
The Complex Structure of Protein AaLpxC from Aquifex aeolicus with ACHN-975 Molecule Suggests an Inhibitory Mechanism at Atomic-Level against Gram-Negative Bacteria

 ,
,
Abstract
1. Introduction
2. Results
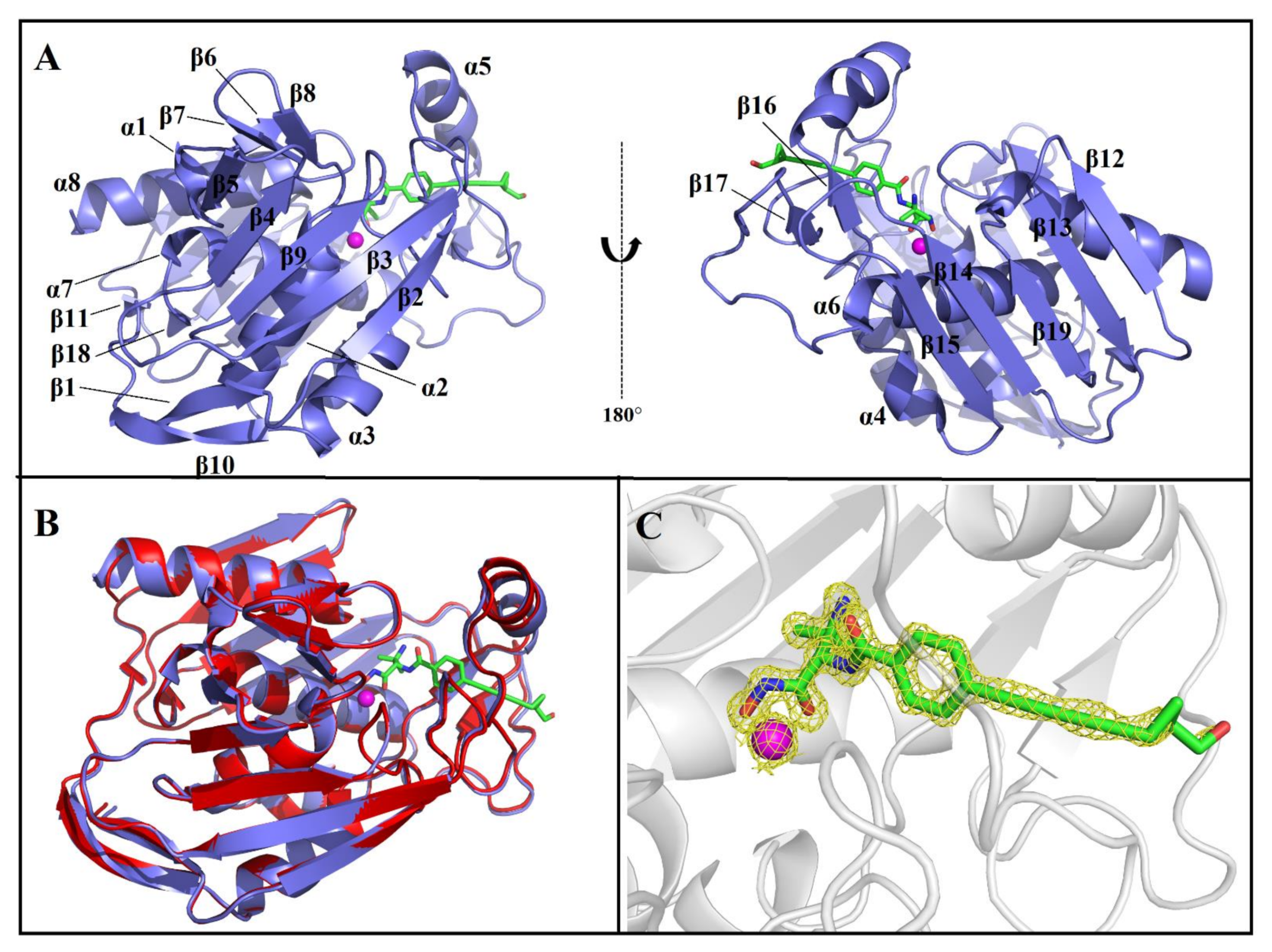
2.1. Overall Structure of AaLpxC/ACHN-975 Complex
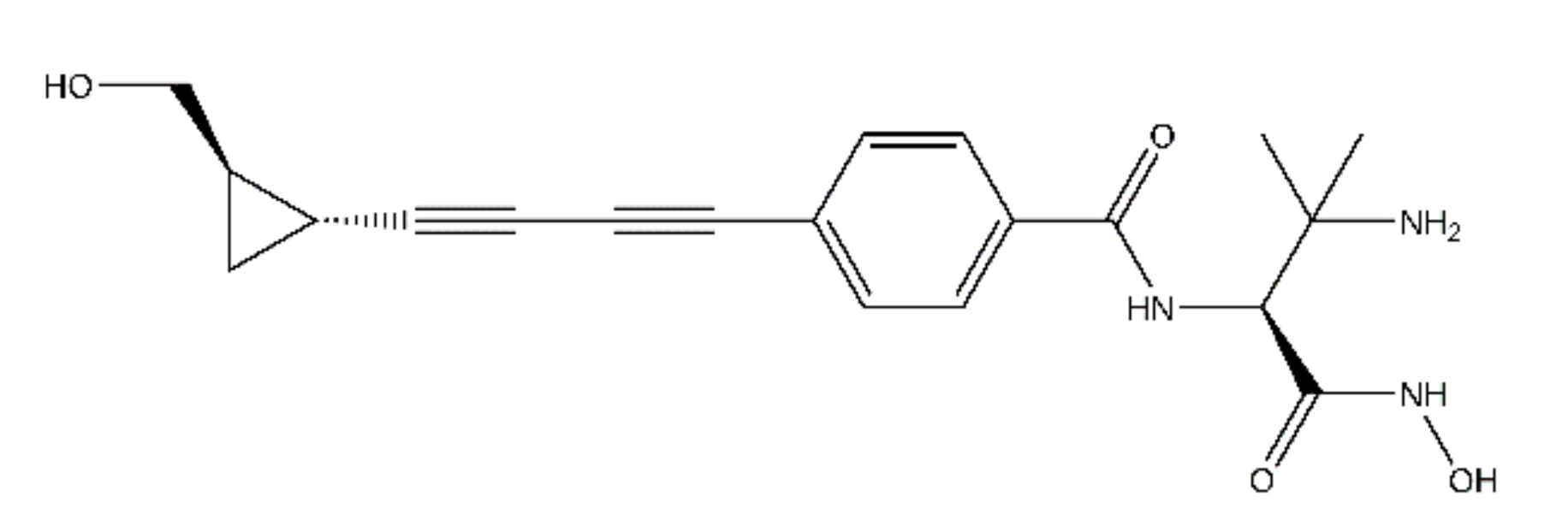
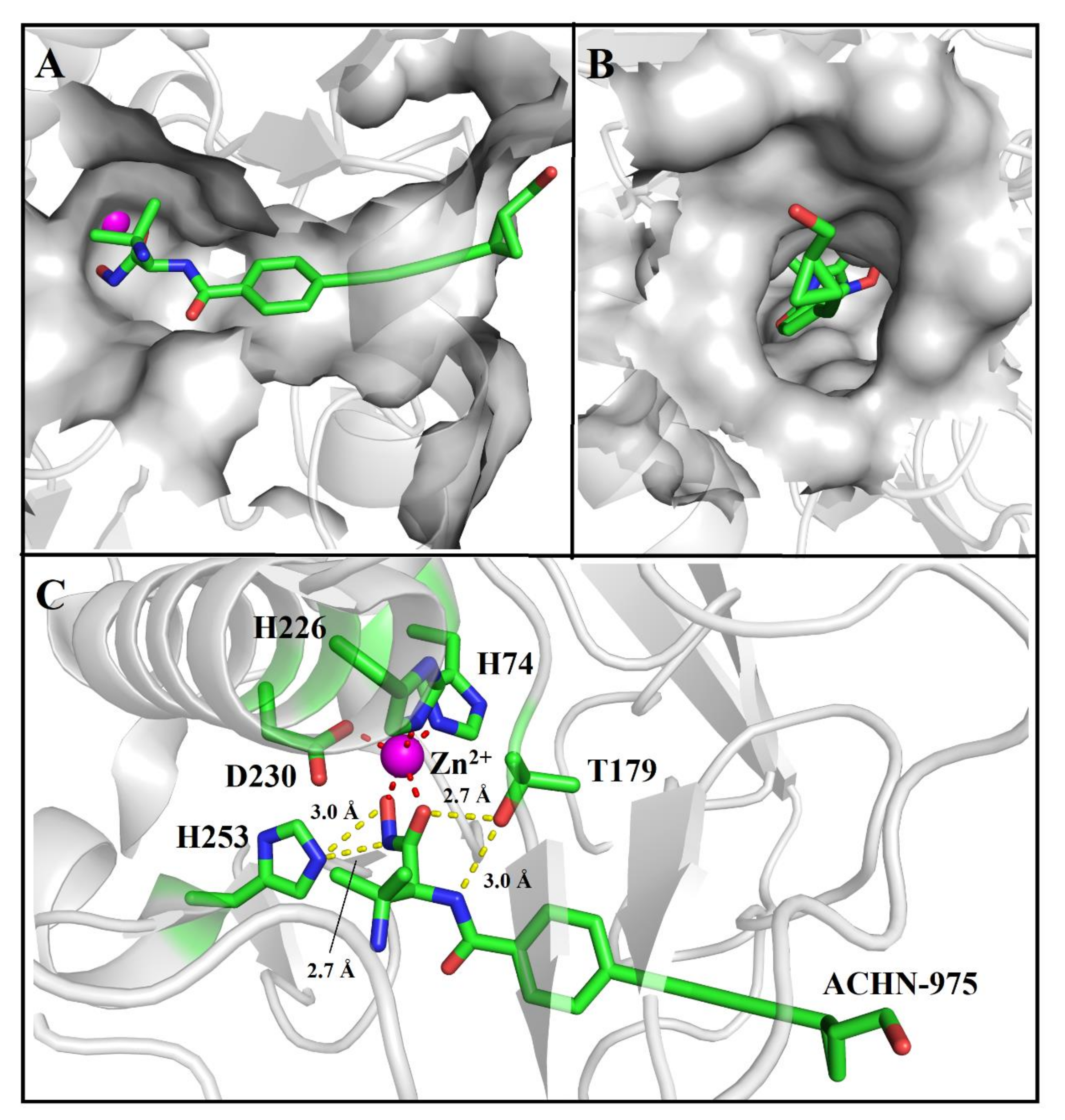
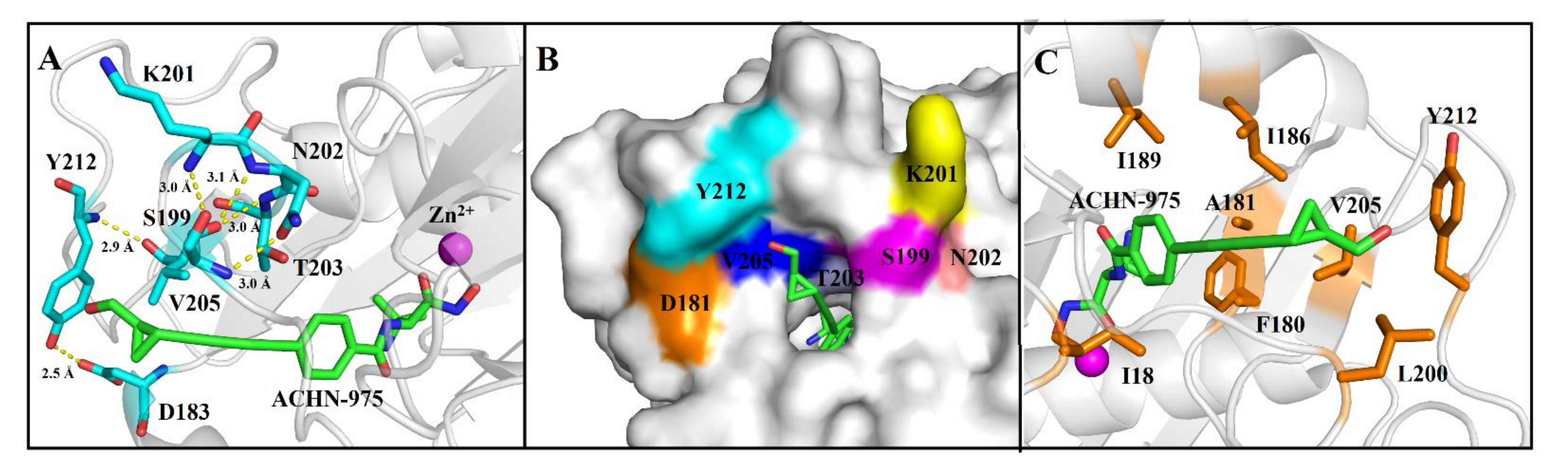
2.2. ACHN-975 Binding
2.3. Charting the Thermodynamic Parameters of the Complexes via ITC
3. Discussion
4. Materials and Methods
4.1. Cloning and Site-Directed Mutagenesis of AaLpxC
4.2. Protein Expression and Purification
4.3. Crystallization and Data Collection
4.4. Structural Determination and Refinement
4.5. Isothermal Titration Calorimetry
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brown, E.D.; Wright, G.D. Antibacterial drug discovery in the resistance era. Nature 2016, 529, 336–343. [Google Scholar] [CrossRef]
- Rice, L.B. Federal funding for the study of antimicrobial resistance in nosocomial pathogens: No ESKAPE. J. Infect. Dis 2008, 197, 1079–1081. [Google Scholar] [CrossRef]
- Rice, L.B. Progress and challenges in implementing the research on ESKAPE pathogens. Infect. Control Hosp. Epidemiol. 2010, 31 (Suppl. 1), S7–S10. [Google Scholar] [CrossRef] [PubMed]
- Vaara, M. Outer membrane permeability barrier to azithromycin, clarithromycin, and roxithromycin in gram-negative enteric bacteria. Antimicrob. Agents Chemother. 1993, 37, 354–356. [Google Scholar] [CrossRef]
- Coggins, B.E.; Li, X.; McClerren, A.L.; Hindsgaul, O.; Raetz, C.R.; Zhou, P. Structure of the LpxC deacetylase with a bound substrate-analog inhibitor. Nat. Struct. Biol. 2003, 10, 645–651. [Google Scholar] [CrossRef] [PubMed]
- Coggins, B.E.; McClerren, A.L.; Jiang, L.; Li, X.; Rudolph, J.; Hindsgaul, O.; Raetz, C.R.; Zhou, P. Refined solution structure of the LpxC-TU-514 complex and pKa analysis of an active site histidine: Insights into the mechanism and inhibitor design. Biochemistry 2005, 44, 1114–1126. [Google Scholar] [CrossRef] [PubMed]
- Lemaitre, N.; Liang, X.; Najeeb, J.; Lee, C.J.; Titecat, M.; Leteurtre, E.; Simonet, M.; Toone, E.J.; Zhou, P.; Sebbane, F. Curative Treatment of Severe Gram-Negative Bacterial Infections by a New Class of Antibiotics Targeting LpxC. mBio 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.B. Confronting the challenges of discovery of novel antibacterial agents. Bioorg. Med. Chem. Lett. 2014, 24, 3683–3689. [Google Scholar] [CrossRef]
- Whittington, D.A.; Rusche, K.M.; Shin, H.; Fierke, C.A.; Christianson, D.W. Crystal structure of LpxC, a zinc-dependent deacetylase essential for endotoxin biosynthesis. Proc. Natl. Acad. Sci. USA 2003, 100, 8146–8150. [Google Scholar] [CrossRef]
- Liang, X.; Lee, C.J.; Chen, X.; Chung, H.S.; Zeng, D.; Raetz, C.R.; Li, Y.; Zhou, P.; Toone, E.J. Syntheses, structures and antibiotic activities of LpxC inhibitors based on the diacetylene scaffold. Bioorg. Med. Chem. 2011, 19, 852–860. [Google Scholar] [CrossRef]
- Mochalkin, I.; Knafels, J.D.; Lightle, S. Crystal structure of LpxC from Pseudomonas aeruginosa complexed with the potent BB-78485 inhibitor. Protein Sci. 2008, 17, 450–457. [Google Scholar] [CrossRef]
- Cole, K.E.; Gattis, S.G.; Angell, H.D.; Fierke, C.A.; Christianson, D.W. Structure of the metal-dependent deacetylase LpxC from Yersinia enterocolitica complexed with the potent inhibitor CHIR-090. Biochemistry 2011, 50, 258–265. [Google Scholar] [CrossRef]
- Barb, A.W.; Jiang, L.; Raetz, C.R.; Zhou, P. Structure of the deacetylase LpxC bound to the antibiotic CHIR-090: Time-dependent inhibition and specificity in ligand binding. Proc. Natl. Acad. Sci. USA 2007, 104, 18433–18438. [Google Scholar] [CrossRef]
- Barb, A.W.; Leavy, T.M.; Robins, L.I.; Guan, Z.; Six, D.A.; Zhou, P.; Hangauer, M.J.; Bertozzi, C.R.; Raetz, C.R. Uridine-based inhibitors as new leads for antibiotics targeting Escherichia coli LpxC. Biochemistry 2009, 48, 3068–3077. [Google Scholar] [CrossRef]
- Kalinin, D.V.; Holl, R. Insights into the Zinc-Dependent Deacetylase LpxC: Biochemical Properties and Inhibitor Design. Curr. Top. Med. Chem. 2016, 16, 2379–2430. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.J.; Liang, X.; Chen, X.; Zeng, D.; Joo, S.H.; Chung, H.S.; Barb, A.W.; Swanson, S.M.; Nicholas, R.A.; Li, Y.; et al. Species-specific and inhibitor-dependent conformations of LpxC: Implications for antibiotic design. Chem. Biol. 2011, 18, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.J.; Liang, X.; Wu, Q.; Najeeb, J.; Zhao, J.; Gopalaswamy, R.; Titecat, M.; Sebbane, F.; Lemaitre, N.; Toone, E.J.; et al. Drug design from the cryptic inhibitor envelope. Nat. Commun. 2016, 7, 10638. [Google Scholar] [CrossRef]
- Liang, X.; Lee, C.J.; Zhao, J.; Toone, E.J.; Zhou, P. Synthesis, structure, and antibiotic activity of aryl-substituted LpxC inhibitors. J. Med. Chem. 2013, 56, 6954–6966. [Google Scholar] [CrossRef] [PubMed]
- Erwin, A.L. Antibacterial Drug Discovery Targeting the Lipopolysaccharide Biosynthetic Enzyme LpxC. Cold Spring Harb. Perspect. Med. 2016, 6, a025304. [Google Scholar] [CrossRef] [PubMed]
- Raetz, C.R.; Whitfield, C. Lipopolysaccharide endotoxins. Annu. Rev. Biochem. 2002, 71, 635–700. [Google Scholar] [CrossRef]
- Barb, A.W.; Zhou, P. Mechanism and inhibition of LpxC: An essential zinc-dependent deacetylase of bacterial lipid A synthesis. Curr. Pharm. Biotechnol. 2008, 9, 9–15. [Google Scholar] [PubMed]
- Buetow, L.; Dawson, A.; Hunter, W.N. The nucleotide-binding site of Aquifex aeolicus LpxC. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2006, 62, 1082–1086. [Google Scholar] [CrossRef] [PubMed]
- Jackman, J.E.; Raetz, C.R.; Fierke, C.A. Site-directed mutagenesis of the bacterial metalloamidase UDP-(3-O-acyl)-N-acetylglucosamine deacetylase (LpxC). Identification of the zinc binding site. Biochemistry 2001, 40, 514–523. [Google Scholar] [CrossRef] [PubMed]
- Mansoor, U.F.; Vitharana, D.; Reddy, P.A.; Daubaras, D.L.; McNicholas, P.; Orth, P.; Black, T.; Siddiqui, M.A. Design and synthesis of potent Gram-negative specific LpxC inhibitors. Bioorg. Med. Chem. Lett. 2011, 21, 1155–1161. [Google Scholar] [CrossRef]
- Cohen, F.; Aggen, J.B.; Andrews, L.D.; Assar, Z.; Boggs, J.; Choi, T.; Dozzo, P.; Easterday, A.N.; Haglund, C.M.; Hildebrandt, D.J.; et al. Optimization of LpxC Inhibitors for Antibacterial Activity and Cardiovascular Safety. ChemMedChem 2019, 14, 1560–1572. [Google Scholar] [CrossRef]
- Wang, Q.; Yu, F.; Cui, Y.; Zhang, K.; Pan, Q.; Zhong, C.; Liu, K.; Zhou, H.; Sun, B.; He, J. Mini-beam modes on standard MX beamline BL17U at SSRF. Rev. Sci. Instrum. 2017, 88, 073301. [Google Scholar] [CrossRef]
- Otwinowski, Z.; Minor, W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997, 276, 307–326. [Google Scholar]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef]
- Adams, P.D.; Grosse-Kunstleve, R.W.; Hung, L.W.; Ioerger, T.R.; McCoy, A.J.; Moriarty, N.W.; Read, R.J.; Sacchettini, J.C.; Sauter, N.K.; Terwilliger, T.C. PHENIX: Building new software for automated crystallographic structure determination. Acta Crystallogr. D Biol. Crystallogr. 2002, 58, 1948–1954. [Google Scholar] [CrossRef]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef]
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014, 42, W320-4. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data Collection | AaLpxC·ACHN-975 |
|---|---|
| Space group | P61 |
| Cell dimensions | |
| a, b, c (Å) | 65.569, 65.569, 131.595 |
| α, β, γ (°) | 90.000, 90.000, 120.000 |
| Resolution (Å) | 50–1.21 (1.25–1.21) |
| Rmerge | 0.084 (0.600) |
| I/ϭ | 35.0 (5.4) |
| Completeness (%) | 95.3 (91.0) |
| Redundancy | 19.6 (19.9) |
| Refinement | |
| Resolution range | 50−1.21 (1.25–1.21) |
| No. of reflections | 92652 (8841) |
| Redundancy | 19.6 (19.9) |
| Rwork/Rfree | 0.1641/0.1811 |
| No. of atoms | |
| Protein | 2157 |
| Ligand/ion | 45 |
| Water | 350 |
| Average B-factors | |
| Protein | 19.35 |
| Ligand | 22.56 |
| Zn2+ | 26.27 |
| Root mean square deviations | |
| Bond lengths (Å) | 0.006 |
| Bond angles (°) | 1.036 |
| Ramachandran | |
| Favored (%) | 96.23 |
| Allowed (%)Outliers (%) | 90.000.00 |
| Protein Data Bank Code | 6IH0 |
| Mutant | Kd (μM) | ∆H (kcal/mol) | ∆G (kcal/mol) | −T∆S (kcal/mol) |
|---|---|---|---|---|
| WT | 0.4 ± 0.1 | −8.2 ± 0.5 | −8.8 | −0.6 |
| I18A | 0.4 ± 0.3 | −6.6 ± 0.9 | −8.8 | −2.2 |
| H19A | 0.4 ± 0.2 | −4.5 ± 0.4 | −8.8 | −4.3 |
| S59A | 0.4 ± 0.1 | −9.1 ± 0.4 | −8.7 | 0.38 |
| E73A | 0.07 ± 0.05 | −7.4 ± 0.5 | −9.7 | −2.4 |
| H74A | NA | NA | NA | NA |
| T179A | NA | NA | NA | NA |
| F180A | 0.09 ± 0.07 | −2.8 ± 0.2 | −9.6 | −6.8 |
| E185A | 0.06 ± 0.03 | −8.1 ± 0.3 | −9.9 | −1.8 |
| I186A | 0.2 ± 0.09 | −5.4 ± 0.40 | −9.3 | −3.9 |
| I189A | 0.1 ± 0.09 | −7.0 ± 0.6 | −9.4 | −2.4 |
| S199A | NA | NA | NA | NA |
| L200A | 0.07 ± 0.02 | −4.8 ± 0.3 | −11.1 | −6.3 |
| T203A | 2.0 ± 1.4 | −7.0 ± 1.6 | −7.8 | −0.8 |
| V205A | NA | NA | NA | NA |
| Y212A | NA | NA | NA | NA |
| H226A | NA | NA | NA | NA |
| D230A | NA | NA | NA | NA |
| K227A | 0.1 ± 0.08 | −6.0 ± 0.49 | −9.5 | −3.4 |
| H253A | NA | NA | NA | NA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fan, S.; Li, D.; Yan, M.; Feng, X.; Lv, G.; Wu, G.; Jin, Y.; Wang, Y.; Yang, Z. The Complex Structure of Protein AaLpxC from Aquifex aeolicus with ACHN-975 Molecule Suggests an Inhibitory Mechanism at Atomic-Level against Gram-Negative Bacteria. Molecules 2021, 26, 1451. https://doi.org/10.3390/molecules26051451
Fan S, Li D, Yan M, Feng X, Lv G, Wu G, Jin Y, Wang Y, Yang Z. The Complex Structure of Protein AaLpxC from Aquifex aeolicus with ACHN-975 Molecule Suggests an Inhibitory Mechanism at Atomic-Level against Gram-Negative Bacteria. Molecules. 2021; 26(5):1451. https://doi.org/10.3390/molecules26051451
Chicago/Turabian StyleFan, Shuai, Danyang Li, Maocai Yan, Xiao Feng, Guangxin Lv, Guangteng Wu, Yuanyuan Jin, Yucheng Wang, and Zhaoyong Yang. 2021. "The Complex Structure of Protein AaLpxC from Aquifex aeolicus with ACHN-975 Molecule Suggests an Inhibitory Mechanism at Atomic-Level against Gram-Negative Bacteria" Molecules 26, no. 5: 1451. https://doi.org/10.3390/molecules26051451
APA StyleFan, S., Li, D., Yan, M., Feng, X., Lv, G., Wu, G., Jin, Y., Wang, Y., & Yang, Z. (2021). The Complex Structure of Protein AaLpxC from Aquifex aeolicus with ACHN-975 Molecule Suggests an Inhibitory Mechanism at Atomic-Level against Gram-Negative Bacteria. Molecules, 26(5), 1451. https://doi.org/10.3390/molecules26051451