
Fluorescent Orthopalladated Complexes of 4-Aryliden-5(4H)-oxazolones from the Kaede Protein: Synthesis and Characterization

 , , ,
, , ,  and
and 
Abstract
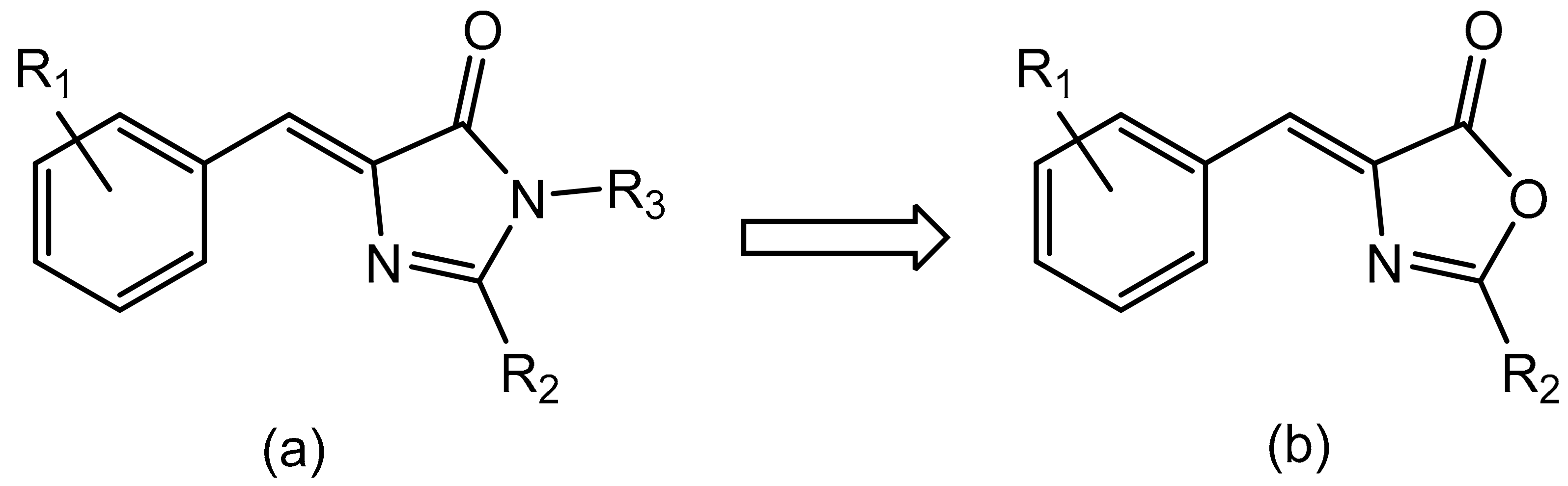
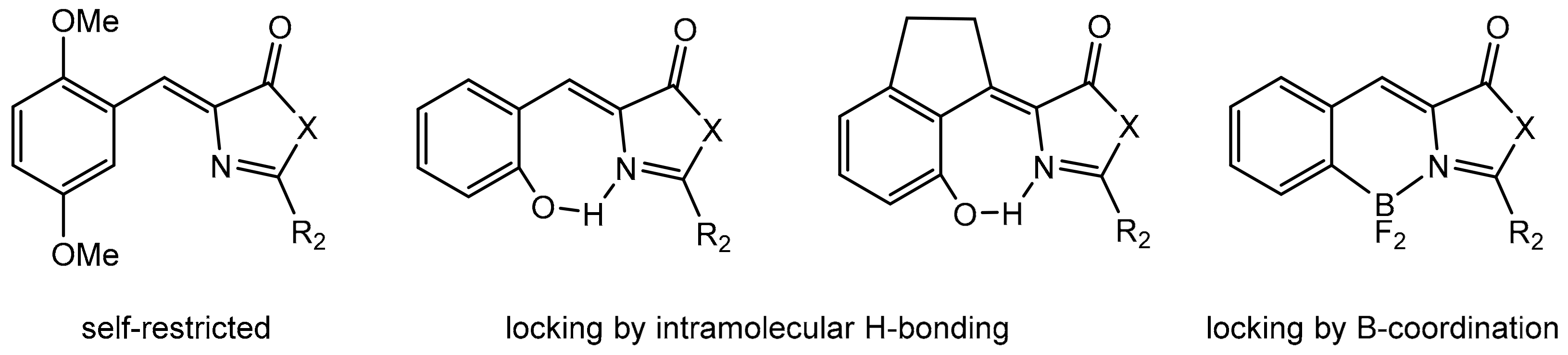
1. Introduction
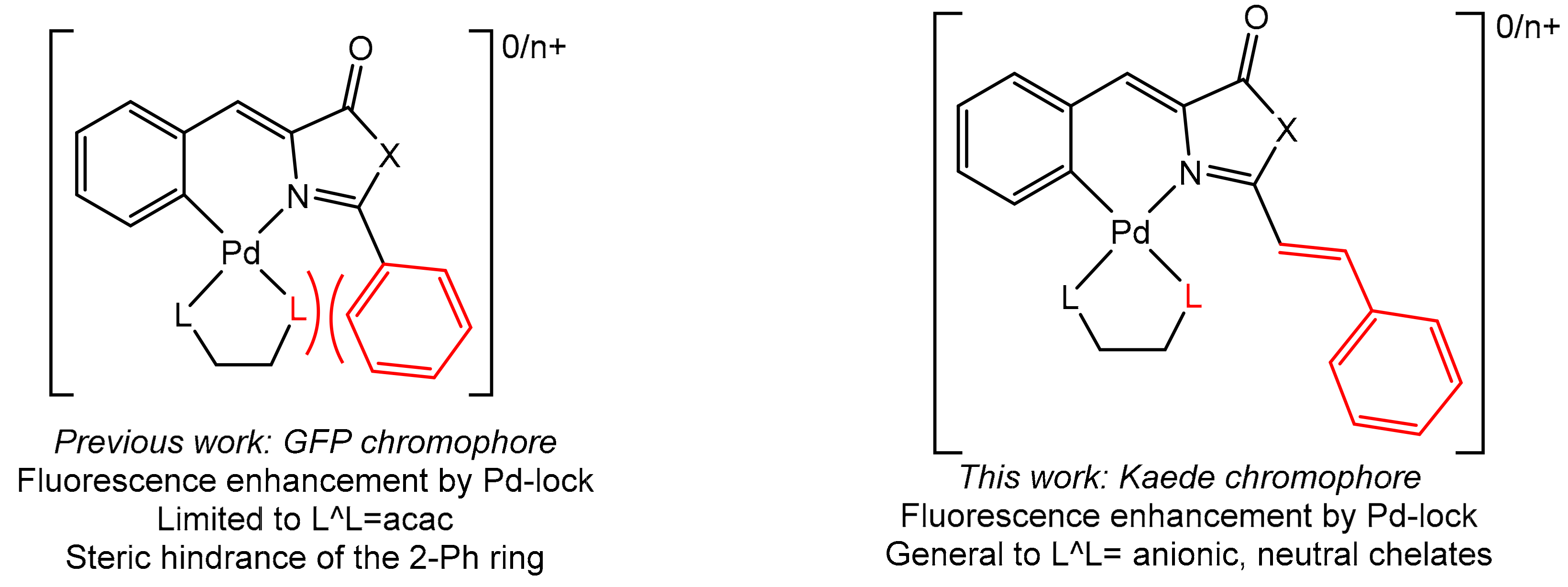
2. Results and Discussion
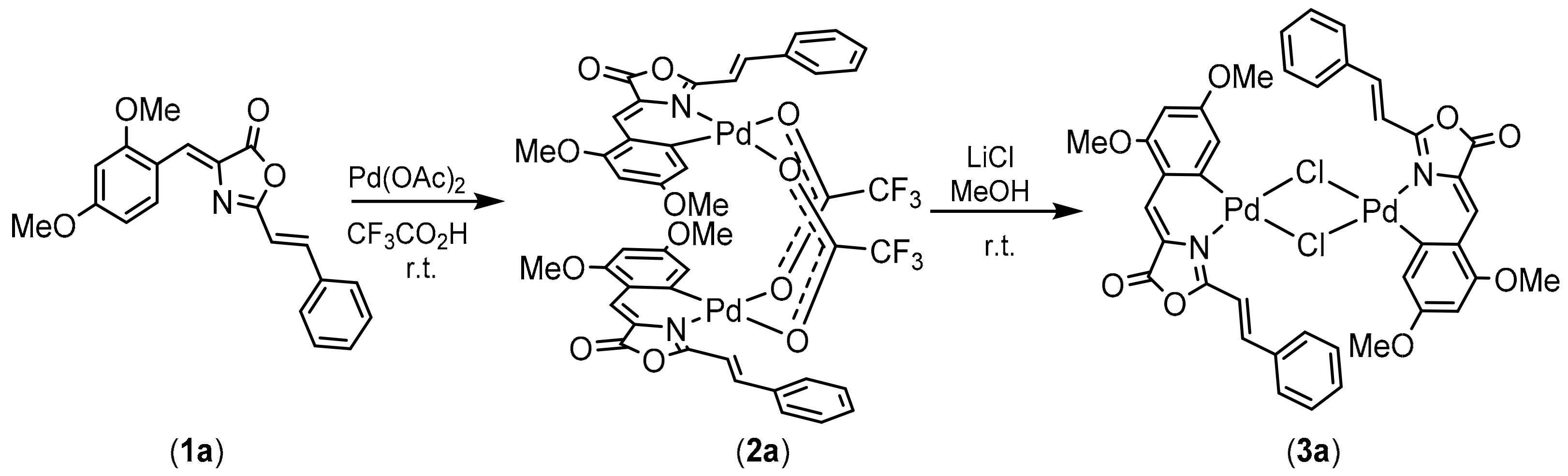
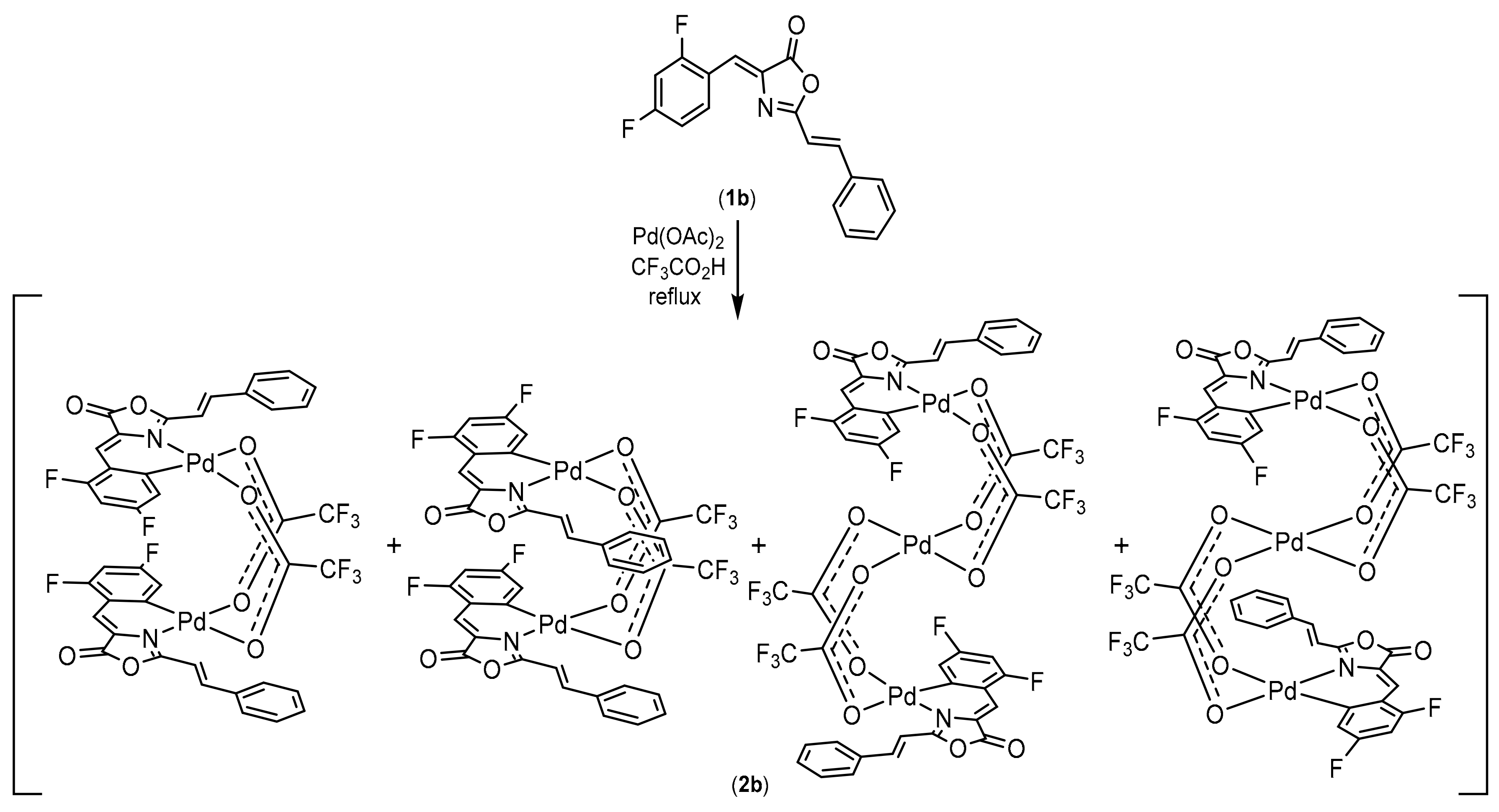
2.1. Synthesis, Characterization and Orthopalladation of Oxazolones 1a and 1b
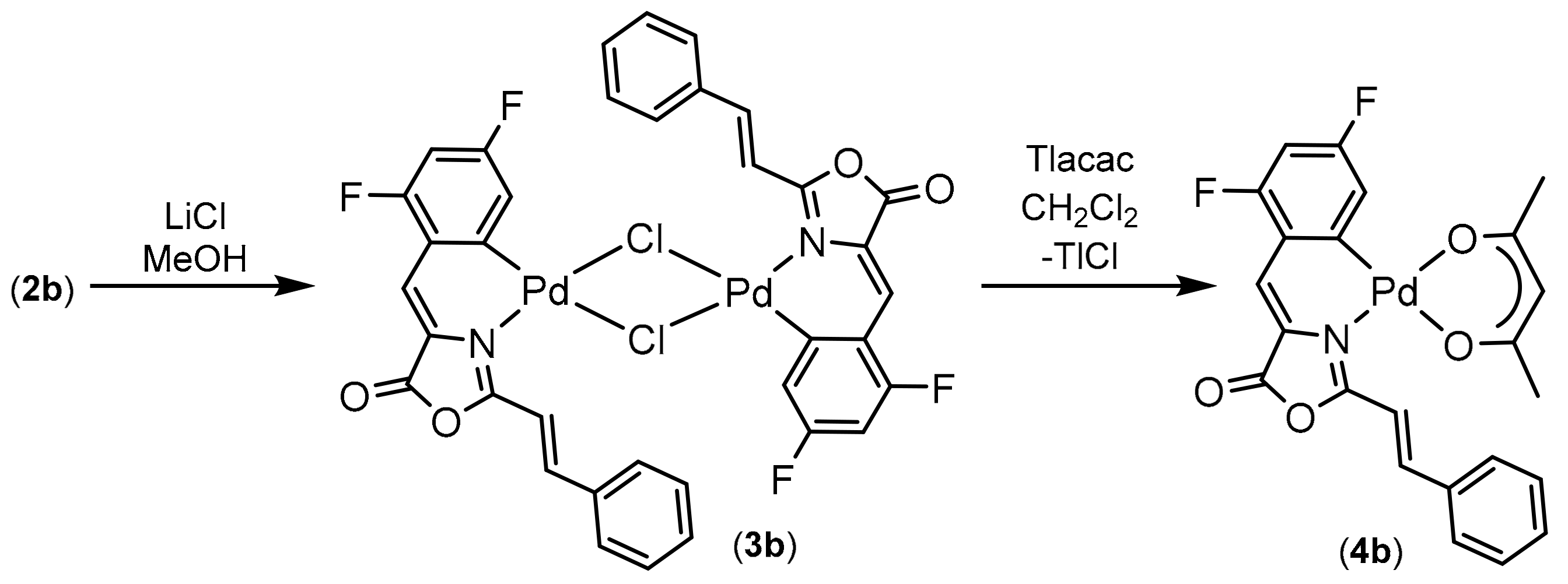
2.2. Synthesis of Complexes with Orthopalladated Oxazolones and Different Ancillary Ligands
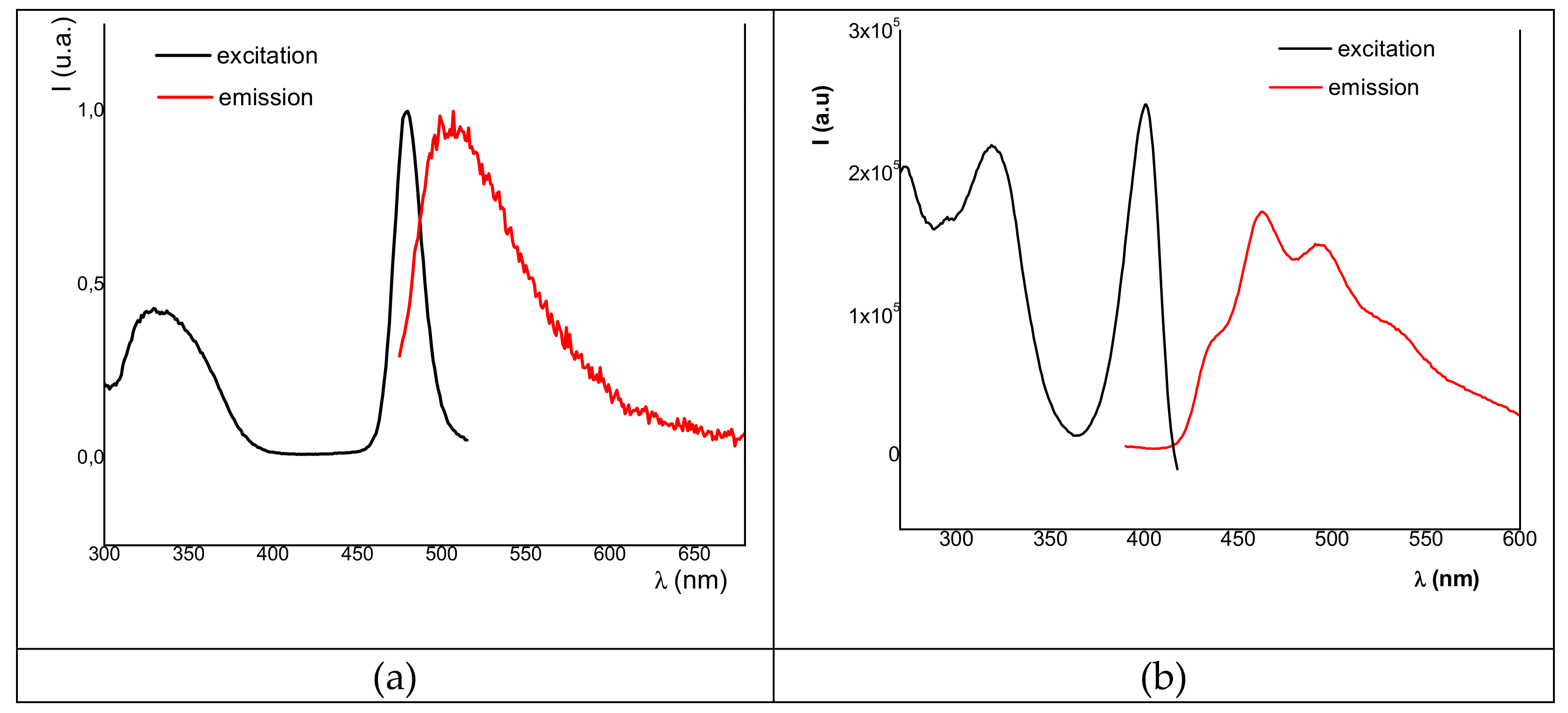
2.3. Fluorescence Studies: Absorption and Excitation–Emission Spectra
3. Materials and Methods
3.1. General Procedures
3.2. X-ray Crystallography
3.3. Synthesis and Characterization of Oxazolones 1a and 1b
3.3.1. Synthesis of 4-((Z)-2,4-Dimethoxybenzylidene)-2-((E)-Styryl)-5(4H)-Oxazolone (1a)
3.3.2. Synthesis of 4-((Z)-2,4-Difluorobenzylidene)-2-((E)-Styryl)-5(4H)-Oxazolone (1b)
3.4. Synthesis and Characterization of the Orthopalladated Dimers with Trifluoroacetate Bridges 2a, 2b
3.4.1. Synthesis of Orthopalladated 2a
3.4.2. Reaction of Oxazolone 1b with Pd(OAc)2: Dinuclear and Trinuclear Derivatives (mixture 2b)
3.5. Synthesis and Characterization of the Orthopalladated Dimers with Chloride Bridges 3a, 3b
3.5.1. Synthesis of Orthopalladated 3a
3.5.2. Synthesis of Orthopalladated 3b
3.6. Synthesis and Characterization of Orthopalladated Complexes with Acetylacetonate 4a and 4b
3.6.1. Synthesis of Orthopalladated 4a
3.6.2. Synthesis of Orthopalladated 4b
3.7. Synthesis and Characterization of Orthopalladated Complex with 8-Hydroxyquinolinate 5a
3.8. Synthesis and Characterization of Orthopalladated Complex with 8-Aminoquinoline 6a
3.9. Synthesis and Characterization of Orthopalladated Complex with 2,2’-Bipyridine 7a
3.10. Synthesis and Characterization of Orthopalladated Complex with 1,10-Phenanthroline 8a
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Edgar, A. Luminescent Materials. In Springer Handbook of Electronic and Photonic Materials; Kasap, S., Capper, P., Eds.; Springer Handbooks; Springer Nature: Cham, Switzerland, 2017. [Google Scholar] [CrossRef]
- Valeur, B.; Berberan-Santos, M.N. Molecular Fluorescence, Principles and Applications, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2013. [Google Scholar]
- Guo, J.; Yu, H.; Cui, T. Applications of fluorescent materials in the detection of alkaline phosphatase activity. J. Biomed. Mater. Res. 2021, 109, 214–226. [Google Scholar] [CrossRef]
- Yang, Z.; Sharma, A.; Qi, J.; Peng, X.; Lee, D.Y.; Hu, R.; Lin, D.; Qu, J.; Kim, J.S. Super-resolution fluorescent materials: An insight into design and bioimaging applications. Chem. Soc. Rev. 2016, 45, 4651–4667. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Ji, X.; Li, Z.; Huang, F. Fluorescent Supramolecular Polymeric Materials. Adv. Mater. 2017, 29, 1606117. [Google Scholar] [CrossRef]
- Xiong, H.; Zheng, H.; Wang, W.; Liang, J.; Wen, W.; Zhang, X.; Wang, S. A convenient purification method for silver nanoclusters and its applications in fluorescent pH sensors for bacterial monitoring. Biosens. Bioelectron. 2016, 86, 164–168. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Yang, C. Blue fluorescent emitters: Design tactics and applications in organic light-emitting diodes. Chem. Soc. Rev. 2013, 42, 4963–4976. [Google Scholar] [CrossRef]
- Shimomura, O. Discovery of Green Fluorescent Protein (GFP) (Nobel Lecture). Angew. Chem. Int. Ed. 2009, 48, 5590–5602. [Google Scholar] [CrossRef]
- Chalfie, M. GFP: Lighting Up Life (Nobel Lecture). Angew. Chem. Int. Ed. 2009, 48, 5603–5611. [Google Scholar] [CrossRef] [PubMed]
- Tsien, R.Y. Constructing and exploiting the fluorescent protein paintbox (Nobel Lecture). Angew. Chem. Int. Ed. 2009, 48, 5612–5626. [Google Scholar] [CrossRef] [PubMed]
- Dedecker, P.; De Schryver, F.C.; Hofkens, J. Fluorescent Proteins: Shine on, You Crazy Diamond. J. Am. Chem. Soc. 2013, 135, 2387–2402. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, C.A.B.; Mariz, I.F.A.; Maçôas, E.M.S.; Afonso, C.A.M. Two-photon absorption properties of push-pull oxazolones derivatives. Dye. Pigment. 2012, 95, 713–722. [Google Scholar] [CrossRef]
- Rodrigues, C.A.B.; Mariz, I.F.A.; Maçôas, E.M.S.; Afonso, C.A.M.; Martinho, J.M.G. Unsaturated oxazolones as nonlinear fluorophores. Dye. Pigment. 2013, 99, 642–652. [Google Scholar] [CrossRef]
- Icli, S.; Icil, H.; Alp, S.; Koc, H.; McKillop, A. NMR, absorption and fluorescence parameters of azlactones. Spectrosc. Lett. 1994, 27, 1115–1128. [Google Scholar] [CrossRef]
- Icli, S.; Doroshenko, A.O.; Alp, S.; Abmanova, N.A.; Egorova, S.I.; Astley, S.T. Structure and Luminescent Properties of the 4-Arylidene-2-Aryl-5-Oxazolones (Azlactones) In Solution and Crystalline State. Spectrosc. Lett. 1999, 32, 553–569. [Google Scholar] [CrossRef]
- Smokal, V.; Kolendo, A.; Krupka, O.; Sahraoui, B. Synthesis, photophysical and photochemical properties of oxazolone derivatives. J. Optoelectron. Adv. Mater. 2008, 10, 607–612. [Google Scholar]
- Blanco-Lomas, M.; Campos, P.J.; Sampedro, D. Benzylidene-Oxazolones as Molecular Photoswitches. Org. Lett. 2012, 14, 4334–4337. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Lomas, M.; Funes-Ardoiz, I.; Campos, P.J.; Sampedro, D. Oxazolone-Based Photoswitches: Synthesis and Properties. Eur. J. Org. Chem. 2013, 6611–6618. [Google Scholar] [CrossRef]
- Funes-Ardoiz, I.; Blanco-Lomas, M.; Campos, P.J.; Sampedro, D. Benzylidene-oxazolones as photoswitches: Photochemistry and theoretical calculations. Tetrahedron 2013, 69, 9766–9771. [Google Scholar] [CrossRef]
- García-Irepa, C.; Marazzi, M.; Frutos, L.M.; Sampedro, D. E/Z Photochemical switches: synthesis, properties and applications. Rsc Adv. 2013, 3, 6241–6266. [Google Scholar] [CrossRef]
- Ertekin, K.; Alp, S.; Karapire, C.; Yenigül, B.; Henden, E.; Içli, S. Fluorescence emission studies of an azlactone derivative embedded in polymer films. An optical sensor for pH measurements. J. Photochem. Photobiol. A Chem. 2000, 137, 155–161. [Google Scholar] [CrossRef]
- Acharya, A.; Bogdanov, A.M.; Grigorienko, B.L.; Bravaya, K.B.; Nemukhin, A.V.; Lukyanov, K.A.; Krylov, A.I. Photoinduced Chemistry in Fluorescent Proteins: Curse or Blessing? Chem. Rev. 2017, 117, 758–795. [Google Scholar] [CrossRef]
- Gozem, S.; Luk, H.L.; Schapiro, I.; Olivucci, M. Theory and Simulation of the Ultrafast Double-Bond Isomerization of Biological Chromophores. Chem. Rev. 2017, 117, 13502–13565. [Google Scholar] [CrossRef]
- Liu, R.S.H. Photoisomerization by hula-twist: A fundamental supramolecular photochemical reaction. Acc. Chem. Res. 2001, 34, 555–562. [Google Scholar] [CrossRef] [PubMed]
- Meech, S.R. Excited State Reactions in Fluorescent Proteins. Chem. Soc. Rev. 2009, 38, 2922–2934. [Google Scholar] [CrossRef] [PubMed]
- Tolbert, L.M.; Baldridge, A.; Kowalik, J.; Solntsev, K.M. Collapse and Recovery of Green Fluorescent Protein Chromophore Emission through Topological Effects. Acc. Chem. Res. 2012, 45, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Follenius-Wund, A.; Bourotte, M.; Schmitt, M.; Iyice, F.; Lami, H.; Bourguignon, J.J.; Haiech, J.; Pigault, C. Fluorescent Derivatives of the GFP Chromophore Give a New Insight into the GFP Fluorescence Process. Biophys. J. 2003, 85, 1839–1850. [Google Scholar] [CrossRef]
- Litvinenko, K.L.; Webber, N.M. Meech, S.R. Internal Conversion in the Chromophore of the Green Fluorescent Protein: Temperature dependence and Isoviscosity Analysis. J. Phys. Chem. A 2003, 107, 2616–2623. [Google Scholar] [CrossRef]
- Mandal, D.; Tahara, T.; Meech, S.R. Excited-State Dynamics in the Green Fluorescent Protein Chromophore. J. Phys. Chem. B 2004, 108, 1102–1108. [Google Scholar] [CrossRef]
- Martin, M.E.; Negri, F.; Olivucci, M. Origin, Nature, and Fate of the Fluorescent State of the Green Fluorescent Protein Chromophore at the CASPT2//CASSCF Resolution. J. Am. Chem. Soc. 2004, 126, 5452–5464. [Google Scholar] [CrossRef] [PubMed]
- Andresen, M.; Wahl, M.C.; Stiel, A.C.; Gräter, F.; Schäfer, L.V.; Trowitzsch, S.; Weber, G.; Eggeling, C.; Grubmüller, H.; Hell, S.W.; et al. Structure and mechanism of the reversible photoswitch of a fluorescent protein. Proc. Natl. Acad. Sci. USA 2005, 102, 13070–13074. [Google Scholar] [CrossRef] [PubMed]
- Stavrov, S.S.; Solntsev, K.M.; Tolbert, L.M.; Huppert, D. Probing the Decay Coordinate of the Green Fluorescent Protein: Arrest of Cis-Trans Isomerization by the Protein Significantly Narrows the Fluorescence Spectra. J. Am. Chem. Soc. 2006, 128, 1540–1546. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.-S.; Huang, G.-J.; Liu, Y.-H.; Peng, S.-M. Photoisomerization of the Green Fluorescence Protein Chromophore and the meta- and para-Amino Analogues. Chem. Commun. 2008, 1344–1346. [Google Scholar] [CrossRef] [PubMed]
- Voliani, V.; Bizzarri, R.; Nifosi, R.; Abbruzzetti, S.; Grandi, E.; Viappiani, C.; Beltram, F. Cis-Trans Photoisomerization of Fluorescent-Protein Chromophore. J. Phys. Chem. B 2008, 112, 10714–10722. [Google Scholar] [CrossRef]
- Ivashkin, P.E.; Yampolsky, I.V.; Lukyanov, K.A. Synthesis and Properties of Chromophores of Fluorescent Proteins. Russ. J. Bioorg. Chem. 2009, 35, 652–669. [Google Scholar] [CrossRef] [PubMed]
- Megley, C.M.; Dickson, L.A.; Maddalo, S.L.; Chandler, G.J.; Zimmer, M. Photophysics and Dihedral Freedom of the Chromophore in Yellow, Blue, and Green Fluorescent Protein. J. Phys. Chem. B 2009, 113, 302–308. [Google Scholar] [CrossRef]
- Rajbongshi, B.K.; Sen, P.; Ramanathan, G. Twisted intramolecular charge transfer in a model green fluorescent protein luminophore analog. Chem. Phys. Lett. 2010, 494, 295–300. [Google Scholar] [CrossRef]
- Conyard, J.; Kondo, M.; Heisler, I.A.; Jones, G.; Baldridge, A.; Tolbert, L.M.; Solntsev, K.M.; Meech, S.R. Chemically Modulating the Photophysics of the GFP Chromophore. J. Phys. Chem. B 2011, 115, 1571–1577. [Google Scholar] [CrossRef]
- Rafiq, S.; Rajbongshi, B.K.; Nair, N.N.; Sen, P.; Ramanathan, G. Excited State Relaxation Dynamics of Model Green Fluorescent Protein Chromophore Analogs: Evidence for Cis−Trans Isomerism. J. Phys. Chem. A 2011, 115, 13733–13742. [Google Scholar] [CrossRef]
- Ai, Y.-J.; Liao, R.-Z.; Fang, W.-H.; Luo, Y. Theoretical Studies on the Isomerization Mechanism of the Ortho-Green Fluorescent Protein Chromophore. Phys. Chem. Chem. Phys. 2012, 14, 13409–13414. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.-W.; Huang, G.-J.; Hsu, H.-Y.; Prabhakar, C.; Lee, Y.-P.; Diau, E.W.-G.; Yang, J.-S. Effects of Hydrogen Bonding on Internal Conversion of GFP-like Chromophores. II. The meta-Amino Systems. J. Phys. Chem. B 2013, 117, 2705–2716. [Google Scholar] [CrossRef]
- Jiang, M.; He, Z.; Zhang, Y.; Sung, H.H.Y.; Lam, J.W.Y.; Peng, Q.; Yan, Y.; Wong, K.S.; Williams, I.D.; Zhao, Y.; et al. Development of benzylidene-methyloxazolone based AIEgens and decipherment of their working mechanism. J. Mater. Chem. C 2017, 5, 7191–7199. [Google Scholar] [CrossRef]
- Conyard, J.; Heisler, I.A.; Chan, Y.; Bulman Page, P.C.; Meech, S.R.; Blancafort, L. A New Twist in the Photophysics of the Gfp Chromophore: A Volume-Conserving Molecular Torsion Couple. Chem. Sci. 2018, 9, 1803–1812. [Google Scholar] [CrossRef]
- Taylor, M.A.; Zhu, L.; Rozanov, N.D.; Stout, K.T.; Chen, C.; Fang, C. Delayed Vibrational Modulation of the Solvated Gfp Chromophore into a Conical Intersection. Phys. Chem. Chem. Phys. 2019, 21, 9728–9739. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Romei, M.G.; Boxer, S.G. Structural Evidence of Photoisomerization Pathways in Fluorescent Proteins. J. Am. Chem. Soc. 2019, 141, 15504–15508. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, T.; Mandal, M.; Gude, V.; Bag, P.P.; Mandal, P.K. Strong electron donation induced differential nonradiative decay pathways for para and meta GFP chromophore analogues. Phys. Chem. Chem. Phys. 2015, 17, 20515–20521. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Burgess, K. Synthesis of Highly Fluorescent GFP-Chromophore Analogues. J. Am. Chem. Soc. 2008, 130, 4089–4096. [Google Scholar] [CrossRef] [PubMed]
- Baldridge, A.; Solntsev, K.M.; Song, C.; Tanioka, T.; Kowalik, J.; Hardcastle, K.; Tolbert, L.M. Inhibition of twisting of a green fluorescent protein-like chromophore by metal complexation. Chem. Commun. 2010, 46, 5686–5688. [Google Scholar] [CrossRef]
- Baranov, M.S.; Lukyanov, K.A.; Borissova, A.O.; Shamir, J.; Kosenkov, D.; Slipchenko, L.V.; Tolbert, L.M.; Yampolsky, I.V.; Solntsev, K.M. Conformationally Locked Chromophores as Models of Excited-State Proton Transfer in Fluorescent Proteins. J. Am. Chem. Soc. 2012, 134, 6025–6032. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.H.; Chen, Y.A.; Tseng, H.W.; Zhang, Z.; Shen, J.Y.; Chuang, W.T.; Lin, T.C.; Lee, C.S.; Hung, W.Y.; Hong, B.C.; et al. Locked ortho- and para-Core Chromophores of Green Fluorescent Protein; Dramatic Emission Enhancement via Structural Constraint. J. Am. Chem. Soc. 2014, 136, 11805–11812. [Google Scholar] [CrossRef] [PubMed]
- Baranov, M.S.; Solntsev, K.M.; Baleeva, N.S.; Mishin, A.S.; Lukyanov, S.A.; Lukyanov, K.A.; Yampolsky, I.V. Red-Shifted Fluorescent Aminated Derivatives of a Conformationally Locked GFP Chromophore. Chem. Eur. J. 2014, 20, 13234–13241. [Google Scholar] [CrossRef] [PubMed]
- Baleeva, N.S.; Myannik, K.A.; Yampolski, I.V.; Baranov, M.S. Bioinspired Fluorescent Dyes Based on a Conformationally Locked Chromophore of the Fluorescent Protein Kaede. Eur. J. Org. Chem. 2015, 5716–5721. [Google Scholar] [CrossRef]
- Liu, X.-Y.; Chang, X.-P.; Xia, S.-H.; Cui, G.; Thiel, W. Excited-State Proton-Transfer-Induced Trapping Enhances the Fluorescence Emission of a Locked GFP Chromophore. J. Chem. Theory Comput. 2016, 12, 753–764. [Google Scholar] [CrossRef] [PubMed]
- Baleeva, N.S.; Tsarkova, A.S.; Baranov, M.S. Conformationally Locked Chromophores of CFP and Sirius Protein. Tetrahedron Lett. 2016, 57, 3043–3045. [Google Scholar] [CrossRef]
- Deng, H.; Yu, C.; Gong, L.; Zhu, X. Self-Restricted Green Fluorescent Protein Chromophore Analogues: Dramatic Emission Enhancement and Remarkable Solvatofluorochromism. J. Phys. Chem. Lett. 2016, 7, 2935–2944. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.; Zhang, Z.; Zhao, Y.; Yu, C.; Gong, L.; Yan, D.; Zhu, X. Self-Restricted oxazolone GFP chromophore for construction of reaction-based fluorescent probe toward dopamine. Mater. Today Chem. 2017, 3, 73–81. [Google Scholar] [CrossRef]
- Baleeva, N.S.; Zaitseva, S.O.; Gorbachev, D.A.; Smirnov, A.Y.; Zagudaylova, M.B.; Baranov, M.S. The Role of N-Substituents in Radiationless Deactivation of Aminated Derivatives of a Locked GFP Chromophore. Eur. J. Org. Chem. 2017, 5219–5224. [Google Scholar] [CrossRef]
- Chen, C.; Liu, W.; Baranov, M.S.; Baleeva, N.S.; Yampolsky, I.V.; Zhu, L.; Wang, Y.; Shamir, A.; Solntsev, K.M.; Fang, C. Unveiling Structural Motions of a HighlyFluorescent Superphotoacid by Locking and Fluorinating the GFP Chromophore in Solution. J. Phys. Chem. Lett. 2017, 8, 5921–5928. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Baranov, M.S.; Zhu, L.; Baleeva, N.S.; Smirnov, A.Y.; Zaitseva, S.O.; Yampolsky, I.V.; Solntsev, K.M.; Fang, C. Designing redder and brighter fluorophores by synergistic tuning of ground and excited states. Chem. Commun. 2019, 55, 2537–2540. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Zhu, L.; Baranov, M.S.; Tang, L.; Baleeva, N.S.; Smirnov, A.Y.; Yampolsky, I.V.; Solntsev, K.M.; Fang, C. Photoinduced Proton Transfer of GFP-Inspired Fluorescent Superphotoacids: Principles and Design. J. Phys. Chem. B 2019, 123, 3804–3821. [Google Scholar] [CrossRef]
- Lin, C.Y.; Romei, M.G.; Oltrogge, L.M.; Mathews, I.I.; Boxer, S.G. Unified Model for Photophysical and Electro-Optical Properties of Green Fluorescent Proteins. J. Am. Chem. Soc. 2019, 141, 15250–15265. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Ahire, K.; Karuso, P. Room Temperature Dual Fluorescence of a Locked GFP Chromophore Analogue. J. Am. Chem. Soc. 2020, 142, 738–749. [Google Scholar] [CrossRef] [PubMed]
- Collado, S.; Pueyo, A.; Baudequin, C.; Bischoff, L.; Jiménez, A.I.; Cativiela, C.; Hourau, C.; Urriolabeitia, E.P. Orthopalladation of GFP-Like Fluorophores Through C–H Bond Activation: Scope and Photophysical Properties. Eur. J. Org. Chem. 2018, 6158–6166. [Google Scholar] [CrossRef]
- Roiban, D.; Serrano, E.; Soler, T.; Grosu, I.; Cativiela, C.; Urriolabeitia, E.P. Unexpected [2 + 2] C–C bond coupling due to photocycloaddition on orthopalladated (Z)-2-aryl-4-arylidene-5(4H)-oxazolones. Chem. Commun. 2009, 4681–4683. [Google Scholar] [CrossRef]
- Roiban, G.D.; Serrano, E.; Soler, T.; Aullón, G.; Grosu, I.; Cativiela, C.; Martínez, M.; Urriolabeitia, E.P. Regioselective Orthopalladation of (Z)-2-Aryl-4-Arylidene-5(4H)-Oxazolones: Scope, Kinetico-Mechanistic, and Density Functional Theory Studies of the C–H Bond Activation. Inorg. Chem. 2011, 50, 8132–8143. [Google Scholar] [CrossRef]
- Serrano, E.; Juan, A.; García-Montero, A.; Soler, T.; Jiménez-Márquez, F.; Cativiela, C.; Gomez, M.V.; Urriolabeitia, E.P. Stereoselective Synthesis of 1,3-Diaminotruxillic Acid Derivatives: An Advantageous Combination of C–H–ortho–Palladation and On-Flow [2+2]–Photocycloaddition in Microreactors. Chem. Eur. J. 2016, 22, 144–152. [Google Scholar] [CrossRef]
- Carrera, C.; Denisi, A.; Cativiela, C.; Urriolabeitia, E.P. Functionalized 1,3-Diaminotruxillic Acids by Pd-Mediated C–H Activation and [2+2]–Photocycloaddition of 5(4H)–Oxazolones. Eur. J. Inorg. Chem. 2019, 3481–3489. [Google Scholar] [CrossRef]
- Urriolabeitia, E.P.; Sánchez, P.; Pop, A.; Silvestru, C.; Laga, E.; Jiménez, A.I.; Cativiela, C. Synthesis of esters of diaminotruxillic bis-amino acids by Pd-mediated photocycloaddition of analogs of the Kaede protein chromophore. Beilstein J. Org. Chem. 2020, 16, 1111–1123. [Google Scholar] [CrossRef] [PubMed]
- Roiban, G.D.; Serrano, E.; Soler, T.; Contel, M.; Grosu, I.; Cativiela, C.; Urriolabeitia, E.P. Ortho-Palladation of (Z)-2-Aryl-4-Arylidene-5(4H)-Oxazolones. Structure and Functionalization. Organometallics 2010, 29, 1428–1435. [Google Scholar] [CrossRef]
- Miyawaki, A. Proteins on the move: Insights gained from fluorescent protein technologies. Nat. Rev. Mol. Cell Biol. 2011, 12, 656–668. [Google Scholar] [CrossRef] [PubMed]
- Ando, R.; Hama, H.; Yamamoto-Hino, M.; Mizuno, H.; Miyawaki, A. An optical marker based on the UV-induced green-to-red photoconversion of a fluorescent protein. Proc. Natl. Acad. Sci. USA 2002, 99, 12651–12656. [Google Scholar] [CrossRef] [PubMed]
- Patterson, G.H.; Lippincott-Schwartz, J. A Photoactivatable GFP for Selective Photolabeling of Proteins and Cells. Science 2002, 297, 1873–1877. [Google Scholar] [CrossRef]
- Plöchl, J. Ueber Phenylglycidasäure (Phenyloxacrylsäure). Chem. Ber. 1883, 16, 2815. [Google Scholar] [CrossRef]
- Plöchl, J. Ueber einige Derivate der Benzoylimidozimmtsäure. Chem. Ber. 1884, 17, 1616. [Google Scholar] [CrossRef]
- Erlenmeyer, E. Ueber die Condensation der Hippursäure mit Phtalsäureanhydrid und mit Benzaldehyd. Justus Liebigs Ann. Der Chem. 1893, 275, 1–8. [Google Scholar]
- Carter, H.E. Azlactones. Org. React. 1946, 3, 198–237. [Google Scholar]
- Filler, R. Advances in Heterocyclic Chemistry; Katritzky, A.R., Ed.; Academic Press: New York, NY, USA, 1954; Chapter 4; p. 75. [Google Scholar]
- Rao, Y.S.; Filler, R. Geometric Isomers of 2-Aryl(Aralkyl)-4-arylidene(alkylidene)-5(4H)-oxazolones. Synthesis 1975, 12, 749–764. [Google Scholar] [CrossRef]
- Rao, Y.S.; Filler, R. Oxazoles. In The Chemistry of Heterocyclic Compounds, Volume 45; Turchi, I.J., Ed.; John Wiley & Sons, Inc.: New York, NY, USA, 1986; Chapter 3; pp. 363–691. [Google Scholar]
- Kim, Y.; Ko, Y.H.; Jung, M.; Selvapalam, N.; Kim, K. A new photo-switchable “on-off” host–guest system. Photochem. Photobiol. Sci. 2011, 10, 1415–1419. [Google Scholar] [CrossRef] [PubMed]
- Prokofev, E.P.; Karpeiskaya, E.I. The proton coupled 13C NMR direct determination of Z-,E-configuration of 4-benzyliden-2-phenyl(methyl)-Δ2-oxazolin-5-ones and products of their solvolysis. Tetrahedron Lett. 1979, 20, 737–740. [Google Scholar] [CrossRef]
- Vana, J.; Lang, J.; Soltesova, M.; Hanusek, J.; Ruzicka, A.; Sedlak, M.; Roithova, J. The role of trinuclear species in a palladium acetate/trifluoroacetic acid catalytic system. Dalton Trans. 2017, 46, 16269–16275. [Google Scholar] [CrossRef]
- Vana, J.; Hanusek, J.; Sedlak, M. Bi and trinuclear complexes in palladium carboxylate-assisted C–H activation reactions. Dalton Trans. 2018, 47, 1378–1382. [Google Scholar] [CrossRef] [PubMed]
- Nakamoto, K. Infrared and Raman Spectra of Inorganic and Coordination Compounds, 5th ed.; John Wiley: New York, NY, USA, 1997. [Google Scholar]
- Guy Orpen, A.; Brammer, L.; Allen, F.H.; Kennard, O.; Watson, D.G.; Taylor, R. Supplement. Tables of bond lengths determined by X-ray and neutron diffraction. Part 2. Organometallic compounds and co-ordination complexes of the d- and f-block metals. J. Chem. Soc. Dalton Trans. 1989, S1–S83. [Google Scholar] [CrossRef]
- SAINT, Version 5.0 ed.; Bruker Analytical X-Ray Systems: Madison, WI, USA, 1998.
- Sheldrick, G.M. SADABS, Program for Absorption and Other Corrections; Göttingen University: Göttingen, Germany, 1996. [Google Scholar]
- Sheldrick, G.M. SHELXT—Integrated Space-Group and Crystal- Structure Determination. Acta Crystallogr. Sect. A Found. Adv. 2015, A71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, C71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pd(1)-N(1) | 2.060(3) | N(1)-Pd(1)-N(3) | 173.70(12) |
| Pd(1)-N(2) | 2.110(3) | N(1)-Pd(1)-C(1) | 98.62(13) |
| Pd(1)-N(3) | 2.006(3) | N(2)-Pd(1)-N(3) | 96.35(11) |
| Pd(1)-C(1) | 2.013(3) | N(2)-Pd(1)-C(1) | 177.06(12) |
| N(1)-Pd(1)-N(2) | 78.55(11) | N(3)-Pd(1)-C(1) | 86.53(12) |
| Compound | λabs,max (nm) | Compound | λabs,max (nm) |
|---|---|---|---|
| 1a | 428 | 1b | 381 |
| 4a | 468 | 4b | 446 |
| 6a | 490 | ||
| 7a | 495 | 8a | 494 |
| Compound | λexc,max (nm) | λemis,max (nm) | Counts | QY |
|---|---|---|---|---|
| 1a | 480, 331 | 505 | 2.7 × 104 | <1% |
| 1b | 420, 326 | 534, 493, 462, 437 | 2.4 × 104 | |
| 4a | 516, 480, 446 | 476, 453 | 1 × 106 | - |
| 4b | 480, 321 | 569 (h), 525 | 3.6 × 104 | - |
| 6a | 539, 469, 371 | 589 | 5 × 106 | 12% 2 |
| 7a | 555, 433, 390 | 599 | 3 × 106 | - |
| 8a | 529, 412 | 564 | 1.6 × 105 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laga, E.; Dalmau, D.; Arregui, S.; Crespo, O.; Jimenez, A.I.; Pop, A.; Silvestru, C.; Urriolabeitia, E.P. Fluorescent Orthopalladated Complexes of 4-Aryliden-5(4H)-oxazolones from the Kaede Protein: Synthesis and Characterization. Molecules 2021, 26, 1238. https://doi.org/10.3390/molecules26051238
Laga E, Dalmau D, Arregui S, Crespo O, Jimenez AI, Pop A, Silvestru C, Urriolabeitia EP. Fluorescent Orthopalladated Complexes of 4-Aryliden-5(4H)-oxazolones from the Kaede Protein: Synthesis and Characterization. Molecules. 2021; 26(5):1238. https://doi.org/10.3390/molecules26051238
Chicago/Turabian StyleLaga, Eduardo, David Dalmau, Sofía Arregui, Olga Crespo, Ana I. Jimenez, Alexandra Pop, Cristian Silvestru, and Esteban P. Urriolabeitia. 2021. "Fluorescent Orthopalladated Complexes of 4-Aryliden-5(4H)-oxazolones from the Kaede Protein: Synthesis and Characterization" Molecules 26, no. 5: 1238. https://doi.org/10.3390/molecules26051238
APA StyleLaga, E., Dalmau, D., Arregui, S., Crespo, O., Jimenez, A. I., Pop, A., Silvestru, C., & Urriolabeitia, E. P. (2021). Fluorescent Orthopalladated Complexes of 4-Aryliden-5(4H)-oxazolones from the Kaede Protein: Synthesis and Characterization. Molecules, 26(5), 1238. https://doi.org/10.3390/molecules26051238