
Exceptionally Long Covalent CC Bonds—A Local Vibrational Mode Study




Abstract
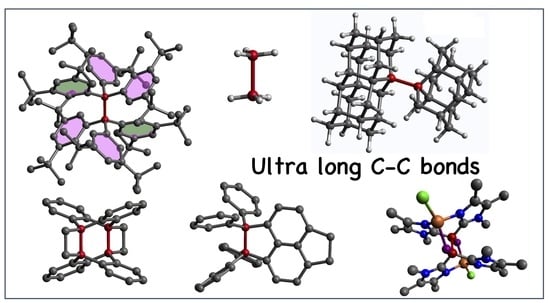
1. Introduction
2. Computational Methods
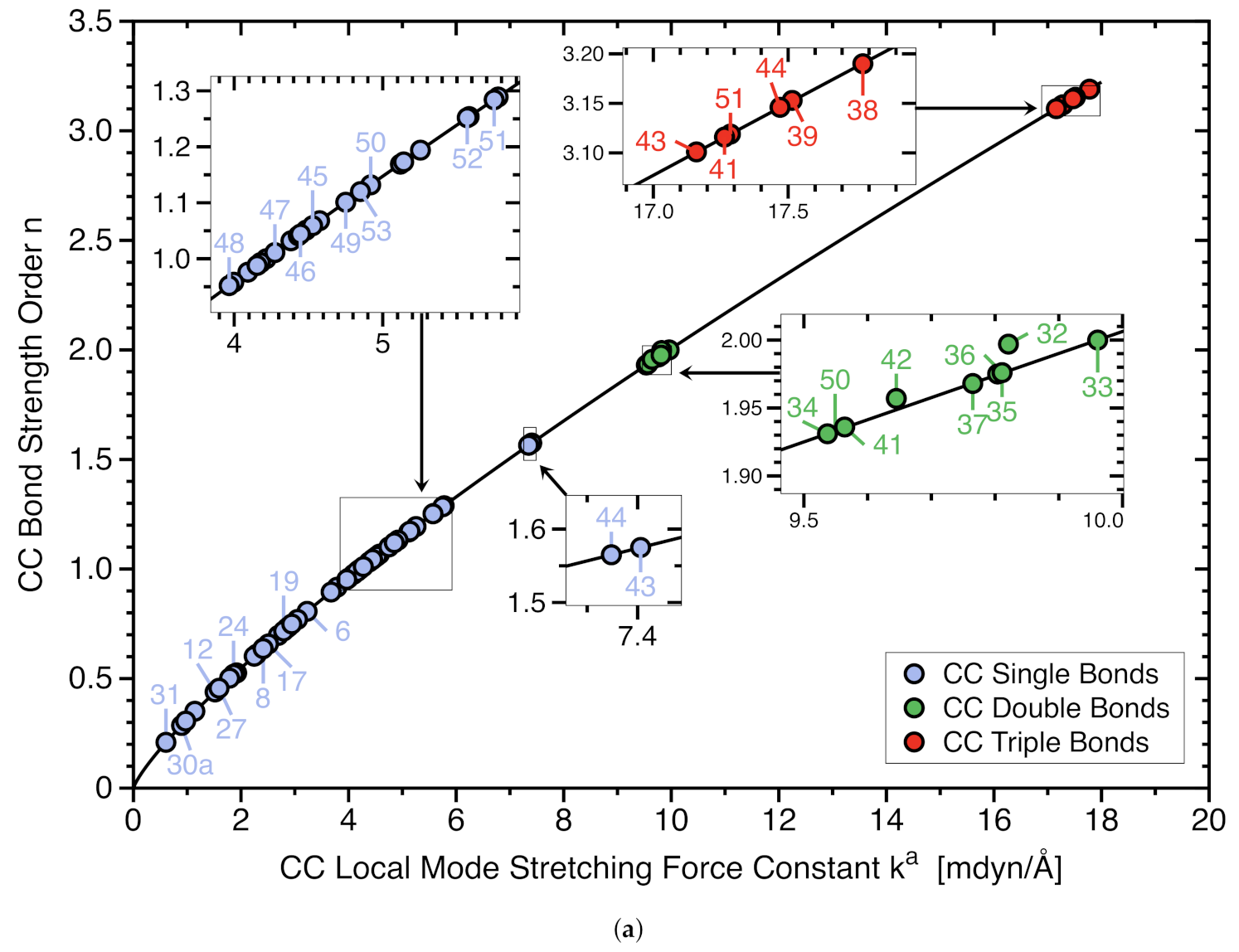
3. Results and Discussion
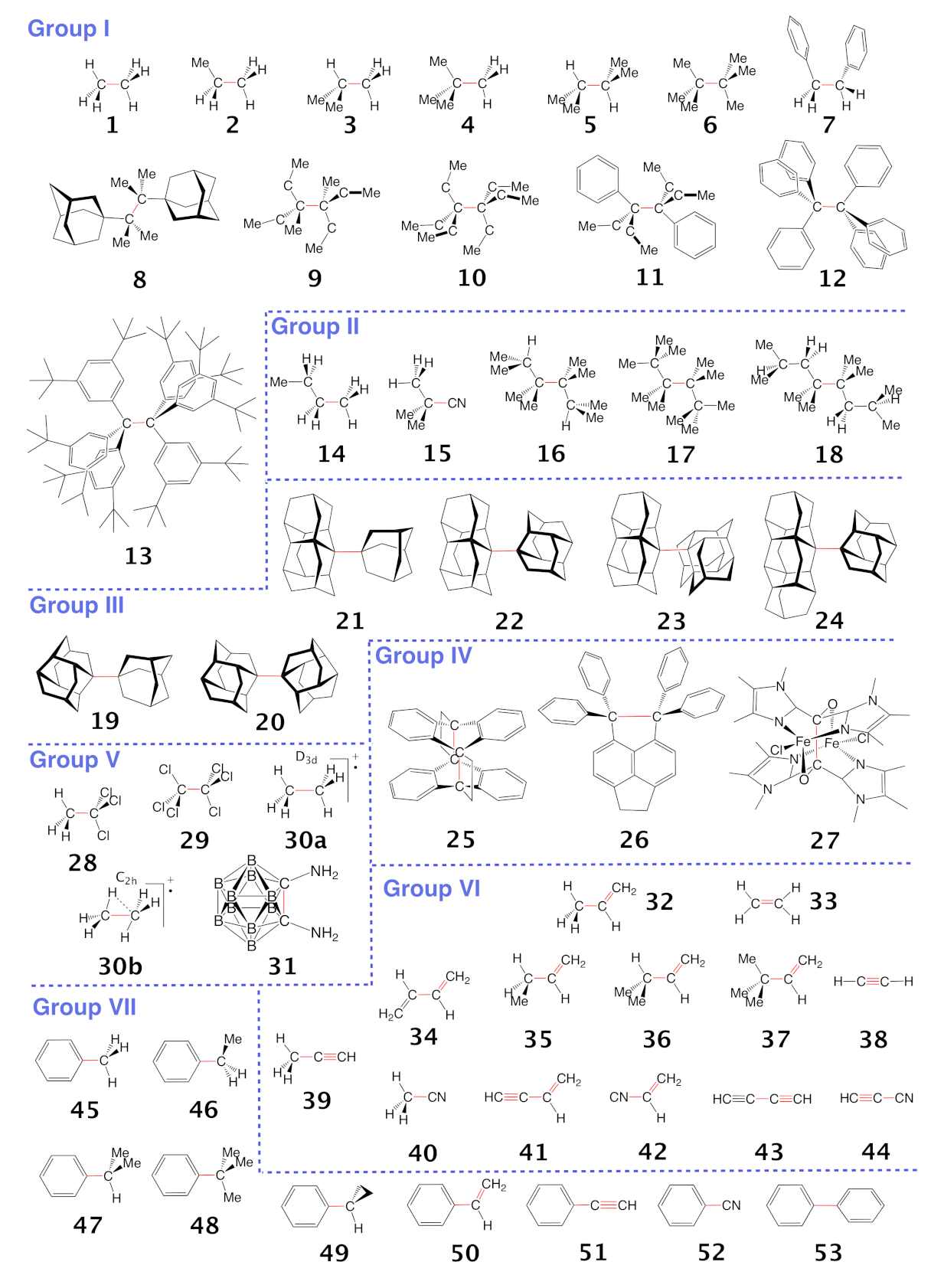
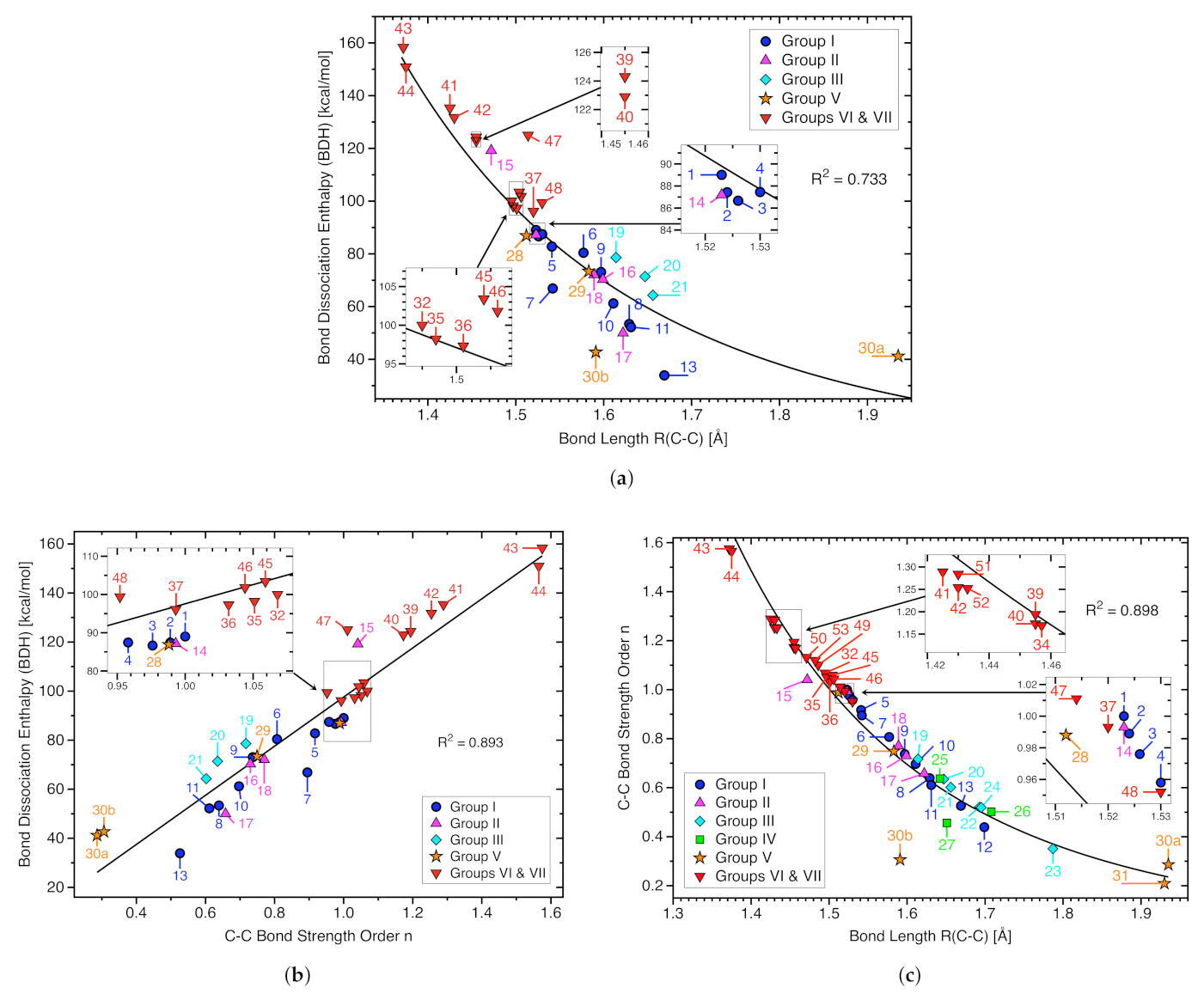
3.1. Bond Dissociation Enthalpies and Bond Lengths as Bond Strength Measure
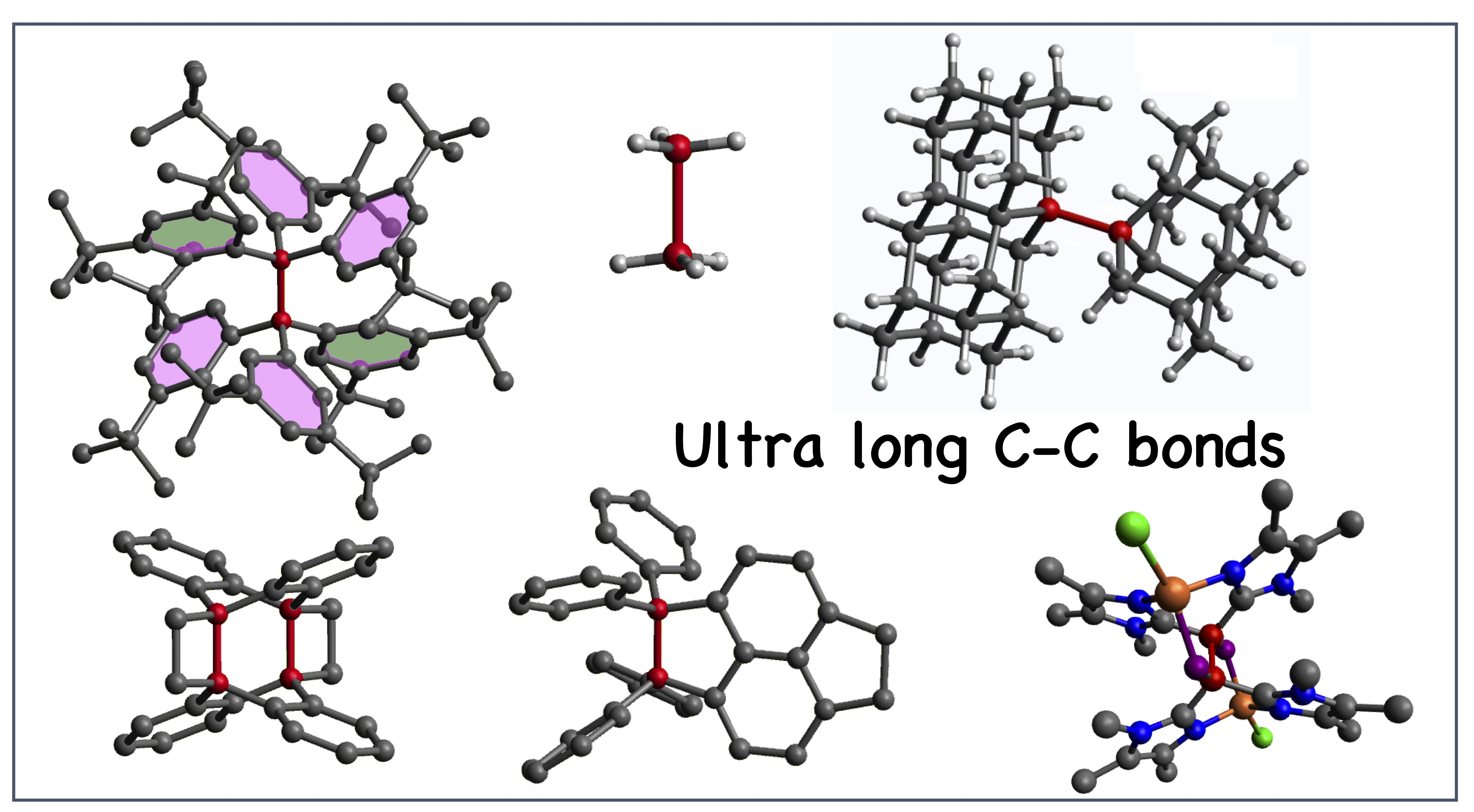
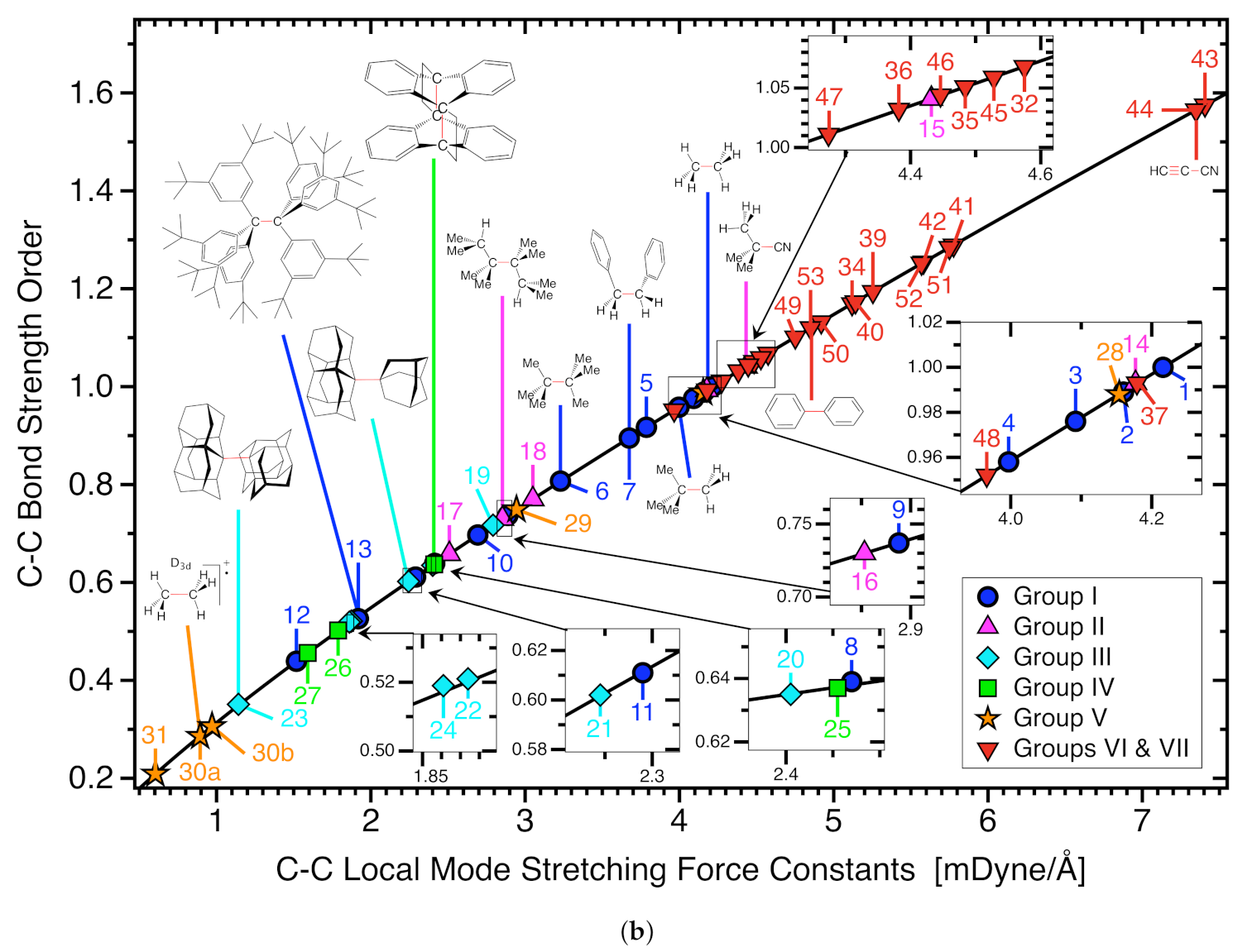
3.2. C−C Bonds of Group I
3.3. C−C Bonds of Group II
3.4. C−C Bonds of Group III
3.5. C−C Bonds of Group IV
3.6. C−C Bonds of Group V
3.7. CC Reference Bonds, Group VI and VII
4. Conclusions and Outlook
- Although steric crowding/strain increases the C−C bond length electronic effects can lead to even longer and weaker CC bonds. The overall weakest C−C bonds are found within Group V where electron deficient bonding is represented by the ethane radical cation in D3d (30a) (BSO n = 0.286) and C2h (30b) (BSO n = 0.306) symmetry and di-N,N-dimethylamino-o-carborane (31) (BSO n = 0.209). However, whereas 31 has the weakest C−C bond, 30a has the longest C−C bond at a value of 1.935 Å compared to the C−C length of 1.930 Å for 31, confirming previous findings that the longer bond is not always the weaker bond.
- A gap beyond 0.1 Å is present between the two longest C−C bonds (30a and 31) and the other CC bonds within this study. This gap is due to the C−C bonds within molecules 30a and 31 being governed by electronic effects. The C−C bond of 30a is heavily affected by its electron deficient nature while the CC bond length of system 31 is influenced by negative hyperconjugation effects.
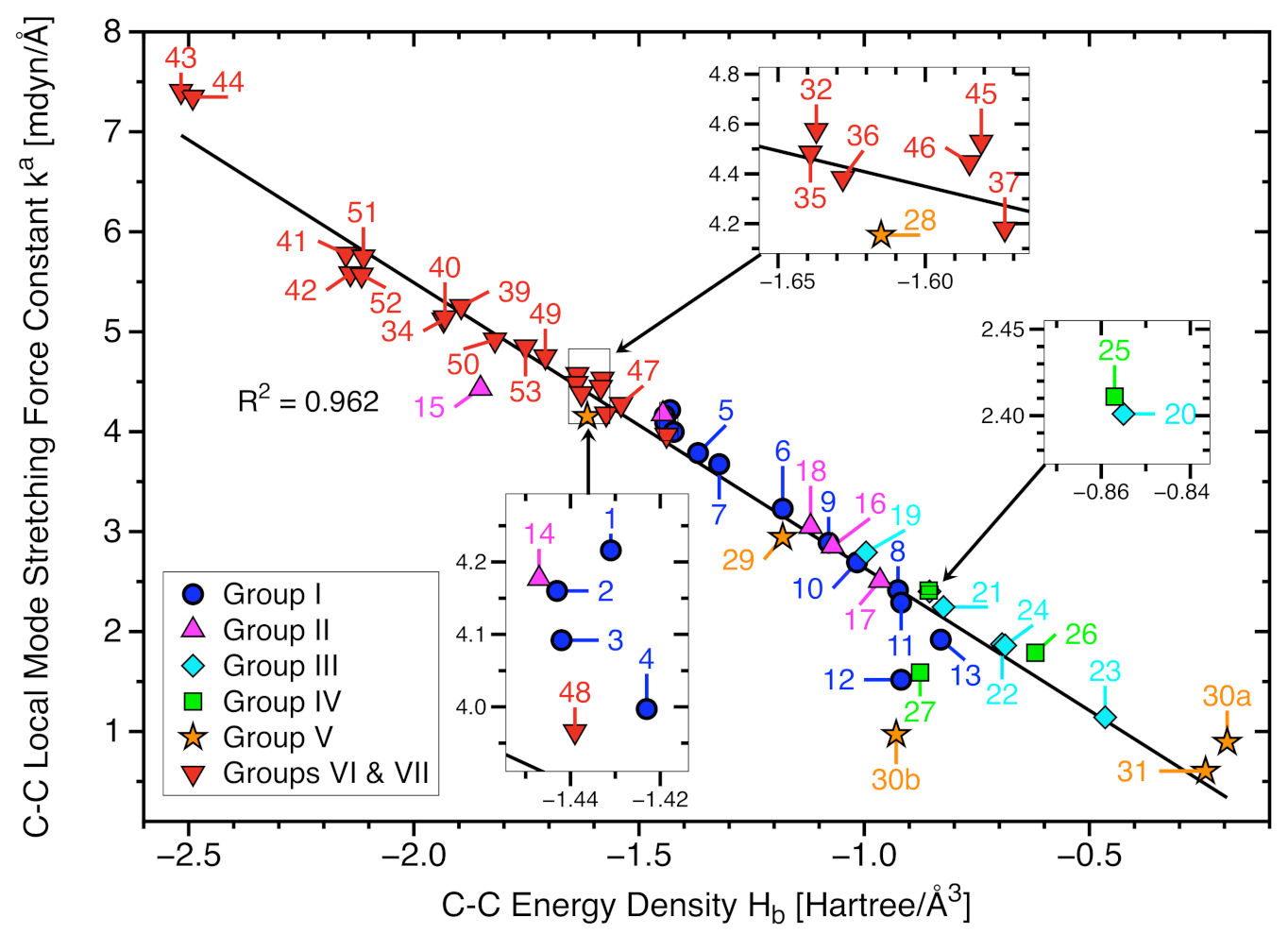
- The covalent character of all C−C bonds has been verified via the negative energy density values taken at the bond critical point r. In most cases is in line with the or BSO n values, reflecting that stronger bonds have more covalent character. However, we also found some exceptions such as the chlorinated ethanes (28 and 29). The results demonstrate that the local mode force constant is a sensitive bond strength measure that considers subtle second order effects that are not considered for the energy density evaluated just at the bond critical point which describes the covalent character, whereas includes counterbalancing steric repulsion effects or C−C bond weakening via delocalization of charge from the Cl lone pair into the (C−C), i.e., the so-called negative hyperconjugation effect, or intramolecular halogen-halogen dispersion interaction.
- Although there is some trend that the BDH value increases with increasing C−C bond strength, the overall correlation is moderate (R = 0.893), revealing that the BDH is an inadequate measure for the intrinsic strength of the CC bond as BDH includes the overall effects of bond breakage. Such concerns in particular crowded molecules with greater possibilities for geometry and electron density reorganization of the fragments such as molecules 7, 13, and 15.
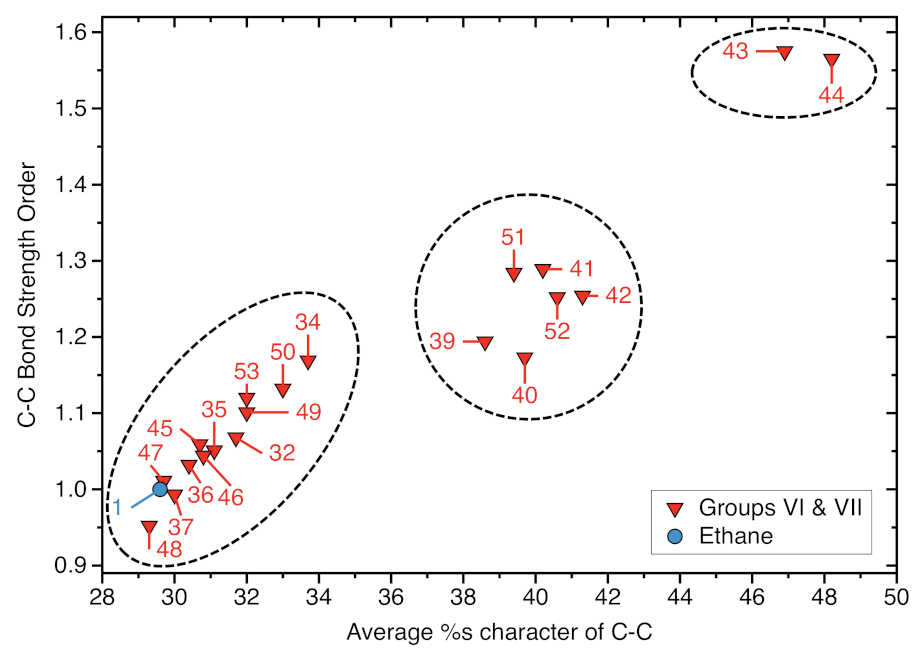
- We found an interesting relationship between the %s-character and the strength of the C−C bond as expressed via the local mode force constant. As the average %s-character of the targeted C−C bond increases the C−C bond strength increases which adds to the %s-character model quantity a physical relevance.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bell, J.A. Chemistry: A Project of the American Chemical Society; W H Freeman & CO: New York, NY, USA, 2004. [Google Scholar]
- Howes, L. The Fight Over the Longest Carbon-Carbon Bond is Redefining What a Bond Is. C&E News 2019, 97, 1–8. [Google Scholar]
- Mandal, N.; Datta, A. Molecular Designs for Expanding the Limits of Ultralong C–C Bonds and Ultrashort H···H Non-bonded Contacts. Chem. Commun. 2020, 56, 15377–15386. [Google Scholar] [CrossRef]
- Wu, Y.; Zhang, J.; Xie, Z. How Long a C–C Bond Can Be? An Example of Extraordinary Long C–C Single Bond in 1,2-Diarylamino-o-carborane. Chin. Chem. Lett. 2019, 30, 1530–1532. [Google Scholar] [CrossRef]
- Li, J.; Pang, R.; Li, Z.; Xiao, G.; Müller, T. Exceptionally Long C–C Single Bonds in Diamino-o-carborane as Induced by Negative Hyperconjugation. Angew. Chem. Int. Ed. 2019, 58, 1397–1401. [Google Scholar] [CrossRef]
- Shimajiri, T.; Suzuki, T.; Ishigaki, Y. Flexible C−C Bonds: Reversible Expansion, Contraction, Formation, and Scission of Extremely Elongated Single Bonds. Angew. Chem. Int. Ed. 2020, 59, 22252–22257. [Google Scholar] [CrossRef] [PubMed]
- Ishigaki, Y.; Shimajiri, T.; Takeda, T.; Katoono, R.; Suzuki, T. Longest C−C Single Bond among Neutral Hydrocarbons with a Bond Length beyond 1.8 Å. Chem. Commun. 2018, 4, 795–806. [Google Scholar] [CrossRef]
- Sethio, D.; Oliveira, V.; Kraka, E. Quantitative Assessment of Tetrel Bonding Utilizing Vibrational Spectroscopy. Molecules 2018, 23, 2763. [Google Scholar] [CrossRef]
- Del Bene, J.; Alkorta, I.; Elguero, J. Carbenes as Electron-Pair Doners to CO2 for C...C Tetrel Bonds and C–C Covalent Bonds. J. Phys. Chem. A 2017, 121, 4039–4047. [Google Scholar] [CrossRef]
- Del Bene, J.; Alkorta, I.; Elguero, J. Carbon-Carbon Bonding Between Nitrogen Heterocyclic Carbenes and CO2. J. Phys. Chem. A 2017, 121, 8136–8146. [Google Scholar] [CrossRef]
- Humason, A.; Zou, W.; Cremer, D. 11,11-Dimethyl-1,6-methano[10]annulene-An Annulene with an Ultralong CC Bond or a Fluxional Molecule? J. Phys. Chem. A 2014, 119, 1666–1682. [Google Scholar] [CrossRef] [PubMed]
- Mandal, N.; Pal, A.K.; Gain, P.; Zohaib, A.; Datta, A. Transition-State-like Planar Structures for Amine Inversion with Ultralong C–C Bonds in Diamino-o-carborane and Diamino-o-dodecahedron. J. Am. Chem. Soc. 2020, 142, 5331–5337. [Google Scholar] [CrossRef] [PubMed]
- Allinger, N.L.; Lii, J.H.; Schaefer, H.F., III. Molecular Mechanics (MM4) Studies on Unusually Long Carbon-Carbon Bond Distances in Hydrocarbons. J. Chem. Theory Comput. 2012, 12, 2774–2778. [Google Scholar] [CrossRef]
- Crabtree, K.N.; Talipov, M.R.; Martinez, O.; O’Connor, G.D.; Khursan, S.L.; McCarthy, M.C. Detection and Structure of HOON: Microwave Spectroscopy Reveals an O−O Bond Exceeding 1.9 Å. Science 2013, 342, 1354–1357. [Google Scholar] [CrossRef]
- Denis, P.A.; Huelmo, C.P. New Trends Along Hydrogen Polyoxides: Unusually Long Oxygen–Oxygen bonds in H2O6 and H2O7. Mol. Phys. 2014, 112, 3047–3056. [Google Scholar] [CrossRef]
- Pupim, C.F.; Catão, A.J.L.; López-Castillo, A. Boron–Nitrogen Dative Bond. J. Mol. Model. 2018, 24, 283. [Google Scholar] [CrossRef]
- Suzuki, T.; Uchimura, Y.; Ishigaki, Y.; Takeda, T.; Katoono, R.; Kawai, H.; Fujiwara, K.; Nagaki, A.; Yoshida, J. Nonadditive Substituent Effects on Expanding Prestrained C–C Bond in Crystal: X-ray Analyses on Unsymmetrically Substituted Tetraarylpyracenes Prepared by a Flow Microreactor Method. Chem. Lett. 2012, 41, 541–543. [Google Scholar] [CrossRef]
- Schreiner, P.R.; Chernish, L.V.; Gunchenko, P.A.; Tikhonchuk, E.Y.; Hausmann, H.; Serafin, M.; Schlecht, S.; Dahl, J.E.P.; Carlson, R.M.K.; Fokin, A.A. Overcoming Lability of Extremely Long Alkane Carbon-Carbon Bonds Through Dispersion Forces. Nature 2011, 477, 308–311. [Google Scholar] [CrossRef]
- Grimme, S.; Schreiner, P.R. Steric Crowding Can Stabilize a Labile Molecule: Solving the Hexaphenylethane Riddle. Angew. Chem. Int. Ed. 2011, 50, 12639–12642. [Google Scholar] [CrossRef]
- Suzuki, T.; Takeda, T.; Kawai, H.; Fujiwara, K. Ultralong C–C Bonds in Hexaphenylethane Derivatives. Pure Appl. Chem. 2008, 80, 547–553. [Google Scholar] [CrossRef]
- Tanaka, K.; Takamoto, N.; Tezuka, Y.; Kato, M.; Toda, F. Preparation and Structural Study of Naphtho- and Anthrocyclobutene Derivatives Which Have Extremely Long C–C bonds. Tetrahedron 2001, 57, 3761–3767. [Google Scholar] [CrossRef]
- De Silva, K.M.N.; Goodman, J.M. What Is the Smallest Saturated Acyclic Alkane that Cannot Be Made? J. Chem. Inf. Model 2005, 45, 81–87. [Google Scholar] [CrossRef]
- Kawai, H.; Takeda, T.; Fujiwara, K.; Wakeshima, M.; Hinatsu, Y.; Suzuki, T. Ultralong Carbon-Carbon Bonds in Dispirobis(10-methylacridan) Derivatives with an Acenaphthene, Pyracene, or Dihydropyracylene Skeleton. Chem. Eur. J. 2008, 14, 5780–5793. [Google Scholar] [CrossRef]
- Kaupp, G.; Boy, J. Overlong C–C Single Bonds. Angew. Chem. Int. Ed. 1997, 36, 48–49. [Google Scholar] [CrossRef]
- Bartell, L.; Boates, T. Structure of the Strained Molecules Hexamethylethane and 1,1,2,2-Tetramethylethane by Gas-Phase Electron Diffraction. J. Mol. Struct. 1976, 32, 379–392. [Google Scholar] [CrossRef][Green Version]
- Szabo, Z.; Thege, I. Some Empirical Correlations on Chemical Bonds. Acta Chem. Acad. Sci. Hung. 1975, 86, 127–145. [Google Scholar]
- Fokin, A.; Chernish, L.; Gunchenko, P.; Tikhonchuk, E.; Hausmann, H.; Serafin, M.; Dahl, J.; Carlson, R.; Schreiner, P. Stable Alkanes Containing Very Long Carbon-Carbon Bonds. J. Am. Chem. Soc. 2012, 134, 13641–13650. [Google Scholar] [CrossRef] [PubMed]
- Plitzko, K.; Rapko, B.; Gollas, B.; Wehrle, G.; Weakley, T.; Pierce, D.T.; Geiger, W.E.; Haddon, R.C.; Boekelheide, V. Bis(η6-hexamethylbenzene)(η6,η6-[2n]cyclophane)diruthenium- (II,II) Complexes and Their Two-Electron Reduction to [2n]Cyclophane Derivatives Having Two Cyclohexadienyl Anion Decks Joined by an Extremely Long Carbon-Carbon Bond. J. Am. Chem. Soc. 1990, 112, 6545–6556. [Google Scholar] [CrossRef]
- Komatsu, K.; Nishinaga, T.; Takeuchi, K.; Lindner, H.J.; Richter, J. A Polycyclic Pentamer of Bicyclo[2.2.2]octene. A Hydrocarbon Molecule with a Long C–C Single Bond Connecting Two Cofacially Disposed Cyclopentadiene Rings. J. Org. Chem. 1994, 59, 7322–7328. [Google Scholar] [CrossRef]
- Slepetz, B.; Kertesz, M. Volume Change during Thermal [4+4] Cycloaddition of [2.2] (9,10)Anthracenophane. J. Am. Chem. Soc. 2013, 135, 13720–13727. [Google Scholar] [CrossRef]
- Takeda, T.; Kawai, H.; Herges, R.; Mucke, E.; Sawai, Y.; Murakoshi, K.; Fujiwara, K.; Suzuki, T. Negligible Diradical Character for the Ultralong C–C Bond in 1,1,2,2-Tetraarylpyracene Derivatives at Room Temperature. Tetrahedron Lett. 2009, 59, 3693–3697. [Google Scholar] [CrossRef]
- Folkertsma, E.; Benthem, S.; Witteman, L.; van Slagnaat, C.A.M.R.; ad Lutz, M.; Klein Gebbink, R.; Moret, M.E. Formation of Exceptionally Weak C–C Bonds by Metal-Templated Pinacole Coupling. Dalton Tans. 2017, 46, 6177–6182. [Google Scholar] [CrossRef]
- Jacovella, U.; Stein, C.; Grütter, M.; Freitag, L.; Lauzin, C.; Reiher, M.; Merkt, F. Structure and Dynamics of the Radical Cation of Ethane Arising from the Jahn-Teller and Pseudo-Jahn-Teller Effects. Phys. Chem. Chem. Phys. 2018, 20, 1072–1081. [Google Scholar] [CrossRef]
- Eriksson, L.A.; Lunell, S. Theoretical Study of Deuterated Ethane Cations. J. Phys. Chem. 1993, 97, 12215–12219. [Google Scholar] [CrossRef]
- Bellville, D.J.; Bauld, N.L. Elongated (One-Electron) Carbon-Carbon Bond in σ and n Organic Cation Radicals. J. Am. Chem. Soc. 1982, 104, 5700–5702. [Google Scholar] [CrossRef]
- Luo, Y.R. Comprehensive Handbook of Chemical Bond Energies; Taylor and Francis: Boca Raton, FL, USA, 2007. [Google Scholar]
- Morse, M.D. Predissociation Measurements of Bond Dissociation Energies. Acc. Chem. Res. 2018, 52, 119–126. [Google Scholar] [CrossRef] [PubMed]
- St. John, P.C.; Guan, Y.; Kim, Y.; Kim, S.; Paton, R.S. Prediction of Organic Homolytic Bond Dissociation Enthalpies at Near Chemical Accuracy with Sub-Second Computational Cost. Nat. Commun. 2020, 11, 2328. [Google Scholar] [CrossRef] [PubMed]
- Cremer, D.; Kraka, E. From Molecular Vibrations to Bonding, Chemical Reactions, and Reaction Mechanism. Curr. Org. Chem. 2010, 14, 1524–1560. [Google Scholar] [CrossRef]
- Kalescky, R.; Kraka, E.; Cremer, D. Are Carbon-Halogen Double and Triple Bonds Possible? Int. J. Quant. Chem. 2014, 114, 1060–1072. [Google Scholar] [CrossRef]
- Kalescky, R.; Zou, W.; Kraka, E.; Cremer, D. Quantitative Assessment of the Multiplicity of Carbon-Halogen Bonds: Carbenium and Halonium Ions with F, Cl, Br, and I. J. Phys. Chem. A 2014, 118, 1948–1963. [Google Scholar] [CrossRef]
- Oliveira, V.; Kraka, E.; Cremer, D. Quantitative Assessment of Halogen Bonding Utilizing Vibrational Spectroscopy. Inorg. Chem. 2016, 56, 488–502. [Google Scholar] [CrossRef]
- Setiawan, D.; Sethio, D.; Cremer, D.; Kraka, E. From Strong to Weak NF Bonds: On the Design of a New Class of Fluorinating Agents. Phys. Chem. Chem. Phys. 2018, 20, 23913–23927. [Google Scholar] [CrossRef] [PubMed]
- Krapp, A.; Bickelhaupt, F.M.; Frenking, G. Orbital Overlap and Chemical Bonding. Chem. Eur. J. 2006, 12, 9196–9216. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Hermann, M.; Schwarz, W.H.E.; Frenking, G. The Lewis Electron-Pair Bonding Model: Modern Energy Decomposition Analysis. Nat. Rev. Chem. 2019, 3, 48–63. [Google Scholar] [CrossRef]
- Zhao, L.; Pan, S.; Holzmann, N.; Schwerdtfeger, P.; Frenking, G. Chemical Bonding and Bonding Models of Main-Group Compounds. Chem. Rev. 2019, 119, 8781–8845. [Google Scholar] [CrossRef] [PubMed]
- Gavezzotti, A. Comparing the Strength of Covalent Bonds, Intermolecular Hydrogen Bonds and Other Intermolecular Interactions for Organic Molecules: X-ray Diffraction Data and Quantum Chemical Calculations. New J. Chem. 2016, 40, 6848–6853. [Google Scholar] [CrossRef]
- Kalescky, R.; Kraka, E.; Cremer, D. Identification of the Strongest Bonds in Chemistry. J. Phys. Chem. A 2013, 117, 8981–8995. [Google Scholar] [CrossRef]
- Kraka, E.; Cremer, D. Weaker Bonds with Shorter Bond Lengths. Rev. Proc. Quim. 2012, 6, 39–42. [Google Scholar] [CrossRef]
- Cremer, D.; Wu, A.; Larsson, A.; Kraka, E. Some Thoughts about Bonds with Bond Lengths, and Force Constants. J. Mol. Model. 2000, 6, 396–412. [Google Scholar] [CrossRef]
- Kaupp, M.; Metz, B.; Stoll, H. Breakdown of Bond Length-Bond Strength Correlation: A Case Study. Angew. Chem. Int. Ed. 2000, 39, 4607–4609. [Google Scholar] [CrossRef]
- Kaupp, M.; Danovich, D.; Shaik, S. Chemistry is About Energy and its Changes: A Critique of Bond-Length/Bond-Strength Correlations. Coord. Chem. Rev. 2017, 344, 355–362. [Google Scholar] [CrossRef]
- Konkoli, Z.; Cremer, D. A New Way of Analyzing Vibrational Spectra. I. Derivation of Adiabatic Internal Modes. Int. J. Quant. Chem. 1998, 67, 1–9. [Google Scholar] [CrossRef]
- Konkoli, Z.; Larsson, J.A.; Cremer, D. A New Way of Analyzing Vibrational Spectra. II. Comparison of Internal Mode Frequencies. Int. J. Quant. Chem. 1998, 67, 11–27. [Google Scholar] [CrossRef]
- Konkoli, Z.; Cremer, D. A New Way of Analyzing Vibrational Spectra. III. Characterization of Normal Vibrational Modes in terms of Internal Vibrational Modes. Int. J. Quant. Chem. 1998, 67, 29–40. [Google Scholar] [CrossRef]
- Konkoli, Z.; Larsson, J.A.; Cremer, D. A New Way of Analyzing Vibrational Spectra. IV. Application and Testing of Adiabatic Modes within the Concept of the Characterization of Normal Modes. Int. J. Quant. Chem. 1998, 67, 41–55. [Google Scholar] [CrossRef]
- Cremer, D.; Larsson, J.A.; Kraka, E. New Developments in the Analysis of Vibrational Spectra on the Use of Adiabatic Internal Vibrational Modes. In Theoretical and Computational Chemistry; Parkanyi, C., Ed.; Elsevier: Amsterdam, The Netherlands, 1998; pp. 259–327. [Google Scholar]
- Kraka, E.; Zou, W.; Tao, Y. Decoding Chemical Information from Vibrational Spectroscopy Data: Local Vibrational Mode Theory. WIREs Comput. Mol. Sci. 2020, 10, 1480. [Google Scholar] [CrossRef]
- Weinhold, F.; Landis, C.R.; Glendening, E.D. What is NBO Analysis and How is it Useful? Int. Rev. Phys. Chem. 2016, 35, 39–440. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory (International Series of Monographs on Chemistry); Clarendon Press: Oxford, UK, 1994. [Google Scholar]
- Popelier, P.L. Atoms in Molecules: An Introduction; Prentice Hall: Upper Saddle River, NJ, USA, 2000. [Google Scholar]
- Chai, J.D.; Head-Gordon, M. Long-Range Corrected Hybrid Density Functionals with Damped Atom–Atom Dispersion Corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef]
- Chai, J.D.; Head-Gordon, M. Systematic Optimization of Long-Range Corrected Hybrid Density Functionals. J. Chem. Phys. 2008, 128, 084106/1–084106/15. [Google Scholar] [CrossRef] [PubMed]
- Dunning, T.H. Gaussian Basis Sets for Use in Correlated Molecular Calculations. I. The Atoms Boron Through Neon and Hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Sato, T.; Nakai, H. Density Functional Method Including Weak Interactions: Dispersion Coefficients Based On the Local Response Approximation. J. Chem. Phys. 2009, 131, 224104/1–224104/12. [Google Scholar] [CrossRef]
- Athar, M.; Das, S.; Jha, P.C.; Jha, A.M. Conformational Equilibrium Study of Calix[4]tetrolarenes Using Density Functional Theory (DFT) and Molecular Dynamics Simulations. Supramol. Chem. 2018, 30, 982–993. [Google Scholar] [CrossRef]
- Hachim, M.E.; Sadik, K.; Byadi, S.; Aboulmouhajir, A. Electronic Investigation and Spectroscopic Analysis Using DFT With the Long-Range Dispersion Correction On the Six lowest Conformers of 2.2.3-Trimethyl Pentane. J. Mol. Model 2020, 26, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Pople, J.A.; Head-Gordon, M.; Raghavachari, K. Quadratic Configuration Interaction. A General Technique for Determining Electron Correlation Energies. J. Chem. Phys. 1987, 87, 5968–5975. [Google Scholar] [CrossRef]
- Kendall, R.A.; Dunning, T.H.; Harrison, R.J. Electron Affinities of the First-Row Atoms Revisited. Systematic Basis Sets and Wave Functions. J. Chem. Phys. 1992, 96, 6796–6806. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Revision B.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Chan, B.; Radom, L. BDE261: A Comprehensive Set of High-Level Theoretical Bond Dissociation Enthalpies. J. Chem. Phys. A 2012, 116, 4975–4986. [Google Scholar] [CrossRef] [PubMed]
- Curtiss, L.; Redfern, P.; Raghavachari, K. Gaussian-4 Theory Using Reduced Order Perturbation Theory. J. Chem. Phys. 2007, 127, 124105/1–124105/8. [Google Scholar] [CrossRef]
- Curtiss, L.; Redfern, P.; Raghavachari, K. Gaussian-4 Theory. J. Chem. Phys. 2007, 126, 084108/1–084108/12. [Google Scholar] [CrossRef] [PubMed]
- Pople, J.; Head-Gordon, M.; Fox, D.; Raghavachari, K.; Curtiss, L. Gaussian-1 Theory: A General Procedure for Prediction of Molecular Energies. J. Chem. Phys. 1989, 90, 5622–5629. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular Interactions From a Natural Bond Orbital, Donor-Acceptor Viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Weinhold, F.; Landis, C.R. Valency and Bonding: A Natural Bond Orbital Donor-Acceptor Perspective; Cambridge University Press: Cambridge, UK, 2005. [Google Scholar]
- Keith, T.A. AIMAll (Version 12.06.03); TK Gristmill Software: Overland Park, KS, USA, 2012. [Google Scholar]
- Cremer, D.; Kraka, E. Chemical Bonds without Bonding Electron Density? Does the Difference Electron-Density Analysis Suffice for a Description of the Chemical Bond? Angew. Chem. Int. Ed. 1984, 23, 627–628. [Google Scholar] [CrossRef]
- Cremer, D.; Kraka, E. A Description of the Chemical Bond in Terms of Local Properties of Electron Density and Energy. Croat. Chem. Acta 1984, 57, 1259–1281. [Google Scholar]
- Kraka, E.; Cremer, D. Chemical Implication of Local Features of the Electron Density Distribution. In Theoretical Models of Chemical Bonding. The Concept of the Chemical Bond; Maksic, Z.B., Ed.; Springer: Heidelberg, Germany, 1990; Volume 2, pp. 453–542. [Google Scholar]
- Zou, W.; Tao, Y.; Freindorf, M.; Makos, M.; Verma, N.; Kraka, E. Local Vibrational Mode Analysis (LModeA); Computational and Theoretical Chemistry Group (CATCO), Southern Methodist University: Dallas, TX, USA, 2020. [Google Scholar]
- Wilson, E.B.; Decius, J.C.; Cross, P.C. Molecular Vibrations. The Theory of Infrared and Raman Vibrational Spectra; McGraw-Hill: New York, NY, USA, 1955. [Google Scholar]
- Woodward, L.A. Introduction to the Theory of Molecular Vibrations and Vibrational Spectroscopy; Oxford University Press: Oxford, UK, 1972. [Google Scholar]
- Wilson, E.B., Jr. A method of obtaining the expanded secular equation for the vibration frequencies of a molecule. J. Chem. Phys. 1939, 7, 1047. [Google Scholar] [CrossRef]
- Califano, S. Vibrational States; Wiley: London, UK, 1976. [Google Scholar]
- Groner, P. Normal Coordinate Analysis; John Wiley: New York, NY, USA, 2006. [Google Scholar]
- Kelley, J.D.; Leventhal, J.J. Normal Modes and Coordinates. In Problems in Classical and Quantum Mechanics; Springer: Berlin/Heidelberg, Germany, 2017; pp. 95–117. [Google Scholar]
- Neto, N. Tensor Formalism in Anharmonic Calculations. Chem. Phys. 1984, 91, 89. [Google Scholar] [CrossRef]
- Stare, J. First-Principle Calculation of Reduced Masses in Vibrational Analysis Using Generalized Internal Coordinates: Some Crucial Aspects and Examples. J. Chem. Inf. Model. 2007, 47, 840–850. [Google Scholar] [CrossRef] [PubMed]
- Zou, W.; Kalescky, R.; Kraka, E.; Cremer, D. Relating Normal Vibrational Modes to Local Vibrational Modes with the Help of an Adiabatic Connection Scheme. J. Chem. Phys. 2012, 137, 084114. [Google Scholar] [CrossRef]
- Kalescky, R.; Kraka, E.; Cremer, D. Local Vibrational Modes of the Formic Acid Dimer-The Strength of the Double H-Bond. Mol. Phys. 2013, 111, 1497–1510. [Google Scholar] [CrossRef]
- Kalescky, R.; Kraka, E.; Cremer, D. New Approach to Tolman’s Electronic Parameter Based on Local Vibrational Modes. Inorg. Chem. 2013, 53, 478–495. [Google Scholar] [CrossRef] [PubMed]
- Kraka, E.; Larsson, J.A.; Cremer, D. Generalization of the Badger Rule Based on the Use of Adiabatic Vibrational Modes. In Computational Spectroscopy; Grunenberg, J., Ed.; Wiley: New York, NY, USA, 2010; pp. 105–149. [Google Scholar]
- Zou, W.; Cremer, D. Properties of Local Vibrational Modes: The Infrared Intensity. Theor. Chem. Acc. 2014, 133, 1451–1466. [Google Scholar] [CrossRef]
- Zou, W.; Cremer, D. C2 in a Box: Determining its Intrinsic Bond Strength for the X1Σ+g Ground State. Chem. Eur. J. 2016, 22, 4087–4097. [Google Scholar] [CrossRef]
- Kalescky, R.; Zou, W.; Kraka, E.; Cremer, D. Local Vibrational Modes of the Water Dimer-Comparison of Theory and Experiment. Chem. Phys. Lett. 2012, 554, 243–247. [Google Scholar] [CrossRef]
- Gunnelin, K.; Glans, P.; Rubensson, J.E.; Såthe, C.; Nordgren, J.; Li, Y.; Gelḿukhanov, F.; Ågren, H. Bond-Length-Dependent Core Hole Localization Observed in Simple Hydrocarbonds. Phys. Rev. Lett. 1999, 83, 1315–1318. [Google Scholar] [CrossRef]
- Afeefy, H.; Liebman, J.; Stein, S. In NIST Computational Chemistry Comparison and Benchmark Database, NIST Standard Reference Database Number 69. 2001. Available online: http:webbook.nist.gov/ (accessed on 19 October 2020).
- Martin, J.; Fernandez, M.; Tortajada, J. Application of Wiberg Indices to Geometry Optimization. C–C Distances. J. Mol. Struct. 1988, 175, 203–208. [Google Scholar] [CrossRef]
- Hilderbrandt, R.; Wieser, J. The Zero Point Average Structure of Isobutane as Determined by Electron Diffraction and Microwave Spectroscopy. J. Mol. Struct. 1973, 15, 27–36. [Google Scholar] [CrossRef]
- Shen, Q. The Molecular Structure of 1,2-Diphenylethane as Determined by Gas-Phase Electron Diffraction. J. Mol. Struct. 1998, 471, 57–61. [Google Scholar] [CrossRef]
- Flamm-ter, M.; Beckhaus, H.D.; Peters, K.; von Schnering, H.G.; Rüchardt, C. 2,3-Di-1-adamantyl-2,3-dimethylbutane; Long Bonds and Low Thermal Stability. Chem. Ber. 1985, 113, 4665–4671. [Google Scholar]
- Zavitsas, A.A. The Relation between Bond Lengths and Dissociation Energies of Carbon-Carbon Bonds. J. Phys. Chem. A 2003, 107, 897–898. [Google Scholar] [CrossRef]
- Winiker, R.; Beckhaus, H.D.; Rüchardt, C. Thermische Stabilität, Spannungsenthalpie und Struktur Symmetrisch Hexaalkylierter Ethane. Chem. Ber. 1980, 113, 3456–3476. [Google Scholar] [CrossRef]
- Kratt, G.; Beckhaus, H.D.; Lindner, H.; Rüchardt, C. Thermolabile Kohlenwasserstoffe, XX. Synthese, Struktur und Spannung Symmetrischer Tetraalkyldiarylethane. Chem. Ber. 1983, 116, 3235–3263. [Google Scholar] [CrossRef]
- Kahr, B.; Van Engen, D.; Mislow, K. Length of the Ethane Bond in Hexaphenylethane and its Derivatives. J. Am. Chem. Soc. 1986, 108, 8305–8307. [Google Scholar] [CrossRef]
- Livingston, R.; Ramachandra Rao, C. The Molecular Structure of Pivalonitrile Electron Diffraction. J. Am. Chem. Soc. 1958, 81, 3584–3586. [Google Scholar] [CrossRef]
- Dougherty, D.; Choi, C.; Kaupp, G.; Buda, A.; Rudziński, J.; Osawa, E. Effects of Substituents on the Length of Central C(sp3)-C(sp3) Bond in Anthracene Photodimers and Related Molecules. J. Chem. Soc. Perkin Trans. 2 1986, 7, 1063–1070. [Google Scholar] [CrossRef]
- Sugie, M.; Kato, M.; Matsumura, C.; Takeo, H. Microwave Spectra and Molecular Structures of 1,2-Dichloroethane, 1,1-Dichloroethane and 1,1,1-Trichloroethane. J. Mol. Struct. 1997, 413–414, 487–494. [Google Scholar] [CrossRef]
- Almenningen, A.; Andersen, B.; Trætteberg, M. An Electron Diffraction Investigation of the Molecular Structure of Hexachloroethane in the Vapour Phase. Acta Chem. Scand. 1964, 18, 603–611. [Google Scholar] [CrossRef][Green Version]
- Bock, C.; Panchenko, Y. An Ab Initio Structural Investigation of 1,3-Butadiene, Isoprene and 2,3-Dimethyl-1,3-butadiene Rotamers. J. Mol. Struct. THEOCHEM 1989, 187, 69–82. [Google Scholar] [CrossRef]
- VanHemelrijk, D.; Van den Enden, L.; Geise, H.; Sellers, H.L.; Schäfer, L. Structure Determination of 1-Butene by Gas Electron Diffraction, Microwave Spectroscopy, Molecular Mechanics, and Molecular Orbital Constrained Electron Diffraction. J. Am. Chem. Soc. 1980, 102, 2189–2195. [Google Scholar] [CrossRef]
- Guirgis, G.; Zheng, C.; Gounev, T.; Durig, J. Conformational Stability, Ab Initio Calculaitons, and ro structural Parameters of 3-Methyl-1-butene and Dimethylvinylsilane. J. Mol. Struct. 2003, 651–653, 771–780. [Google Scholar] [CrossRef]
- Kraśnicki, A.; Zbigniew, K.; Drouin, B.; Pearson, J. Terahertz Spectroscopy of Isotopic Acrylonitrile. J. Mol. Spectrosc. 2011, 1006, 20–27. [Google Scholar] [CrossRef]
- Thorwirth, C.; Harding, M.E.; Muders, D.; Gauss, J. The Empirical Equilibrium Structure of Diacetylene. J. Mol. Spect. 2008, 251, 220–223. [Google Scholar] [CrossRef][Green Version]
- Kang, L.; Novick, S. The Microwave Spectra of the Weakly Bound Complex Between Carbon Monoxide and Cyanoacetylene, OC H-C≡C–C≡N. J. Mol. Spectrosc. 2012, 276–277, 10–13. [Google Scholar] [CrossRef]
- Amir-Ebrahimi, V.; Choplin, A. Microwave Spectrum of the 13C-Ring-Monosubstituted Toluenes and Structure of Toluene. J. Mol. Spectrosc. 1982, 89, 42–52. [Google Scholar] [CrossRef]
- Scharfenberg, P.; Rozsondai, B.; Hargittai, I. Conformation and Structure of Ethylbenzene in the Vapour Phase. Z. Naturforsch. 1980, 35a, 431–436. [Google Scholar] [CrossRef]
- Vilkov, L.; Sadova, N.; Mochalov, S. The Structure of Cumene and Phenylcyclobutane Molecules, as Studied by Means of Electron Diffraction Patterns. Dokl. Akad. Nauk SSSR 1968, 179, 896–899. [Google Scholar]
- Campanelli, A.; Ramondo, F.; Domenicano, A.; Hargittai, I. Molecular Structure and Conformation of Tert-Butylbenzene: A Concert. Study Gas-Phase Electron Diffr. Theor. Calc. J. Phys. Chem. 1994, 98, 11046–11052. [Google Scholar] [CrossRef]
- Shen, Q.; Wells, C.; Trætteberg, M.; Bohn, R.; Willis, A.; Knee, J. Molecular Structure and Conformation of Cyclopropylbenzene as Determined by Ab Initio Molecular Orbital Calculations, Pulsed Jet Fourier Transform Microwave Spectroscopic, and Gas-Phase Electron Diffraction Investigations. J. Org. Chem. 2001, 66, 5840–5845. [Google Scholar] [CrossRef]
- Lide, D. Structure of the Isobutane Molecule; Change of Dipole Moment on Isotopic Substitution. J. Chem. Phys. 1960, 33, 1519–1522. [Google Scholar] [CrossRef]
- Cochran, J.; Hagen, K.; Paulen, G.; Shen, Q.; Tom, S.; Trætteberg, M.; Wells, C. On the Planarity of Styrene and its Derivatives: The Molecular Structures of Styrene and (Z)-β-Bromostyrene as Determined by Ab Initio Calculations and Gas-Phase Electron Diffraction. J. Mol. Spectrosc. 1997, 413–414, 313–326. [Google Scholar] [CrossRef]
- Rudolph, H.; Demaison, J.; Császár, A. Accurate Determination of the Deformation of the Benzene Ring Upon Substitution: Equilibrium Structures of Benzonitrile and Phenylacetylene. J. Chem. Phys. A 2013, 117, 12969–12982. [Google Scholar] [CrossRef] [PubMed]
- Karle, I.; Brockway, L. The Structures of Biphenyl, O-Terphenyl Tetraphenylene. J. Am. Chem. Soc. 1942, 66, 1974–1979. [Google Scholar] [CrossRef]
- Isea, R. What is the maximum stretching for a C–C single bond? J. Mol. Struct. 2001, 540, 131–138. [Google Scholar] [CrossRef]
- Rösel, S.; Balestrieri, C.; Schreiner, P.R. Sizing the Role of London Dispersion in the Dissociation of All-meta Tert-Butyl Hexaphenylethane. Chem. Sci. 2017, 8, 405–410. [Google Scholar] [CrossRef]
- Fokin, A.A.; Zhuk, T.S.; Blomeyer, S.; Perez, C.; .Chernish, L.V.; Pashenko, A.E.; Antony, J.; Vishnevskiy, Y.V.; Berger, R.J.F.; Grimme, S.; et al. Intramolecular London Dispersion Interaction Effects on Gas-Phase and Solid-State Structures of Diamondoid Dimers. J. Am. Chem. Soc. 2017, 139, 16696–16707. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.; Schreiner, P. London Dispersion in Molecular Chemistry—Reconsidering Steric Effects. Angew. Chem. Int. Ed. 2015, 54, 12274–12296. [Google Scholar] [CrossRef]
- Folkertsma, E.; Benthem, S.H.; Jastrzebski, J.T.; Lutz, M.; Moret, M.E.; Gebbink, R.J.K. 1,2-Addition of Diethylzinc to a Bis(Imidazolyl)ketone Ligand. Eur. J. Inorg. Chem. 2018, 2018, 1167–1175. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Z.; Pham, H.D.M.; Li, J.; Li, C.C.; Castillo-Pazos, D.J.; Khaliullin, R.Z.; Li, C.J. Light-enabled metal-free pinacol coupling by hydrazine. Chem. Sci. 2019, 10, 10937–10943. [Google Scholar] [CrossRef] [PubMed]
- Schneider, W.F.; T Nance, B.I.; Wallington, T.J. Bond Strength Trends in Halogenated Methanols: Evidence for Negative Hyperconjugation? J. Am. Chem. Soc. 1995, 117, 478–485. [Google Scholar] [CrossRef]
- Wiberg, K.B.; Rablen, P.R. Origin of the Stability of Carbon Tetrafluoride: Negative Hyperconjugation Reexamined. J. Am. Chem. Soc. 1993, 115, 614–625. [Google Scholar] [CrossRef]
- Johansson, M.P.; Swart, M. Intramolecular halogen–halogen bonds? Phys. Chem. Chem. Phys. 2013, 15, 614–625. [Google Scholar] [CrossRef]
- Lunell, S.; Huang, M.B. Theoretical Confirmation of the E.S.R. Spectrum of the Ethane Cation. J. Chem. Soc. Chem. Commun. 1989, 15, 1031–1033. [Google Scholar] [CrossRef]
- Huang, M.B.; Lunell, S. Equilibrium Structure and Hyperfine Parameters of the Ethane Cation. Chem. Phys. 1990, 147, 85–90. [Google Scholar] [CrossRef]
- Tsuzuki, S.; Uchimaru, T. Accuracy of Intermolecular Interaction Energies, Particularly Those of Hetero-atom Containing Molecules Obtained by DFT Calculations with Grimme’s D2, D3 and D3BJ Dispersion Corrections. Phys. Chem. Chem. Phys. 2020, 22, 11543–11553. [Google Scholar] [CrossRef]
- Klein, D. Organic Chemistry; Wiley: Hoboken, NJ, USA, 2015. [Google Scholar]
- Bakowicz, J.; Turowska-Tyrk, I. Structural Transformations in Crystals Induced by Radiation and Pressure. Part 10. The Crystallographic Picture of Photochemical Behaviour of bi(anthracene-9,10-dimethylene) under High Pressure. Crystals 2020, 10, 1031. [Google Scholar] [CrossRef]
- Wang, D.; Yan, Y.; Zhou, D.; Liu, Y. Evolution of Crystal and Electronic Structures of Magnesium Dicarbide at High Pressure. Nature Sci. Rep. 2020, 5, 17815/1–17815/8. [Google Scholar] [CrossRef] [PubMed]
- Neves, W.Q.; Alencar, R.S.; Ferreira, R.S.; Torres-Dias, A.C.; Andrade, N.F.; San-Miguel, A.; Kim, Y.A.; Endo, M.; Kim, D.W.; Muramatsu, H.; et al. Effects of Pressure on the Structural and Electronic Properties of Linear Carbon Chains Encapsulated in Double Wall Carbon Nanotubes. Carbon 2018, 133, 446–456. [Google Scholar] [CrossRef]
- Chen, M.; Christmann, A.M.; Muniz, A.R.; Ramasubramaniam, A.; Maroudas, D. Molecular-Dynamics Analysis of Nanoindentation of Graphene Nanomeshes: Implications for 2D Mechanical Metamaterials. ACS Appl. Nano Mater. 2020, 3, 3613–3624. [Google Scholar] [CrossRef]
- Rojas, W.Y.; Winter, A.D.; Grote, J.; Kim, S.S.; Naik, R.R.; Williams, A.D.; Weiland, C.; Principe, E.; Fischer, D.A.; Banerjee, S.; et al. Strain and Bond Length Dynamics upon Growth and Transfer of Graphene by NEXAFS Spectroscopy from First-Principles and Experiment. Langmuir 2018, 34, 1783–1794. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| # | sym | bond | BDH | BDH | BSO n | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | D3d | C−C | 1.523 | 1.536 [97] | 89.0 | 89.7 [98] | 4.216 | 1092 | 1.000 | 1.659 | −1.431 |
| 2 | C2v | C−C | 1.524 | 1.528 [99] | 87.4 | 87.2 [98] | 4.160 | 1085 | 0.989 | 1.671 | −1.443 |
| 3 | C3v | C−C | 1.526 | 1.535 [100] | 86.7 | 88.9 [98] | 4.092 | 1076 | 0.976 | 1.675 | −1.442 |
| 4 | T | C−C | 1.530 | 1.539 [26] | 87.5 | 86.0 [98] | 3.997 | 1063 | 0.958 | 1.669 | −1.423 |
| 5 | C2h | C−C | 1.541 | 1.544 [25] | 82.8 | 86.6 [98] | 3.786 | 1035 | 0.917 | 1.645 | −1.369 |
| 6 | D3 | C−C | 1.577 | 1.582 [25] | 80.4 | 76.0 [98] | 3.229 | 956 | 0.807 | 1.544 | −1.181 |
| 7 | C2 | C−C | 1.542 | 1.550 [101] | 66.9 | 66.6 [98] | 3.675 | 1020 | 0.895 | 1.601 | −1.322 |
| 8 | C2 | C−C | 1.629 | 1.677 [18] | 53.4 | 43.7 [102] | 2.414 | 826 | 0.639 | 1.387 | −1.925 |
| 9 | C1 | C−C | 1.597 | 1.601 [103] | 73.1 | 60.2 [104] | 2.888 | 904 | 0.737 | 1.481 | −1.079 |
| 10 | C2 | C−C | 1.611 | 1.635 [103] | 61.2 | 51.0 [104] | 2.693 | 873 | 0.697 | 1.443 | −1.016 |
| 11 | C2 | C−C | 1.631 | 1.635 [105] | 52.2 | 44.7 [105] | 2.290 | 805 | 0.611 | 1.371 | −1.918 |
| 12 | D2 | C−C | 1.699 | - | 16.6 | - | 1.518 | 678 | 0.439 | 1.188 | −1.663 |
| 13 | S6 | C−C | 1.669 | 1.670 [106] | 33.9 | - | 1.919 | 737 | 0.526 | 1.275 | −1.830 |
| 14 | C2h | C−C | 1.523 | 1.539 [26] | 86.0 | 87.2 [98] | 4.177 | 1087 | 0.993 | 1.672 | −1.447 |
| 15 | C3v | C−CN | 1.472 | 1.460 [107] | 119.2 | 115.8 [98] | 4.432 | 1120 | 1.041 | 1.787 | −1.852 |
| C−C | 1.535 | - | - | - | 3.915 | 1052 | 0.942 | 1.644 | −1.388 | ||
| 16 | C2 | C−C | 1.599 | 1.601 [103] | 70.3 | 62.2 [106] | 2.853 | 898 | 0.730 | 1.477 | −1.070 |
| 17 | C2 | C−C | 1.622 | 1.630 [18] | 50.0 | 44.0 [106] | 2.509 | 842 | 0.658 | 1.410 | −1.965 |
| 18 | C2 | C−C | 1.589 | 1.606 [103] | 72.1 | 57.8 [106] | 3.050 | 929 | 0.770 | 1.506 | −1.119 |
| 19 | C1 | C−C | 1.614 | 1.660 [19] | 78.6 | - | 2.792 | 889 | 0.717 | 1.427 | −1.996 |
| 20 | C1 | C−C | 1.647 | 1.647 [18] | 71.4 | ≈ 71 [18] | 2.401 | 824 | 0.635 | 1.331 | −1.855 |
| 21 | C1 | C−C | 1.656 | 1.659 [18] | 64.3 | - | 2.245 | 797 | 0.602 | 1.310 | −1.824 |
| 22 | C1 | C−C | 1.693 | 1.704 [18] | - | - | 1.874 | 728 | 0.521 | 1.218 | −1.694 |
| 23 | C1 | C−C | 1.787 | - | - | - | 1.142 | 568 | 0.351 | 1.014 | −1.465 |
| 24 | C1 | C−C | 1.695 | 1.707 [18] | - | - | 1.861 | 726 | 0.519 | 1.211 | −1.687 |
| 25 | D2h | C−C | 1.642 | 1.640 [108] | - | - | 2.411 | 826 | 0.637 | 1.317 | −1.857 |
| 26 | C1 | C−C | 1.708 | 1.754 [31] | - | - | 1.788 | 711 | 0.502 | 1.153 | −1.620 |
| 27 | C2h | C−C | 1.651 | - | - | - | 1.591 | 671 | 0.456 | 1.347 | −1.876 |
| 28 | C3v | C−C | 1.512 | 1.516 [109] | 86.9 | 88.3 [98] | 4.154 | 1084 | 0.988 | 1.747 | −1.615 |
| 29 | D3d | C−C | 1.583 | 1.564 [110] | 73.4 | 70.1 [98] | 2.944 | 913 | 0.749 | 1.575 | −1.181 |
| 30a | D3d | C−C | 1.935 | - | 41.2 | - | 0.894 | 503 | 0.286 | 0.523 | −1.194 |
| 30b | C2h | C−C | 1.591 | - | 42.7 | - | 0.971 | 524 | 0.306 | 1.272 | −1.929 |
| 31 | C2v | C−C | 1.930 | - | - | - | 0.604 | 413 | 0.209 | 0.742 | −1.242 |
| 32 | Cs | C−C | 1.495 | 1.501 [99] | 100.0 | 100.9 [98] | 4.575 | 1138 | 1.068 | 1.770 | −1.637 |
| C=C | 1.324 | 1.336 [99] | - | - | 9.821 | 1667 | 1.997 | 2.444 | −1.197 | ||
| 33 | D2h | C=C | 1.322 | 1.339 [97] | 173.9 | 172.2 [98] | 9.961 | 1679 | 2.000 | 2.449 | −1.214 |
| 34 | C2h | C−C | 1.457 | 1.467 [111] | - | 116.0 [98] | 5.119 | 1203 | 1.169 | 1.920 | −1.934 |
| C=C | 1.329 | 1.349 [111] | - | - | 9.537 | 1642 | 1.931 | 2.426 | −1.145 | ||
| 35 | C1 | C−C | 1.497 | 1.502 [112] | 98.2 | 99.6 [98] | 4.484 | 1126 | 1.051 | 1.776 | −1.639 |
| C=C | 1.324 | 1.340 [112] | - | - | 9.804 | 1665 | 1.975 | 2.443 | −1.195 | ||
| 36 | Cs | C−C | 1.501 | 1.500 [113] | 97.3 | 99.7 [98] | 4.382 | 1113 | 1.032 | 1.775 | −1.628 |
| C=C | 1.324 | 1.341 [113] | - | - | 9.811 | 1666 | 1.976 | 2.443 | −1.198 | ||
| 37 | Cs | C−C | 1.520 | 1.522 [103] | 96.1 | 97.5 [98] | 4.179 | 1087 | 0.993 | 1.749 | −1.573 |
| C=C | 1.324 | - | - | - | 9.765 | 1662 | 1.968 | 2.438 | −1.188 | ||
| 38 | D∞h | C≡C | 1.194 | 1.208 [97] | 228.1 | 229.9 [98] | 17.777 | 2243 | 3.190 | 2.894 | −1.700 |
| 39 | C3v | C−C | 1.455 | 1.450 [26] | 124.3 | 123.5 [98] | 5.254 | 1219 | 1.194 | 1.844 | −1.895 |
| C≡C | 1.196 | 1.207 [26] | - | - | 17.515 | 2226 | 3.153 | 2.862 | −1.742 | ||
| 40 | C3v | C−C | 1.455 | 1.458 [99] | 122.9 | 121.1 [98] | 5.141 | 1206 | 1.173 | 1.828 | −1.931 |
| 41 | Cs | C−C | 1.425 | 1.431 [26] | 135.3 | 133.6 [98] | 5.777 | 1278 | 1.289 | 1.974 | −1.151 |
| C=C | 1.329 | - | - | - | 9.564 | 1645 | 1.936 | 2.418 | −1.137 | ||
| C≡C | 1.199 | - | - | - | 17.264 | 2210 | 3.116 | 2.860 | −1.699 | ||
| 42 | Cs | C−C | 1.430 | 1.429 [114] | 131.7 | 132.1 [98] | 5.582 | 1256 | 1.254 | 1.938 | −1.141 |
| C=C | 1.327 | 1.339 [114] | - | - | 9.645 | 1656 | 1.957 | 2.428 | −1.166 | ||
| 43 | D∞h | C−C | 1.372 | 1.383 [26] | 158.3 | 155.0 [98] | 7.406 | 1447 | 1.575 | 2.142 | −1.517 |
| C≡C | 1.199 | 1.209 [115] | - | - | 17.160 | 2203 | 3.101 | 2.858 | −1.684 | ||
| 44 | C∞v | C−C | 1.375 | 1.379 [116] | 150.9 | 152.4 [98] | 7.348 | 1442 | 1.565 | 2.122 | −1.491 |
| C≡C | 1.196 | 1.204 [116] | - | - | 17.470 | 2223 | 3.146 | 2.878 | −1.708 | ||
| 45 | Cs | C−C | 1.504 | 1.512 [117] | 103.4 | 103.9 [98] | 4.528 | 1132 | 1.059 | 1.745 | −1.581 |
| 46 | Cs | C−C | 1.506 | 1.524 [118] | 101.8 | 102.3 [98] | 4.446 | 1121 | 1.044 | 1.751 | −1.585 |
| 47 | Cs | C−C | 1.514 | 1.500 [119] | 125.0 | 102.1 [98] | 4.274 | 1100 | 1.011 | 1.732 | −1.540 |
| 48 | Cs | C−C | 1.530 | 1.524 [120] | 99.4 | 97.4 [98] | 3.966 | 1059 | 0.952 | 1.681 | −1.439 |
| 49 | Cs | C−C | 1.486 | 1.520 [121] | - | 111.9 [122] | 4.751 | 1159 | 1.101 | 1.815 | −1.708 |
| 50 | Cs | C−C | 1.471 | 1.475 [123] | - | 116.9 [98] | 4.919 | 1180 | 1.132 | 1.868 | −1.820 |
| C=C | 1.327 | - | - | - | 9.549 | 1644 | 1.933 | 2.427 | −1.154 | ||
| 51 | C2v | C−C | 1.430 | 1.436 [124] | - | 140.7 [98] | 5.750 | 1275 | 1.284 | 1.961 | −1.112 |
| C≡C | 1.198 | - | - | - | 17.286 | 2211 | 3.119 | 2.860 | −1.707 | ||
| 52 | C2v | C−C | 1.433 | 1.438 [124] | - | 132.7 [98] | 5.569 | 1255 | 1.252 | 1.929 | −1.116 |
| 53 | D2 | C−C | 1.482 | 1.480 [125] | - | 118.0 [98] | 4.850 | 1171 | 1.120 | 1.840 | −1.752 |
| # | % s (C1) | % s (C2) | Sum of % s Character | Av. of % s Character |
|---|---|---|---|---|
| 1 | 29.6 (sp) | 29.6 (sp) | 59.2 | 29.6 |
| 33 | 40.7 (sp) | 40.7 (sp) | 81.4 | 40.7 |
| 38 | 52.3 (sp) | 52.3 (sp) | 104.6 | 52.3 |
| 32 | 30.2 (sp) | 33.2 (sp) | 63.4 | 31.7 |
| 34 | 33.7 (sp) | 33.7 (sp) | 67.4 | 33.7 |
| 35 | 28.8 (sp) | 33.4 (sp) | 62.2 | 31.1 |
| 36 | 27.2 (sp) | 33.7 (sp) | 60.8 | 30.4 |
| 37 | 25.6 (sp) | 34.4 (sp) | 60.0 | 30.0 |
| 39 | 29.5 (sp) | 47.7 (sp) | 77.1 | 38.6 |
| 40 | 26.8 (sp) | 52.4 (sp) | 79.3 | 39.7 |
| 41 | 47.9 (sp) | 32.5 (sp) | 80.4 | 40.2 |
| 42 | 52.2 (sp) | 30.3 (sp) | 82.5 | 41.3 |
| 43 | 46.9 (sp) | 46.9 (sp) | 93.8 | 46.9 |
| 44 | 45.7 (sp) | 50.7 (sp) | 96.4 | 48.2 |
| 45 | 31.1 (sp) | 30.3 (sp) | 61.4 | 30.7 |
| 46 | 31.5 (sp) | 30.0 (sp) | 61.5 | 30.8 |
| 47 | 31.9 (sp) | 27.5 (sp) | 59.4 | 29.7 |
| 48 | 32.5 (sp) | 26.0 (sp) | 58.5 | 29.3 |
| 49 | 31.7 (sp) | 32.3 (sp) | 64.0 | 32.0 |
| 50 | 32.0 (sp) | 34.0 (sp) | 66.0 | 33.0 |
| 51 | 30.6 (sp) | 48.0 (sp) | 78.7 | 39.4 |
| 52 | 28.6 (sp) | 52.5 (sp) | 81.1 | 40.6 |
| 53 | 32.0 (sp) | 32.0 (sp) | 64.0 | 32.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Delgado, A.A.A.; Humason, A.; Kalescky, R.; Freindorf, M.; Kraka, E. Exceptionally Long Covalent CC Bonds—A Local Vibrational Mode Study. Molecules 2021, 26, 950. https://doi.org/10.3390/molecules26040950
Delgado AAA, Humason A, Kalescky R, Freindorf M, Kraka E. Exceptionally Long Covalent CC Bonds—A Local Vibrational Mode Study. Molecules. 2021; 26(4):950. https://doi.org/10.3390/molecules26040950
Chicago/Turabian StyleDelgado, Alexis Antoinette Ann, Alan Humason, Robert Kalescky, Marek Freindorf, and Elfi Kraka. 2021. "Exceptionally Long Covalent CC Bonds—A Local Vibrational Mode Study" Molecules 26, no. 4: 950. https://doi.org/10.3390/molecules26040950
APA StyleDelgado, A. A. A., Humason, A., Kalescky, R., Freindorf, M., & Kraka, E. (2021). Exceptionally Long Covalent CC Bonds—A Local Vibrational Mode Study. Molecules, 26(4), 950. https://doi.org/10.3390/molecules26040950