
Arterial Blood Pressure Variability and Other Vascular Factors Contribution to the Cognitive Decline in Parkinson’s Disease

 ,
,  and
and
Abstract
1. Introduction
2. Blood Pressure Variability
3. The Role of Levodopa in Neurocirculatory Abnormalities and Cognitive Decline in Parkinson’s Disease
4. White Matter Hyperintensities
5. Metabolic and Vascular Risk Factors
6. Genetic Factors
6.1. COMT
6.2. APOE
6.3. VEGF
6.4. RAA System Genes
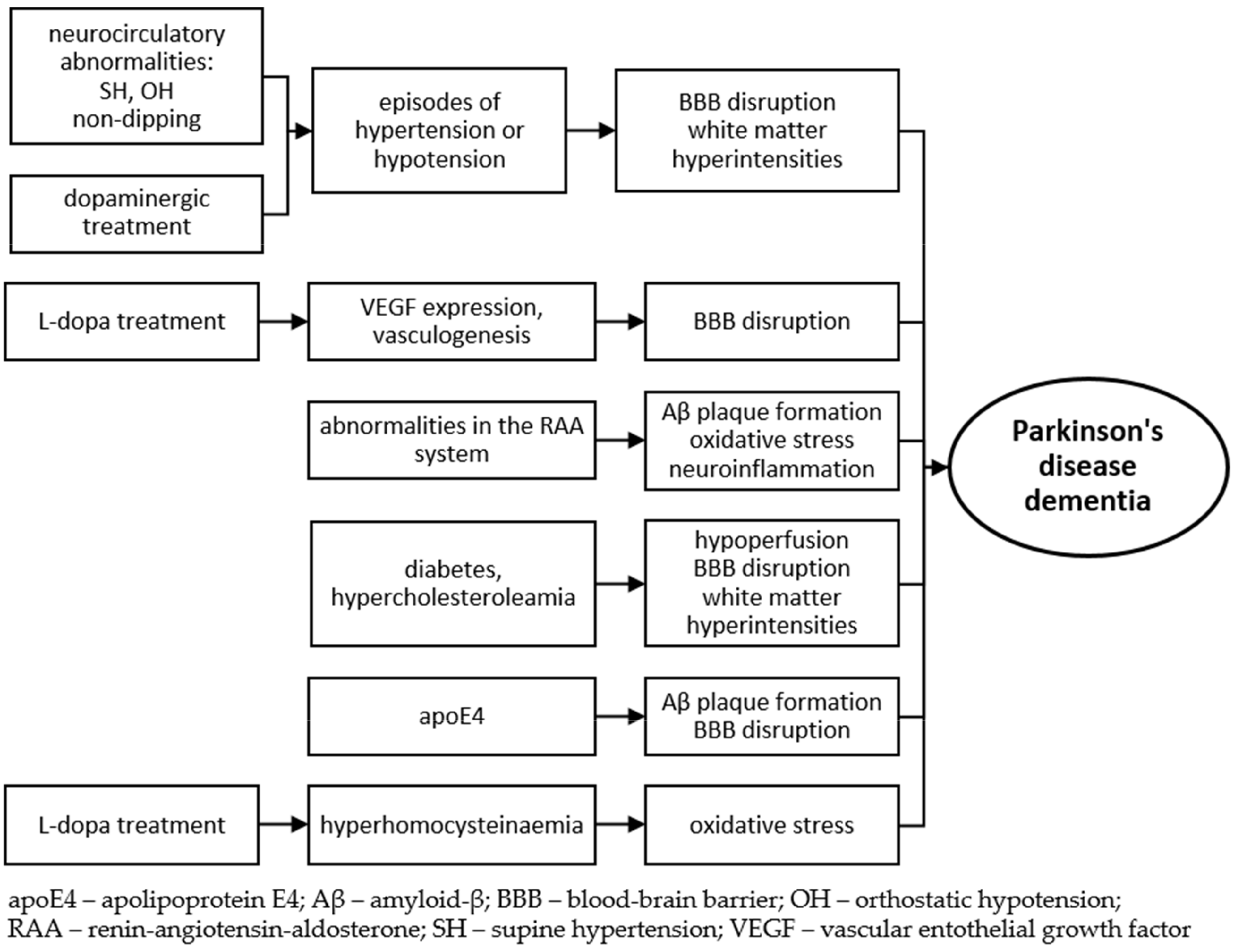
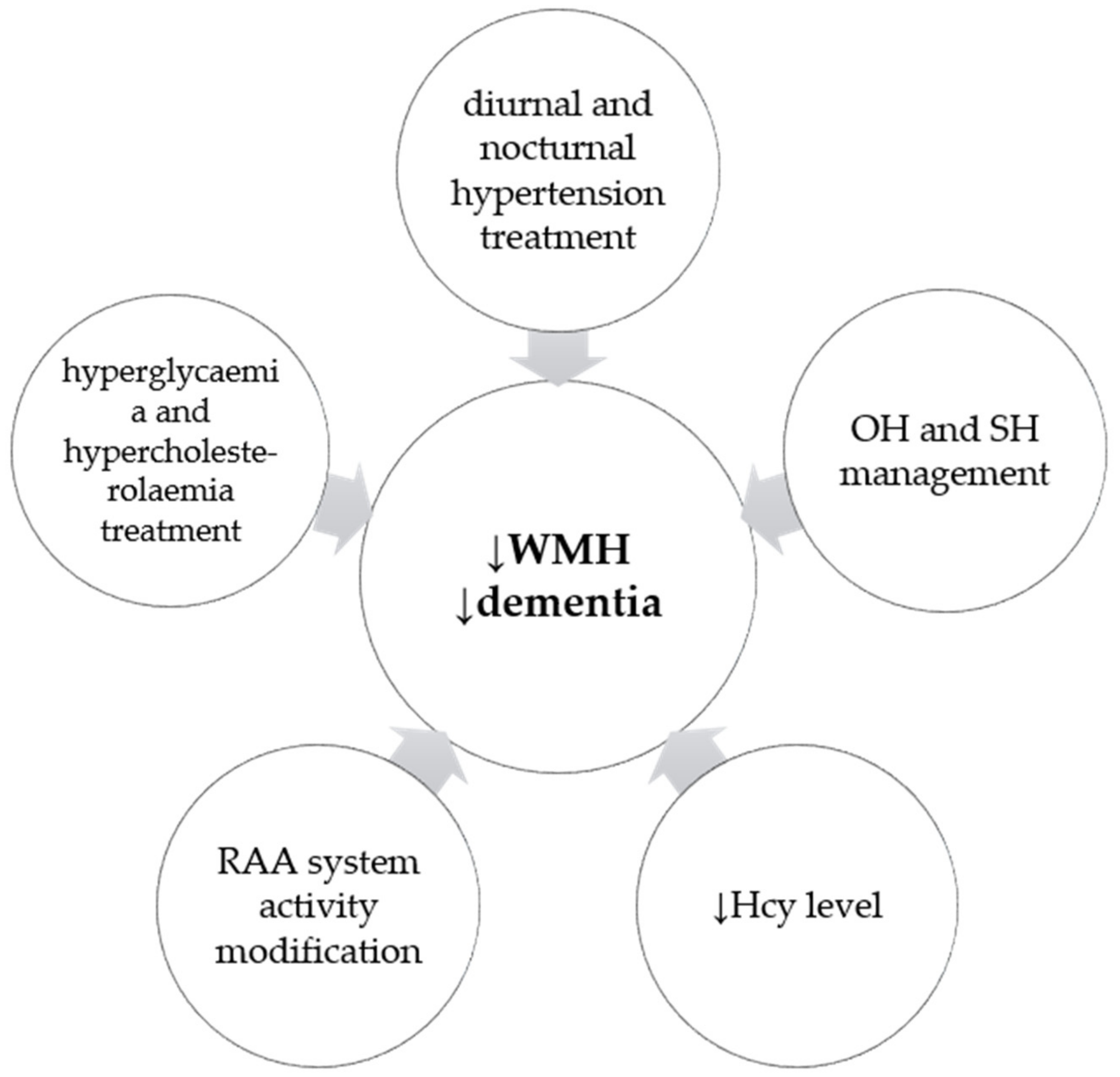
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Poewe, W.; Gauthier, S.; Aarsland, D.; Leverenz, J.B.; Barone, P.; Weintraub, D.; Tolosa, E.; Dubois, B. Diagnosis and management of Parkinson’s disease dementia. Int. J. Clin. Pract. 2008, 62, 1581–1587. [Google Scholar] [CrossRef][Green Version]
- Emre, M. Dementia associated with Parkinson’s disease. Lancet Neurol. 2003, 2, 229–237. [Google Scholar] [CrossRef]
- Aarsland, D.; Andersen, K.; Larsen, J.P.; Lolk, A.; Kragh-Sørensen, P. Prevalence and characteristics of dementia in Parkinson disease: An 8-year prospective study. Arch. Neurol. 2003, 60, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Zhu, K.; van Hilten, J.J.; Marinus, J. Predictors of dementia in Parkinson’s disease; findings from a 5-year prospective study using the SCOPA-COG. Parkinsonism Relat. Disord. 2014, 20, 980–985. [Google Scholar] [CrossRef]
- Horvath, J.; Herrmann, F.R.; Burkhard, P.R.; Bouras, C.; Kövari, E. Neuropathology of dementia in a large cohort of patients with Parkinson’s disease. Parkinsonism Relat. Disord. 2013, 19, 864–868. [Google Scholar] [CrossRef] [PubMed]
- Hanganu, A.; Bedetti, C.; Jubault, T.; Gagnon, J.F.; Mejia-Constain, B.; Degroot, C.; Lafontaine, A.L.; Chouinard, S.; Monchi, O. Mild cognitive impairment in patients with Parkinson’s disease is associated with increased cortical degeneration. Mov. Disord. 2013, 28, 1360–1369. [Google Scholar] [CrossRef]
- Parati, G.; Stergiou, G.S.; Dolan, E.; Bilo, G. Blood pressure variability: Clinical relevance and application. J. Clin. Hypertens. 2018, 20, 1133–1137. [Google Scholar] [CrossRef]
- Pathak, A.; Senard, J.M. Blood pressure disorders during Parkinson’s disease: Epidemiology, pathophysiology and management. Expert Rev. Neurother. 2006, 6, 1173–1180. [Google Scholar] [CrossRef]
- Jain, S.; Goldstein, D.S. Cardiovascular dysautonomia in Parkinson disease: From pathophysiology to pathogenesis. Neurobiol. Dis. 2012, 46, 572–580. [Google Scholar] [CrossRef] [PubMed]
- Biaggioni, I.; Arthur Hewitt, L.; Rowse, G.J.; Kaufmann, H. Integrated analysis of droxidopa trials for neurogenic orthostatic hypotension. BMC Neurol. 2017, 17, 90. [Google Scholar] [CrossRef]
- Milazzo, V.; Di Stefano, C.; Vallelonga, F.; Sobrero, G.; Zibetti, M.; Romagnolo, A.; Merola, A.; Milan, A.; Espay, A.J.; Lopiano, L.; et al. Reverse blood pressure dipping as marker of dysautonomia in Parkinson disease. Parkinsonism Relat. Disord. 2018, 56, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Fabbian, F.; Smolensky, M.H.; Tiseo, R.; Pala, M.; Manfredini, R.; Portaluppi, F. Dipper and non-dipper blood pressure 24-hour patterns: Circadian rhythm-dependent physiologic and pathophysiologic mechanisms. Chronobiol. Int. 2013, 30, 17–30. [Google Scholar] [CrossRef]
- Agarwal, R. Regulation of circadian blood pressure: From mice to astronauts. Curr. Opin. Nephrol. Hypertens. 2010, 19, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Kuwajima, I.; Suzuki, Y.; Shimosawa, T.; Kanemaru, A.; Hoshino, S.; Kuramoto, K. Diminished nocturnal decline in blood pressure in elderly hypertensive patients with left ventricular hypertrophy. Am. Heart. J. 1992, 123, 1307–1311. [Google Scholar] [CrossRef]
- Fanciulli, A.; Strano, S.; Ndayisaba, J.P.; Goebel, G.; Gioffrè, L.; Rizzo, M.; Colosimo, C.; Caltagirone, C.; Poewe, W.; Wenning, G.K.; et al. Detecting nocturnal hypertension in Parkinson’s disease and multiple system atrophy: Proposal of a decision-support algorithm. J. Neurol. 2014, 261, 1291–1299. [Google Scholar] [CrossRef]
- Berganzo, K.; Díez-Arrola, B.; Tijero, B.; Somme, J.; Lezcano, E.; Llorens, V.; Ugarriza, I.; Ciordia, R.; Gómez-Esteban, J.C.; Zarranz, J.J. Nocturnal hypertension and dysautonomia in patients with Parkinson’s disease: Are they related? J. Neurol. 2013, 260, 1752–1756. [Google Scholar] [CrossRef]
- Kim, J.S.; Oh, Y.S.; Lee, K.S.; Kim, Y.I.; Yang, D.W.; Goldstein, D.S. Association of cognitive dysfunction with neurocirculatory abnormalities in early Parkinson disease. Neurology 2012, 79, 1323–1331. [Google Scholar] [CrossRef]
- Anang, J.B.; Gagnon, J.F.; Bertrand, J.A.; Romenets, S.R.; Latreille, V.; Panisset, M.; Montplaisir, J.; Postuma, R.B. Predictors of dementia in Parkinson disease: A prospective cohort study. Neurology 2014, 83, 1253–1260. [Google Scholar] [CrossRef]
- Udow, S.J.; Robertson, A.D.; MacIntosh, B.J.; Espay, A.J.; Rowe, J.B.; Lang, A.E.; Masellis, M. ‘Under pressure’: Is there a link between orthostatic hypotension and cognitive impairment in α-synucleinopathies? J. Neurol. Neurosurg. Psychiatry 2016, 87, 1311–1321. [Google Scholar] [CrossRef]
- Tanaka, R.; Shimo, Y.; Yamashiro, K.; Ogawa, T.; Nishioka, K.; Oyama, G.; Umemura, A.; Hattori, N. Association between abnormal nocturnal blood pressure profile and dementia in Parkinson’s disease. Parkinsonism Relat. Disord. 2018, 46, 24–29. [Google Scholar] [CrossRef]
- Calabresi, P.; Di Filippo, M.; Ghiglieri, V.; Tambasco, N.; Picconi, B. Levodopa-induced dyskinesias in patients with Parkinson’s disease: Filling the bench-to-bedside gap. Lancet Neurol. 2010, 9, 1106–1117. [Google Scholar] [CrossRef]
- Westin, J.E.; Lindgren, H.S.; Gardi, J.; Nyengaard, J.R.; Brundin, P.; Mohapel, P.; Cenci, M.A. Endothelial Proliferation and Increased Blood-Brain Barrier Permeability in the Basal Ganglia in a Rat Model of 3,4-Dihydroxyphenyl-L-Alanine-Induced Dyskinesia. J. Neurosci. 2006, 26, 9448–9461. [Google Scholar] [CrossRef] [PubMed]
- Ohlin, K.E.; Francardo, V.; Lindgren, H.S.; Sillivan, S.E.; O’Sullivan, S.S.; Luksik, A.S.; Vassoler, F.M.; Lees, A.J.; Konradi, C.; Cenci, M.A. Vascular endothelial growth factor is upregulated by L-dopa in the parkinsonian brain: Implications for the development of dyskinesia. Brain 2011, 134, 2339–2357. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.H.; Lerner, R.P.; Eidelberg, D. Effects of levodopa on regional cerebral metabolism and blood flow. Mov. Disord. 2015, 30, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.S.; Chung, S.J.; Lee, Y.H.; Lee, H.S.; Ye, B.S.; Sohn, Y.H.; Lee, P.H. Levodopa-induced dyskinesia is closely linked to progression of frontal dysfunction in PD. Neurology 2019, 92, e1468–e1478. [Google Scholar] [CrossRef] [PubMed]
- Espay, A.J.; LeWitt, P.A.; Hauser, R.A.; Merola, A.; Masellis, M.; Lang, A.E. Neurogenic orthostatic hypotension and supine hypertension in Parkinson’s disease and related synucleinopathies: Prioritisation of treatment targets. Lancet Neurol. 2016, 15, 954–966. [Google Scholar] [CrossRef]
- Goldstein, D.S.; Eldadah, B.A.; Holmes, C.; Pechnik, S.; Moak, J.; Saleem, A.; Sharabi, Y. Neurocirculatory abnormalities in Parkinson disease with orthostatic hypotension: Independence from levodopa treatment. Hypertension 2005, 46, 1333–1339. [Google Scholar] [CrossRef]
- Wolf, J.P.; Bouhaddi, M.; Louisy, F.; Mikehiev, A.; Mourot, L.; Cappelle, S.; Vuillier, F.; Andre, P.; Rumbach, L.; Regnard, J. Side-effects of L-dopa on venous tone in Parkinson’s disease: A leg-weighing assessment. Clin. Sci. 2006, 110, 369–377. [Google Scholar] [CrossRef]
- Fanciulli, A.; Göbel, G.; Ndayisaba, J.P.; Granata, R.; Duerr, S.; Strano, S.; Colosimo, C.; Poewe, W.; Pontieri, F.E.; Wenning, G.K. Supine hypertension in Parkinson’s disease and multiple system atrophy. Clin. Auton. Res. 2016, 26, 97–105. [Google Scholar] [CrossRef]
- Montastruc, F.; Moulis, F.; Araujo, M.; Chebane, L.; Rascol, O.; Montastruc, J.L. Ergot and non-ergot dopamine agonists and heart failure in patients with Parkinson’s disease. Eur. J. Clin. Pharmacol. 2017, 73, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Torti, M.; Bravi, D.; Vacca, L.; Stocchi, F. Are All Dopamine Agonists Essentially the Same? Drugs 2019, 79, 693–703. [Google Scholar] [CrossRef]
- Oka, H.; Nakahara, A.; Umehara, T. Rotigotine Improves Abnormal Circadian Rhythm of Blood Pressure in Parkinson’s Disease. Eur. Neurol. 2018, 79, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, Y.; Nakamura, Y.; Cao, X.; Ohara, H.; Yamazaki, Y.; Murayama, N.; Sugiyama, Y.; Izumi-Nakaseko, H.; Ando, K.; Yamazaki, H.; et al. Intravenous Administration of Apomorphine Does NOT Induce Long QT Syndrome: Experimental Evidence from In Vivo Canine Models. Basic Clin. Pharmacol. Toxicol. 2015, 116, 468–475. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Kawamoto, A.; Matsubayashi, K.; Nishinaga, M.; Kimura, S.; Ozawa, T. Diurnal blood pressure variations and silent cerebrovascular damage in elderly patients with hypertension. J. Hypertens. 1992, 10, 875–878. [Google Scholar]
- Bohnen, N.I.; Albin, R.L. White matter lesions in Parkinson disease. Nat. Rev. Neurol. 2011, 7, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Bohnen, N.I.; Müller, M.L.; Zarzhevsky, N.; Koeppe, R.A.; Bogan, C.W.; Kilbourn, M.R.; Frey, K.A.; Albin, R.L. Leucoaraiosis, nigrostriatal denervation and motor symptoms in Parkinson’s disease. Brain 2011, 134, 2358–2365. [Google Scholar] [CrossRef]
- Siennicki-Lantz, A.; Reinprecht, F.; Axelsson, J.; Elmståhl, S. Cerebral perfusion in the elderly with nocturnal blood pressure fall. Eur. J. Neurol. 2007, 14, 715–720. [Google Scholar] [CrossRef]
- Axelsson, J.; Reinprecht, F.; Siennicki-Lantz, A.; Elmståhl, S. Lower cognitive performance in 81-year-old men with greater nocturnal blood pressure dipping. Int. J. Gen. Med. 2009, 1, 69–75. [Google Scholar] [CrossRef]
- Oh, Y.S.; Kim, J.S.; Lee, K.S. Orthostatic and supine blood pressures are associated with white matter hyperintensities in Parkinson disease. J. Mov. Disord. 2013, 6, 23–27. [Google Scholar] [CrossRef]
- Janelidze, S.; Lindqvist, D.; Francardo, V.; Hall, S.; Zetterberg, H.; Blennow, K.; Adler, C.H.; Beach, T.G.; Serrano, G.E.; van Westen, D.; et al. Increased CSF biomarkers of angiogenesis in Parkinson disease. Neurology 2015, 85, 1834–1842. [Google Scholar] [CrossRef] [PubMed]
- Sławek, J.; Roszmann, A.; Robowski, P.; Dubaniewicz, M.; Sitek, E.J.; Honczarenko, K.; Gorzkowska, A.; Budrewicz, S.; Mak, M.; Gołąb-Janowska, M.; et al. The impact of MRI white matter hyperintensities on dementia in Parkinson’s disease in relation to the homocysteine level and other vascular risk factors. Neurodegener. Dis. 2013, 12, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Oh, Y.S.; Kim, J.S.; Yang, D.W.; Koo, J.S.; Kim, Y.I.; Jung, H.O.; Lee, K.S. Nighttime blood pressure and white matter hyperintensities in patients with Parkinson disease. Chronobiol. Int. 2013, 30, 811–817. [Google Scholar] [CrossRef] [PubMed]
- Debette, S.; Markus, H.S. The clinical importance of white matter hyperintensities on brain magnetic resonance imaging: Systematic review and meta-analysis. BMJ 2010, 341, c3666. [Google Scholar] [CrossRef]
- Kandiah, N.; Mak, E.; Ng, A.; Huang, S.; Au, W.L.; Sitoh, Y.Y.; Tan, L.C. Cerebral white matter hyperintensity in Parkinson’s disease: A major risk factor for mild cognitive impairment. Parkinsonism Relat. Disord. 2013, 19, 680–683. [Google Scholar] [CrossRef]
- Sunwoo, M.K.; Jeon, S.; Ham, J.H.; Hong, J.Y.; Lee, J.E.; Lee, J.M.; Sohn, Y.H.; Lee, P.H. The burden of white matter hyperintensities is a predictor of progressive mild cognitive impairment in patients with Parkinson’s disease. Eur. J. Neurol. 2014, 21, 922-e50. [Google Scholar] [CrossRef] [PubMed]
- Compta, Y.; Buongiorno, M.; Bargalló, N.; Valldeoriola, F.; Muñoz, E.; Tolosa, E.; Ríos, J.; Cámara, A.; Fernández, M.; Martí, M.J. White matter hyperintensities, cerebrospinal amyloid-β and dementia in Parkinson’s disease. J. Neurol. Sci. 2016, 367, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Oka, H.; Umehara, T.; Nakahara, A.; Matsuno, H. Comparisons of cardiovascular dysautonomia and cognitive impairment between de novo Parkinson’s disease and de novo dementia with Lewy bodies. BMC Neurol. 2020, 20, 350. [Google Scholar] [CrossRef]
- Sforza, M.; Assogna, F.; Rinaldi, D.; Sette, G.; Tagliente, S.; Pontieri, F. Orthostatic hypotension acutely impairs executive functions in Parkinson’s disease. Neurol. Sci. 2018, 39, 1459–1462. [Google Scholar] [CrossRef]
- Centi, J.; Freeman, R.; Gibbons, C.H.; Neargarder, S.; Canova, A.O.; Cronin-Golomb, A. Effects of orthostatic hypotension on cognition in Parkinson disease. Neurology 2017, 88, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Pilleri, M.; Facchini, S.; Gasparoli, E.; Biundo, R.; Bernardi, L.; Marchetti, M.; Formento, P.; Antonini, A. Cognitive and MRI correlates of orthostatic hypotension in Parkinson’s disease. J. Neurol. 2013, 260, 253–259. [Google Scholar] [CrossRef]
- Allcock, L.M.; Kenny, R.A.; Mosimann, U.P.; Tordoff, S.; Wesnes, K.A.; Hildreth, A.J.; Burn, D.J. Orthostatic hypotension in Parkinson’s disease: Association with cognitive decline? Int. J. Geriatr. Psychiatry 2006, 21, 778–783. [Google Scholar] [CrossRef]
- Nicoletti, A.; Luca, A.; Baschi, R.; Cicero, C.E.; Mostile, G.; Davì, M.; La Bianca, G.; Restivo, V.; Zappia, M.; Monastero, R. Vascular risk factors, white matter lesions and cognitive impairment in Parkinson’s disease: The PACOS longitudinal study. J. Neurol. 2021, 268, 549–558. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Wen, M.C.; Ng, S.Y.; Hartono, S.; Chia, N.S.; Choi, X.; Tay, K.Y.; Au, W.L.; Chan, L.L.; Tan, E.K.; et al. Periventricular white matter hyperintensity burden and cognitive impairment in early Parkinson’s disease. Eur. J. Neurol. 2020, 27, 959–966. [Google Scholar] [CrossRef]
- Ramirez, J.; Dilliott, A.A.; Binns, M.A.; Breen, D.P.; Evans, E.C.; Beaton, D.; McLaughlin, P.M.; Kwan, D.; Holmes, M.F.; Ozzoude, M.; et al. Parkinson’s Disease, NOTCH3 Genetic Variants, and White Matter Hyperintensities. Mov. Disord. 2020, 35, 2090–2095. [Google Scholar] [CrossRef] [PubMed]
- Dadar, M.; Gee, M.; Shuaib, A.; Duchesne, S.; Camicioli, R. Cognitive and motor correlates of grey and white matter pathology in Parkinson’s disease. Neuroimage Clin. 2020, 27, 102353. [Google Scholar] [CrossRef]
- Linortner, P.; McDaniel, C.; Shahid, M.; Levine, T.F.; Tian, L.; Cholerton, B.; Poston, K.L. White Matter Hyperintensities Related to Parkinson’s Disease Executive Function. Mov. Disord. Clin. Pract. 2020, 7, 629–638. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Lee, W.J.; Chung, S.J.; Yoo, H.S.; Jung, J.H.; Baik, K.; Sohn, Y.H.; Seong, J.K.; Lee, P.H. Microstructural Connectivity is More Related to Cognition than Conventional MRI in Parkinson’s Disease. J. Parkinsons Dis. 2020. [Google Scholar] [CrossRef]
- Chahine, L.M.; Dos Santos, C.; Fullard, M.; Scordia, C.; Weintraub, D.; Erus, G.; Rosenthal, L.; Davatzikos, C.; McMillan, C.T. Modifiable vascular risk factors, white matter disease and cognition in early Parkinson’s disease. Eur. J. Neurol. 2019, 26, 246-e18. [Google Scholar] [CrossRef] [PubMed]
- Hanning, U.; Teuber, A.; Lang, E.; Trenkwalder, C.; Mollenhauer, B.; Minnerup, H. White matter hyperintensities are not associated with cognitive decline in early Parkinson’s disease—The DeNoPa cohort. Parkinsonism Relat. Disord. 2019, 69, 61–67. [Google Scholar] [CrossRef]
- Pozorski, V.; Oh, J.M.; Okonkwo, O.; Krislov, S.; Barzgari, A.; Theisen, F.; Sojkova, J.; Bendlin, B.B.; Johnson, S.C.; Gallagher, C.L. Cross-sectional and longitudinal associations between total and regional white matter hyperintensity volume and cognitive and motor function in Parkinson’s disease. Neuroimage Clin. 2019, 23, 101870. [Google Scholar] [CrossRef] [PubMed]
- Stojkovic, T.; Stefanova, E.; Soldatovic, I.; Markovic, V.; Stankovic, I.; Petrovic, I.; Agosta, F.; Galantucci, S.; Filippi, M.; Kostic, V. Exploring the relationship between motor impairment, vascular burden and cognition in Parkinson’s disease. J. Neurol. 2018, 265, 1320–1327. [Google Scholar] [CrossRef] [PubMed]
- Dadar, M.; Zeighami, Y.; Yau, Y.; Fereshtehnejad, S.M.; Maranzano, J.; Postuma, R.B.; Dagher, A.; Collins, D.L. White matter hyperintensities are linked to future cognitive decline in de novo Parkinson’s disease patients. Neuroimage Clin. 2018, 20, 892–900. [Google Scholar] [CrossRef] [PubMed]
- Ham, J.H.; Lee, J.J.; Sunwoo, M.K.; Hong, J.Y.; Sohn, Y.H.; Lee, P.H. Effect of olfactory impairment and white matter hyperintensities on cognition in Parkinson’s disease. Parkinsonism Relat. Disord. 2016, 24, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Mak, E.; Dwyer, M.G.; Ramasamy, D.P.; Au, W.L.; Tan, L.C.; Zivadinov, R.; Kandiah, N. White Matter Hyperintensities and Mild Cognitive Impairment in Parkinson’s Disease. J. Neuroimaging 2015, 25, 754–760. [Google Scholar] [CrossRef]
- Shin, J.; Choi, S.; Lee, J.E.; Lee, H.S.; Sohn, Y.H.; Lee, P.H. Subcortical white matter hyperintensities within the cholinergic pathways of Parkinson’s disease patients according to cognitive status. J. Neurol. Neurosurg. Psychiatry 2012, 83, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Kim, J.S.; Yoo, J.Y.; Song, I.U.; Kim, B.S.; Jung, S.L.; Yang, D.W.; Kim, Y.I.; Jeong, D.S.; Lee, K.S. Influence of white matter hyperintensities on the cognition of patients with Parkinson disease. Alzheimer Dis. Assoc. Disord. 2010, 24, 227–233. [Google Scholar] [CrossRef]
- Dalaker, T.O.; Larsen, J.P.; Dwyer, M.G.; Aarsland, D.; Beyer, M.K.; Alves, G.; Bronnick, K.; Tysnes, O.B.; Zivadinov, R. White matter hyperintensities do not impact cognitive function in patients with newly diagnosed Parkinson’s disease. Neuroimage 2009, 47, 2083–2089. [Google Scholar] [CrossRef] [PubMed]
- Beyer, M.K.; Aarsland, D.; Greve, O.J.; Larsen, J.P. Visual rating of white matter hyperintensities in Parkinson’s disease. Mov. Disord. 2006, 21, 223–229. [Google Scholar] [CrossRef]
- Dadar, M.; Fereshtehnejad, S.M.; Zeighami, Y.; Dagher, A.; Postuma, R.B.; Collins, D.L. White Matter Hyperintensities Mediate Impact of Dysautonomia on Cognition in Parkinson’s Disease. Mov. Disord. Clin. Pract. 2020, 7, 639–647. [Google Scholar] [CrossRef]
- Doiron, M.; Langlois, M.; Dupré, N.; Simard, M. The influence of vascular risk factors on cognitive function in early Parkinson’s disease. Int. J. Geriatr. Psychiatry 2018, 33, 288–297. [Google Scholar] [CrossRef]
- Hu, G.; Jousilahti, P.; Bidel, S.; Antikainen, R.; Tuomilehto, J. Type 2 diabetes and the risk of Parkinson’s disease. Diabetes Care 2007, 30, 842–847. [Google Scholar] [CrossRef] [PubMed]
- Simon, K.C.; Chen, H.; Schwarzschild, M.; Ascherio, A. Hypertension, hypercholesterolemia, diabetes, and risk of Parkinson disease. Neurology 2007, 69, 1688–1695. [Google Scholar] [CrossRef]
- Cereda, E.; Barichella, M.; Pedrolli, C.; Klersy, C.; Cassani, E.; Caccialanza, R.; Pezzoli, G. Diabetes and risk of Parkinson’s disease: A systematic review and meta-analysis. Diabetes Care 2011, 34, 2614–2623. [Google Scholar] [CrossRef]
- Xu, Y.; Yang, J.; Shang, H. Meta-analysis of risk factors for Parkinson’s disease dementia. Transl. Neurodegener. 2016, 5, 11. [Google Scholar] [CrossRef] [PubMed]
- Sławek, J.; Białecka, M. Homocysteine and Dementia. In Diet and Nutrition in Dementia and Cognitive Decline; Martin, C.R., Preedy, V.R., Eds.; Academic Press: Cambridge, MA, USA, 2015; Chapter 57; pp. 611–621. [Google Scholar] [CrossRef]
- Nyholm, D. Duodopa® treatment for advanced Parkinson’s disease: A review of efficacy and safety. Parkinsonism Relat. Disord. 2012, 18, 916–929. [Google Scholar] [CrossRef] [PubMed]
- Klostermann, F.; Jugel, C.; Müller, T.; Marzinzik, F. Malnutritional neuropathy under intestinal levodopa infusion. J. Neural. Transm. 2012, 119, 369–372. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Feng, H.; Peng, S.; Xiao, J.; Zhang, J. Association of plasma homocysteine, vitamin B12 and folate levels with cognitive function in Parkinson’s disease: A meta-analysis. Neurosci. Lett. 2017, 636, 190–195. [Google Scholar] [CrossRef]
- Labandeira-Garcia, J.L.; Rodriguez-Pallares, J.; Dominguez-Meijide, A.; Valenzuela, R.; Villar-Cheda, B.; Rodríguez-Perez, A.I. Dopamine-Angiotensin interactions in the basal ganglia and their relevance for Parkinson’s disease. Mov. Disord. 2013, 28, 1337–1342. [Google Scholar] [CrossRef] [PubMed]
- Milsted, A.; Barna, B.P.; Ransohoff, R.M.; Brosnihan, K.B.; Ferrario, C.M. Astrocyte cultures derived from human brain tissue express angiotensinogen mRNA. Proc. Natl. Acad. Sci. USA 1990, 87, 5720–5723. [Google Scholar] [CrossRef] [PubMed]
- Labandeira-García, J.L.; Garrido-Gil, P.; Rodriguez-Pallares, J.; Valenzuela, R.; Borrajo, A.; Rodríguez-Perez, A.I. Brain renin-angiotensin system and dopaminergic cell vulnerability. Front. Neuroanat. 2014, 8, 67. [Google Scholar] [CrossRef]
- Villar-Cheda, B.; Valenzuela, R.; Rodriguez-Perez, A.I.; Guerra, M.J.; Labandeira-Garcia, J.L. Aging-related changes in the nigral angiotensin system enhances proinflammatory and pro-oxidative markers and 6-OHDA-induced dopaminergic degeneration. Neurobiol. Aging 2012, 33, 204.e1–204.e11. [Google Scholar] [CrossRef]
- Janelidze, S.; Hertze, J.; Nägga, K.; Nilsson, K.; Nilsson, C.; Swedish BioFINDER Study Group; Wennström, M.; van Westen, D.; Blennow, K.; Zetterberg, H.; et al. Increased blood-brain barrier permeability is associated with dementia and diabetes but not amyloid pathology or APOE genotype. Neurobiol. Aging 2017, 51, 104–112. [Google Scholar] [CrossRef]
- Faucheux, B.A.; Bonnet, A.M.; Agid, Y.; Hirsch, E.C. Blood vessels change in the mesencephalon of patients with Parkinson’s disease. Lancet 1999, 353, 981–982. [Google Scholar] [CrossRef]
- Yasuhara, T.; Shingo, T.; Muraoka, K.; wen Ji, Y.; Kameda, M.; Takeuchi, A.; Yano, A.; Nishio, S.; Matsui, T.; Miyoshi, Y.; et al. The differences between high and low-dose administration of VEGF to dopaminergic neurons of in vitro and in vivo Parkinson’s disease model. Brain Res. 2005, 1038, 1–10. [Google Scholar] [CrossRef]
- Muñoz, A.; Garrido-Gil, P.; Dominguez-Meijide, A.; Labandeira-Garcia, J.L. Angiotensin type 1 receptor blockage reduces l-dopa-induced dyskinesia in the 6-OHDA model of Parkinson’s disease. Involvement of vascular endothelial growth factor and interleukin-1β. Exp. Neurol. 2014, 261, 720–732. [Google Scholar] [CrossRef] [PubMed]
- Van Dalen, J.W.; Marcum, Z.A.; Gray, S.L.; Barthold, D.; Moll van Charante, E.P.; van Gool, W.A.; Crane, P.K.; Larson, E.B.; Richard, E. Association of Angiotensin II-Stimulating Antihypertensive Use and Dementia Risk: Post Hoc Analysis of the PreDIVA Trial. Neurology 2021, 96, e67–e80. [Google Scholar] [CrossRef]
- Kehoe, P.G. The Coming of Age of the Angiotensin Hypothesis in Alzheimer’s Disease: Progress Toward Disease Prevention and Treatment? J. Alzheimers Dis. 2018, 62, 1443–1466. [Google Scholar] [CrossRef]
- Lim, E.W.; Aarsland, D.; Ffytche, D.; Taddei, R.N.; van Wamelen, D.J.; Wan, Y.M.; Tan, E.K.; Ray Chaudhuri, K.; Kings Parcog groupMDS Nonmotor Study Group. Amyloid-β and Parkinson’s disease. J. Neurol. 2019, 266, 2605–2619. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, S.; Wang, H.F.; Wang, X.; Li, J.; Xing, C.M. The association of renin-angiotensin system blockade use with the risks of cognitive impairment of aging and Alzheimer’s disease: A meta-analysis. J. Clin. Neurosci. 2016, 33, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Lipska, B.K.; Halim, N.; Ma, Q.D.; Matsumoto, M.; Melhem, S.; Kolachana, B.S.; Hyde, T.M.; Herman, M.M.; Apud, J.; et al. Functional analysis of genetic variation in catechol-O-methyltransferase (COMT): Effects on mRNA, protein, and enzyme activity in postmortem human brain. Am. J. Hum. Genet. 2004, 75, 807–821. [Google Scholar] [CrossRef] [PubMed]
- Tunbridge, E.M.; Harrison, P.J.; Warden, D.R.; Johnston, C.; Refsum, H.; Smith, A.D. Polymorphisms in the catechol-O-methyltransferase (COMT) gene influence plasma total homocysteine levels. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2008, 147B, 996–999. [Google Scholar] [CrossRef]
- Lamberti, P.; Zoccolella, S.; Iliceto, G.; Armenise, E.; Fraddosio, A.; de Mari, M.; Livrea, P. Effects of levodopa and COMT inhibitors on plasma homocysteine in Parkinson’s disease patients. Mov. Disord. 2005, 20, 69–72. [Google Scholar] [CrossRef]
- Huertas, I.; Jesús, S.; García-Gómez, F.J.; Lojo, J.A.; Bernal-Bernal, I.; Bonilla-Toribio, M.; Martín-Rodriguez, J.F.; García-Solís, D.; Gómez-Garre, P.; Mir, P. Genetic factors influencing frontostriatal dysfunction and the development of dementia in Parkinson’s disease. PLoS ONE 2017, 12, e0175560. [Google Scholar] [CrossRef]
- Bäckström, D.; Eriksson Domellöf, M.; Granåsen, G.; Linder, J.; Mayans, S.; Elgh, E.; Zetterberg, H.; Blennow, K.; Forsgren, L. Polymorphisms in dopamine-associated genes and cognitive decline in Parkinson’s disease. Acta Neurol. Scand. 2018, 137, 91–98. [Google Scholar] [CrossRef]
- Williams-Gray, C.H.; Evans, J.R.; Goris, A.; Foltynie, T.; Ban, M.; Robbins, T.W.; Brayne, C.; Kolachana, B.S.; Weinberger, D.R.; Sawcer, S.J.; et al. The distinct cognitive syndromes of Parkinson’s disease: 5 year follow-up of the CamPaIGN cohort. Brain 2009, 132, 2958–2969. [Google Scholar] [CrossRef]
- Białecka, M.; Kurzawski, M.; Roszmann, A.; Robowski, P.; Sitek, E.J.; Honczarenko, K.; Gorzkowska, A.; Budrewicz, S.; Mak, M.; Jarosz, M.; et al. Association of COMT, MTHFR, and SLC19A1(RFC-1) polymorphisms with homocysteine blood levels and cognitive impairment in Parkinson’s disease. Pharmacogenet. Genom. 2012, 22, 716–724. [Google Scholar] [CrossRef]
- Mata, I.F.; Leverenz, J.B.; Weintraub, D.; Trojanowski, J.Q.; Hurtig, H.I.; Van Deerlin, V.M.; Ritz, B.; Rausch, R.; Rhodes, S.L.; Factor, S.A.; et al. APOE, MAPT, and SNCA genes and cognitive performance in Parkinson disease. JAMA Neurol. 2014, 71, 1405–1412. [Google Scholar] [CrossRef]
- Zlokovic, B.V. Cerebrovascular effects of apolipoprotein E: Implications for Alzheimer disease. JAMA Neurol. 2013, 70, 440–444. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Chen, P.C.; Poole, C. APOE-[epsilon]2 allele associated with higher prevalence of sporadic Parkinson disease. Neurology 2004, 62, 2198–2202. [Google Scholar] [CrossRef] [PubMed]
- Williams-Gray, C.H.; Goris, A.; Saiki, M.; Foltynie, T.; Compston, D.A.; Sawcer, S.J.; Barker, R.A. Apolipoprotein E genotype as a risk factor for susceptibility to and dementia in Parkinson’s disease. J. Neurol. 2009, 256, 493–498. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.J.; Hauser, M.A.; Scott, W.K.; Martin, E.R.; Booze, M.W.; Qin, X.J.; Walter, J.W.; Nance, M.A.; Hubble, J.P.; Koller, W.C.; et al. Apolipoprotein E controls the risk and age at onset of Parkinson disease. Neurology 2004, 62, 2005–2009. [Google Scholar] [CrossRef]
- Pankratz, N.; Byder, L.; Halter, C.; Rudolph, A.; Shults, C.W.; Conneally, P.M.; Foroud, T.; Nichols, W.C. Presence of an APOE4 allele results in significantly earlier onset of Parkinson’s disease and a higher risk with dementia. Mov. Disord. 2006, 21, 45–49. [Google Scholar] [CrossRef]
- Alata, W.; Ye, Y.; St-Amour, I.; Vandal, M.; Calon, F. Human apolipoprotein E ε4 expression impairs cerebral vascularization and blood-brain barrier function in mice. J. Cereb. Blood Flow Metab. 2015, 35, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Bell, R.D.; Winkler, E.A.; Singh, I.; Sagare, A.P.; Deane, R.; Wu, Z.; Holtzman, D.M.; Betsholtz, C.; Armulik, A.; Sallstrom, J.; et al. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature 2012, 485, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Pierzchlińska, A.; Białecka, M.; Kurzawski, M.; Sławek, J. The impact of Apolipoprotein E alleles on cognitive performance in patients with Parkinson’s disease. Neurol. Neurochir. Pol. 2018, 52, 477–482. [Google Scholar] [CrossRef]
- Pang, S.; Li, J.; Zhang, Y.; Chen, J. Meta-Analysis of the Relationship between the APOE Gene and the Onset of Parkinson’s Disease Dementia. Parkinsons Dis. 2018, 2018, 9497147. [Google Scholar] [CrossRef] [PubMed]
- Koukourakis, M.I.; Papazoglou, D.; Giatromanolaki, A.; Bougioukas, G.; Maltezos, E.; Sivridis, E. VEGF gene sequence variation defines VEGF gene expression status and angiogenic activity in non-small cell lung cancer. Lung Cancer 2004, 46, 293–298. [Google Scholar] [CrossRef]
- Renner, W.; Kotschan, S.; Hoffmann, C.; Obermayer-Pietsch, B.; Pilger, E. A common 936 C/T mutation in the gene for vascular endothelial growth factor is associated with vascular endothelial growth factor plasma levels. J. Vasc. Res. 2000, 37, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Mihci, E.; Ozkaynak, S.S.; Sallakci, N.; Kizilay, F.; Yavuzer, U. VEGF polymorphisms and serum VEGF levels in Parkinson’s disease. Neurosci. Lett. 2011, 494, 1–5. [Google Scholar] [CrossRef]
- Del Bo, R.; Scarlato, M.; Ghezzi, S.; Martinelli Boneschi, F.; Fenoglio, C.; Galbiati, S.; Virgilio, R.; Galimberti, D.; Galimberti, G.; Crimi, M.; et al. Vascular endothelial growth factor gene variability is associated with increased risk for AD. Ann. Neurol. 2005, 57, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, X.A.; Alvarez, I.; Aleixandre, M.; Linares, C.; Muresanu, D.; Winter, S.; Moessler, H. Severity-Related Increase and Cognitive Correlates of Serum VEGF Levels in Alzheimer’s Disease ApoE4 Carriers. J. Alzheimers Dis. 2018, 63, 1003–1013. [Google Scholar] [CrossRef]
- Takeuchi, H.; Tomita, H.; Taki, Y.; Kikuchi, Y.; Ono, C.; Yu, Z.; Sekiguchi, A.; Nouchi, R.; Kotozaki, Y.; Nakagawa, S.; et al. The VEGF gene polymorphism impacts brain volume and arterial blood volume. Hum. Brain Mapp. 2017, 38, 3516–3526. [Google Scholar] [CrossRef]
- Donzuso, G.; Monastero, R.; Cicero, C.E.; Luca, A.; Mostile, G.; Giuliano, L.; Baschi, R.; Caccamo, M.; Gagliardo, C.; Palmucci, S.; et al. Neuroanatomical changes in early Parkinson’s disease with mild cognitive impairment: A VBM study; the Parkinson’s Disease Cognitive Impairment Study (PaCoS). Neurol. Sci. 2021. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhang, Y.; Han, X.; Li, X.; Xue, L.; Xie, A. Association of VEGF gene polymorphisms with sporadic Parkinson’s disease in Chinese Han population. Neurol. Sci. 2016, 37, 1923–1929. [Google Scholar] [CrossRef]
- Moore, A.M.; Mahoney, E.; Dumitrescu, L.; De Jager, P.L.; Koran, M.; Petyuk, V.A.; Robinson, R.A.; Ruderfer, D.M.; Cox, N.J.; Schneider, J.A.; et al. APOE ε4-specific associations of VEGF gene family expression with cognitive aging and Alzheimer’s disease. Neurobiol. Aging 2020, 87, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Perez-Lloret, S.; Otero-Losada, M.; Toblli, J.E.; Capani, F. Renin-angiotensin system as a potential target for new therapeutic approaches in Parkinson’s disease. Expert Opin. Investig. Drugs 2017, 26, 1163–1173. [Google Scholar] [CrossRef]
- Rigat, B.; Hubert, C.; Corvol, P.; Soubrier, R. PCR detection of the insertion/deletion polymorphism of the human angiotensin converting enzyme gene (DCP1) (dipeptidyl carboxypeptidase 1). Nucleic Acids Res. 1992, 20, 1433. [Google Scholar] [CrossRef] [PubMed]
- Bonnardeaux, A.; Davies, E.; Jeunemaitre, X.; Féry, I.; Charru, A.; Clauser, E.; Tiret, L.; Cambien, F.; Corvol, P.; Soubrier, F. Angiotensin II type 1 receptor gene polymorphisms in human essential hypertension. Hypertension 1994, 24, 63–69. [Google Scholar] [CrossRef]
- Schmieder, R.E.; Erdmann, J.; Delles, C.; Jacobi, J.; Fleck, E.; Hilgers, K.; Regitz-Zagrosek, V. Effect of the angiotensin II type 2-receptor gene (+1675 G/A) on left ventricular structure in humans. J. Am. Coll. Cardiol. 2001, 37, 175–182. [Google Scholar] [CrossRef]
- Caulfield, M.; Lavender, P.; Farrall, M.; Munroe, P.; Lawson, M.; Turner, P.; Clark, A.J. Linkage of the angiotensinogen gene to essential hypertension. N. Engl. J. Med. 1994, 330, 1629–1633. [Google Scholar] [CrossRef]
- Mellick, G.D.; Buchanan, D.D.; McCann, S.J.; Davis, D.R.; Le Couteur, D.G.; Chan, D.; Johnson, A.G. The ACE deletion polymorphism is not associated with Parkinson’s disease. Eur. Neurol. 1999, 41, 103–106. [Google Scholar] [CrossRef]
- Pascale, E.; Purcaro, C.; Passarelli, E.; Guglielmi, R.; Vestri, A.R.; Passarelli, F.; Meco, G. Genetic polymorphism of Angiotensin-Converting Enzyme is not associated with the development of Parkinson’s disease and of L-dopa-induced adverse effects. J. Neurol. Sci. 2009, 276, 18–21. [Google Scholar] [CrossRef]
- Lin, J.J.; Yueh, K.C.; Chang, D.C.; Lin, S.Z. Association between genetic polymorphism of angiotensin-converting enzyme gene and Parkinson’s disease. J. Neurol. Sci. 2002, 199, 25–29. [Google Scholar] [CrossRef]
- Gustafson, D.R.; Melchior, L.; Eriksson, E.; Sundh, V.; Blennow, K.; Skoog, I. The ACE Insertion Deletion polymorphism relates to dementia by metabolic phenotype, APOEepsilon4, and age of dementia onset. Neurobiol. Aging 2010, 31, 910–916. [Google Scholar] [CrossRef]
- Zettergren, A.; Kern, S.; Gustafson, D.; Gudmundsson, P.; Sigström, R.; Östling, S.; Eriksson, E.; Zetterberg, H.; Blennow, K.; Skoog, I. The ACE Gene Is Associated with Late-Life Major Depression and Age at Dementia Onset in a Population-Based Cohort. Am. J. Geriatr. Psychiatry 2017, 25, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Zannas, A.S.; McQuoid, D.R.; Payne, M.E.; MacFall, J.R.; Ashley-Koch, A.; Steffens, D.C.; Potter, G.G.; Taylor, W.D. Association of gene variants of the renin-angiotensin system with accelerated hippocampal volume loss and cognitive decline in old age. Am. J. Psychiatry 2014, 171, 1214–1221. [Google Scholar] [CrossRef] [PubMed]
- Purandare, N.; Oude Voshaar, R.C.; Davidson, Y.; Gibbons, L.; Hardicre, J.; Byrne, J.; McCollum, C.; Jackson, A.; Burns, A.; Mann, D.M. Deletion/insertion polymorphism of the angiotensin-converting enzyme gene and white matter hyperintensities in dementia: A pilot study. J. Am. Geriatr. Soc. 2006, 54, 1395–1400. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Jin, F.; Yang, Z.; Lu, Z.; Kan, R.; Li, S.; Zheng, C.; Wang, L. The insertion polymorphism in angiotensin-converting enzyme gene associated with the APOE epsilon 4 allele increases the risk of late-onset Alzheimer disease. J. Mol. Neurosci. 2006, 30, 267–271. [Google Scholar] [CrossRef]
- Assareh, A.A.; Mather, K.A.; Crawford, J.D.; Wen, W.; Anstey, K.J.; Easteal, S.; Tan, X.; Mack, H.A.; Kwok, J.B.; Schofield, P.R.; et al. Renin-angiotensin system genetic polymorphisms and brain white matter lesions in older Australians. Am. J. Hypertens. 2014, 27, 1191–1198. [Google Scholar] [CrossRef] [PubMed]
- Rigat, B.; Hubert, C.; Alhenc-Gelas, F.; Cambien, F.; Corvol, P.; Soubrier, F. An insertion/deletion polymorphism in the angiotensin I-converting enzyme gene accounting for half the variance of serum enzyme levels. J. Clin. Investig. 1990, 86, 1343–1346. [Google Scholar] [CrossRef]
- Rai, V. Methylenetetrahydrofolate Reductase (MTHFR) C677T Polymorphism and Alzheimer Disease Risk: A Meta-Analysis. Mol. Neurobiol. 2017, 54, 1173–1186. [Google Scholar] [CrossRef] [PubMed]
- Wright, S.M.; Jensen, S.L.; Cockriel, K.L.; Davis, B.; Tschanz, J.T.; Munger, R.G.; Corcoran, C.D.; Kauwe, J. Association study of rs3846662 with Alzheimer’s disease in a population-based cohort: The Cache County Study. Neurobiol. Aging 2019, 84, 242.e1–242.e6. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Blood Pressure Variability | |||
|---|---|---|---|
| Authors (year), Type of the Study | Subjects | Factor | Outcome |
| Oka et al. (2020) [47], retrospective study | PD = 75 (de novo) DLB = 24 | Circadian blood pressure variability. Supine hypertension. | Better performance assessed with MMSE positively associated with the percentage of nocturnal BP fall in PD. No correlation with SH. Nocturnal BP fall (%) positively associated with better performance in Frontal Assessment Battery. SH negatively correlated with Frontal Assessment Battery. |
| Sforza et al. (2018) [48], retrospective study | PD = 28 | Orthostatic hypotension. | In upright position, PD-OH(+) performed worse at the Stroop’s test word reading time and number of errors at the interference section compared to PD-OH(−). |
| Tanaka et al. (2018) [20], retrospective study | PD-NCI = 110 PDD = 27 | Circadian blood pressure variability. Orthostatic hypotension. | The riser pattern associated with dementia. Coexistence of the riser pattern and OH more associated with dementia than the riser pattern alone. |
| Centi et al. (2017) [49], retrospective study | PD-OH(+) = 18 PD-OH(−) = 19 controls = 18 | Orthostatic hypotension. | Upright posture correlated with the deficits in sustained attention and response inhibition, reduced semantic fluency and verbal memory in the PD-OH(+). OH correlated with the deficits in executive function, memory and visuospatial function. |
| Anang et al. (2014) [18], prospective study | PD = 80 (dementia free at baseline) | Orthostatic hypotension. | OH strongly associated with dementia risk. |
| Pilleri et al. (2013) [50], retrospective study | PD = 48 | Orthostatic hypotension. | PD-OH(+) performed significantly worse in sustained attention, visuospatial and verbal memory, compared with PD-OH(−). |
| Kim et al. (2012) [17], retrospective study | PD-NCI = 25 PD-MCI = 48 PDD = 14 | Circadian blood pressure variability, supine hypertension and orthostatic hypotension. | The OH group had more severe impairment in verbal immediate/delayed memory. Dementia significantly more prevalent in patients having OH, SH or OH + SH. Non-dipping not associated with cognitive impairment. |
| Allcock et al. (2006) [51], retrospective study | PD-OH(+) = 87 PD-OH(−) = 88 | Orthostatic hypotension. | OH(+) subjects worse in sustained attention and visual episodic memory. OH not associated with the MMSE score, the prevalence of dementia, or the simple and choice reaction times, working memory or long term memory. |
| White Matter Hyperintensities | |||
| Authors (Year), Type of the Study | Subjects | Factor | Outcome |
| Nicoletti et al. (2021) [52], prospective study | PD-NCI = 84 PD-MCI = 55 | WMH | WMH was a predictor of PDD development. |
| Huang et al. (2020) [53], retrospective study | Early PD: PD-NCI = 81 PD-MCI = 94 | WMH | PD-MCI associated with the periventricular WMH but not with total WMH. Periventricular WMH associated with worse executive function and visuospatial function. |
| Ramirez et al. (2020) [54], retrospective study | PD = 139 | WMH | WMH negatively associated with global cognition. |
| Dadar et al. (2020) [55], prospective study | PD = 50 controls = 45 | WMH | No correlation between WMH and total Dementia Rating Scale. Greater WMH burden in patients diagnosed with dementia at 36 months. |
| Linortner et al. (2020) [56], retrospective study | PD = 85 controls = 18 | WMH | Dementia and executive impairment significantly more prevalent in PD patients with WMH than without WMH. WMH associated with worse performance in Symbol Digit Modalities and Stroop tests. |
| Lee et al. (2020) [57], retrospective study | PD = 136 (de novo) | WMH | Performance in language function, frontal/executive and visual memory associated with the severity of WMH. |
| Chahine et al. (2019) [58], prospective study | PD = 141 controls = 63 | WMH | Annual rate of change in global cognition correlated with WMH. Higher temporal WMH associated with greater decline over time in verbal memory. |
| Hanning et al. (2019) [59], prospective study | Drug-naïve: PD-NCI = 79 PD-MCI = 29 controls = 107 | WMH (volume and CHIPS score) | No association between global or localised WMH and cognitive decline, both cross-sectional and longitudinal. |
| Pozorski et al. (2019) [60], prospective study | PD = 29 controls = 42 | WMH | Greater regional and global WMH at baseline more strongly associated with lower executive function in PD than in controls. Increased regional WMH more strongly associated with impaired memory performance in PD relative to controls. Longitudinally, no associations between WMH and cognitive change. |
| Stojkovic et al. (2018) [61], retrospective study | PD-NCI = 49 PD-MCI = 61 PDD = 23 | WMH | PDD patients had significantly greater whole brain WMH than PD-NCI subjects. |
| Dadar et al. (2018) [62], prospective study | PD = 365 (de novo) controls = 174 | WMH | PD subjects with greater WMH had significantly more severe cognitive decline than PD subjects with low WMH load or controls with high WMH load. |
| Ham et al. (2016) [63], retrospective study | PD = 171 (non-demented) | WMH | Total WMH and deep WMH associated with worse performance in semantic fluency. |
| Mak et al. (2015) [64], retrospective study | PD-NCI = 65 PD-MCI = 25 | WMH | Greater total and periventricular WMH in PD-MCI than in PD-NCI. Spatial distribution of WMH associated with global cognition, performance on the Frontal Assessment Battery and Fruit Fluency. |
| Sunwoo et al. (2014) [45], prospective study | PD-NCI = 46 PD-MCI = 65 | WMH (volume and CHIPS score) | The progression from PD-MCI to PDD correlated with WMH volume and CHIPS score. In PD-MCI patients WMH volume and CHIPS score associated with longitudinal decline in general cognition, semantic fluency and Stroop test scores. |
| Kandiah et al. (2013) [44], prospective study | PD-NCI = 67 PD-MCI = 24 | WMH | PD-MCI patients had significantly greater volume of periventricular and deep subcortical WMH than PD-NCI. Regional WMH significantly greater among PD-MCI in the frontal, parietal and occipital regions. |
| Sławek et al. (2013) [41], retrospective study | PD-NCI = 135 PDD = 57 controls = 184 | WMH | WMH significantly greater in PDD than PD-NCI group. |
| Kim et al. (2012) [17], retrospective study | PD-NCI = 25 PD-MCI = 48 PDD = 14 | WMH (CHIPS score) | The severity of WMH in the periventricular and subcortical white matter higher in PDD than in PD-NCI or PD-MCI. No difference in WMH between PD-NCI and PD-MCI. |
| Shin et al. (2012) [65], retrospective study | PD-NCI = 44 PD-MCI = 87 PDD = 40 | WMH (CHIPS score) | The CHIPS score significantly higher in PDD than in PD-NCI or PD-MCI. WMH negatively associated with performance in MMSE. The CHIPS score correlated with the performance in contrasting programme and forward digit span tests. |
| Lee et al. (2010) [66], retrospective study | PD-NCI = 11 PD-MCI = 25 PDD = 35 | WMH | Greater total and periventricular WMH in the PDD group compared to PD-MCI and PD-NCI groups. No difference in WMH between PD-MCI and PD-NCI groups. |
| Dalaker et al. (2009) [67], retrospective study | Drug-naïve: PD-NCI = 133 PD-MCI = 30 controls = 102 | WMH | No differences between the groups in total volume or spatial distribution of WMH. No correlation between WMH and cognitive functions. |
| Beyer et al. (2006) [68], retrospective study | PD-NCI = 19 PDD = 16 controls = 20 | WMH | PDD group had significantly more WMH in deep white matter and periventricular areas than the PD-NCI group. |
| Interaction between WMH and BP Variability | |||
| Authors (year), type of the study | Subjects | Factor | Outcome |
| Dadar et al. (2020) [69], prospective study | PD = 365 (de novo) controls = 174 | WMH Orthostatic hypotension. | A correlation between WMH burden and worse Montreal Cognitive Assessment (MoCA) score in PD over time. WMH linked with diastolic OH. Direct effect of diastolic OH on the rate of cognitive decline via WMH burden. |
| Oh et al. (2013) [39], retrospective study | PD = 117 | WMH Orthostatic hypotension, supine hypertension. | Orthostatic hypotension and supine hypertension correlated with WMH score. |
| Oh et al. (2013) [42], retrospective study | Drug-naïve: PD = 129 | WMH Circadian blood pressure variability. | Nocturnal hypertension associated with WMH in the basal ganglia. No influence of the non-dipping pattern on WMH. Nighttime systolic BP closely correlated with WMH. |
| Kim et al. (2012) [17], retrospective study | PD-NCI = 25 PD-MCI = 48 PDD = 14 | WMH (CHIPS score) Circadian blood pressure variability, supine hypertension and orthostatic hypotension. | WMH significantly more severe in patients having OH, SH or OH + SH. No difference in WMH between the dippers and non-dippers. |
| Gene | Name of the Protein | Function of the Protein | Role in Cognitive Decline | References |
|---|---|---|---|---|
| COMT | catechol-O-methyltransferase | metabolism of catecholamines and l-dopa, involved in Hcy synthesis | polymorphism associated with Hcy overproduction | [92] |
| APOE | apolipoprotein E | component of several lipoproteins | APOE4 variant linked to Aβ accumulation and BBB disruption | [105] |
| VEGF | vascular endothelial growth factor | angiogenic activity, essential in vasculogenesis. | microvascular pathologies, BBB disruption | [23] |
| MTHFR | methylenetetrahydrofolate reductase | Hcy and folate metabolism | polymorphism correlated with HHcy | [132] |
| ACE | angiotensin converting enzyme | catalyzes AII synthesis; its inhibitors lower BP | Aβ degradation; polymorphism associated with WMH | [87,130] |
| AGT | angiotensinogen | precursor of all components within the RAA system | polymorphism associated with WMH | [130] |
| AGTR1 | angiotensin receptor type 1 | AII receptor | oxidative stress and neuroinflammation; polymorphisms associated with hippocampal atrophy | [79,127] |
| AGTR2 | angiotensin receptor type 2 | AII receptor | possible neuroprotection | [87] |
| HMGCR | HMG-CoA reductase | rate-controlling enzyme in the cholesterol synthesis pathway | polymorphism decreases cholesterol production | [133] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pierzchlińska, A.; Kwaśniak-Butowska, M.; Sławek, J.; Droździk, M.; Białecka, M. Arterial Blood Pressure Variability and Other Vascular Factors Contribution to the Cognitive Decline in Parkinson’s Disease. Molecules 2021, 26, 1523. https://doi.org/10.3390/molecules26061523
Pierzchlińska A, Kwaśniak-Butowska M, Sławek J, Droździk M, Białecka M. Arterial Blood Pressure Variability and Other Vascular Factors Contribution to the Cognitive Decline in Parkinson’s Disease. Molecules. 2021; 26(6):1523. https://doi.org/10.3390/molecules26061523
Chicago/Turabian StylePierzchlińska, Anna, Magdalena Kwaśniak-Butowska, Jarosław Sławek, Marek Droździk, and Monika Białecka. 2021. "Arterial Blood Pressure Variability and Other Vascular Factors Contribution to the Cognitive Decline in Parkinson’s Disease" Molecules 26, no. 6: 1523. https://doi.org/10.3390/molecules26061523
APA StylePierzchlińska, A., Kwaśniak-Butowska, M., Sławek, J., Droździk, M., & Białecka, M. (2021). Arterial Blood Pressure Variability and Other Vascular Factors Contribution to the Cognitive Decline in Parkinson’s Disease. Molecules, 26(6), 1523. https://doi.org/10.3390/molecules26061523