
Design, Synthesis, Molecular Modeling and Antitumor Evaluation of Novel Indolyl-Pyrimidine Derivatives with EGFR Inhibitory Activity



Abstract
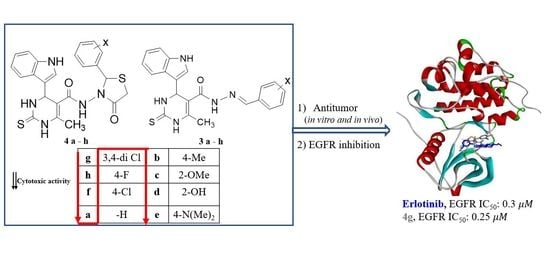
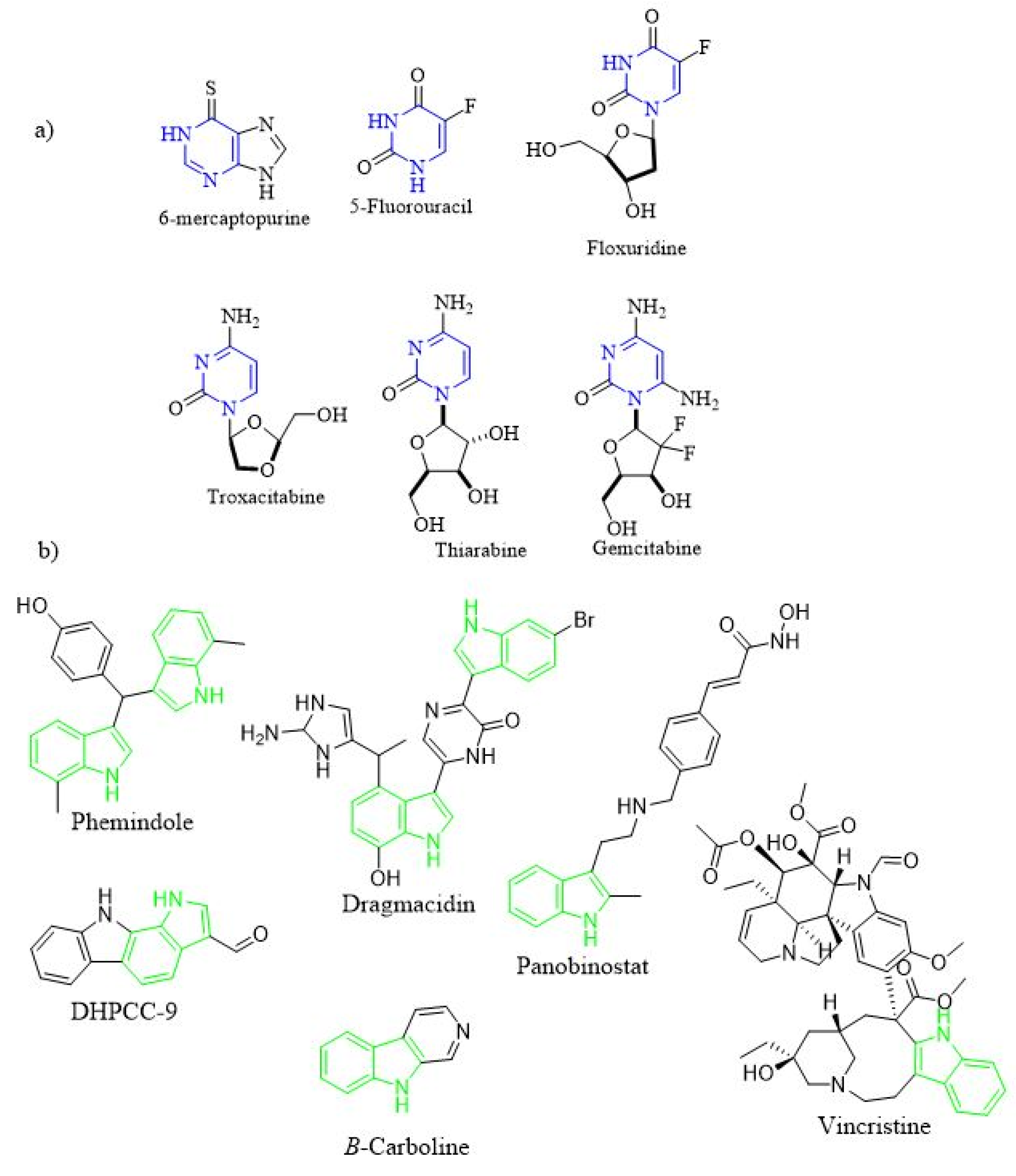
1. Introduction
2. Results and Discussion
2.1. Chemistry
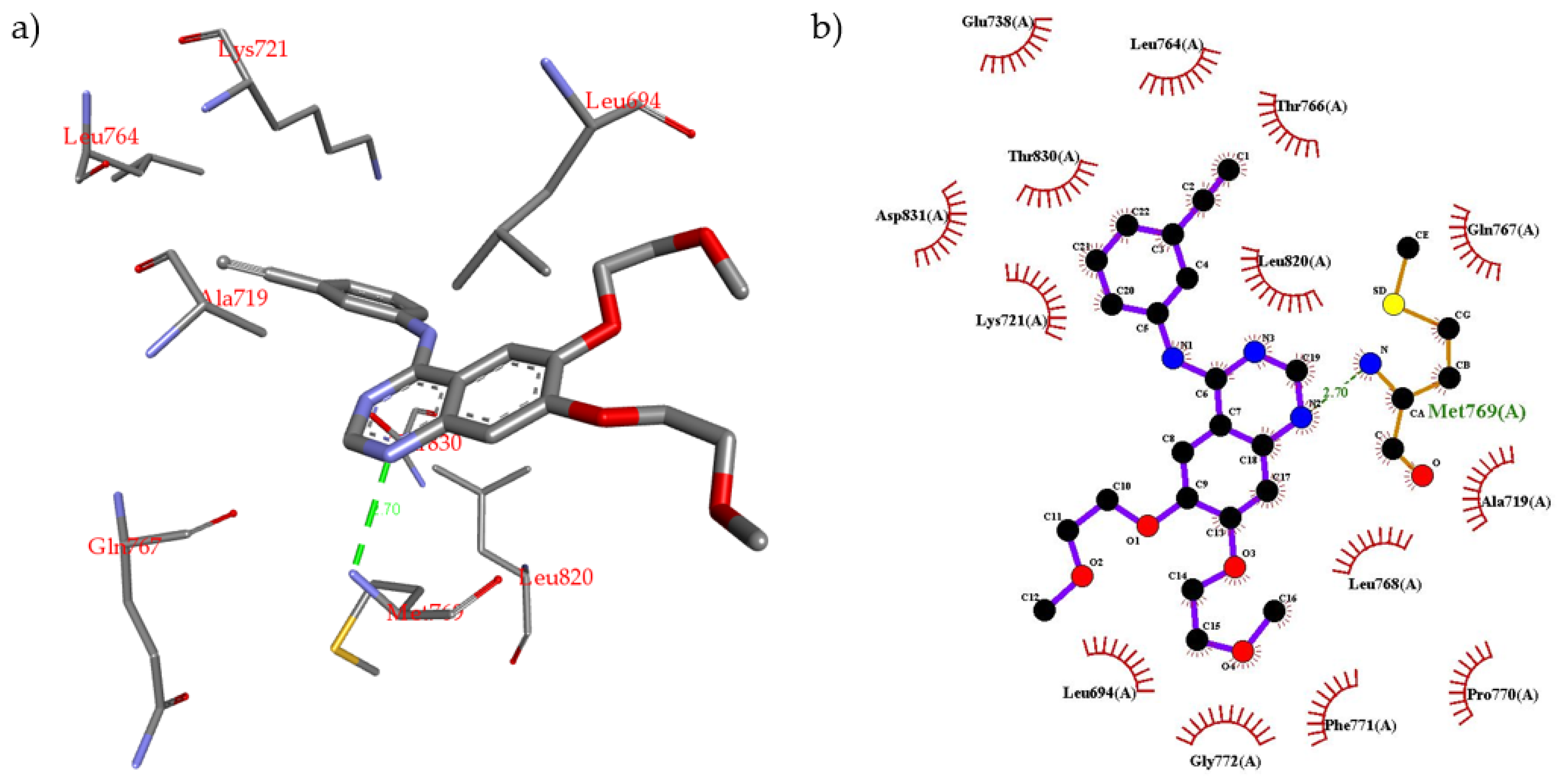
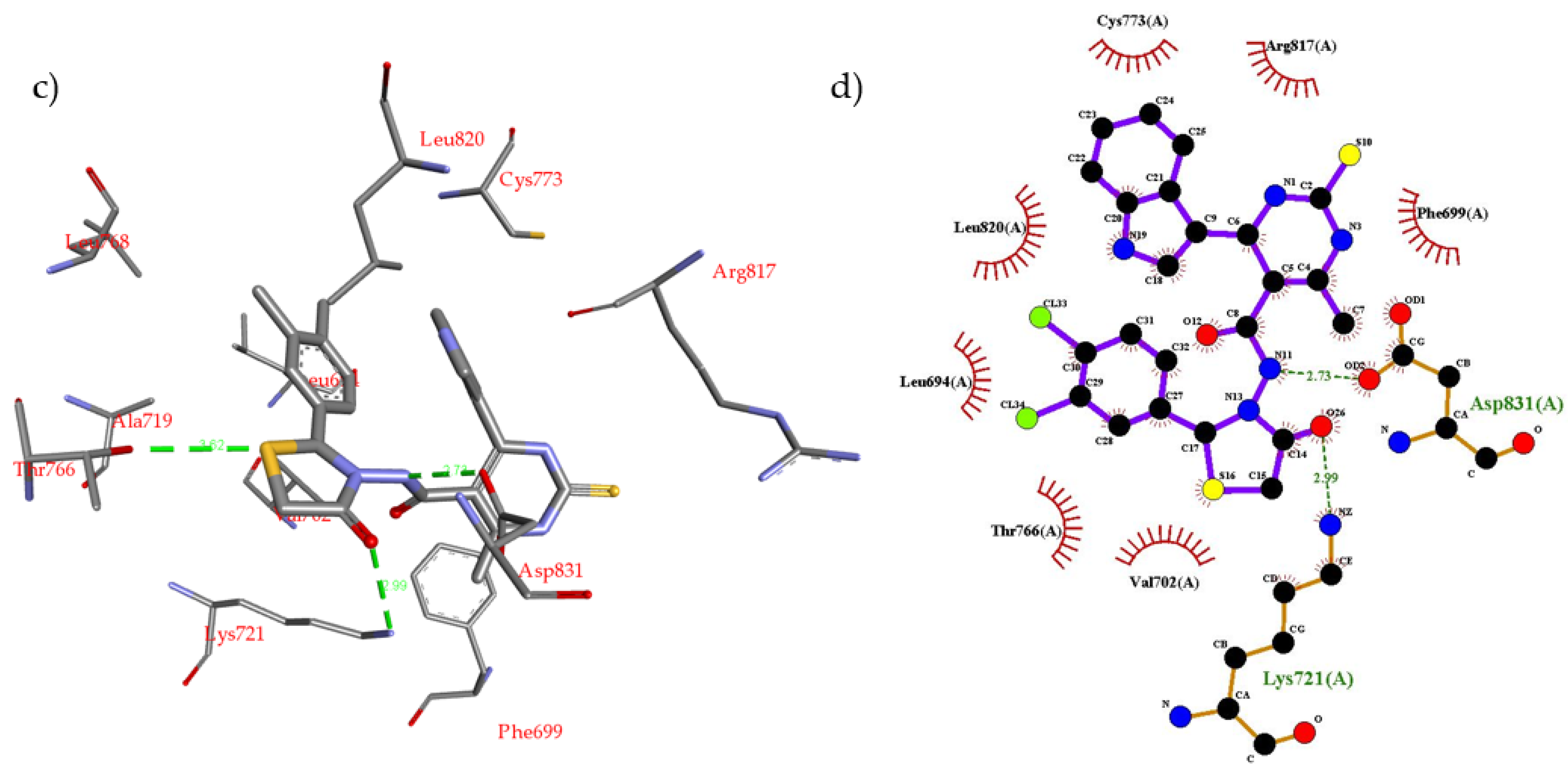
2.2. Molecular Modeling
2.3. Biological Screening
2.3.1. In Vitro Antiproliferative Activity
2.3.2. In Vivo Antitumor Evaluation
2.3.3. In Vitro EGFR Inhibition Assay
3. Materials and Methods
3.1. Instruments
3.2. Chemistry
3.2.1. Ethyl 4-(1H-indol-3-yl)-6-methyl-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-Carboxylate (1)
3.2.2. 4-(1H-indol-3-yl)-6-methyl-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carbohydrazide (2)
3.2.3. General Procedure for the Preparation of Compounds (3a–h)
4-(1H-indol-3-yl)-6-methyl-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carboxylic acid (4-methyl-benzylidene)-hydrazide (3b)
4-(1H-indol-3-yl)-6-methyl-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carboxylic acid (2-methoy- benzylidene)-hydrazide (3c)
4-(1H-indol-3-yl)-6-methyl-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carboxylic acid (4-dimethylamino-benzylidene)-hydrazide (3e)
4-(1H-indol-3-yl)-6-methyl-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carboxylic acid (3,4-dichloro- benzylidene)-hydrazide (3g)
3.2.4. General Procedure for the Preparation of Compounds (4a–h)
4-(1H-indol-3-yl)-6-methyl-N-(4-oxo-2-phenylthiazolidin-3-yl)-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carboxamide (4a)
4-(1H-indol-3-yl)-6-methyl-N-(4-oxo-2-(p-tolyl)thiazolidin-3-yl)-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carboxamide (4b)
4-(1H-indol-3-yl)-N-(2-(2-methoxyphenyl)-4-oxothiazolidin-3-yl)-6-methyl-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carboxamide (4c)
N-(2-(2-hydroxyphenyl)-4-oxothiazolidin-3-yl)-4-(1H-indol-3-yl)-6-methyl-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carboxamide (4d)
N-(2-(4-(dimethylamino)phenyl)-4-oxothiazolidin-3-yl)-4-(1H-indol-3-yl)-6-methyl-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carboxamide (4e)
N-(2-(4-chlorophenyl)-4-oxothiazolidin-3-yl)-4-(1H-indol-3-yl)-6-methyl-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carboxamide (4f)
N-(2-(3,4-dichlorophenyl)-4-oxothiazolidin-3-yl)-4-(1H-indol-3-yl)-6-methyl-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carboxamide (4g)
N-(2-(4-fluorophenyl)-4-oxothiazolidin-3-yl)-4-(1H-indol-3-yl)-6-methyl-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carboxamide (4h)
3.3. Biological Evaluation
3.3.1. In Vitro Cytotoxicity Assay
3.3.2. In Vivo Antitumor Assay
3.3.3. In Vitro EGFR Inhibitory Assay
3.3.4. Data Analysis
3.4. Molecular Modeling Procedure
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| MCF-7 | Michigan Cancer Foundation-7 cell line |
| HepG2 | Hepatoma G2 cell line |
| HCT116 | human colorectal carcinoma cell line |
| 5-FU | 5-Fluorouracil |
| EGFR | Epidermal Growth Factor Receptor |
| SAR | Structure-Activity Relationship |
| HCl | Hydrochloric acid |
| MS | Mass Spectrometry |
| IR | Infrared Spectroscopy |
| NMR | Nuclear Magnetic Resonance |
| IC50 | Concentrations of tested compounds that give about 50% inhibition of cell viability |
| EAC | Ehrlich ascites carcinoma |
References
- Said, A.M.; Parker, M.W.; Vander Kooi, C.W. Design, synthesis, and evaluation of a novel benzamidine-based inhibitor of VEGF-C binding to Neuropilin-2. Bioorg. Chem. 2020, 100, 103856. [Google Scholar] [CrossRef] [PubMed]
- Seyfried, T.N.; Huysentruyt, L.C. On the origin of cancer metastasis. Crit Rev. Oncog. 2013, 18, 43–73. [Google Scholar] [CrossRef] [PubMed]
- Guan, X. Cancer metastases: Challenges and opportunities. Acta Pharm. Sin. B 2015, 5, 402–418. [Google Scholar] [CrossRef] [PubMed]
- Dillekås, H.; Rogers, M.S.; Straume, O. Are 90% of deaths from cancer caused by metastases? Cancer Med. 2019, 8, 5574–5576. [Google Scholar] [CrossRef] [PubMed]
- Baskar, R.; Lee, K.A.; Yeo, R.; Yeoh, K.W. Cancer and radiation therapy: Current advances and future directions. Int. J. Med. Sci. 2012, 9, 193–199. [Google Scholar] [CrossRef]
- Parsa, N. Environmental factors inducing human cancers. Iran. J. Public Health 2012, 41, 1–9. [Google Scholar]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA A Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed]
- Thakor, A.S.; Gambhir, S.S. Nanooncology: The future of cancer diagnosis and therapy. CA Cancer J. Clin. 2013, 63, 395–418. [Google Scholar] [CrossRef]
- Jain, K.S.; Arya, N.; Inamdar, N.N.; Auti, P.B.; Unawane, S.A.; Puranik, H.H.; Sanap, M.S.; Inamke, A.D.; Mahale, V.J.; Prajapati, C.S.; et al. The Chemistry and Bio-Medicinal Significance of Pyrimidines & Condensed Pyrimidines. Curr. Top. Med. Chem. 2016, 16, 3133–3174. [Google Scholar] [CrossRef]
- Mabkhoot, Y.N. Synthesis and chemical characterisation of new bis-thieno [2,3-b]thiophene derivatives. Molecules 2010, 15, 3329–3337. [Google Scholar] [CrossRef] [PubMed]
- Sayed, A.R.; Gomha, S.M.; Abdelrazek, F.M.; Farghaly, M.S.; Hassan, S.A.; Metz, P. Design, efficient synthesis and molecular docking of some novel thiazolyl-pyrazole derivatives as anticancer agents. BMC Chem. 2019, 13, 116. [Google Scholar] [CrossRef] [PubMed]
- Abu-Melha, S.; Edrees, M.M.; Salem, H.H.; Kheder, N.A.; Gomha, S.M.; Abdelaziz, M.R. Synthesis and Biological Evaluation of Some Novel Thiazole-Based Heterocycles as Potential Anticancer and Antimicrobial Agents. Molecules 2019, 24, 539. [Google Scholar] [CrossRef] [PubMed]
- Abdel-aziz, H.M.; Gomha, S.M.; El-Sayed, A.A.; Mabkhot, Y.N.; Alsayari, A.; Muhsinah, A.B. Facile synthesis and antiproliferative activity of new 3-cyanopyridines. BMC Chem. 2019, 13, 137. [Google Scholar] [CrossRef]
- Ali, E.M.H.; Abdel-Maksoud, M.S.; Oh, C.H. Thieno[2,3-d]pyrimidine as a promising scaffold in medicinal chemistry: Recent advances. Bioorg. Med. Chem. 2019, 27, 1159–1194. [Google Scholar] [CrossRef]
- Abdelhamid, A.O.; Abdelall, E.K.A.; Abdel-Riheem, N.A.; Ahmed, S.A. Synthesis and Antimicrobial Activity of Some New 5-Arylazothiazole, Pyrazolo[1,5-a] Pyrimidine, [1,2,4]Triazolo[4,3-a]Pyrimidine, and Pyrimido[1,2-a]Benzimidazole Derivatives Containing the Thiazole Moiety. Phosphorus Sulfur Silicon Relat. Elem. 2010, 185, 709–718. [Google Scholar] [CrossRef]
- Liu, H.B.; Gao, W.W.; Tangadanchu, V.K.R.; Zhou, C.H.; Geng, R.X. Novel aminopyrimidinyl benzimidazoles as potentially antimicrobial agents: Design, synthesis and biological evaluation. Eur. J. Med. Chem. 2018, 143, 66–84. [Google Scholar] [CrossRef]
- Ahmed, N.M.; Youns, M.; Soltan, M.K.; Said, A.M. Design, synthesis, molecular modelling, and biological evaluation of novel substituted pyrimidine derivatives as potential anticancer agents for hepatocellular carcinoma. J. Enzym. Inhib. Med. Chem. 2019, 34, 1110–1120. [Google Scholar] [CrossRef]
- Xie, F.; Zhao, H.; Zhao, L.; Lou, L.; Hu, Y. Synthesis and biological evaluation of novel 2,4,5-substituted pyrimidine derivatives for anticancer activity. Bioorg. Med. Chem. Lett. 2009, 19, 275–278. [Google Scholar] [CrossRef]
- Nagarapu, L.; Vanaparthi, S.; Bantu, R.; Ganesh Kumar, C. Synthesis of novel benzo[4,5]thiazolo[1,2-a]pyrimidine-3-carboxylate derivatives and biological evaluation as potential anticancer agents. Eur. J. Med. Chem. 2013, 69, 817–822. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wu, J.; Zhang, L.; Luo, M.; Sun, D. Synthesis and Antifungal Activities of Some Novel Pyrimidine Derivatives. Molecules 2011, 16, 5618–5628. [Google Scholar] [CrossRef]
- Awad, S.M.; Zohny, Y.M.; Ali, S.A.; Mahgoub, S.; Said, A.M. Design, Synthesis, Molecular Modeling, and Biological Evaluation of Novel Thiouracil Derivatives as Potential Antithyroid Agents. Molecules 2018, 23. [Google Scholar] [CrossRef]
- Dragún, M.; Rada, B.; Novotný, L.; Beránek, J. Antiviral activities of pyrimidine nucleoside analogues: Some structure—Activity relationships. Acta Virol. 1990, 34, 321–329. [Google Scholar] [PubMed]
- Hoffmann, H.-H.; Kunz, A.; Simon, V.A.; Palese, P.; Shaw, M.L. Broad-spectrum antiviral that interferes with de novo pyrimidine biosynthesis. Proc. Natl. Acad. Sci. USA 2011, 108, 5777–5782. [Google Scholar] [CrossRef] [PubMed]
- Krečmerová, M.; Dračínský, M.; Snoeck, R.; Balzarini, J.; Pomeisl, K.; Andrei, G. New prodrugs of two pyrimidine acyclic nucleoside phosphonates: Synthesis and antiviral activity. Bioorg. Med. Chem. 2017, 25, 4637–4648. [Google Scholar] [CrossRef]
- Shaquiquzzaman, M.; Khan, S.A.; Amir, M.; Alam, M.M. Synthesis, anticonvulsant and neurotoxicity evaluation of some new pyrimidine-5-carbonitrile derivatives. Saudi Pharm J. 2012, 20, 149–154. [Google Scholar] [CrossRef]
- Mohamed, M.S.; Awad, S.M.; Sayed, A.I. Synthesis of certain pyrimidine derivatives as antimicrobial agents and anti-inflammatory agents. Molecules 2010, 15, 1882–1890. [Google Scholar] [CrossRef] [PubMed]
- Inoyama, D.; Paget, S.D.; Russo, R.; Kandasamy, S.; Kumar, P.; Singleton, E.; Occi, J.; Tuckman, M.; Zimmerman, M.D.; Ho, H.P.; et al. Novel Pyrimidines as Antitubercular Agents. Antimicrob. Agents Chemother. 2018, 62. [Google Scholar] [CrossRef]
- Suryawanshi, S.N.; Kumar, S.; Shivahare, R.; Pandey, S.; Tiwari, A.; Gupta, S. Design, synthesis and biological evaluation of aryl pyrimidine derivatives as potential leishmanicidal agents. Bioorg. Med. Chem. Lett. 2013, 23, 5235–5238. [Google Scholar] [CrossRef]
- Iman, M.; Davood, A.; Khamesipour, A. Design of antimalarial agents based on pyrimidine derivatives as methionine aminopeptidase 1b inhibitor: Molecular docking, quantitative structure activity relationships, and molecular dynamics simulation studies. J. Chin. Chem. Soc. 2020, 67, 880–890. [Google Scholar] [CrossRef]
- Partridge, F.A.; Forman, R.; Willis, N.J.; Bataille, C.J.R.; Murphy, E.A.; Brown, A.E.; Heyer-Chauhan, N.; Marinič, B.; Sowood, D.J.C.; Wynne, G.M.; et al. 2,4-Diaminothieno[3,2-d]pyrimidines, a new class of anthelmintic with activity against adult and egg stages of whipworm. PLoS Negl. Trop. Dis. 2018, 12, e0006487. [Google Scholar] [CrossRef] [PubMed]
- Romeo, R.; Iannazzo, D.; Veltri, L.; Gabriele, B.; Macchi, B.; Frezza, C.; Marino-Merlo, F.; Giofrè, S.V. Pyrimidine 2,4-Diones in the Design of New HIV RT Inhibitors. Molecules 2019, 24, 1718. [Google Scholar] [CrossRef]
- Liu, P.; Yang, Y.; Tang, Y.; Yang, T.; Sang, Z.; Liu, Z.; Zhang, T.; Luo, Y. Design and synthesis of novel pyrimidine derivatives as potent antitubercular agents. Eur. J. Med. Chem. 2019, 163, 169–182. [Google Scholar] [CrossRef]
- Park, D.I.; Dournes, C.; Sillaber, I.; Uhr, M.; Asara, J.M.; Gassen, N.C.; Rein, T.; Ising, M.; Webhofer, C.; Filiou, M.D.; et al. Purine and pyrimidine metabolism: Convergent evidence on chronic antidepressant treatment response in mice and humans. Sci. Rep. 2016, 6, 35317. [Google Scholar] [CrossRef] [PubMed]
- Funayama, S.; Cordell, G.A. (Eds.) Chapter 11—Alkaloids Derived from Nucleic Acids and Related Compounds. In Alkaloids; Academic Press: Boston, MA, USA, 2015; pp. 193–208. [Google Scholar] [CrossRef]
- Daly, M.M.; Allfrey, V.G.; Mirsky, A.E. Purine and pyrimidine contents of some desoxypentose nucleic acids. J. Gen. Physiol. 1950, 33, 497–510. [Google Scholar] [CrossRef]
- Cerecedo, L.R. Physiology of Pyrimidines. Nature 1940, 146, 274. [Google Scholar] [CrossRef]
- Sondhi, S.M.; Goyal, R.N.; Lahoti, A.M.; Singh, N.; Shukla, R.; Raghubir, R. Synthesis and biological evaluation of 2-thiopyrimidine derivatives. Bioorg. Med. Chem. 2005, 13, 3185–3195. [Google Scholar] [CrossRef] [PubMed]
- Parker, W.B. Enzymology of purine and pyrimidine antimetabolites used in the treatment of cancer. Chem. Rev. 2009, 109, 2880–2893. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, Y.; Maeda, Y.; Sato, H.; Nakano, M.; Mellor, G.W. Rational design of 4-amino-5,6-diaryl-furo[2,3-d]pyrimidines as potent glycogen synthase kinase-3 inhibitors. Bioorg. Med. Chem. Lett. 2008, 18, 1967–1971. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.C.; Lee, Y.R.; Yang, B.S.; Shin, K.J.; Kim, D.J.; Chung, B.Y.; Yoo, K.H. Synthesis and biological evaluations of pyrazolo[3,4-d]pyrimidines as cyclin-dependent kinase 2 inhibitors. Eur. J. Med. Chem. 2003, 38, 525–532. [Google Scholar] [CrossRef]
- Joshi, G.; Nayyar, H.; Kalra, S.; Sharma, P.; Munshi, A.; Singh, S.; Kumar, R. Pyrimidine containing epidermal growth factor receptor kinase inhibitors: Synthesis and biological evaluation. Chem. Biol. Drug Des. 2017, 90, 995–1006. [Google Scholar] [CrossRef]
- Dadashpour, S.; Emami, S. Indole in the target-based design of anticancer agents: A versatile scaffold with diverse mechanisms. Eur. J. Med. Chem. 2018, 150, 9–29. [Google Scholar] [CrossRef] [PubMed]
- Prakash, B.; Amuthavalli, A.; Edison, D.; Sivaramkumar, M.S.; Velmurugan, R. Novel indole derivatives as potential anticancer agents: Design, synthesis and biological screening. Med. Chem. Res. 2018, 27, 321–331. [Google Scholar] [CrossRef]
- Putt, K.S.; Chen, G.W.; Pearson, J.M.; Sandhorst, J.S.; Hoagland, M.S.; Kwon, J.T.; Hwang, S.K.; Jin, H.; Churchwell, M.I.; Cho, M.H.; et al. Small-molecule activation of procaspase-3 to caspase-3 as a personalized anticancer strategy. Nat. Chem. Biol. 2006, 2, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Seshacharyulu, P.; Ponnusamy, M.P.; Haridas, D.; Jain, M.; Ganti, A.K.; Batra, S.K. Targeting the EGFR signaling pathway in cancer therapy. Expert Opin. Targets 2012, 16, 15–31. [Google Scholar] [CrossRef]
- Chen, Y.; Wu, J.; Wang, A.; Qi, Z.; Jiang, T.; Chen, C.; Zou, F.; Hu, C.; Wang, W.; Wu, H.; et al. Discovery of N-(5-((5-chloro-4-((2-(isopropylsulfonyl)phenyl)amino)pyrimidin-2-yl)amino)-4-methoxy-2-(4-methyl-1,4-diazepan-1-yl)phenyl)acrylamide (CHMFL-ALK/EGFR-050) as a potent ALK/EGFR dual kinase inhibitor capable of overcoming a variety of ALK/EGFR associated drug resistant mutants in NSCLC. Eur. J. Med. Chem. 2017, 139, 674–697. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Ren, H.; Zhao, M.; Chong, Y.; Zhao, W.; He, Y.; Zhao, Y.; Zhang, H.; Qi, C. Development of a series of novel 4-anlinoquinazoline derivatives possessing quinazoline skeleton: Design, synthesis, EGFR kinase inhibitory efficacy, and evaluation of anticancer activities in vitro. Eur. J. Med. Chem. 2017, 138, 669–688. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Nair, S.K.; Murray, B.W. Recent progress on third generation covalent EGFR inhibitors. Bioorg. Med. Chem Lett. 2016, 26, 1861–1868. [Google Scholar] [CrossRef]
- Desai, N.C.; Bhatt, M.J. Catalytic synthesis and antimicrobial activity of N-(3-chloro-2-oxo-4-phenylazetidin-1-yl)-4-(1H-indol-3-yl)-6-methyl-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carboxamides. Heterocycl. Commun. 2016, 22, 131–136. [Google Scholar] [CrossRef]
- Viveka, S.; Nagaraja, G.K.; Shama, P.; Basavarajaswamy, G.; Rao, K.P.; Yanjarappa Sreenivasa, M. One pot synthesis of thiazolo[2,3-b]dihydropyrimidinone possessing pyrazole moiety and evaluation of their anti-inflammatory and antimicrobial activities. Med. Chem. Res. 2018, 27, 171–185. [Google Scholar] [CrossRef]
- AbdElhameid, M.K.; Labib, M.B.; Negmeldin, A.T.; Al-Shorbagy, M.; Mohammed, M.R. Design, synthesis, and screening of ortho-amino thiophene carboxamide derivatives on hepatocellular carcinomaas VEGFR-2Inhibitors. J. Enzym. Inhib Med. Chem. 2018, 33, 1472–1493. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Stamos, J.; Sliwkowski, M.X.; Eigenbrot, C. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J. Biol. Chem. 2002, 277, 46265–46272. [Google Scholar] [CrossRef]
- Nasr, T.; Bondock, S.; Youns, M. Anticancer activity of new coumarin substituted hydrazide-hydrazone derivatives. Eur. J. Med. Chem. 2014, 76, 539–548. [Google Scholar] [CrossRef]
- Ahmed, O.M.; Ahmed, R.R. Anti-proliferative and apoptotic efficacy of diallyl disulfide on Ehrlich ascites carcinoma. Hepatoma Res. 2015, 1, 67–74. [Google Scholar] [CrossRef][Green Version]
- Zhao, P.; Yang, X.; Qi, S.; Liu, H.; Jiang, H.; Hoppmann, S.; Cao, Q.; Chua, M.S.; So, S.K.; Cheng, Z. Molecular imaging of hepatocellular carcinoma xenografts with epidermal growth factor receptor targeted affibody probes. Biomed. Res. Int. 2013, 2013, 759057. [Google Scholar] [CrossRef]
- Moerkens, M.; Zhang, Y.; Wester, L.; van de Water, B.; Meerman, J.H. Epidermal growth factor receptor signalling in human breast cancer cells operates parallel to estrogen receptor α signalling and results in tamoxifen insensitive proliferation. BMC Cancer 2014, 14, 283. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.L.; Qu, X.J.; Russell, P.J.; Goldstein, D. Regulation of epidermal growth factor receptor in human colon cancer cell lines by interferon alpha. Gut 2004, 53, 123–129. [Google Scholar] [CrossRef]
- Maher, M.; Kassab, A.E.; Zaher, A.F.; Mahmoud, Z. Novel pyrazolo[3,4-d]pyrimidines: Design, synthesis, anticancer activity, dual EGFR/ErbB2 receptor tyrosine kinases inhibitory activity, effects on cell cycle profile and caspase-3-mediated apoptosis. J. Enzym. Inhib Med. Chem. 2019, 34, 532–546. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
Sample Availability: Sample availability of the compounds 4a–h is very limited from the authors. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | * Cytotoxic Activity IC50 a (μM) | |||
|---|---|---|---|---|
| MCF-7 | HCT-116 | HepG2 | WI-38 | |
| 1 | 26.8 ± 2.20 | 27.2 ± 1.11 | 28.8 ± 2.25 | 30.0 ± 1.20 |
| 2 | 21.6 ± 1.8 | 22.2 ± 1.5 | 25.5 ± 2.17 | 40.50 ± 2.25 |
| 3a | 17.0 ± 1.80 | 15.2 ± 2.32 | 19.2 ± 1.50 | 38.23 ± 2.10 |
| 3b | 18.50 ± 2.11 | 15.6 ± 3.15 | 19.4 ± 2.19 | 39.13 ± 1.30 |
| 3c | 18.9 ± 2.00 | 16.3 ± 4.16 | 19.9 ± 2.06 | 37.42 ± 1.49 |
| 3d | 19.2± 2.35 | 16.6 ± 1.27 | 20.2 ± 2.33 | 35.26 ± 2.42 |
| 3e | 19.8 ± 2.70 | 16.2 ± 1.15 | 20.4 ± 2.30 | 40.01 ± 2.31 |
| 3f | 10.7 ± 2.30 | 10.20 ± 1.18 | 10.6 ± 2.32 | 29.82 ± 1.51 |
| 3g | 9.0 ± 2.40 | 8.5 ± 2.30 | 10.0 ± 2.46 | 30.01 ± 3.72 |
| 3h | 10.2 ± 2.01 | 10.11 ± 2.45 | 10.4 ± 2.08 | 28.10 ± 4.22 |
| 4a | 11.2 ± 4.31 | 12.3 ± 2.17 | 17.0 ± 4.30 | 38.03 ± 5.51 |
| 4b | 11.6 ± 3.22 | 12.6 ± 1.05 | 18.0 ± 3.20 | 39.8 ± 4.51 |
| 4c | 12.3 ± 4.08 | 13.2 ± 1.03 | 18.5 ± 4.38 | 29.5 ± 4.02 |
| 4d | 12.6 ± 1.70 | 13.6 ± 1.09 | 18.7 ± 1.20 | 29.9 ± 3.63 |
| 4e | 14.2 ± 0.74 | 13.7 ± 0.55 | 18.62 ± 0.94 | 20.24 ± 5.52 |
| 4f | 8.01 ± 1.83 | 8.02 ± 0.26 | 8.9 ± 1.73 | 19.33 ± 5.87 |
| 4g | 5.1 ± 1.14 | 5.02± 1.19 | 6.6 ± 1.40 | 16.32 ± 3.21 |
| 4h | 6.6 ± 1.28 | 7.02 ± 0.46 | 7.5 ± 1.29 | 18.12 ± 2.06 |
| 5-FU | 5.38 ± 0.24 | 7.88 ± 0.2 | 5.34 ± 0.4 | 5.70 ± 1.50 |
| erlotinib | 6.65 ± 0.82 | 7.49 ± 0.65 | nd | 22.50 ± 0.65 |
| Group | MST (day) a | % ILS a | Tumor Volume (mL) a | Viable Tumor Cell Count (106/mL) a |
|---|---|---|---|---|
| Normal | nd | nd | nd | Nd |
| EAC only | 16.5 | nd | 8.01 | 80.25 |
| 3g | 30 | 110.0 | 2.97 | 48.00 |
| 4f | 37 | 130.0 | 2.50 | 40.70 |
| 4g | 46 | 230.0 | 0.89 | 21.70 |
| 4h | 40 | 180.0 | 1.18 | 28.50 |
| 5-FU | 53.0 | 265.5 | 0.80 | 19.07 |
| Compound | % Inhibition of EGFR | IC50 (µM) a |
|---|---|---|
| 3g | 53 | 0.50 ± 0.04 |
| 4f | 71 | 0.38 ± 0.02 |
| 4g | 79 | 0.25 ± 0.01 |
| 4h | 70 | 0.39 ± 0.02 |
| erlotinib | 81 | 0.30 ± 0.01 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmed, N.M.; Youns, M.M.; Soltan, M.K.; Said, A.M. Design, Synthesis, Molecular Modeling and Antitumor Evaluation of Novel Indolyl-Pyrimidine Derivatives with EGFR Inhibitory Activity. Molecules 2021, 26, 1838. https://doi.org/10.3390/molecules26071838
Ahmed NM, Youns MM, Soltan MK, Said AM. Design, Synthesis, Molecular Modeling and Antitumor Evaluation of Novel Indolyl-Pyrimidine Derivatives with EGFR Inhibitory Activity. Molecules. 2021; 26(7):1838. https://doi.org/10.3390/molecules26071838
Chicago/Turabian StyleAhmed, Naglaa M., Mahmoud M. Youns, Moustafa K. Soltan, and Ahmed M. Said. 2021. "Design, Synthesis, Molecular Modeling and Antitumor Evaluation of Novel Indolyl-Pyrimidine Derivatives with EGFR Inhibitory Activity" Molecules 26, no. 7: 1838. https://doi.org/10.3390/molecules26071838
APA StyleAhmed, N. M., Youns, M. M., Soltan, M. K., & Said, A. M. (2021). Design, Synthesis, Molecular Modeling and Antitumor Evaluation of Novel Indolyl-Pyrimidine Derivatives with EGFR Inhibitory Activity. Molecules, 26(7), 1838. https://doi.org/10.3390/molecules26071838