
Synthesis of Computationally Designed 2,5(6)-Benzimidazole Derivatives via Pd-Catalyzed Reactions for Potential E. coli DNA Gyrase B Inhibition




Abstract
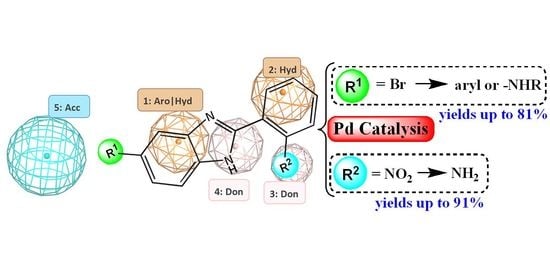
1. Introduction
2. Results and Discussion
2.1. Computer-Aided Design of Benzimidazole Derivatives with Potential E. coli DNA GyrB Inhibitory Activity
2.2. Synthetic Methods for the Preparation of 2-Aminophenyl-5(6)-Substituted Benzimidazole Derivatives
3. Materials and Methods
3.1. Generation of the Pharmacophore Model
3.2. Docking Studies on E. coli DNA GyrB
3.3. General Synthetic Methods
Procedures for the Synthesis of Products
- 5(6)-bromo-2-(2-nitrophenyl)-1H-benzimidazole (1):

- In a round-bottom flask, 4-bromo-1,2-benzenediamine (2.5 g; 13.4 mmol), 2-nitrobenzaldehyde (2.23 g; 14.8 mmol), and 250 mg of Montmorillonite K10 were mixed in ethanol (30 mL). The reaction was stirred at room temperature for 4 h. The mixture was filtered, the solvent was evaporated, and the dark orange slurry obtained was dissolved in ethyl acetate, followed by the addition of silica powder and the evaporation of the solvent to dryness. The resulting solid was loaded in a silica column, and the crude was purified dichloromethane/ethyl acetate 10:1 (Rf = 0.33). The product was obtained as a yellow solid in 62% yield (2.64 g). Characterization data in accordance to literature [45]. 1H NMR (400 MHz, DMSO-d6) ppm: 14.50–12.00 (brs, 1H1), 8.05 (dd, J = 8.0, 1.3 Hz, 1H3′), 7.97 (dd, J = 7.8, 1.5 Hz, 1H6′), 7.88 (td, J = 7.6, 1.3 Hz, 1H5′), 7.82 (d, J = 1.9 Hz, 1H4/7), 7.78 (td, J = 7.8, 1.5 Hz, 1H4′), 7.59 (d, J = 8.5 Hz, 1H7/4), 7.39 (dd, J = 8.5, 2.0 Hz, 1H6/5); 13C NMR (101 MHz, DMSO-d6) ppm: 148.9 (C2′), 148.7 (C2), 132.8 (C5′), 131.2 (C4′), 131.1 (C6′), 125.4 (C6/5), 124.4 (C3′), 123.9 (C1′), 118.1 (C4/7), 116.5 (C7/4), 114.7 (C5/6); HRMS (ESI-TOF) calcd. for C13H9BrN3O2: [M + H]+: 317.9878, found 317.9877.

- 1-benzyl-5(6)-bromo-2-(2-nitrophenyl)-1H-benzimidazole (2): In an oven-dried Schlenk tube coupled with a condenser, 5-bromo-2-(2-nitrophenyl)-1H-benzimidazole (2.5 g, 7.86 mmol) was dissolved in dry THF (5 mL). The system was degassed with 5x vacuum/argon cycles. To the stirring solution at 0 °C, NaH (60% in mineral oil) (226 mg, 9.43 mmol) was slowly added under an argon flow. After the evolution of the hydrogen gas, benzyl bromide (1.12 mL, 9.43 mmol) and a catalytic amount of tetra-n-butylammonium iodide were added. The mixture was heated at reflux temperature (70 °C) for 2 h. After cooling, the reaction was quenched with methanol, and the solvent was evaporated. The crude was purified by column chromatography using dichloromethane/ethyl acetate 1:20 (Rf = 0.40). The isomer mixture was obtained as a yellow solid in 87% yield (2.8 g). 1H NMR (400 MHz, CDCl3) (1:1 mixture of regioisomers) ppm: 8.17–8.10 (m, 1HAr), 7.89 (d, J = 1.8 Hz, 0.5HAr), 7.65–7.59 (m, 2.5HAr), 7.44–7.33 (m, 2HAr), 7.30 (dd, J = 8.6, 1.7 Hz, 0.5HAr), 7.22–7.16 (m, 3HAr), 7.03 (d, J = 1.8 Hz, 0.5HAr), 6.96–6.90 (m, 2HAr), 5.03 (m, 2Hbenzyl); 13C NMR (101 MHz, CDCl3) (1:1 mixture of regioisomers) ppm: 150.9, 150.5, 148.8, 144.1, 141.8, 136.2, 135.1, 135.0, 134.1, 133.6, 133.5, 132.73, 132.70, 131.58, 131.56, 129.2, 129.1, 128.41, 128.38, 126.8, 126.76, 126.72, 126.4, 125.38, 125.4, 125.2, 125.1, 123.1, 121.5, 117.0, 116.0, 113.8, 112.1, 48.7, 48.6; HRMS (ESI-TOF) calcd. for C20H15BrN3O2: [M + H]+: 408.0348, found 408.0345.
- General procedure for synthesis, isolation and characterization of 1-boc-5-bromo-2-(2-nitrophenyl)-benzimidazole (3a) and 1-boc-6-bromo-2-(2-nitrophenyl)-benzimidazole (3b): In a round-bottom flask, 5-bromo-2-(2-nitrophenyl)-1H-benzimidazole (1.66 g, 5.22 mmol), di-tert-butyl dicarbonate (2.28 g, 10.44 mmol) and DMAP (638 mg, 5.22 mmol) were mixed in dichloromethane (30 mL), and the reaction was left stirring at room temperature for 24 h. The solvent was evaporated, and the product was purified by column chromatography using hexane/ethyl acetate 4:1 as eluent (Rf = 0.40 for 3a Rf = 0.27 for 3b). Two pure fractions of light yellow solids (isomers 3a and 3b) were obtained in a 1:1 ratio with an overall yield of 89% (1.95 g).
- 1-boc-5-bromo-2-(2-nitrophenyl)-benzimidazole (3a):

- 1H NMR (400 MHz, CDCl3) ppm: 8.30 (dd, J = 8.2, 1.3 Hz, 1H3′), 7.95 (d, J = 8.7 Hz, 1H7), 7.92 (d, J = 1.9 Hz, 1H4), 7.79 (td, J = 7.5, 1.3 Hz, 1H5′), 7.70 (td, J = 7.9, 1.5 Hz, 1H4′), 7.64 (dd, J = 7.5, 1.6 Hz, 1H6′), 7.54 (dd, J = 8.7, 1.9 Hz, 1H6), 1.37 (s, 9H3″–5″). 13C NMR (101 MHz, CDCl3) ppm: 151.0 (C2), 147.8 (C1″), 147.7 (C2′), 144.0 (C3a), 133.9 (C5′), 132.2 (C7a), 132.1 (C6′), 130.8 (C4′), 128.8 (C1′), 128.7 (C6), 124.7 (C3′), 123.4 (C4), 117.7 (C5), 116.7 (C7), 86.4 (C2″), 27.7 (C3″,C4″,C5″); Assignment of regioisomer was done using 2D-NMR, see Supplementary Materials: Figure S18;

- 1-boc-6-bromo-2-(2-nitrophenyl)-benzimidazole (3b):

- 1H NMR (400 MHz, CDCl3) ppm: 8.30 (dd, J = 8.2, 1.3 Hz, 1H3′), 8.28 (d, J = 1.9 Hz, 1H7), 7.79 (td, J = 7.5, 1.3 Hz, 1H5′), 7.70 (td, J = 7.9, 1.5 Hz, 1H4′), 7.65 (dd, J = 7.5, 1.5 Hz, 1H6′), 7.64 (d, J = 8.5, 1H4), 7.52 (dd, J = 8.5, 1.9 Hz, 1H5), 1.37 (s, 9H3″–5″). 13C NMR (101 MHz, CDCl3) ppm: 150.2, 147.6, 147.6, 141.6, 133.9, 133.8, 132.0, 130.7, 128.7, 127.9, 124.6, 121.4, 119.0, 118.5, 86.4, 27.5; HRMS (ESI-TOF) calcd. for C18H17BrN3O4: [M + H]+: 418.0402, found 418.0403. Assignment of regioisomer was done using 2D-NMR, see Supplementary Materials: Figure S23.

- 1-benzyl-5(6)-(3,4,5-trimethoxyphenyl)-2-(2-nitrophenyl)-benzimidazole (4): In a Schlenk tube, 1-benzyl-5(6)-bromo-2-(2-nitrophenyl)-benzimidazole (500 mg, 1.22 mmol) and 3,4,5-trimethoxyphenylboronic acid (526 mg, 2.48 mmol) were dissolved in a mixture of THF/H2O 4:1 (15 mL). Under an argon flow, Pd(OAc)2 (27 mg; 0.12 mmol), PPh3 (80 mg; 0.30 mmol) and K2CO3 (380 mg, 9.2 mmol) were added and the reaction was heated to 70 °C for 16 h. After cooling, ethyl acetate was added to the reaction, and the insoluble solid was filtered. The organic phase was then washed with a 1 M solution of NaOH (5x), followed by water (2x). After drying with Na2SO4 and filtering, the solvent was evaporated. Then, a column chromatography in silica gel was performed using dichloromethane/ethyl acetate 5:1 as eluent (Rf = 0.38). The isomer mixture was obtained as a yellow solid in 66% yield (399 mg). 1H NMR (400 MHz, CDCl3) (1:1 mixture of regioisomers) ppm: 8.20–8.11 (m, 1HAr), 7.94 (d, J = 1.6 Hz, 0.5HAr), 7.79 (d, J = 8.4 Hz, 0.5HAr), 7.67–7.58 (m, 2HAr), 7.48–7.40 (m, 2HAr), 7.27–7.18 (m, 4HAr), 7.02–6.95 (m, 2HAr), 6.76 (s, 1HAr), 6.63 (s, 1HAr), 5.19 (s, 1Hbenzyl), 5.17 (s, 1Hbenzyl), 3.87–3.80 (m, 9HOMe); 13C NMR (101 MHz, CDCl3) (1:1 mixture of regioisomers) ppm: 153.57, 153.55, 150.4, 150.3, 148.9, 137.71, 137.67, 137.5, 137.1, 135.6, 135.4, 135.3, 134.5, 133.54, 133.51, 132.83, 132.77, 131.54, 131.46, 129.12, 129.08, 128.3, 128.2, 126.9, 126.8, 125.7, 125.2, 125.1, 123.6, 122.9, 120.2, 118.4, 111.0, 109.3, 105.0, 104.9, 61.1, 56.3, 48.7; HRMS (ESI-TOF) calcd. for C29H26N3O5: [M + H]+: 496.1872, found 496.1870.
- 1-benzyl-5(6)-(3-fluoro-4-(methoxycarbonyl)phenyl)-2-(2-nitrophenyl)-benzimidazole (5): In a Schlenk tube, 1-benzyl-5(6)-bromo-2-(2-nitrophenyl)-benzimidazole (300 mg, 0.74 mmol) and 3-fluoro-4-(methoxycarbonyl)phenylboronic acid (293 mg, 1.48 mmol) were dissolved in a mixture of THF/H2O 4:1 (8 mL). Under an argon flow, Pd(OAc)2 (16 mg; 0.074 mmol), PPh3 (48 mg; 0.18 mmol), and K2CO3 (100 mg, 1.38 mmol) were added and the reaction was heated to 70 °C for 16 h. After cooling, ethyl acetate was added to the reaction, and the insoluble solid was filtered. The organic phase was then washed with water (2x). After drying with Na2SO4 and filtering, the solvent was evaporated. Then, a column chromatography in silica gel was performed using dichloromethane/ethyl acetate 5:1 as eluent (Rf = 0.38). The isomer mixture was obtained as a yellow solid in 80% yield (286 mg). 1H NMR (400 MHz, acetone-d6) (1:1 mixture of regioisomers) ppm: 8.28–8.20 (m, 1HAr), 8.06 (d, J = 1.7 Hz, 0.5HAr), 8.04–7.95 (m, 1HAr), 7.95 (d, J = 1.7 Hz, 0.5HAr), 7.90–7.83 (m, 2HAr), 7.80 (d, J = 8.5 Hz, 0.5HAr), 7.76–7.52 (m, 4.5HAr), 7.33–7.23 (m, 3HAr), 7.20–7.15 (m, 2HAr), 5.28 (s, 1Hbenzyl), 5.25 (s, 1Hbenzyl), 3.94 (s, 1.5HOMe), 3.93 (s, 1.5HOMe); 13C NMR (101 MHz, acetone-d6) (1:1 mixture of regioisomers) ppm: 165.0, 164.9, 164.9, 164.9, 164.3, 164.2, 161.7, 161.6, 152.0, 151.9, 150.4, 150.3, 149.3, 149.2, 149.1, 149.0, 144.9, 144.9, 137.2, 137.1, 137.1, 136.9, 134.4, 134.3, 134.3, 134.2, 133.8, 133.8, 133.3, 133.2, 133.2, 132.3, 132.3, 129.6, 129.5, 128.6, 128.6, 127.9, 127.9, 126.5, 126.4, 125.8, 125.7, 123.6, 123.6, 123.4, 122.6, 121.2, 119.4, 117.6, 117.5, 117.5, 117.4, 116.1, 116.1, 115.9, 115.8, 112.5, 110.7, 52.5, 52.5, 49.0, 48.8; HRMS (ESI-TOF) calcd. for C28H21FN3O4: [M + H]+: 482.1516, found 482.1513.
- 1-benzyl-N-(4-(methylsulfonyl)phenyl)-2-(2-nitrophenyl)-1-benzimidazol-5(6)-amine (6): In a Schlenk tube, Pd(OAc)2 (0.074 mmol, 16.5 mg), XPhos (0.11 mmol, 53 mg), 1-benzyl-5(6)-bromo-2-(2-nitrophenyl)-benzimidazole (300 mg, 0.74 mmol), 4-(methylsulfonyl)aniline (151 mg, 0.88 mmol) and Cs2CO3 (1.47 mmol, 479 mg) were added to dry dioxane (5 mL) and the temperature was set to 100 °C. The reaction was carried out at reflux temperature for 16 h. After cooling, the crude mixture was filtered and the solid was washed with acetone. The filtrate was evaporated and a column chromatography in silica gel was performed using dichloromethane/ethyl acetate 1:1 as eluent (Rf = 0.29). After drying, a brown solid was obtained in 78% yield (282 mg). 1H NMR (400 MHz, acetone-d6): (1:1 mixture of regioisomers) ppm: 8.25–8.10 (m, 2HAr), 7.90–7.80 (m, 2HAr), 7.80–7.55 (m, 4HAr), 7.42 (d, J = 8.6 Hz, 0.5HAr), 7.36–7.16 (m, 6HAr), 7.16–7.08 (m, 1.5HAr), 7.04 (d, J = 8.9 Hz, 1HAr), 5.46 (s, 1Hbenzyl), 5.44 (s, 1Hbenzyl), 3.01 (s, 3HMe). 13C NMR (101 MHz, acetone-d6) (1:1 mixture of regioisomers) ppm: 151.6, 151.3, 150.8, 150.6, 150.4, 145.1, 141.0, 137.6, 137.3, 136.9, 136.8, 134.21, 134.15, 133.4, 133.3, 133.2, 132.2, 132.1, 130.8, 130.4, 130.1, 130.0, 129.62, 129.57, 128.6, 127.91, 127.87, 126.60, 126.56, 125.73, 125.70, 121.4, 119.9, 118.4, 114.5, 114.2, 114.1, 113.7, 112.5, 104.1, 48.9, 44.94, 44.88; HRMS (ESI-TOF) calcd. for C27H23N4O4S: [M + H]+: 499.1440, found 499.1434.
- 1-benzyl-N-(3-(methylsulfonyl)phenyl)-2-(2-nitrophenyl)-benzimidazol-5(6)-amine (7): In a Schlenk tube, Pd(OAc)2 (0.074 mmol, 16.5 mg), XPhos (0.11 mmol, 53 mg), 1-benzyl-5(6)-bromo-2-(2-nitrophenyl)-benzimidazole (300 mg, 0.74 mmol), 3-(methylsulfonyl)aniline (151 mg, 0.88 mmol) and Cs2CO3 (1.47 mmol, 479 mg) were added to dry dioxane (5 mL) and the temperature was set to 100 °C. The reaction was carried out at reflux temperature for 16 h. After cooling, the crude mixture was filtered and the solid was washed with acetone. The filtrate was evaporated and a column chromatography in silica gel was performed using dichloromethane/ethyl acetate 1:1 as eluent (Rf = 0.23). After drying, a brown solid was obtained in 81% yield (300 mg). 1H NMR (400 MHz, acetone-d6) (1:1 mixture of regioisomers) ppm: 8.12 (s, 1HAr), 7.77 (dd, J = 7.6, 1.7 Hz, 1HAr), 7.45–7.36 (m, 2HAr), 7.31 (dd, J = 7.3, 1.8 Hz, 1HAr), 7.04–6.95 (m, 4HAr), 6.85–6.75 (m, 5HAr), 6.70–6.64 (m, 3HAr), 4.95 (m, 2Hbenzyl), 2.70 (s, 3HMe); 13C NMR (101 MHz, acetone-d6) (1:1 mixture of regioisomers) ppm: 149.7, 149.0, 146.3, 143.5, 141.8, 136.9, 136.3, 133.6, 132.2, 131.6, 131.2, 130.3, 128.6, 127.7, 127.0, 124.94, 124.93, 118.7, 117.6, 116.0, 111.9, 111.6, 109.9, 47.6, 43.7; HRMS (ESI-TOF) calcd. for C27H23N4O4S: [M + H]+: 499.1440, found 499.1436.
- 1-boc-5-(3,4,5-trimethoxyphenyl)-2-(2-nitrophenyl)-benzimidazole (8a): In a Schlenk tube, 1-boc-5-bromo-2-(2-nitrophenyl)-benzimidazole (300 mg, 0.72 mmol) and 3,4,5-trimethoxyphenylboronic acid (229 mg, 1.08 mmol) were dissolved in a mixture of THF/H2O 4:1 (5 mL). Under an argon flow, Pd(OAc)2 (16 mg; 0.72 mmol), PPh3 (47 mg; 0.18 mmol), and K2CO3 (89 mg, 2.2 mmol) were added and the reaction was heated to 70 °C for 16 h. After cooling, ethyl acetate was added to the reaction, and the insoluble solid was filtered. The organic phase was then washed with a 1 M solution of NaOH (5x), followed by water (2x). After drying with Na2SO4 and filtering, the solvent was evaporated. Then, a column chromatography in silica gel was performed using a gradient mixture as eluent, starting with dichloromethane and ending in dichloromethane/ethyl acetate 20:1 (Rf = 0.50). The light yellow solid was obtained in 72% yield (260 mg). 1H NMR (400 MHz, acetone-d6) ppm: 8.37–8.34 (m, 1HAr), 8.14 (d, J = 8.6 Hz, 1HAr), 8.00–7.95 (m, 2HAr), 7.91–7.85 (m, 2HAr), 7.78 (dd, J = 8.6, 1.8 Hz, 1HAr), 7.04 (s, 2HAr), 3.96 (s, 6HOMe), 3.79 (s, 3HOMe), 1.39 (s, 9Hboc); 13C NMR (101 MHz, acetone-d6) ppm: 154.9, 151.3, 148.93, 148.91, 144.6, 139.1, 139.0, 137.6, 135.0, 133.5, 133.3, 131.8, 130.1, 125.5, 125.4, 119.2, 116.2, 105.8, 86.5, 60.73, 60.65, 56.7, 27.7; HRMS (ESI-TOF) calcd. for C27H28N3O7: [M + H]+: 506.1927, found 506.1924.
- 1-boc-5-(3-fluoro-4-(methoxycarbonyl)phenyl)-2-(2-nitrophenyl)-benzimidazole (9a): In a Schlenk tube, 1-boc-5-bromo-2-(2-nitrophenyl)-benzimidazole (300 mg, 0.74 mmol) and 3-fluoro-4-(methoxycarbonyl)phenylboronic acid (170 mg, 0.86 mmol) were dissolved in a mixture of THF/H2O 4:1 (5 mL). Under an argon flow, Pd(OAc)2 (16 mg; 0.074 mmol), PPh3 (48 mg; 0.18 mmol), and K2CO3 (298 mg, 2.15 mmol) were added and the reaction was heated to 70 °C for 16 h. After cooling, ethyl acetate was added to the reaction, and the insoluble solid was filtered. The organic phase was then washed with water (2x). After drying with Na2SO4 and filtering, the solvent was evaporated. Then, a column chromatography in silica gel was performed using dichloromethane as eluent (Rf = 0.36). The product was obtained as a yellow solid in 66% yield (235 mg). 1H NMR (400 MHz, acetone-d6) ppm: 8.32 (d, J = 1.8 Hz, 1HAr), 8.24 (d, J = 8.6 Hz, 1HAr), 7.94 (t, J = 7.9 Hz, 1HAr), 7.86 (td, J = 7.5, 1.3 Hz, 1HAr), 7.80–7.66 (m, 4HAr), 7.59 (dd, J = 8.2, 1.8 Hz, 1HAr), 7.52 (dd, J = 12.3, 1.8 Hz, 1HAr), 3.80 (s, 6HOMe), 1.26 (s, 9Hboc). 13C NMR (101 MHz, acetone-d6) ppm: 165.0 (d, J = 3.7 Hz), 163.1 (d, J = 258.1 Hz), 152.0, 148.87 (d, J = 8.8 Hz), 148.84, 148.79, 144.5, 136.8 (d, J = 2.0 Hz), 135.1, 134.8, 133.7 (d, J = 1.7 Hz), 133.3, 131.9, 129.9, 125.4, 124.8, 123.9 (d, J = 3.4 Hz), 121.6, 118.2 (d, J = 10.6 Hz), 116.4 (d, J = 23.6 Hz), 115.0, 86.8, 52.6, 27.7; HRMS (ESI-TOF) calcd. for C26H23FN3O6: [M + H]+: 492.1571, found 492.1564.
- 5(6)-(3,4,5-trimethoxyphenyl)-2-(2-nitrophenyl)-1H-benzimidazole (10): In a round-bottom flask, 1-boc-5-(3,4,5-trimethoxyphenyl)-2-(2-nitrophenyl)-1H-benzimidazole (0.28 mmol, 140 mg) was dissolved in a mixture of DCM/TFA 1:1 (2.0 mL). After 3 h, the acid was neutralized by a slow addition of a saturated aqueous solution of NaHCO3. Dichloromethane was added and the organic phase was washed 2x with a saturated solution of NaHCO3 and then 2x with H2O, followed by drying anhydrous Na2SO4, filtering and solvent evaporation. The product was purified by column chromatography in silica gel using DCM/Ethyl acetate 1:2 as eluent (Rf = 0.37). After drying, a yellow solid was obtained in 99% yield (112 mg). 1H NMR (400 MHz, DMSO-d6) ppm: 13.09 (brs, 1HNH), 8.04 (dd, J = 8.1, 1.2 Hz, 1HAr), 8.00 (dd, J = 7.8, 1.4 Hz, 1HAr), 7.91–7.87 (m, 2HAr), 7.77 (td, J = 7.8, 1.4 Hz, 1HAr), 7.67 (d, J = 8.4 Hz, 1HAr), 7.59 (dd, J = 8.5, 1.7 Hz, 1HAr), 6.97 (s, 2HAr), 3.89 (s, 6HOMe), 3.70 (s, 3Hboc); 13C NMR (101 MHz, DMSO-d6) ppm: 153.2, 149.0, 148.0, 136.80, 136.78, 135.3, 132.7, 131.0, 130.9, 124.3, 124.1, 122.2, 104.4, 60.1, 56.0; HRMS (ESI-TOF) calcd. for C22H20N3O5: [M + H]+: 406.1403, found 406.1404.
- 5(6)-(3-fluoro-4-(methoxycarbonyl)phenyl)-2-(2-nitrophenyl)-1H-benzimidazole (11): In a round-bottom flask, 1-boc-5-(3-fluoro-4-(methoxycarbonyl)phenyl)-2-(2-nitrophenyl)-benzimidazole (0.42 mmol, 208 mg) was dissolved in DCM (2.0 mL) and TFA (0.4 mL) was added. After 5 h, the acid was neutralized by a slow addition of a saturated aqueous solution of NaHCO3. Dicloromethane was added and the organic phase was washed 2x with a saturated solution of NaHCO3 and then 2x with H2O, followed by drying anhydrous Na2SO4, filtering and solvent evaporation. The product was purified by column chromatography in silica gel using DCM/Ethyl acetate 6:1 as eluent (Rf = 0.27). After drying, a yellow solid was obtained in 65% yield (107 mg). 1H NMR (400 MHz, acetone-d6) ppm: 12.20–12.15 (m, 1HNH), 8.09–7.93 (m, 4HAr), 7.88 (td, J = 7.6, 1.4 Hz, 1HAr), 7.83–7.59 (m, 5HAr), 3.92 (s, 3HOMe); 13C NMR (101 MHz, acetone-d6) ppm: 165.0 (d, J = 3.8 Hz), 163.0 (d, J = 258.1 Hz), 150.5, 149.9, 149.3, 133.5, 133.4 (d, J = 1.7 Hz), 132.1, 131.8, 125.8, 125.3, 123.6 (d, J = 3.2 Hz), 117.4, 116.0 (d, J = 23.4 Hz), 52.5; HRMS (ESI-TOF) calcd. for C21H15FN3O4: [M + H]+: 392.1047, found 392.1042.
- N-(3-(methylsulfonyl)phenyl)-2-(2-nitrophenyl)-1H-benzimidazol-5(6)-amine (12): In a Schlenk tube, Pd(OAc)2 (0.072 mmol, 16.1 mg), XPhos (0.11 mmol, 52 mg), 1-benzyl-5(6)-bromo-2-(2-nitrophenyl)-benzimidazole (300 mg, 0.72 mmol), 3-(methylsulfonyl)aniline (147 mg, 0.86 mmol) and Cs2CO3 (1.43 mmol, 467 mg) were added to dry dioxane (5 mL) and the temperature was set to 100 °C. The reaction was carried out at reflux temperature for 16 h. After cooling, the crude mixture was filtered and the solid was washed with acetone. The filtrate was evaporated and dissolved in a mixture of DCM/TFA 1:1 (4.0 mL). After 2 h, the acid was neutralized by a slow addition of a saturated aqueous solution of NaHCO3. Ethyl acetate was added to the mixture, and the organic phase was washed 2x with a saturated solution of NaHCO3 and then 2x with H2O. The organic phase was dried with anhydrous Na2SO4, filtered, and evaporated. A purification by column chromatography in silica gel was performed using dichloromethane/ethyl acetate 1:1 as eluent (Rf = 0.38). After drying, a brown solid was obtained in 48% yield (140 mg). 1H NMR (400 MHz, acetone-d6) ppm: 11.95 (brs, 1HNH), 8.02 (dd, J = 7.7, 1.5 Hz, 1HAr), 7.98 (dd, J = 8.1, 1.3 Hz, 1HAr), 7.90–7.80 (m, 2HAr), 7.74 (m, 1HAr), 7.65–7.55 (m, 2HAr), 7.47 (s, 1HAr), 7.44 (d, J = 7.9 Hz, 1HAr), 7.35 (d, J = 8.2 Hz, 1HAr), 7.31 (d, J = 7.7 Hz, 1HAr), 7.16 (d, J = 8.6 Hz, 1HAr), 3.08 (s, 3HMe); 13C NMR (101 MHz, acetone-d6) ppm: 150.4, 143.4, 133.3, 131.8, 131.4, 131.1, 125.9, 125.2, 117.8, 113.7, 44.3; HRMS (ESI-TOF) calcd. for C20H17N4O4S: [M + H]+: 409.0971, found 409.0964.
- 2-(2-aminophenyl)-5(6)-(3,4,5-trimethoxyphenyl)-1H-benzimidazole (13): In a round-bottom flask, 5(6)-(3,4,5-trimethoxyphenyl)-2-(2-nitrophenyl)-1H-benzimidazole (50 mg, 0.125 mmol), hydrazine monohydrate (45 μL, 1.25 mmol) and Pd/C 5% (10 mg) were mixed in methanol (1.0 mL). The solution was stirred for 30 min at reflux temperature (70 °C). The resulting solution was filtrated and the solvent evaporated. A white solid was obtained in 91% yield (43 mg). 1H NMR (400 MHz, CD3OD) ppm: 7.77 (brs, 1HAr), 7.72 (dd, J = 7.8, 1.3 Hz, 1HAr), 7.61 (brs, 1HAr), 7.48 (dd, J = 8.3, 1.7 Hz, 1HAr), 7.19 (ddd, J = 8.4, 7.2, 1.5 Hz, 1HAr), 6.88 (dd, J = 8.2, 1.2 Hz, 1HAr), 6.75 (ddd, J = 8.2, 7.1, 1.2 Hz, 1HAr), 3.92 (s, 6HOMe), 3.81 (s, 3HOMe); 13C NMR (101 MHz, CD3OD) ppm: 154.8, 154.7, 149.1, 139.7, 138.4, 131.8, 128.6, 123.1, 117.9, 117.7, 113.2, 105.8, 61.2, 56.7; HRMS (ESI-TOF) calcd. for C22H22N3O3: [M + H]+: 376.1661, found 376.1655.
- 2-(2-aminophenyl)-5(6)-(3-fluoro-4-(methoxycarbonyl)phenyl)-1H-benzimidazole (14): In a round-bottom flask, 5(6)-(3-fluoro-4-(methoxycarbonyl)phenyl)-2-(2-nitrophenyl)-1H-benzimidazole (20 mg, 0.051 mmol), hydrazine monohydrate (25 μL, 0.75 mmol) and Pd/C 5% (2 mg) were mixed in methanol (2.0 mL). The solution was stirred for 1 h at reflux temperature (70 °C). The product precipitated on the reaction mixture, and thus the crude was filtrated and the filtrate was dissolved in DMF. After evaporation of the solvent, a yellow solid was obtained in 56% yield (15.7 mg). 1H NMR (400 MHz, DMSO-d6) ppm: 12.83 (brs, 1HNH), 8.13–7.54 (m, 7HAr), 7.29 (brs, 2HAr), 7.18 (t, J = 7.7 Hz, 1HAr), 6.85 (d, J = 8.2 Hz, 1HAr), 6.67 (t, J = 7.5, 1HAr), 3.88 (s, 3HOMe); 13C NMR (101 MHz, DMSO-d6) ppm: 164.0, 161.8 (d, J = 295 Hz), 154.0 (d, J = 24 Hz), 148.4, 143.7, 134.3, 132.3, 130.7, 127.4, 122.6, 121.9, 121.1, 118.6, 116.2, 115.0, 111.3, 109.7 (d, J = 6.8 Hz), 52.3; HRMS (ESI-TOF) calcd. for C21H16FN3O2: [M + H]+: 362.1305, found 362.1310.
- 2-(2-aminophenyl)-N-(3-(methylsulfonyl)phenyl)-1H-benzimidazol-5(6)-amine (15): In a round-bottom flask, N-(3-(methylsulfonyl)phenyl)-2-(2-nitrophenyl)-1H-benzimidazol-5(6)-amine (40 mg, 0.098 mmol), hydrazine monohydrate (50 μL, 1.5 mmol) and Pd/C 5% (8 mg) were mixed in methanol (2.0 mL). The solution was stirred for 30 min at reflux temperature (70 °C). The resulting solution was filtrated and the solvent evaporated. A white solid was obtained in 80% yield (30 mg). 1H NMR (400 MHz, CD3OD) ppm: 7.68 (dd, J = 7.9, 1.5 Hz, 1HAr), 7.54 (m, 2HAr), 7.39 (m, 2HAr), 7.27 (m, 2HAr), 7.18 (m, 1HAr), 7.07 (dd, J = 8.6, 2.1 Hz, 1HAr), 6.87 (dd, J = 8.2, 1.2 Hz, 1HAr), 6.74 (td, J = 7.6, 1.2 Hz, 1HAr), 3.08 (s, 3HMe); 13C NMR (101 MHz, CD3OD) ppm: 154.4, 148.9, 148.5, 142.8, 131.6, 131.3, 128.5, 120.7, 117.9, 117.8, 117.4, 113.5, 113.3, 44.4; HRMS (ESI-TOF) calcd. for C20H19N4O2S: [M + H]+: 379.1229, found 379.1228.
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vitaku, E.; Smith, D.T.; Njardarson, J.T. Analysis of the structural diversity, substitution patterns, and frequency of nitrogen heterocycles among U.S. FDA approved pharmaceuticals. J. Med. Chem. 2014, 57, 10257–10274. [Google Scholar] [CrossRef]
- Luca, L.D. Naturally occurring and synthetic imidazoles: Their chemistry and their biological activities. Curr. Med. Chem. 2006, 13, 1–23. [Google Scholar]
- Jakopin, Z.; Ilas, J.; Barancokova, M.; Brvar, M.; Tammela, P.; Sollner Dolenc, M.; Tomasic, T.; Kikelj, D. Discovery of substituted oxadiazoles as a novel scaffold for DNA gyrase inhibitors. Eur. J. Med. Chem. 2017, 130, 171–184. [Google Scholar] [CrossRef]
- Gaba, M.; Mohan, C. Development of drugs based on imidazole and benzimidazole bioactive heterocycles: Recent advances and future directions. Med. Chem. Res. 2016, 25, 173–210. [Google Scholar] [CrossRef]
- Tahlan, S.; Kumar, S.; Narasimhan, B. Antimicrobial potential of 1h-benzo[d]imidazole scaffold: A review. BMC Chemistry 2019, 13, 18. [Google Scholar] [CrossRef]
- Cho, J.C.; Crotty, M.P.; Pardo, J. Ridinilazole: A novel antimicrobial for clostridium difficile infection. Ann. Gastroenterol. 2019, 32, 134–140. [Google Scholar] [CrossRef]
- World Health Organization. Global Priority List of Antibiotic Resistant Bacteria to Guide Research, Discovery, and Development of New Antibiotics; World Health Organization: Geneva, Switzerland, 2017; pp. 1–7. [Google Scholar]
- Saíz-Urra, L.; Pérez, M.Á.C.; Froeyen, M. Thermodynamic computational approach to capture molecular recognition in the binding of different inhibitors to the DNA gyrase b subunit from escherichia coli. J. Mol. Model. 2013, 19, 3187–3200. [Google Scholar] [CrossRef]
- Tomašič, T.; Barančoková, M.; Zidar, N.; Ilaš, J.; Tammela, P.; Kikelj, D. Design, synthesis, and biological evaluation of 1-ethyl-3-(thiazol-2-yl)urea derivatives as escherichia coli DNA gyrase inhibitors. Arch. Pharm. 2018, 351, 1700333-n/a. [Google Scholar] [CrossRef]
- Gjorgjieva, M.; Tomašič, T.; Barančokova, M.; Katsamakas, S.; Ilaš, J.; Tammela, P.; Peterlin Mašič, L.; Kikelj, D. Discovery of benzothiazole scaffold-based DNA gyrase b inhibitors. J. Med. Chem. 2016, 59, 8941–8954. [Google Scholar] [CrossRef]
- Chemical Computing Group ULC. Molecular Operating Environment (MOE). Available online: https://www.chemcomp.com/Products.htm (accessed on 7 January 2021).
- Alaqeel, S.I. Synthetic approaches to benzimidazoles from o-phenylenediamine: A literature review. J. Saudi Chem. Soc. 2017, 21, 229–237. [Google Scholar] [CrossRef]
- Sreerama, R.; Barnali, M.; Balamurali, M.M.; Kaushik, C. Synthesis and medicinal applications of benzimidazoles: An overview. Curr. Org. Synth. 2017, 14, 40–60. [Google Scholar]
- Jyoti, S.; Pradeep, K.S.; Ravi, B.; Anand, K.H. Synthetic approaches towards benzimidazoles by the reaction of o-phenylenediamine with aldehydes using a variety of catalysts: A review. Curr. Org. Chem. 2018, 22, 2280–2299. [Google Scholar]
- Faheem, M.; Rathaur, A.; Pandey, A.; Kumar Singh, V.; Tiwari, A.K. A review on the modern synthetic approach of benzimidazole candidate. ChemistrySelect 2020, 5, 3981–3994. [Google Scholar] [CrossRef]
- Zheng, Y.; Zheng, M.; Ling, X.; Liu, Y.; Xue, Y.; An, L.; Gu, N.; Jin, M. Design, synthesis, quantum chemical studies and biological activity evaluation of pyrazole–benzimidazole derivatives as potent aurora a/b kinase inhibitors. Bioorg. Med. Chem. Lett. 2013, 23, 3523–3530. [Google Scholar] [CrossRef]
- Starčević, K.; Kralj, M.; Ester, K.; Sabol, I.; Grce, M.; Pavelić, K.; Karminski-Zamola, G. Synthesis, antiviral and antitumor activity of 2-substituted-5-amidino-benzimidazoles. Biorg. Med. Chem. 2007, 15, 4419–4426. [Google Scholar] [CrossRef]
- Joy, M.; Karuvalam, R.; Haridas, K.; Sajith, A.; Pakkath, R.; Bhaskaran, S.; Padusha, M.; Bakulev, V. Suzuki-miyaura coupling under microwave enhanced conditions: Synthesis of 2-(hetero)aryl benzimidazoles. Arkivoc 2020, 2019, 431–445. [Google Scholar]
- Ezquerra, J.; Lamas, C.; Pastor, A.; García-Navío, J.; Vaquero, J.J. Suzuki-type cross-coupling reaction of 1-benzyl-2-iodo-1h-benzimidazoles with aryl boronic acids: A regioselective route to n-alkylated 6-alkoxy-2-aryl-1h-benzimidazoles. Tetrahedron 1997, 53, 12755–12764. [Google Scholar] [CrossRef]
- Düfert, M.A.; Billingsley, K.L.; Buchwald, S.L. Suzuki-miyaura cross-coupling of unprotected, nitrogen-rich heterocycles: Substrate scope and mechanistic investigation. J. Am. Chem. Soc. 2013, 135, 12877–12885. [Google Scholar] [CrossRef]
- Martin, A.D.; Siamaki, A.R.; Belecki, K.; Gupton, B.F. A convergent approach to the total synthesis of telmisartan via a suzuki cross-coupling reaction between two functionalized benzimidazoles. J. Org. Chem. 2015, 80, 1915–1919. [Google Scholar] [CrossRef]
- Savall, B.M.; Fontimayor, J.R. Synthesis of 2-arylbenzimidazoles via microwave suzuki–miyaura reaction of unprotected 2-chlorobenzimidazoles. Tetrahedron Lett. 2008, 49, 6667–6669. [Google Scholar] [CrossRef]
- Hooper, M.W.; Utsunomiya, M.; Hartwig, J.F. Scope and mechanism of palladium-catalyzed amination of five-membered heterocyclic halides. J. Org. Chem. 2003, 68, 2861–2873. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, I.; Taguchi, N.; Yoneda, Y.; Yamashita, M.; Ohta, S. Highly effective procedure for introduction of amino group into the 2-position of imidazole ring. Heterocycles 1996, 43, 1375–1379. [Google Scholar] [CrossRef]
- Coon, T.; Moree, W.J.; Li, B.; Yu, J.; Zamani-Kord, S.; Malany, S.; Santos, M.A.; Hernandez, L.M.; Petroski, R.E.; Sun, A.; et al. Brain-penetrating 2-aminobenzimidazole h1-antihistamines for the treatment of insomnia. Bioorg. Med. Chem. Lett. 2009, 19, 4380–4384. [Google Scholar] [CrossRef] [PubMed]
- Sang, W.; Gavi, A.J.; Yu, B.-Y.; Cheng, H.; Yuan, Y.; Wu, Y.; Lommens, P.; Chen, C.; Verpoort, F. Palladium-catalyzed ligand-free c-n coupling reactions: Selective diheteroarylation of amines with 2-halobenzimidazoles. Chem. Asian J. 2020, 15, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Gergely, M.; Bényei, A.; Kollár, L. 2-aminobenzimidazole and -benzoxazole as n-nucleophile in palladium-catalysed aminocarbonylation. Tetrahedron 2020, 76, 131079. [Google Scholar] [CrossRef]
- Cheung, M.; Emmitte, K.A.; Salovich, J.M. Benzimidazole Thiophene Compounds as PLK Inhibitors. Patent No. WO2007/030361A2, 15 March 2007. [Google Scholar]
- Jain, P.; Yi, S.; Flaherty, P.T. Suzuki-miyaura cross-coupling of potassium organoborates with 6-sulfonate benzimidazoles using microwave irradiation. J. Heterocycl. Chem. 2013, 50, E166–E173. [Google Scholar] [CrossRef]
- Kasai, S.; Igawa, H.; Takahashi, M.; Maekawa, T.; Kakegawa, K.; Yasuma, T.; Kina, A.; Aida, J.; Khamrai, U.; Kundu, M. Benzimidazole Derivatives as MCH Receptor Antagonists. Patent No. WO/2013/105676, 18 July 2013. [Google Scholar]
- Chang, S.-Y.; Lin, G.-T.; Cheng, Y.-C.; Huang, J.-J.; Chang, C.-L.; Lin, C.-F.; Lee, J.-H.; Chiu, T.-L.; Leung, M.-K. Construction of highly efficient carbazol-9-yl-substituted benzimidazole bipolar hosts for blue phosphorescent light-emitting diodes: Isomer and device performance relationships. ACS Appl. Mater. Interfaces 2018, 10, 42723–42732. [Google Scholar] [CrossRef]
- López-Rodríguez, M.a.L.; Benhamú, B.; Ayala, D.; Rominguera, J.L.; Murcia, M.; Ramos, J.A.; Viso, A. Pd(0) amination of benzimidazoles as an efficient method towards new (benzimidazolyl)piperazines with high affinity for the 5-ht1a receptor. Tetrahedron 2000, 56, 3245–3253. [Google Scholar] [CrossRef]
- Boydston, A.J.; Vu, P.D.; Dykhno, O.L.; Chang, V.; Wyatt, A.R.; Stockett, A.S.; Ritschdorff, E.T.; Shear, J.B.; Bielawski, C.W. Modular fluorescent benzobis(imidazolium) salts: Syntheses, photophysical analyses, and applications. J. Am. Chem. Soc. 2008, 130, 3143–3156. [Google Scholar] [CrossRef]
- Chen, Y.-C.; Faver, J.C.; Ku, A.F.; Miklossy, G.; Riehle, K.; Bohren, K.M.; Ucisik, M.N.; Matzuk, M.M.; Yu, Z.; Simmons, N. C–n coupling of DNA-conjugated (hetero)aryl bromides and chlorides for DNA-encoded chemical library synthesis. Bioconjugate Chem. 2020, 31, 770–780. [Google Scholar] [CrossRef]
- Durcik, M.; Tammela, P.; Barančoková, M.; Tomašič, T.; Ilaš, J.; Kikelj, D.; Zidar, N. Synthesis and evaluation of n-phenylpyrrolamides as DNA gyrase b inhibitors. ChemMedChem 2018, 13, 186–198. [Google Scholar] [CrossRef]
- Ostrov, D.A.; Hernández Prada, J.A.; Corsino, P.E.; Finton, K.A.; Le, N.; Rowe, T.C. Discovery of novel DNA gyrase inhibitors by high-throughput virtual screening. Antimicrob. Agents Chemother. 2007, 51, 3688–3698. [Google Scholar] [CrossRef] [PubMed]
- Eakin, A.E.; Green, O.; Hales, N.; Walkup, G.K.; Bist, S.; Singh, A.; Mullen, G.; Bryant, J.; Embrey, K.; Gao, N.; et al. Pyrrolamide DNA gyrase inhibitors: Fragment-based nuclear magnetic resonance screening to identify antibacterial agents. Antimicrob. Agents Chemother. 2012, 56, 1240–1246. [Google Scholar] [CrossRef] [PubMed]
- Cotman, A.E.; Trampuž, M.; Brvar, M.; Kikelj, D.; Ilaš, J.; Peterlin-Mašič, L.; Montalvão, S.; Tammela, P.; Frlan, R. Design, synthesis, and evaluation of novel tyrosine-based DNA gyrase b inhibitors. Archiv Pharmazie 2017, 350, 1700087-n/a. [Google Scholar] [CrossRef]
- Basarab, G.S.; Manchester, J.I.; Bist, S.; Boriack-Sjodin, P.A.; Dangel, B.; Illingworth, R.; Sherer, B.A.; Sriram, S.; Uria-Nickelsen, M.; Eakin, A.E. Fragment-to-hit-to-lead discovery of a novel pyridylurea scaffold of atp competitive dual targeting type ii topoisomerase inhibiting antibacterial agents. J. Med. Chem. 2013, 56, 8712–8735. [Google Scholar] [CrossRef]
- Ronkin, S.M.; Badia, M.; Bellon, S.; Grillot, A.-L.; Gross, C.H.; Grossman, T.H.; Mani, N.; Parsons, J.D.; Stamos, D.; Trudeau, M.; et al. Discovery of pyrazolthiazoles as novel and potent inhibitors of bacterial gyrase. Bioorg. Med. Chem. Lett. 2010, 20, 2828–2831. [Google Scholar] [CrossRef]
- Trzoss, M.; Bensen, D.C.; Li, X.; Chen, Z.; Lam, T.; Zhang, J.; Creighton, C.J.; Cunningham, M.L.; Kwan, B.; Stidham, M.; et al. Pyrrolopyrimidine inhibitors of DNA gyrase b (gyrb) and topoisomerase iv (pare), part ii: Development of inhibitors with broad spectrum, gram-negative antibacterial activity. Bioorg. Med. Chem. Lett. 2013, 23, 1537–1543. [Google Scholar] [CrossRef] [PubMed]
- Tari, L.W.; Li, X.; Trzoss, M.; Bensen, D.C.; Chen, Z.; Lam, T.; Zhang, J.; Lee, S.J.; Hough, G.; Phillipson, D.; et al. Tricyclic gyrb/pare (tribe) inhibitors: A new class of broad-spectrum dual-targeting antibacterial agents. PLoS ONE 2013, 8, e84409. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef]
- Mann, J.; Baron, A.; Opoku-Boahen, Y.; Johansson, E.; Parkinson, G.; Kelland, L.R.; Neidle, S. A new class of symmetric bisbenzimidazole-based DNA minor groove-binding agents showing antitumor activity. J. Med. Chem. 2001, 44, 138–144. [Google Scholar] [CrossRef]
- Pessoa-Mahana, D.; Espinosa-Bustos, C.; Mella-Raipán, J.; Canales-Pacheco, J.; Pessoa-Mahana, H. Microwave-assisted synthesis and regioisomeric structural elucidation of novel benzimidazo[1,2d][1,4]benzodiazepinone derivatives. Arkivoc 2009, 7, 131–140. [Google Scholar] [CrossRef]
- Yadagiri, B.; Lown, J.W. Convenient routes to substituted benzimidazoles and imidazolo[4,5-b]pyridines using nitrobenzene as oxidant. Synth. Commun. 1990, 20, 955–963. [Google Scholar] [CrossRef]
- Miyaura, N.; Suzuki, A. Palladium-catalyzed cross-coupling reactions of organoboron compounds. Chem. Rev. 1995, 95, 2457–2483. [Google Scholar] [CrossRef]
- Fernández, F.; Cordero, B.; Durand, J.; Muller, G.; Malbosc, F.; Kihn, Y.; Teuma, E.; Gómez, M. Palladium catalyzed suzuki c–c couplings in an ionic liquid: Nanoparticles responsible for the catalytic activity. Dalton Trans. 2007, 5572–5581. [Google Scholar] [CrossRef] [PubMed]
- Hooshmand, S.E.; Heidari, B.; Sedghi, R.; Varma, R.S. Recent advances in the suzuki–miyaura cross-coupling reaction using efficient catalysts in eco-friendly media. Green Chem. 2019, 21, 381–405. [Google Scholar] [CrossRef]
- Ruiz-Castillo, P.; Buchwald, S.L. Applications of palladium-catalyzed c–n cross-coupling reactions. Chem. Rev. 2016, 116, 12564–12649. [Google Scholar] [CrossRef] [PubMed]
- Surry, D.S.; Buchwald, S.L. Dialkylbiaryl phosphines in pd-catalyzed amination: A user’s guide. Chem. Sci. 2011, 2, 27–50. [Google Scholar] [CrossRef]
- King, A.O.; Larsen, R.D.; Negishi, E. Palladium-catalyzed heterogeneous hydrogenation. In Handbook of Organopalladium Chemistry for Organic Synthesis; Negishi, E., Ed.; Wiley: New York, NY, USA, 2002; pp. 2719–2752. [Google Scholar]
- Greene, T.W.; Wuts, P.G.M. Protection for the amino group (3rd edition). In Protective Groups in Organic Synthesis; John Wiley & Sons, Inc.: New York, NY, USA, 1999; pp. 494–653. [Google Scholar]
Sample Availability: Samples of the compounds are available from the authors. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| # | Structure | ChemScore | Predicted log IC50 (nM) |
|---|---|---|---|
| 1 |  | 33.7 | −0.025 ± 0.56 |
| 2 |  | 28.7 | 2.7 ± 1.0 |
| 3 |  | 27.5 | 3.4 ± 1.1 |
 | |||||
|---|---|---|---|---|---|
| # | T (°C) | Time (h) | Solvent | Catalyst | Isolated Yield–1 (%) |
| 1 | 180 | 8 | Nitrobenzene | - | 48 |
| 2 | 80 | 3 | Ethanol | - | 47 |
| 3 | 25 | 6 | Ethanol | - | 50 |
| 4 | 25 | 4 | Ethanol | Montmorillonite K10 | 62 |
| 5 | 80 | 2 | Ethanol | Montmorillonite K10 | 54 |
 | ||||
|---|---|---|---|---|
| # | Phosphine | Solvent | Sub/Cat | Conv (%) |
| 1 | 15 mol% BINAP | Toluene | 10 | trace |
| 2 | 15 mol% DPEphos | Toluene | 10 | trace |
| 3 | 15 mol% XPhos | Toluene | 10 | 91 |
| 4 | 15 mol% BINAP | Dioxane | 10 | trace |
| 5 | 15 mol% DPEphos | Dioxane | 10 | trace |
| 6 | 15 mol% XPhos | Dioxane | 10 | 100 (78)b) |
| 7c) | 15 mol% XPhos | Dioxane | 10 | 79 |
| 8 | 7.5 mol% XPhos | Dioxane | 20 | 57 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aroso, R.T.; Guedes, R.C.; Pereira, M.M. Synthesis of Computationally Designed 2,5(6)-Benzimidazole Derivatives via Pd-Catalyzed Reactions for Potential E. coli DNA Gyrase B Inhibition. Molecules 2021, 26, 1326. https://doi.org/10.3390/molecules26051326
Aroso RT, Guedes RC, Pereira MM. Synthesis of Computationally Designed 2,5(6)-Benzimidazole Derivatives via Pd-Catalyzed Reactions for Potential E. coli DNA Gyrase B Inhibition. Molecules. 2021; 26(5):1326. https://doi.org/10.3390/molecules26051326
Chicago/Turabian StyleAroso, Rafael T., Rita C. Guedes, and Mariette M. Pereira. 2021. "Synthesis of Computationally Designed 2,5(6)-Benzimidazole Derivatives via Pd-Catalyzed Reactions for Potential E. coli DNA Gyrase B Inhibition" Molecules 26, no. 5: 1326. https://doi.org/10.3390/molecules26051326
APA StyleAroso, R. T., Guedes, R. C., & Pereira, M. M. (2021). Synthesis of Computationally Designed 2,5(6)-Benzimidazole Derivatives via Pd-Catalyzed Reactions for Potential E. coli DNA Gyrase B Inhibition. Molecules, 26(5), 1326. https://doi.org/10.3390/molecules26051326