chem. 1970, 5, 185–199. [Google Scholar]
- Mayer, I. Löwdin population analysis is not rotationally invariant. Chem. Phys. Lett. 2004, 393, 209–212. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Ertural, C.; Steinberg, S.; Dronskowski, R. Development of a robust tool to extract Mulliken and Löwdin charges from plane waves and its application to solid-state materials. RSC Adv. 2019, 9, 29821–29830. [Google Scholar] [CrossRef]
- Nelson, R.; Ertural, C.; George, J.; Deringer, V.L.; Hautier, G.; Dronskowski, R. LOBSTER: Local orbital projections, atomic charges, and chemical-bonding analysis from projector-augmented-wave-based density-functional theory. J. Comput. Chem. 2020, 41, 1931–1940. [Google Scholar] [CrossRef]
- Maintz, S.; Deringer, V.L.; Tchougréeff, A.L.; Dronskowski, R. Analytic projection from plane-wave and PAW wavefunctions and application to chemical-bonding analysis in solids. J. Comput. Chem. 2013, 34, 2557–2567. [Google Scholar] [CrossRef]
- Momany, F.A. Determination of partial atomic charges from ab initio molecular electrostatic potentials. Application to formamide, methanol, and formic acid. J. Phys. Chem. 1978, 82, 592–601. [Google Scholar] [CrossRef]
- Cox, S.R.; Williams, D.E. Representation of the molecular electrostatic potential by a net atomic charge model. J. Comput. Chem. 1981, 2, 304–323. [Google Scholar] [CrossRef]
- Singh, U.C.; Kollman, P.A. An approach to computing electrostatic charges for molecules. J. Comput. Chem. 1984, 5, 129–145. [Google Scholar] [CrossRef]
- Chirlian, L.E.; Francl, M.M. Atomic charges derived from electrostatic potentials: A detailed study. J. Comput. Chem. 1987, 8, 894–905. [Google Scholar] [CrossRef]
- Breneman, C.M.; Wiberg, K.B. Determining atom-centered monopoles from molecular electrostatic potentials. The need for high sampling density in formamide conformational analysis. J. Comput. Chem. 1990, 11, 361–373. [Google Scholar] [CrossRef]
- Bayly, C.I.; Cieplak, P.; Cornell, W.; Kollman, P.A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: The RESP model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Laio, A.; VandeVondele, J.; Rothlisberger, U. D-RESP: Dynamically Generated Electrostatic Potential Derived Charges from Quantum Mechanics/Molecular Mechanics Simulations. J. Phys. Chem. B 2002, 106, 7300–7307. [Google Scholar] [CrossRef]
- Kirchner, B.; Hutter, J. Solvent effects on electronic properties from Wannier functions in a dimethyl sulfoxide/water mixture. J. Chem. Phys. 2004, 121, 5133. [Google Scholar] [CrossRef]
- Golze, D.; Hutter, J.; Iannuzzi, M. Wetting of Water on Hexagonal Boron Nitride@Rh(111): A QM/MM Model Based on Atomic Charges Derived for Nano-Structured Substrates. Phys. Chem. Chem. Phys. 2015, 17, 14307–14316. [Google Scholar] [CrossRef]
- Cioslowski, J. A new population analysis based on atomic polar tensors. J. Am. Chem. Soc. 1989, 111, 8333–8336. [Google Scholar] [CrossRef]
- Blöchl, P.E. Electrostatic decoupling of periodic images of plane-wave-expanded densities and derived atomic point charges. J. Chem. Phys. 1995, 103, 7422–7428. [Google Scholar] [CrossRef]
- Gilbert, A.T.B.; Gill, P.M. A point-charge model for electrostatic potentials based on a local projection of multipole moments. Mol. Simul. 2006, 32, 1249–1253. [Google Scholar] [CrossRef]
- Hirshfeld, F. Bonded-atom fragments for describing molecular charge densities. Theor. Chim. Acta 1977, 44, 129–138. [Google Scholar] [CrossRef]
- Bultinck, P.; Van Alsenoy, C.; Ayers, P.W.; Carbó-Dorca, R. Critical analysis and extension of the Hirshfeld atoms in molecules. J. Chem. Phys. 2007, 126, 144111. [Google Scholar] [CrossRef] [PubMed]
- Lillestolen, T.C.; Wheatley, R.J. Redefining the atom: Atomic charge densities produced by an iterative stockholder approach. Chem. Commun. 2008, 5909–5911. [Google Scholar] [CrossRef]
- Manz, T.A.; Sholl, D.S. Improved Atoms-in-Molecule Charge Partitioning Functional for Simultaneously Reproducing the Electrostatic Potential and Chemical States in Periodic and Nonperiodic Materials. J. Chem. Theory Comput. 2012, 8, 2844–2867. [Google Scholar] [CrossRef]
- Geldof, D.; Krishtal, A.; Blockhuys, F.; Van Alsenoy, C. FOHI-D: An iterative Hirshfeld procedure including atomic dipoles. J. Chem. Phys. 2014, 140, 144104. [Google Scholar] [CrossRef]
- Yáñez, M.; Stewart, R.F.; Pople, J.A. The projection of molecular charge density into spherical atoms. I. Density basis functions for first-row atoms. Acta Cryst. 1978, A34, 641–648. [Google Scholar]
- Gill, P.M.W. Extraction of Stewart Atoms from Electron Densities. J. Phys. Chem. 1996, 100, 15421–15427. [Google Scholar] [CrossRef][Green Version]
- Bader, R.F.W. A quantum theory of molecular structure and its applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar] [CrossRef]
- Delle Site, L.; Alavi, A.; Lynden-Bell, R.M. The electrostatic properties of water molecules in condensed phases: An ab initio study. Mol. Phys. 1999, 96, 1683–1693. [Google Scholar] [CrossRef]
- Voronoi, G. Nouvelles applications des paramètres continus à la théorie des formes quadratiques. Deuxième mémoire. Recherches sur les parallélloèdres primitifs. J. Reine Angew. Math. 1908, 134, 198. [Google Scholar] [CrossRef]
- Medvedev, N.N. The algorithm for three-dimensional voronoi polyhedra. J. Comput. Phys. 1986, 67, 223–229. [Google Scholar] [CrossRef]
- Becke, A.D. A multicenter numerical integration scheme for polyatomic molecules. J. Chem. Phys. 1988, 88, 2547–2553. [Google Scholar] [CrossRef]
- Pollak, M.; Rein, R. Molecular-Orbital Studies of Intermolecular Interaction Energies. II. Approximations Concerned with Coulomb Interactions and Comparison of the Two London Schemes. J. Chem. Phys. 1967, 47, 2045–2052. [Google Scholar] [CrossRef]
- Politzer, P.; Harris, R.R. Properties of atoms in molecules. I. Proposed definition of the charge on an atom in a molecule. J. Am. Chem. Soc. 1970, 92, 6451–6454. [Google Scholar] [CrossRef]
- Batista, E.R.; Xantheas, S.S.; Jónsson, H. Multipole moments of water molecules in clusters and ice Ih from first principles calculations. J. Chem. Phys. 1999, 111, 6011–6015. [Google Scholar] [CrossRef]
- Rousseau, B.; Peeters, A.; Alsenoy, C.V. Atomic charges from modified Voronoi polyhedra. J. Mol. Struct. THEOCHEM 2001, 538, 235–238. [Google Scholar] [CrossRef]
- Swart, M.; Van Duijnen, P.T. Atomic radii in molecules for use in a polarizable force field. Int. J. Quantum Chem. 2011, 111, 1763–1772. [Google Scholar] [CrossRef]
- Richards, F.M. The interpretation of protein structures: Total volume, group volume distributions and packing density. J. Mol. Biol. 1974, 82, 1–14. [Google Scholar] [CrossRef]
- Fonseca Guerra, C.; Handgraaf, J.W.; Baerends, E.J.; Bickelhaupt, F.M. Voronoi deformation density (VDD) charges: Assessment of the Mulliken, Bader, Hirshfeld, Weinhold, and VDD methods for charge analysis. J. Comput. Chem. 2004, 25, 189–210. [Google Scholar] [CrossRef]
- Gellatly, B.; Finney, J. Calculation of protein volumes: An alternative to the Voronoi procedure. J. Mol. Biol. 1982, 161, 305–322. [Google Scholar] [CrossRef]
- Thomas, M.; Brehm, M.; Kirchner, B. Voronoi dipole moments for the simulation of bulk phase vibrational spectra. Phys. Chem. Chem. Phys. 2015, 17, 3207–3213. [Google Scholar] [CrossRef]
- Bondi, A. van der Waals Volumes and Radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Rowland, R.S.; Taylor, R. Intermolecular Nonbonded Contact Distances in Organic Crystal Structures: Comparison with Distances Expected from van der Waals Radii. J. Phys. Chem. 1996, 100, 7384–7391. [Google Scholar] [CrossRef]
- Mantina, M.; Chamberlin, A.C.; Valero, R.; Cramer, C.J.; Truhlar, D.G. Consistent van der Waals Radii for the Whole Main Group. J. Phys. Chem. A 2009, 113, 5806–5812. [Google Scholar] [CrossRef]
- Thomas, M.; Brehm, M.; Fligg, R.; Vöhringer, P.; Kirchner, B. Computing vibrational spectra from ab initio molecular dynamics. Phys. Chem. Chem. Phys. 2013, 15, 6608–6622. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Brehm, M.; Hollóczki, O.; Kelemen, Z.; Nyulászi, L.; Pasinszki, T.; Kirchner, B. Simulating the Vibrational Spectra of Ionic Liquid Systems: 1-Ethyl-3-Methylimidazolium Acetate and its Mixtures. J. Chem. Phys. 2014, 141, 024510. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Kirchner, B. Classical Magnetic Dipole Moments for the Simulation of Vibrational Circular Dichroism by ab Initio Molecular Dynamics. J. Phys. Chem. Lett. 2016, 7, 509–513. [Google Scholar] [CrossRef] [PubMed]
- Brehm, M.; Thomas, M. Computing Bulk Phase Raman Optical Activity Spectra from ab initio Molecular Dynamics Simulations. J. Phys. Chem. Lett. 2017, 8, 3409–3414. [Google Scholar] [CrossRef]
- Brehm, M.; Thomas, M. Computing Bulk Phase Resonance Raman Spectra from ab initio Molecular Dynamics and Real-Time TDDFT. J. Chem. Theory Comput. 2019, 15, 3901–3905. [Google Scholar] [CrossRef] [PubMed]
- Elgabarty, H.; Khaliullin, R.Z.; Kühne, T.D. Covalency of hydrogen bonds in liquid water can be probed by proton nuclear magnetic resonance experiments. Nat. Commun. 2015, 6, 8318. [Google Scholar] [CrossRef]
- Zhang, C.; Khaliullin, R.Z.; Bovi, D.; Guidoni, L.; Kühne, T.D. Vibrational Signature of Water Molecules in Asymmetric Hydrogen Bonding Environments. J. Phys. Chem. Lett. 2013, 4, 3245–3250. [Google Scholar] [CrossRef]
- Müller-Plathe, F.; van Gunsteren, W.F. Computer simulation of a polymer electrolyte: Lithium iodide in amorphous poly(ethylene oxide). J. Chem. Phys. 1995, 103, 4745. [Google Scholar] [CrossRef]
- Youngs, T.G.A.; Hardacre, C. Application of Static Charge Transfer within an Ionic-Liquid Force Field and Its Effect on Structure and Dynamics. ChemPhysChem 2008, 9, 1548–1558. [Google Scholar] [CrossRef]
- Rigby, J.; Izgorodina, E.I. Assessment of atomic partial charge schemes for polarisation and charge transfer effects in ionic liquids. Phys. Chem. Chem. Phys. 2013, 15, 1632–1646. [Google Scholar] [CrossRef]
- Hollóczki, O.; Malberg, F.; Welton, T.; Kirchner, B. On the origin of ionicity in ionic liquids. Ion pairing versus charge transfer. Phys. Chem. Chem. Phys. 2014, 16, 16880–16890. [Google Scholar] [CrossRef]
- Bühl, M.; Chaumont, A.; Schurhammer, R.; Wipff, G. Ab Initio Molecular Dynamics of Liquid 1,3-Dimethylimidazolium Chloride. J. Phys. Chem. B 2005, 109, 18591–18599. [Google Scholar] [CrossRef]
- Chaban, V. Polarizability versus mobility: Atomistic force field for ionic liquids. Phys. Chem. Chem. Phys. 2011, 13, 16055–16062. [Google Scholar] [CrossRef] [PubMed]
- Brehm, M.; Pulst, M.; Kressler, J.; Sebastiani, D. Triazolium-Based Ionic Liquids—A Novel Class of Cellulose Solvents. J. Phys. Chem. B 2019, 123, 3994–4003. [Google Scholar] [CrossRef] [PubMed]
- Schröder, C. Comparing reduced partial charge models with polarizable simulations of ionic liquids. Phys. Chem. Chem. Phys. 2012, 14, 3089–3102. [Google Scholar] [CrossRef] [PubMed]
- Morrow, T.I.; Maginn, E.J. Molecular Dynamics Study of the Ionic Liquid 1-n-Butyl-3-methylimidazolium Hexafluorophosphate. J. Phys. Chem. B 2002, 106, 12807–12813. [Google Scholar] [CrossRef]
- Schmidt, J.; Krekeler, C.; Dommert, F.; Zhao, Y.; Berger, R.; Site, L.D.; Holm, C. Ionic Charge Reduction and Atomic Partial Charges from First-Principles Calculations of 1,3-Dimethylimidazolium Chloride. J. Phys. Chem. B 2010, 114, 6150–6155. [Google Scholar] [CrossRef]
- Giernoth, R.; Bröhl, A.; Brehm, M.; Lingscheid, Y. Interactions in Ionic Liquids probed by in situ NMR Spectroscopy. J. Mol. Liq. 2014, 192, 55–58. [Google Scholar] [CrossRef]
- Mondal, A.; Balasubramanian, S. Quantitative Prediction of Physical Properties of Imidazolium Based Room Temperature Ionic Liquids through Determination of Condensed Phase Site Charges: A Refined Force Field. J. Phys. Chem. B 2014, 118, 3409–3422. [Google Scholar] [CrossRef]
- Schulz, A.; Jacob, C.R. Description of intermolecular charge transfer with subsystem density-functional theory. J. Chem. Phys. 2019, 151, 131103. [Google Scholar] [CrossRef]
- Jacob, C.R.; Neugebauer, J. Subsystem density-functional theory. WIREs Comput. Mol. Sci. 2014, 4, 325–362. [Google Scholar] [CrossRef]
- Brehm, M.; Thomas, M.; Gehrke, S.; Kirchner, B. TRAVIS—A free analyzer for trajectories from molecular simulation. J. Chem. Phys. 2020, 152, 164105. [Google Scholar] [CrossRef]
- Brehm, M.; Kirchner, B. TRAVIS - A free Analyzer and Visualizer for Monte Carlo and Molecular Dynamics Trajectories. J. Chem. Inf. Model. 2011, 51, 2007–2023. [Google Scholar] [CrossRef]
- Rycroft, C.H.; Grest, G.S.; Landry, J.W.; Bazant, M.Z. Analysis of Granular Flow in a Pebble-Bed Nuclear Reactor. Phys. Rev. E 2006, 74, 021306. [Google Scholar] [CrossRef]
- Rycroft, C.H. Voro++: A Three-Dimensional Voronoi Cell Library in C++. Chaos 2009, 19, 041111. [Google Scholar] [CrossRef]
- Fletcher, R.; Reeves, C. Function minimization by conjugate gradients. Comput. J. 1964, 7, 149–154. [Google Scholar] [CrossRef]
- Kiefer, J. Sequential minimax search for a maximum. Proc. Am. Math. Soc. 1953, 4, 502–506. [Google Scholar] [CrossRef]
- Avriel, M.; Wilde, D.J. Optimality proof for the symmetric Fibonacci search technique. Fibonacci Q. 1966, 4, 265–269. [Google Scholar]
- Polak, E.; Ribiére, G. Note sur la convergence de directions conjugées. Rev. Fr. Informat Rech. Opertionelle 1969, 3e, 35–43. [Google Scholar]
- Polyak, B.T. The conjugate gradient method in extreme problems. USSR Comp. Math. Math. Phys. 1969, 9, 94–112. [Google Scholar] [CrossRef]
- Cordero, B.; Gómez, V.; Platero-Prats, A.E.; Revés, M.; Echeverría, J.; Cremades, E.; Barragán, F.; Alvarez, S. Covalent Radii Revisited. Dalton Trans. 2008, 21, 2832–2838. [Google Scholar] [CrossRef]
- Brehm, M.; Weber, H.; Pensado, A.; Stark, A.; Kirchner, B. Proton Transfer and Polarity Changes in Ionic Liquid-Water Mixtures: A Perspective on Hydrogen Bonds from ab initio Molecular Dynamics at the Example of 1-Ethyl-3-Methylimidazolium Acetate-Water Mixtures—Part 1. Phys. Chem. Chem. Phys. 2012, 14, 5030–5044. [Google Scholar] [CrossRef]
- Brehm, M.; Weber, H.; Pensado, A.; Stark, A.; Kirchner, B. Liquid Structure and Cluster Formation in Ionic Liquid/Water Mixtures—An Extensive ab initio Molecular Dynamics Study on 1-Ethyl-3-Methylimidazolium Acetate/Water Mixtures—Part 2. Z. Phys. Chem. 2013, 227, 177–203. [Google Scholar] [CrossRef]
- Brehm, M.; Sebastiani, D. Simulating Structure and Dynamics in Small Droplets of 1-Ethyl-3-Methylimidazolium Acetate. J. Chem. Phys. 2018, 148, 193802. [Google Scholar] [CrossRef]
- Malberg, F.; Brehm, M.; Hollóczki, O.; Pensado, A.S.; Kirchner, B. Understanding the Evaporation of Ionic Liquids using the Example of 1-Ethyl-3-Methylimidazolium Ethylsulfate. Phys. Chem. Chem. Phys. 2013, 15, 18424–18436. [Google Scholar] [CrossRef]
- Stark, A.; Brehm, M.; Brüssel, M.; Lehmann, S.B.C.; Pensado, A.S.; Schöppke, M.; Kirchner, B. A Theoretical and Experimental Chemist’s Joint View on Hydrogen Bonding in Ionic Liquids and Their Binary Mixtures. Top. Curr. Chem. 2014, 351, 149–187. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Grigera, J.R.; Straatsma, T.P. The Missing Term in Effective Pair Potentials. J. Phys. Chem. 1987, 91, 6269–6271. [Google Scholar] [CrossRef]
- VandeVondele, J.; Hutter, J. Gaussian basis sets for accurate calculations on molecular systems in gas and condensed phases. J. Chem. Phys. 2007, 127, 114105. [Google Scholar] [CrossRef]
- CP2k Developers Group. CP2k: Open Source Molecular Dynamics. Available online: http://www.cp2k.org (accessed on 18 October 2020).
- Hutter, J.; Iannuzzi, M.; Schiffmann, F.; VandeVondele, J. cp2k: Atomistic simulations of condensed matter systems. WIREs Comput. Mol. Sci. 2014, 4, 15–25. [Google Scholar] [CrossRef]
- Kühne, T.D.; Iannuzzi, M.; Ben, M.; Rybkin, V.V.; Seewald, P.; Stein, F.; Laino, T.; Khaliullin, R.Z.; Schütt, O.; Schiffmann, F.; et al. CP2K: An Electronic Structure and Molecular Dynamics Software Package-Quickstep: Efficient and Accurate Electronic Structure Calculations. J. Chem. Phys. 2020, 152, 194103. [Google Scholar] [CrossRef]
- VandeVondele, J.; Krack, M.; Mohamed, F.; Parrinello, M.; Chassaing, T.; Hutter, J. Quickstep: Fast and accurate density functional calculations using a mixed Gaussian and plane waves approach. Comput. Phys. Commun. 2005, 167, 103–128. [Google Scholar] [CrossRef]
- VandeVondele, J.; Hutter, J. An efficient orbital transformation method for electronic structure calculations. J. Chem. Phys. 2003, 118, 4365–4369. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. B 1964, 136, 864. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, 1133. [Google Scholar] [CrossRef]
- Becke, A. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Smith, D.G.A.; Burns, L.A.; Patkowski, K.; Sherrill, C.D. Revised Damping Parameters for the D3 Dispersion Correction to Density Functional Theory. J. Phys. Chem. Lett. 2016, 7, 2197–2203. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, S. A Consistent and Accurate ab initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Goedecker, S.; Teter, M.; Hutter, J. Separable dual-space Gaussian pseudopotentials. Phys. Rev. B 1996, 54, 1703–1710. [Google Scholar] [CrossRef]
- Hartwigsen, C.; Goedecker, S.; Hutter, J. Relativistic separable dual-space Gaussian pseudopotentials from H to Rn. Phys. Rev. B 1998, 58, 3641–3662. [Google Scholar] [CrossRef]
- Krack, M. Pseudopotentials for H to Kr optimized for gradient-corrected exchange-correlation functionals. Theor. Chem. Acc. 2005, 114, 145–152. [Google Scholar] [CrossRef]
- Nose, S. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 1984, 81, 511–519. [Google Scholar] [CrossRef]
- Nose, S. A molecular dynamics method for simulations in the canonical ensemble. Mol. Phys. 1984, 52, 255–268. [Google Scholar] [CrossRef]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef] [PubMed]
- Martyna, G.; Klein, M.; Tuckerman, M. Nosé-Hoover chains: The canonical ensemble via continuous dynamics. J. Chem. Phys. 1992, 97, 2635–2643. [Google Scholar] [CrossRef]
- Martínez, L.; Andrade, R.; Birgin, E.G.; Martínez, J.M. Packmol: A package for building initial configurations for molecular dynamics simulations. J. Comput. Chem. 2009, 30, 2157–2164. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Plimpton, S. Fast Parallel Algorithms for Short-Range Molecular Dynamics. J. Comp. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef]
- Brehm, M.; Thomas, M. An Efficient Lossless Compression Algorithm for Trajectories of Atom Positions and Volumetric Data. J. Chem. Inf. Model. 2018, 58, 2092–2107. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graphics 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Stone, J. An Efficient Library for Parallel Ray Tracing and Animation. Master’s Thesis, Computer Science Department, University of Missouri-Rolla, Rolla, MO, USA, 1998. [Google Scholar]
- Williams, T.; Kelley, C.; Bröker, H.B.; Campbell, J.; Cunningham, R.; Denholm, D.; Elber, G.; Fearick, R.; Grammes, C.; Zellner, J.; et al. Gnuplot 4.6: An Interactive Plotting Program. 2013. Available online: http://gnuplot.sourceforge.net/ (accessed on 18 October 2020).
- Ligneres, V.L.; Carter, E.A. An Introduction to Orbital Free Density Functional Theory. In Handbook of Materials Modeling; Yip, S., Ed.; Springer: Dordrecht, The Netherlands, 2005; pp. 137–148. [Google Scholar]
- Elstner, M.; Porezag, D.; Jungnickel, G.; Elsner, J.; Haugk, M.; Frauenheim, T.; Suhai, S.; Seifert, G. Self-consistent-charge density-functional tight-binding method for simulations of complex materials properties. Phys. Rev. B 1998, 58, 7260–7268. [Google Scholar] [CrossRef]
- Yang, Y.; Yu, H.; York, D.; Cui, Q.; Elstner, M. Extension of the Self-Consistent-Charge Density-Functional Tight-Binding Method: Third-Order Expansion of the Density Functional Theory Total Energy and Introduction of a Modified Effective Coulomb Interaction. J. Phys. Chem. A 2007, 111, 10861–10873. [Google Scholar] [CrossRef]
- Liu, X.; Seiffert, L.; Fennel, T.; Kühn, O. A DFT-based tight-binding approach to the self-consistent description of molecule metal-nanoparticle interactions. J. Phys. B 2019, 52, 185101. [Google Scholar] [CrossRef]
- Bannwarth, C.; Ehlert, S.; Grimme, S. GFN2-xTB—An Accurate and Broadly Parametrized Self-Consistent Tight-Binding Quantum Chemical Method with Multipole Electrostatics and Density-Dependent Dispersion Contributions. J. Chem. Theor. Comput. 2019, 15, 1652–1671. [Google Scholar] [CrossRef]
- Bannwarth, C.; Caldeweyher, E.; Ehlert, S.; Hansen, A.; Pracht, P.; Seibert, J.; Spicher, S.; Grimme, S. Extended tight-binding quantum chemistry methods. WIREs Comput. Mol. Sci. 2021, 11, e1493. [Google Scholar] [CrossRef]
- Møller, C.; Plesset, M.S. Note on an Approximation Treatment for Many-Electron Systems. Phys. Rev. 1934, 46, 618–622. [Google Scholar] [CrossRef]
- Head-Gordon, M.; Pople, J.A.; Frisch, M.J. MP2 energy evaluation by direct methods. Chem. Phys. Lett. 1988, 153, 503–506. [Google Scholar] [CrossRef]
- Kümmel, H.G. A biography of the coupled cluster method. In Recent Progress in Many-Body Theories; Bishop, R.F., Brandes, T., Gernoth, K.A., Walet, N.R., Xian, Y., Eds.; World Scientific Publishing: Singapore, 2002; pp. 334–348. [Google Scholar]
- Saitow, M.; Becker, U.; Riplinger, C.; Valeev, E.F.; Neese, F. A new near-linear scaling, efficient and accurate, open-shell domain-based local pair natural orbital coupled cluster singles and doubles theory. J. Chem. Phys. 2017, 146, 164105. [Google Scholar] [CrossRef]
- Liakos, D.G.; Guo, Y.; Neese, F. Comprehensive Benchmark Results for the Domain Based Local Pair Natural Orbital Coupled Cluster Method (DLPNO-CCSD(T)) for Closed- and Open-Shell Systems. J. Phys. Chem. A 2020, 124, 90–100. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | Composition | Cell/pm | Density/g cm | Temp./K | Grid Resolution |
|---|---|---|---|---|---|
| Benzene | 32 benzene | 1690 | 0.860 | 350 | |
| Methanol | 48 methanol | 1515 | 0.735 | 350 | |
| Phenol | 32 phenol | 1688 | 1.040 | 400 | |
| IL | 36 [EMIm]+ 36 [OAc]− | 2121 | 1.066 | 350 | |
| ILW | 27 [EMIm]+ 27 [OAc]− 81 H2O | 2158 | 1.000 | 350 |
| Molecule | Charge | Std. Dev. |
|---|---|---|
| Benzene | 0 | 0.010 |
| Methanol | 0 | 0.013 |
| Phenol | 0 | 0.012 |
| IL | ||
| [EMIm] | 0.846 | 0.016 |
| [OAc] | −0.846 | 0.011 |
| ILW | ||
| [EMIm] | 0.859 | 0.017 |
| [OAc] | −0.817 | 0.015 |
| Water | −0.014 | 0.015 |
| Atom | Charge | Std. Dev. | ||
|---|---|---|---|---|
| Benzene | ||||
| C | 0.141 | 0.007 | 173.7 | 72.9 |
| H | −0.141 | 0.010 | 107.6 | 41.8 |
| Methanol | ||||
| C | 0.764 | 0.008 | 172.5 | 76.4 |
| HC | −0.169 | 0.010 | 112.9 | 51.7 |
| O | −0.609 | 0.023 | 152.9 | 81.1 |
| HO | 0.350 | 0.026 | 104.0 | 28.8 |
| Phenol | ||||
| C1 | 0.248 | 0.014 | 170.1 | 73.8 |
| C2 | 0.084 | 0.013 | 172.8 | 73.9 |
| C3 | 0.154 | 0.010 | 171.6 | 72.9 |
| C4 | 0.122 | 0.012 | 173.2 | 72.8 |
| H2 | −0.115 | 0.011 | 107.9 | 41.3 |
| H3 | −0.134 | 0.011 | 111.5 | 41.5 |
| H4 | −0.130 | 0.011 | 111.4 | 40.4 |
| O | −0.447 | 0.025 | 152.9 | 78.0 |
| HO | 0.229 | 0.024 | 104.3 | 39.2 |
| Atom | IL | ILW | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Charge | Std. Dev. | Charge | Std. Dev. | ||||||
| [EMIm] | |||||||||
| N1 | −0.306 | 0.011 | 167.3 | 82.7 | −0.316 | 0.011 | 162.2 | 80.8 | |
| C2 | 0.270 | 0.015 | 165.2 | 79.5 | 0.350 | 0.015 | 160.1 | 76.9 | |
| N3 | −0.223 | 0.013 | 163.7 | 80.3 | −0.294 | 0.011 | 168.3 | 80.1 | |
| C4 | 0.149 | 0.014 | 169.1 | 78.0 | 0.254 | 0.014 | 168.1 | 74.8 | |
| C5 | 0.148 | 0.013 | 169.1 | 78.7 | 0.219 | 0.013 | 171.9 | 75.7 | |
| C6 | 0.468 | 0.009 | 172.3 | 73.5 | 0.541 | 0.008 | 170.3 | 71.7 | |
| C7 | 0.518 | 0.007 | 171.4 | 71.4 | 0.455 | 0.007 | 178.0 | 71.9 | |
| C8 | 0.655 | 0.010 | 165.2 | 72.0 | 0.688 | 0.008 | 163.5 | 71.4 | |
| H2 | 0.092 | 0.025 | 107.3 | 37.3 | 0.055 | 0.022 | 108.0 | 36.3 | |
| H4 | 0.041 | 0.022 | 106.9 | 38.2 | −0.025 | 0.018 | 105.1 | 40.8 | |
| H5 | −0.093 | 0.019 | 107.3 | 39.8 | −0.124 | 0.015 | 109.8 | 40.8 | |
| H6 | −0.092 | 0.020 | 107.3 | 39.8 | −0.123 | 0.015 | 109.8 | 40.8 | |
| H7 | −0.156 | 0.015 | 109.6 | 41.6 | −0.139 | 0.014 | 105.9 | 40.2 | |
| H8 | −0.115 | 0.019 | 108.9 | 41.7 | −0.125 | 0.014 | 110.1 | 42.1 | |
| [OAc] | |||||||||
| C1’ | 0.761 | 0.015 | 162.4 | 73.4 | 0.882 | 0.018 | 165.2 | 72.7 | |
| C2’ | 0.482 | 0.008 | 175.2 | 71.7 | 0.414 | 0.008 | 175.2 | 73.5 | |
| H’ | −0.162 | 0.011 | 109.8 | 41.1 | −0.138 | 0.011 | 109.1 | 41.8 | |
| O’ | −0.801 | 0.020 | 155.5 | 83.1 | −0.849 | 0.024 | 155.6 | 85.0 | |
| Water | |||||||||
| OW | −0.553 | 0.024 | 155.8 | 79.2 | |||||
| HW | 0.269 | 0.025 | 108.1 | 36.0 | |||||
| Atom | SZV | DZVP | TZVPP | |||
|---|---|---|---|---|---|---|
| Charge | Std. Dev. | Charge | Std. Dev. | Charge | Std. Dev. | |
| [EMIm] | 0.948 | 0.015 | 0.859 | 0.017 | 0.841 | 0.018 |
| N1 | −0.331 | 0.010 | −0.316 | 0.011 | −0.313 | 0.011 |
| C2 | 0.352 | 0.015 | 0.350 | 0.015 | 0.382 | 0.015 |
| N3 | −0.280 | 0.011 | −0.294 | 0.011 | −0.307 | 0.011 |
| C4 | 0.241 | 0.014 | 0.254 | 0.014 | 0.257 | 0.014 |
| C5 | 0.158 | 0.014 | 0.219 | 0.013 | 0.220 | 0.013 |
| C6 | 0.395 | 0.009 | 0.541 | 0.008 | 0.515 | 0.008 |
| C7 | 0.461 | 0.008 | 0.455 | 0.007 | 0.513 | 0.007 |
| C8 | 0.619 | 0.009 | 0.688 | 0.008 | 0.705 | 0.008 |
| H2 | 0.072 | 0.022 | 0.055 | 0.022 | 0.038 | 0.021 |
| H4 | 0.031 | 0.019 | −0.025 | 0.018 | −0.033 | 0.018 |
| H5 | −0.057 | 0.014 | −0.124 | 0.015 | −0.119 | 0.015 |
| H6 | −0.057 | 0.014 | −0.123 | 0.015 | −0.118 | 0.015 |
| H7 | −0.128 | 0.013 | −0.139 | 0.014 | −0.157 | 0.014 |
| H8 | −0.093 | 0.013 | −0.125 | 0.014 | −0.131 | 0.014 |
| [OAc] | −0.909 | 0.016 | −0.817 | 0.015 | −0.797 | 0.015 |
| C1’ | 0.711 | 0.018 | 0.882 | 0.018 | 0.791 | 0.015 |
| C2’ | 0.378 | 0.009 | 0.414 | 0.008 | 0.506 | 0.008 |
| H’ | −0.132 | 0.010 | −0.138 | 0.011 | −0.164 | 0.011 |
| O’ | −0.800 | 0.028 | −0.849 | 0.024 | −0.801 | 0.024 |
| Water | −0.013 | 0.014 | −0.014 | 0.015 | −0.015 | 0.015 |
| OW | −0.565 | 0.027 | −0.553 | 0.024 | −0.553 | 0.023 |
| HW | 0.276 | 0.029 | 0.269 | 0.025 | 0.269 | 0.025 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brehm, M.; Thomas, M. Optimized Atomic Partial Charges and Radii Defined by Radical Voronoi Tessellation of Bulk Phase Simulations. Molecules 2021, 26, 1875. https://doi.org/10.3390/molecules26071875
Brehm M, Thomas M. Optimized Atomic Partial Charges and Radii Defined by Radical Voronoi Tessellation of Bulk Phase Simulations. Molecules. 2021; 26(7):1875. https://doi.org/10.3390/molecules26071875
Chicago/Turabian StyleBrehm, Martin, and Martin Thomas. 2021. "Optimized Atomic Partial Charges and Radii Defined by Radical Voronoi Tessellation of Bulk Phase Simulations" Molecules 26, no. 7: 1875. https://doi.org/10.3390/molecules26071875
APA StyleBrehm, M., & Thomas, M. (2021). Optimized Atomic Partial Charges and Radii Defined by Radical Voronoi Tessellation of Bulk Phase Simulations. Molecules, 26(7), 1875. https://doi.org/10.3390/molecules26071875