
Calculation of the Vapour Pressure of Organic Molecules by Means of a Group-Additivity Method and Their Resultant Gibbs Free Energy and Entropy of Vaporization at 298.15 K



Abstract
1. Introduction
2. Method
3. Sources of Vapor-Pressure Data
4. Results
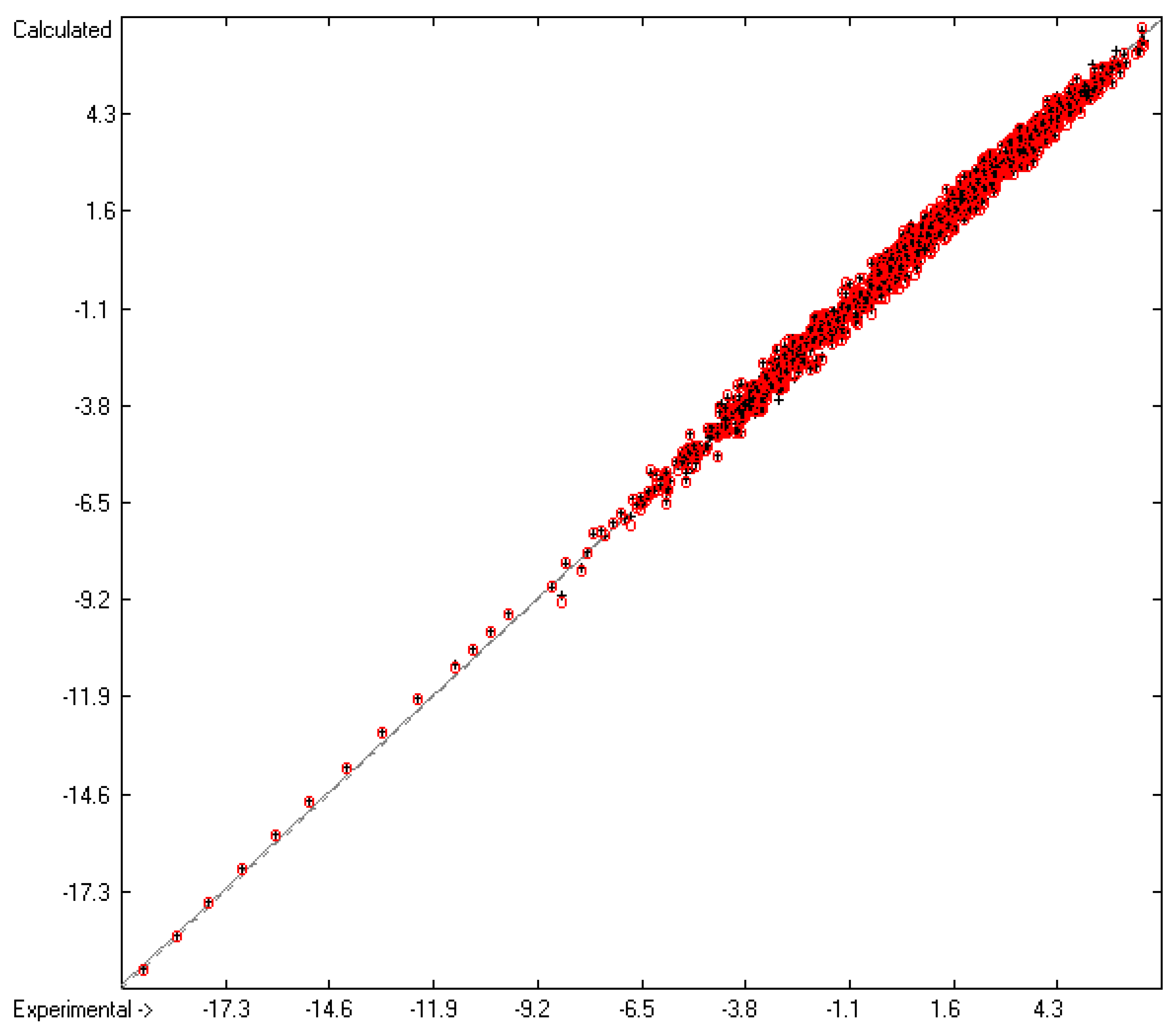
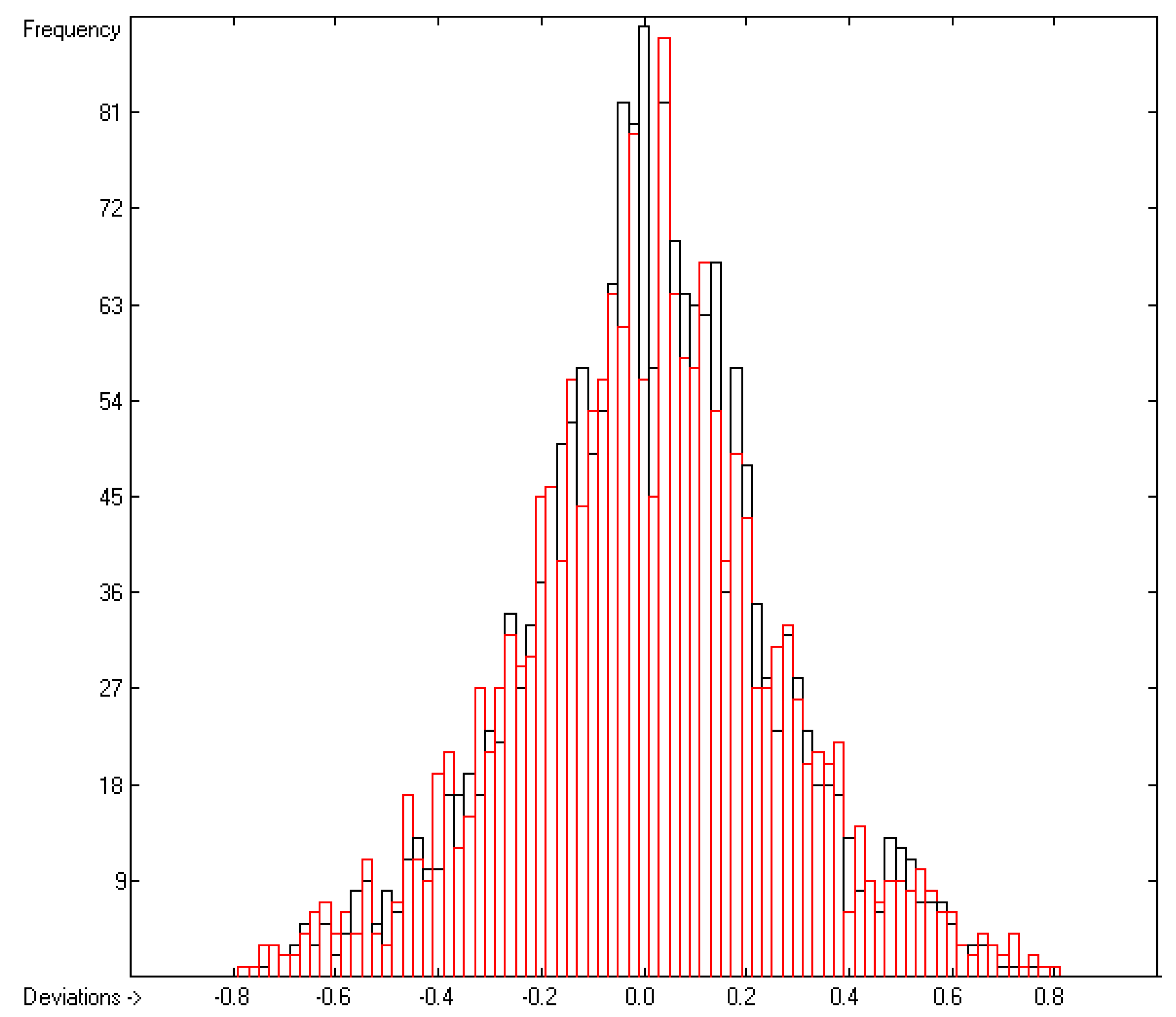
4.1. Vapour Pressure
4.2. Gibbs Free Energy of Vaporization
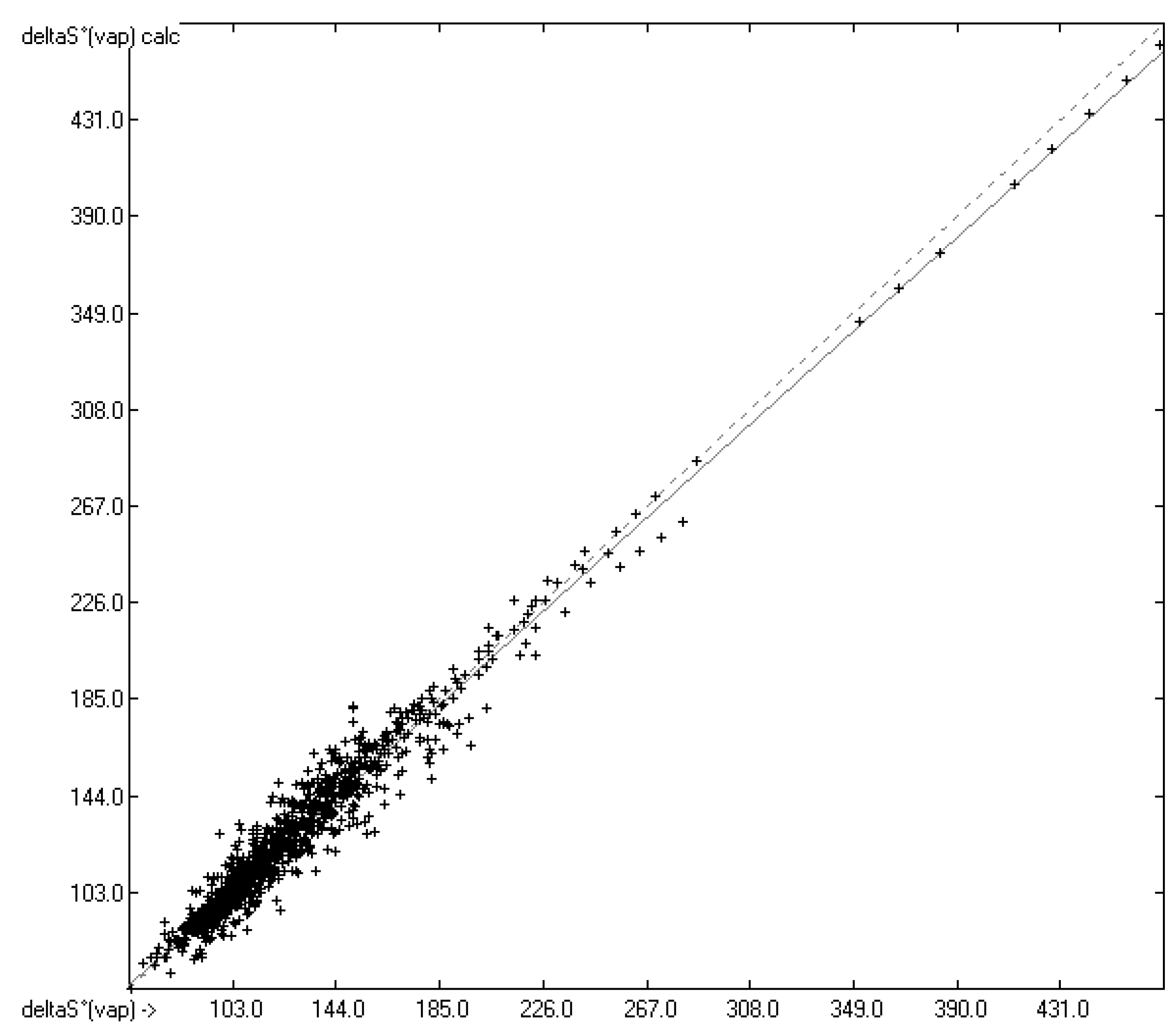
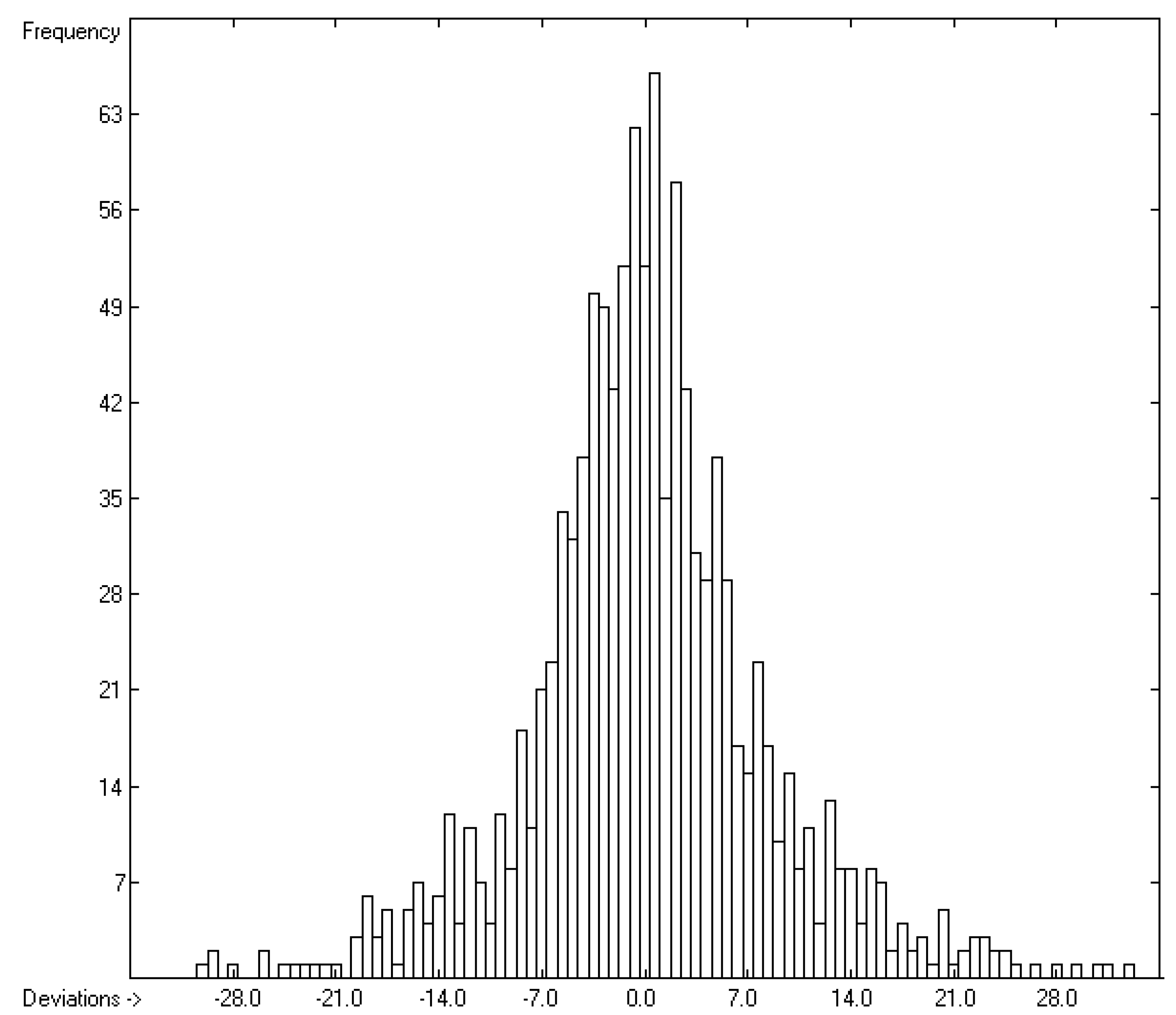
4.3. Standard Entropy of Vaporization
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Pope III, C.A.; Dockery, D.W. Health Effects of Fine Particulate Air Pollution: Lines that Connect. J. Air Waste Manag. Assoc. 2006, 56, 709–742. [Google Scholar] [CrossRef]
- Heal, M.R.; Kumar, P.; Harrison, R.M. Particles, air quality, policy and health. Chem. Soc. Rev. 2012, 41, 6606–6630. [Google Scholar] [CrossRef]
- Hundy, G.F.; Trott, A.R.; Welch, T.C. Chapter 16—Food Refrigeration—Product by Product. In Refrigeration, Air Conditioning and Heat Pumps (Fifth ed.); Elsevier: Amsterdam, The Netherland, 2016; pp. 253–271. [Google Scholar] [CrossRef]
- Santos, L.M.N.B.F.; Lobo Ferreira, A.I.M.C.; Stejfa, V.; Rodrigues, A.S.M.C.; Rocha, M.A.A.; Torres, M.C.; Tavares, F.M.S.; Carpinteiro, F.S. Development of the Knudsen effusion methodology for vapour pressure measurements of low volatile liquids and solids based on a quartz crystal microbalance. J. Chem. Thermodynamics 2018, 126, 171–186. [Google Scholar] [CrossRef]
- Antoine, C. Tension des vapeurs: Nouvelle relation entre les tension et les temperatures. Comptes Rendus 1888, 107, 681–684. [Google Scholar]
- Mackay, D.; Shiu, W.Y.; Ma, K.-C.; Lee, S.C. Handbook of Physical-Chemical Properties and Environmental Fate for Organic Chemicals, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2006. [Google Scholar]
- O’Meara, S.; Booth, A.M.; Barley, M.H.; Topping, D.; McFiggans, G. An assessment of vapour pressure estimation methods. Phys. Chem. Chem. Phys. 2014, 16, 19453. [Google Scholar] [CrossRef] [PubMed]
- Dearden, J.C. Quantitative structure-property relationships for prediction of boiling point, vapor pressure and melting point. Toxicol. Chem. 2003, 22, 1696–1709. [Google Scholar] [CrossRef]
- Vetere, A; Methods for Predicting and Correlating the Vapour Pressures of Pure Compounds. Fluid Phase Equil. 1988, 43, 191–203. [CrossRef]
- Kühne, R.; Ebert, R.-U.; Schüürmann, G. Estimation of Vapour Pressures for Hydrocarbons and Halogenated Hydrocarbons from Chemical Structure by a Neural Network. Chemosphere 1997, 34, 671–686. [Google Scholar] [CrossRef]
- Moller, B.; Rarey, J.; Ramjugernath, D. Estimation of the vapour pressure of non-electrolyte organic compounds via group contributions and group interactions. J. Mol. Liquids 2008, 143, 52–63. [Google Scholar] [CrossRef]
- Tu, C.-H. Group-contribution method for the estimation of vapor pressures. Fluid Phase Equil. 1994, 99, 105–120. [Google Scholar] [CrossRef]
- Goll, E.S.; Jurs, P.C. Prediction of Vapor Pressures of Hydrocarbons and Halohydrocarbons from Molecular Structure with a Computational Neural Network Model. J. Chem. Inf. Comput. Sci. 1999, 39, 1081–1089. [Google Scholar] [CrossRef]
- McClelland, H.E.; Jurs, P.C. Quantitative Structure-Property Relationships for the Prediction of Vapor Pressures of Organic Compounds from Molecular Structures. J. Chem. Inf. Comput. Sci. 2000, 40, 967–975. [Google Scholar] [CrossRef]
- Paul, P.K.C. Prediction of vapour pressure using descriptors derived from molecular dynamics. Org. Biomol. Chem. 2005, 3, 1176–1179. [Google Scholar] [CrossRef]
- Abraham, M.H.; Acree, W.E., Jr. Estimation of vapor pressure of liquid and solid organic and organometallic compounds at 298.15K. Fluid Phase Equil. 2020, 519, 112595. [Google Scholar] [CrossRef]
- Naef, R. A generally applicable computer algorithm based on the group additivity method for the calculation of seven molecular descriptors: Heat of combustion, LogPO/W, LogS, refractivity, polarizability, toxicity and LogBB of organic compounds; scope and limits of applicability. Molecules 2015, 20, 18279–18351. [Google Scholar] [CrossRef] [PubMed]
- Naef, R.; Acree, W.E. Calculation of five thermodynamic molecular descriptors by means of a general computer algorithm based on the group-additivity method: Standard enthalpies of vaporization, sublimation and solvation, and entropy of fusion of ordinary organic molecules and total phase-change entropy of liquid crystals. Molecules 2017, 22, 1059. [Google Scholar] [CrossRef]
- Naef, R.; Acree, W.E. Application of a general computer algorithm based on the group-additivity method for the calculation of two molecular descriptors at both ends of dilution: Liquid viscosity and activity coefficient in water at infinite dilution. Molecules 2018, 23, 5. [Google Scholar] [CrossRef] [PubMed]
- Naef, R.; Acree, W.E., Jr. Calculation of the surface tension of ordinary organic and ionic liquids by means of a generally applicable computer algorithm based on the group-additivity method. Molecules 2018, 23, 1224. [Google Scholar] [CrossRef] [PubMed]
- Naef, R. Calculation of the Isobaric Heat Capacities of the Liquid and Solid Phase of Organic Compounds at 298.15 K by Means of the Group-Additivity Method. Molecules 2020, 25, 1147. [Google Scholar] [CrossRef]
- Hardtwig, E. Fehler- und Ausgleichsrechnung. Hochschultaschenbücher 262/262a; Bibliographisches Institut AG: Mannheim, Germany, 1968. [Google Scholar]
- McDonald, R.A.; Shrader, S.A.; Stull, D.R. Vapor Pressures and Freezing Points of 30 Organics. J. Chem. Eng. Data 1959, 4, 311–313. [Google Scholar] [CrossRef]
- Quina, F.H.; Carroll, F.A.; Cheuy, D.M. A Linear Solvation Energy Relationship to Predict Vapor Pressure from Molecular Structure. J. Braz. Chem. Soc. 2005, 16, 1010–1016. [Google Scholar] [CrossRef][Green Version]
- Katritzky, A.R.; Slavov, S.H.; Dobchev, D.A.; Karelson, M. Rapid QSPR model development technique for prediction of vapor pressure of organic compounds. Comp. Chem. Eng. 2007, 31, 1123–1130. [Google Scholar] [CrossRef]
- Poling, B.E.; Thomson, G.H.; Friend, D.G.; Rowley, R.L.; Wilding, W.V. Physical and Chemical Data. In Perry’s Chemical Engineers’ Handbook, 8th ed.; The McGraw-Hill Companies: New York, NY, USA, 2008. [Google Scholar] [CrossRef]
- Lepori, L.; Matteoli, E.; Gianni, P. Vapor Pressure and Its Temperature Dependence of 28 Organic Compounds: Cyclic Amines, Cyclic Ethers, and Cyclic and Open Chain Secondary Alcohols. J. Chem. Eng. Data 2017, 62, 194–203. [Google Scholar] [CrossRef]
- Costa, J.C.S.; Mendes, A.; Santos, L.M.B.F. Chain Length Dependence of the Thermodynamic Properties of n-Alkanes and their Monosubstituted Derivatives. J. Chem. Eng. Data 2018, 63, 1–20. [Google Scholar] [CrossRef]
- Costa, J.C.S.; Santos, L.M.B.F. Chain-Length Dependence of the Thermodynamic Behavior of Homologous α,ω-Disubstituted Alkanes. J. Chem. Eng. Data 2019, 64, 2229–2246. [Google Scholar] [CrossRef]
- Dang, C.; Bannan, T.; Shelley, P.; Priestley, M.; Worrall, S.D.; Waters, J.; Coe, H.; Percival, C.J.; Topping, D. The effect of structure and isomerism on the vapor pressures of organic molecules and its potential atmospheric relevance. Aerosol Sci. Tech. 2019, 53, 1040–1055. [Google Scholar] [CrossRef]
- Noyes, R.M.; Noyes, W.A.; Steinmetz, H. Vapor Pressures of cis and trans Disubstituted Ethylenes. J. Am. Chem. Soc. 1950, 72, 33–34. [Google Scholar] [CrossRef]
- Nass, K.; Lenoir, D.; Kettrup, A. Calculation of the Thermodynamic Properties of Polycyclic Aromatic Hydrocarbons by an Incremental Procedure. Angew. Chem Int. Ed. Engl. 1995, 34, 1735–1736. [Google Scholar] [CrossRef]
- Verevkin, S.P. Thermochemical Properties of Diphenylalkanes. J. Chem. Eng. Data 1999, 44, 175–179. [Google Scholar] [CrossRef]
- Mokbel, I.; Ruzicka, K.; Majer, V.; Ruzicka, V.; Ribeiro, M.; Jose, J.; Zabransky, M. Vapor pressures and thermal data for three high-boiling compounds of petroleum interest: 1-phenyldodecane, (5α)-cholestane, adamantane. Fluid Phase Equil. 2000, 169, 191–207. [Google Scholar] [CrossRef]
- Lei, Y.D.; Chankalal, R.; Chan, A.; Wania, F. Supercooled Liquid Vapor Pressures of the Polycyclic Aromatic Hydrocarbon. J. Chem. Eng. Data 2002, 47, 801–806. [Google Scholar] [CrossRef]
- Chickos, J.S.; Hanshaw, W. Vapor Pressures and Vaporization Enthalpies of the n-Alkanes from C21 to C30 at T = 298.15 K by Correlation Gas Chromatography. J. Chem. Eng. Data 2004, 49, 77–85. [Google Scholar] [CrossRef]
- Haftka, J.J.H.; Parsons, J.R.; Govers, H.A.J. Supercooled liquid vapour pressures and related thermodynamic properties of polycyclic aromatic hydrocarbons determined by gas chromatography. J. Chromatogr. A 2006, 1135, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Chickos, J.; Wang, T.; Sharma, E. Hypothetical Thermodynamic Properties: Vapor Pressures and Vaporization Enthalpies of the Even n-Alkanes from C40 to C76 at T = 298.15 K by Correlation-Gas Chromatography. Are the Vaporization Enthalpies a Linear Function of Carbon Number? J. Chem. Eng. Data 2008, 53, 481–491. [Google Scholar] [CrossRef]
- Chickos, J.; Lipkind, D. Hypothetical Thermodynamic Properties: Vapor Pressures and Vaporization Enthalpies of the Even n-Alkanes from C78 to C92 at T = 298.15 K by Correlation-Gas Chromatography. J. Chem. Eng. Data 2008, 53, 2432–2440. [Google Scholar] [CrossRef]
- Hanshaw, W.; Nutt, M.; Chickos, J.S. Hypothetical Thermodynamic Properties. Subcooled Vaporization Enthalpies and Vapor Pressures of Polyaromatic Hydrocarbons. J. Chem. Eng. Data 2008, 53, 1903–1913. [Google Scholar] [CrossRef]
- Stejfa, V.; Fulem, M.; Ruzicka, K.; Cervinka, C.; Rocha, M.A.A.; Santos, L.M.N.B.F.; Schröder, B. Thermodynamic study of selected monoterpenes. J. Chem. Thermodyn. 2013, 60, 117–125. [Google Scholar] [CrossRef]
- Stejfa, V.; Fulem, M.; Ruzicka, K.; Cervinka, C. Thermodynamic study of selected monoterpenes II. J. Chem. Thermodyn. 2014, 79, 272–279. [Google Scholar] [CrossRef]
- Stejfa, V.; Fulem, M.; Ruzicka, K.; Cervinka, C. Thermodynamic study of selected monoterpenes III. J. Chem. Thermodyn. 2014, 79, 280–289. [Google Scholar] [CrossRef]
- Zaitsaua, D.H.; Emel’yanenko, V.N.; Pimerzin, A.A.; Verevkin, S.P. Benchmark properties of biphenyl as a liquid organic hydrogen carrier: Evaluation of thermochemical data with complementary experimental and computational methods. J. Chem. Thermodyn. 2018, 122, 1–12. [Google Scholar] [CrossRef]
- Orf, M.; Kurian, M.; Espinosa, L.; Nelson, C.; Simmons, D.; Chickos, J. Thermochemical properties of sesquiterpenes in natural products by correlation gas chromatography: Application to bergamotene oil. J. Chem. Thermodyn. 2018, 126, 128–136. [Google Scholar] [CrossRef]
- Zafar, A.; Chickos, J. The vapor pressure and vaporization enthalpy of squalene and squalene by correlation gas chromatography. J. Chem. Thermodyn. 2019, 135, 192–197. [Google Scholar] [CrossRef]
- Siripoltangman, N.; Chickos, J. Vapor pressure and vaporization enthalpy studies of the major components of ginger, α-zingiberene, β-sesquiphellandrene and (-) ar curcumene by correlation chromatography. J. Chem. Thermodyn. 2019, 138, 107–115. [Google Scholar] [CrossRef]
- Fischer-Lodike, C.; Zafar, A.; Chickos, J. The vapor pressure and vaporization enthalpy of pristane and phytane by correlation gas chromatography. J. Chem. Thermodyn. 2020, 141, 105931. [Google Scholar] [CrossRef]
- Stejfa, V.; Fulem, M.; Ruzicka, K. Thermodynamic study of selected monoterpenes IV. Chem. Thermodyn. 2020, 144, 106013. [Google Scholar] [CrossRef]
- Barton, D.; Chickos, J. The vapor pressure and vaporization enthalpy of (-) β-Elemene and (-) β-Bisabolene By correlation gas chromatography. J. Chem. Thermodyn. 2020, 148, 106139. [Google Scholar] [CrossRef]
- Stejfa, V.; Mahnel, T.; Skorepova, E.; Rohli cek, J.; Eigner, V.; Schröder, B.; Ruzicka, K.; Fulem, M. A combined thermodynamic and crystallographic study of 1,3-diisopropylnaphthalene. J. Chem. Thermodyn. 2020, 150, 106193. [Google Scholar] [CrossRef]
- Pimenova, S.M.; Lukyanova, V.A.; Druzhinina, A.I.; Dorofeeva, O.V. Thermodynamic properties of 1,3,3-trimethylcyclopropene. J. Chem. Thermodyn. 2020, 151, 106240. [Google Scholar] [CrossRef]
- N’Guimbi, J.; Berro, C.; Mokbel, I.; Rauzy, E.; Jose, J. Experimental vapour pressures of 13 secondary and tertiary alcohols—correlation and prediction by a group contribution method. Fluid Phase Equil. 1999, 162, 143–158. [Google Scholar] [CrossRef]
- Verevkin, S.P. Determination of vapor pressures and enthalpies of vaporization of 1,2-alkanediols. Fluid Phase Equil. 2004, 224, 23–29. [Google Scholar] [CrossRef]
- Umnahanant, P.; Kweskin, S.; Nichols, G.; Dunn, M.J.; Smart-Ebinne, H.; Chickos, J.S. Vaporization Enthalpies of the α,ω-Alkanediols by Correlation Gas Chromatography. J. Chem. Eng. Data 2006, 51, 2246–2254. [Google Scholar] [CrossRef]
- Stejfa, V.; Bazyleva, A.; Fulem, M.; Rohli cek, J.; Skorepova, E.; Ruzicka, K.; Blokhin, A.V. Polymorphism and thermophysical properties of l- and dl-menthol. J. Chem. Thermodyn. 2018, 131, 524–543. [Google Scholar] [CrossRef] [PubMed]
- Albinsaad, M.; Scott, N.I.; Chickos, J. Vapor Pressures and Vaporization Enthalpies of 1-Nonadecanol. Isophytol, 2 Z- and 2 E-Phytol, and (2E,7R,11R) phytol by Correlation Gas Chromatography. J. Chem. Thermodyn. 2020, 153, 106307. [Google Scholar] [CrossRef]
- Pokorny, V.; Stejfa, V.; Klajmon, M.; Fulem, M.; Ruzicka, K. Vapor Pressures and Thermophysical Properties of 1-Heptanol, 1-Octanol, 1-Nonanol, and 1-Decanol: Data Reconciliation and PC-SAFT Modeling. J. Chem. Eng. Data 2020, 66, 805–821. [Google Scholar] [CrossRef]
- Verevkin, S.P. Thermochemistry of phenols: Quantification of the ortho-, para-, and meta-interactions in tert-alkyl substituted phenols. J. Chem. Thermodyn. 1999, 31, 559–585. [Google Scholar] [CrossRef]
- Freitas, V.L.S.; Lima, A.C.M.O.; Sapei, E.; Ribeiro da Silva, M.D.M.C. Comprehensive thermophysical and thermochemical studies of vanillyl alcohol. J. Chem. Thermodyn. 2016, 102, 287–292. [Google Scholar] [CrossRef]
- del Rio, A.; Coto, B.; Pando, C.; Renuncio, J.A.R. Vapor–liquid equilibria and excess properties of octane + 1,1-dimethylpropyl methyl ether (TAME) mixtures. Fluid Phase Equil. 2002, 200, 41–51. [Google Scholar] [CrossRef]
- Verevkin, S.P.; Krasnykh, E.L.; Vasiltsova, T.V.; Heintz, A. Determination of Ambient Temperature Vapor Pressures and Vaporization Enthalpies of Branched Ethers. J. Chem. Eng. Data 2003, 48, 591–599. [Google Scholar] [CrossRef]
- Efimova, A.A.; Druzhinina, A.I.; Varushchenko, R.M.; Dorofeeva, O.V.; Krasnykh, E.L. Phase Equilibria and Thermodynamic Properties of Some Branched Alkyl Ethers. J. Chem. Eng. Data 2009, 54, 2457–2469. [Google Scholar] [CrossRef]
- Verevkin, S.P.; Siewert, R.; Emel’yanenko, V.N.; Bara, J.E.; Cao, H.; Pimerzin, A.A. Diphenyl Ether Derivatives as Potential Liquid Organic Hydrogen Carriers: Thermochemical and Computational Study. J. Chem. Eng. Data 2020, 65, 1108–1116. [Google Scholar] [CrossRef]
- Jiang, H.; Li, H.; Wang, C.; Tan, T.; Han, S. (Vapour + liquid) equilibria for (2,2-dimethoxypropane + methanol) and (2,2-dimethoxypropane + acetone). J. Chem. Thermodyn. 2003, 35, 1567–1572. [Google Scholar] [CrossRef]
- Verevkin, S.P.; Konnova, M.E.; Zherikova, K.V.; Pimerzin, A.A. Thermodynamics of glycerol and diols valorisation via reactive systems of acetals synthesis. Fluid Phase Equil. 2020, 510, 112503. [Google Scholar] [CrossRef]
- de Kruif, C.G.; Oonk, H.A.J. Enthalpies of vaporization and vapour pressures of seven aliphatic carboxylic acids. J. Chem. Tlermodyn. 1979, 11, 287–290. [Google Scholar] [CrossRef]
- Ribeiro da Silva, M.A.V.; Monte, M.J.S.; Ribeiro, J.R. Vapour pressures and the enthalpies and entropies of sublimation of five dicarboxylic acids. J. Chem. Thermodyn. 1999, 31, 1093–1107. [Google Scholar] [CrossRef]
- Lagoa, A.L.C.; Diogo, H.P.; Minas da Piedade, M.E.; Amaral, L.M.P.F.; Guedes, R.C.; Costa Cabral, B.J.; Kulikov, D.V.; Verevkin, S.P.; Siedler, M.; Epple, M. Energetics of the C-Cl Bond in CH3CH(Cl)COOH. Enthalpy of Formation of (S)-(-)-2-Chloropropionic Acid and of the 1-Carboxyethyl Radical. J. Phys. Chem. A 2002, 106, 9855–9861. [Google Scholar] [CrossRef]
- Bruns, E.A.; Greaves, J.; Finlayson-Pitts, B.J. Measurement of Vapor Pressures and Heats of Sublimation of Dicarboxylic Acids Using Atmospheric Solids Analysis Probe Mass Spectrometry. J. Phys. Chem. A 2012, 116, 5900–5909. [Google Scholar] [CrossRef] [PubMed]
- Huisman, A.J.; Krieger, U.K.; Zuend, A.; Marcolli, C.; Peter, T. Vapor pressures of substituted polycarboxylic acids are much lower than previously reported. Atmos. Chem. Phys. 2013, 13, 6647–6662. [Google Scholar] [CrossRef]
- Devore, J.A.; O’Neal, H.E. Heats of Formation of the Acetyl Halides and of the Acetyl Radical. J. Phys. Chem. 1969, 73, 2644–2648. [Google Scholar] [CrossRef]
- Bureau, N.; Mokbel, J.J.I.; de Hemptinne, J.-C. Vapour pressure measurements and prediction for heavy esters. J. Chem. Thermodyn. 2001, 33, 1485–1498. [Google Scholar] [CrossRef]
- Chickos, J.S.; Zhao, H.; Nichols, G. The vaporization enthalpies and vapor pressures of fatty acid methyl esters C18, C21 to C23, and C25 to C29 by correlation – gas chromatography. Thermochim. Acta 2004, 424, 111–121. [Google Scholar] [CrossRef]
- Rohac, V.; Ruzicka, K.; Ruzicka, V.; Zaitsau, D.H.; Kabo, G.J.; Diky, V.; Aim, K. Vapour pressure of diethyl phthalate. J. Chem. Thermodyn. 2004, 36, 929–937. [Google Scholar] [CrossRef]
- Vasiltsova, T.V.; Verevkin, S.P.; Bich, E.; Heintz, A.; Bogel-Lukasik, R.; Domanska, U. Thermodynamic Properties of Mixtures Containing Ionic Liquids. 7. Activity Coefficients of Aliphatic and Aromatic Esters and Benzylamine in 1-Methyl-3-ethylimidazolium Bis(trifluoromethylsulfonyl) Imide Using the Transpiration Method. J. Chem. Eng. Data 2006, 51, 213–218. [Google Scholar] [CrossRef]
- Lipp, S.V.; Krasnykh, E.L. Vapor Pressures and Enthalpies of Vaporization of a Series of the Symmetric Linear n-Alkyl Esters of Dicarboxylic Acids. J. Chem. Eng. Data 2011, 56, 800–810. [Google Scholar] [CrossRef]
- Gobble, C.; Chickos, J. Vapor Pressures and Vaporization Enthalpies of a Series of Dialkyl Phthalates by Correlation Gas Chromatography. J. Chem. Eng. Data 2014, 59, 1353–1365. [Google Scholar] [CrossRef]
- Kozlovskiy, M.; Gobble, C.; Chickos, J. Vapor pressures and vaporization enthalpies of a series of esters used in flavors by correlation gas chromatography. J. Chem. Thermodyn. 2015, 86, 65–74. [Google Scholar] [CrossRef]
- Ishak, H.; Stephan, J.; Karam, R.; Goutaudier, C.; Mokbel, I.; Saliba, C.; Saab, J. Aqueous solubility, vapor pressure and octanol-water partition coefficient of two phthalate isomers dibutyl phthalate and di-isobutyl phthalate contaminants of recycled food packages. Fluid Phase Equil. 2016, 427, 362–370. [Google Scholar] [CrossRef]
- Simmons, D.; Chickos, J. Enthalpy of vaporization and vapor pressure of whiskey lactone and menthalactone by correlation gas chromatography. J. Chem. Thermodyn. 2017, 110, 65–70. [Google Scholar] [CrossRef]
- Emel’yanenko, V.N.; Altuntepe, E.; Held, C.; Pimerzin, A.A.; Verevkin, S.P. Renewable platform chemicals: Thermochemical study of levulinic acid esters. Thermochim. Acta 2018, 659, 213–221. [Google Scholar] [CrossRef]
- Verevkin, S.P.; Emel’yanenko, V.N.; Pimerzin, A.A.; Yermalayeu, A.V. How much different are thermochemical properties of enantiomers and their racemates? Thermochemical properties of enantiopure and racemate of methyl- and butyl lactates. J. Chem. Phys. 2018, 149, 054506. [Google Scholar] [CrossRef]
- Emel’yanenko, V.N.; Yermalayeu, A.V.; Portnova, S.V.; Pimerzin, A.A.; Verevkin, S.P. Renewable platform chemicals: Evaluation of thermochemical data of alkyl lactates with complementary experimental and computational methods. J. Chem. Thermodyn. 2019, 128, 55–67. [Google Scholar] [CrossRef]
- Portnova, S.V.; Yamshchikova, Y.F.; Krasnykh, E.L.; Nikitin, E.D.; Popov, A.P.; Faizullin, M.Z. Vapor Pressure, Vaporization Enthalpies, Critical Parameters, and Heat Capacities of Alkyl Glycolates. J. Chem. Eng. Data 2020, 65, 2566–2577. [Google Scholar] [CrossRef]
- Siewert, R.; Zaitsau, D.H.; Emel’yanenko, V.N.; Verevkin, S.P. Biomass Valorization: Thermodynamics of the Guerbet Condensation Reaction. J. Chem. Eng. Data 2019, 64, 4904–4914. [Google Scholar] [CrossRef]
- Francesconi, R.; Comelli, F. Vapor-Liquid Equilibria, Excess Molar Enthalpies, and Excess Molar Volumes of Dialkyl Carbonates + Methyl tert-Butyl Ether at 298.15 K. J. Chem. Eng. Data 1997, 42, 697–701. [Google Scholar] [CrossRef]
- Chernyak, Y.; Clements, J.H. Vapor Pressure and Liquid Heat Capacity of Alkylene Carbonates. J. Chem. Eng. Data 2004, 49, 1180–1184. [Google Scholar] [CrossRef]
- Kozlova, S.A.; Emel’yanenko, V.N.; Georgieva, M.; Verevkin, S.P.; Chernyak, Y.; Schäffner, B.; Börner, A. Vapour pressure and enthalpy of vaporization of aliphatic dialkyl carbonates. J. Chem. Thermodyn. 2008, 40, 1136–1140. [Google Scholar] [CrossRef]
- Verevkin, S.P.; Toktonov, A.V.; Chernyak, Y.; Schäffner, B.; Börner, A. Vapour pressure and enthalpy of vaporization of cyclic alkylene carbonates. Fluid Phase Equil. 2008, 268, 1–6. [Google Scholar] [CrossRef]
- Pokorny, V.; Stejfa, V.; Fulem, M.; Cervinka, C.; Ruzicka, K. Vapor Pressures and Thermophysical Properties of Ethylene Carbonate, Propylene Carbonate, γ-Valerolactone, and γ-Butyrolactone. J. Chem. Eng. Data 2017, 62, 4174–4186. [Google Scholar] [CrossRef]
- Jimenez, P.; Roux, M.V.; Davalos, J.Z.; Martin-Luengo, M.A.; Abboud, J.-L. Structural effects on the thermochemical properties of cycloalkanones II. Enthalpy of combustion, vapour pressures, enthalpy of sublimation, and standard molar enthalpy of formation in the gaseous phase of cyclopentadecanone. J. Chem. Thermodyn. 1997, 29, 1281–1288. [Google Scholar] [CrossRef]
- Zaitsau, D.H.; Verevkin, S.P.; Sazonova, A.Y. Vapor pressures and vaporization enthalpies of 5-nonanone, linalool and 6-methyl-5-hepten-2-one. Data evaluation. Fluid Phase Equil. 2015, 386, 140–148. [Google Scholar] [CrossRef]
- Almeida, A.R.R.P.; Monte, M.J.S. Vapour pressures and phase transition properties of four substituted acetophenones. J. Chem. Thermodyn. 2017, 107, 42–50. [Google Scholar] [CrossRef]
- Indritz, D.; Stone, J.; Williams, F. Vapor Pressure of Di-tert-butyl Peroxide. J. Chem. Eng. Data 1978, 23, 6–7. [Google Scholar] [CrossRef]
- Oxley, J.C.; Smith, J.L.; Luo, W.; Brady, J. Determining the Vapor Pressures of Diacetone Diperoxide (DADP), and Hexamethylene Triperoxide Diamine (HMTD). Propellants Explos. Pyrotech. 2009, 34, 539–543. [Google Scholar] [CrossRef]
- Messerly, J.F.; Finke, H.L.; Osborn, A.G.; Douslin, D.D. Low-temperature calorimetric and vaporpressure studies on alkanediamines. J. Chem. Thermodyn. 1975, 7, 1029–1046. [Google Scholar] [CrossRef]
- Verevkin, S.P.; Beckhaus, H.-D.; Schüle, U.; Rüchardt, C. Experimental Enthalpies of Formation and Strain of the Methylated l-Amino-2-phenylethanes. Struct. Chem. 1998, 9, 1–7. [Google Scholar] [CrossRef]
- Verevkin, S.P.; Georgieva, M.; Melkhanova, S.V. Vapor Pressures and Phase Transitions of a Series of the Aminonaphthalenes. J. Chem. Eng. Data 2007, 52, 286–290. [Google Scholar] [CrossRef]
- Razzouk, A.; Hajiaji, A.; Mokbel, I.; Mougin, P.; Jose, J. Experimental vapor pressures of 1,2-bis(dimethylamino)ethane, 1-methylmorpholine, 1,2-bis(2-aminoethoxy)ethane and N-benzylethanolamine between 273.18 and 364.97 K. Fluid Phase Equil. 2009, 282, 11–13. [Google Scholar] [CrossRef]
- Efimova, A.A.; Emel’yanenko, V.N.; Verevkin, S.P.; Chernyak, Y. Vapour pressure and enthalpy of vaporization of aliphatic poly-amines. J. Chem. Thermodyn. 2010, 42, 330–336. [Google Scholar] [CrossRef]
- Gobble, C.; Rath, N.; Chickos, J. The Vaporization Enthalpies and Vapor Pressures of Some Primary Amines of Pharmaceutical Importance by Correlation Gas Chromatography. J. Chem. Eng. Data 2013, 58, 2600–2609. [Google Scholar] [CrossRef]
- Fulem, M.; Ruzicka, K.; Cervinka, C.; Bazyleva, A.; Della Gatta, G. Thermodynamic study of alkane-α,ω-diamines – Evidence of odd–even pattern of sublimation properties. Fluid Phase Equil. 2014, 371, 93–105. [Google Scholar] [CrossRef]
- Bouzina, Z.; Negadi, A.; Mokbel, I.; Jose, J.; Negadi, L. Phase equilibrium properties of binary mixtures containing 1,3-pentanediamine (or 1,5-diamino-2-methylpentane) and water at several temperatures. J. Chem. Thermodyn. 2015, 84, 81–86. [Google Scholar] [CrossRef]
- Bouzina, Z.; Dergal, F.; Mokbel, I.; Negadi, A.; Saab, J. Liquid-vapor equilibria of pure and aqueous solutions of diethylenetriamine or dipropylenetriamine. Fluid Phase Equil. 2016, 414, 164e169. [Google Scholar] [CrossRef]
- Emel’yanenko, V.N.; Turovtsev, V.V.; Orlov, Y.D.; Fedina, Y.A. Vapour pressure and enthalpy of vaporization of cyclic imines. Thermochim. Acta 2019, 682, 178049. [Google Scholar] [CrossRef]
- Zaitseva, K.V.; Zaitsau, D.H.; Varfolomeev, M.A.; Verevkin, S.P. Vapour pressures and enthalpies of vaporisation of alkyl formamides. Fluid Phase Equil. 2019, 494, 228–238. [Google Scholar] [CrossRef]
- Zaitseva, K.V.; Zaitsau, D.H.; Varfolomeev, M.A.; Verevkin, S.P. Vapour pressures and enthalpies of vaporisation of N alkyl acetamides. J. Mol. Liquids 2019, 293, 111453. [Google Scholar] [CrossRef]
- Zaitseva, K.V.; Zaitsau, D.H.; Varfolomeev, M.A.; Verevkin, S.P. Vapour pressures and enthalpies of vaporisation of N,N-di-alkyl-acetamides. Fluid Phase Equil. 2019, 499, 112241. [Google Scholar] [CrossRef]
- Stejfa, V.; Chun, S.; Pokorny, V.; Fulem, M.; Ruzicka, K. Thermodynamic study of acetamides. J. Mol. Liquids 2020, 319, 114019. [Google Scholar] [CrossRef]
- Verevkin, S.P.; Emel’yanenko, V.N.; Algarra, M.; Lopez-Romero, J.M.; Aguiar, F.; Rodriguez-Borges, J.E. Esteves da Silva, Vapor pressures and enthalpies of vaporization of azides. J. Chem. Thermodyn. 2011, 43, 1652–1659. [Google Scholar] [CrossRef]
- Emel’yanenko, V.N.; Algarra, M.; Esteves da Silva, J.C.G.; Hierrezuelo, J.; Lopez-Romero, J.M.; Verevkin, S.P. Thermochemistry of organic azides revisited. Thermochim. Acta 2014, 597, 78–84. [Google Scholar] [CrossRef]
- Scott, D.W.; Oliver, G.D.; Gross, M.E.; Hubbard, W.N.; Huffman, H.M. Hydrazine: Heat Capacity, Heats of Fusion and Vaporization, Vapor Pressure, Entropy and Thermodynamic Functions. J. Am. Chem. Soc. 1949, 71, 2293–2297. [Google Scholar] [CrossRef]
- Aston, J.G.; Fink, H.L.; Janz, G.J.; Russell, K.E. The Heat Capacity, Heats of Fusion and Vaporization, Vapor Pressures, Entropy and Thermodynamic Functions of Methylhydrazine. J. Am. Chem. Soc. 1951, 73, 1939–1943. [Google Scholar] [CrossRef]
- Aston, J.G.; Janz, G.J.; Russell, K.E. The Heat Capacity, Heats of Fusion and Vaporization, Vapor Pressures and Entropy of Symmetrical Dimethylhydrazine. J. Am. Chem. Soc. 1951, 73, 1943–1945. [Google Scholar] [CrossRef]
- Ahmad, S.; Giesen, R.; Lucas, K. Vapor-Liquid Equilibrium Studies for Systems Containing n-Butylisocyanate at Temperatures between 323.15 K and 371.15 K. J. Chem. Eng. Data 2004, 49, 826–831. [Google Scholar] [CrossRef]
- Ramos, L.A.; Ulic, S.E.; Romano, R.M.; Vishnevskiy, Y.V.; Mitzel, N.W.; Beckers, H.; Willner, H.; Tong, S.; Ge, M.; Della Vedova, C.O. Chlorodifluoroacetyl Isothiocyanate, ClF2CC(O)NCS: Preparation and Structural and Spectroscopic Studies. J. Phys. Chem. A 2013, 117, 5597–5606. [Google Scholar] [CrossRef] [PubMed]
- Davis, P.S.; Kilpatrick, J.E. Entropy, related thermodynamic properties, and structure of methylisocyanate. J. Chem. Thermodyn. 2013, 58, 134–141. [Google Scholar] [CrossRef]
- Xu, M.; Wu, L.; Zhang, J. Measurement of vapor-liquid equilibrium data and azeotropic analysis for 2,4-toluene diisocyanate+entrainer. CIESC 2016. [Google Scholar] [CrossRef]
- Woodman, A.L.; Murbach, W.J.; Kaufman, M.H. Vapor Pressure and Viscosity Relationships for a Homologous Series of α,ι-Dinitriles. J. Phys. Chem. 1960, 64, 658–660. [Google Scholar] [CrossRef]
- Cao, Y.; Dai, X.; Song, H.; Yao, S.; Lan, X. Vapor Pressure Measurement and Isobaric Vapor−Liquid Equilibria for Binary Mixtures of ZE-2-Methyl-2-butenenitrile and 2-Methyl-3-butenenitrile. J. Chem. Eng. Data 2016, 61, 1573–1577. [Google Scholar] [CrossRef]
- Toops Jr., E.E. Physical Properties of eight High-Purity Nitroparaffins. J. Phys. Chem. 1956, 60, 304–306. [Google Scholar] [CrossRef]
- Verevkin, S.P. Thermochemistry of nitro compounds. Experimental standard enthalpies of formation and improved group-additivity values. Thermochim. Acta 1997, 307, 17–25. [Google Scholar] [CrossRef]
- Verevkin, S.P.; Heintz, A. Thermochemistry of substituted benzenes: Quantification of ortho-, para-, meta-, and buttress interactions in alkyl-substituted nitrobenzenes. J. Chem. Thermodyn. 2000, 32, 1169–1182. [Google Scholar] [CrossRef]
- Ribeiro da Silva, M.A.V.; Lima, L.M.S.S.; Amaral, L.M.P.F.; Ferreira, A.I.M.C.L.; Gomes, J.R.B. Standard molar enthalpies of formation, vapour pressures, and enthalpies of sublimation of 2-chloro-4-nitroaniline and 2-chloro-5-nitroaniline. J. Chem. Thermodyn. 2003, 35, 1343–1359. [Google Scholar] [CrossRef]
- Ribeiro da Silva, M.A.V.; Monte, M.J.S.; Ferreira, A.I.M.C.L.; Oliveira, J.A.S.A.; Cimas, A. Experimental and Computational Thermodynamic Study of Three Monofluoronitrobenzene Isomers. J. Phys. Chem. B 2010, 114, 7909–7919. [Google Scholar] [CrossRef] [PubMed]
- Thompson, H.W.; Dainton, F.S. The Photochemistry of Alkyl Nitrites. Trans. Faraday Soc. 1937, 33, 1546–1555. [Google Scholar] [CrossRef]
- Fischer, R.G.; Ballschmiter, K. Prediction of the environmental distribution of alkyl dinitrates—Chromatographic determination of vapor pressure p0, water solubility SH2O, gas-water partition coefficient KGW (Henry’s law constant) and octanol-water partition coefficient KOW. Fresenius, J. Anal. Chem. 1998, 360, 769–776. [Google Scholar] [CrossRef]
- Klein, R.G. Calculations and Measurements on the Volatility of Nitrosamines and their Aqueous Solutions. Toxicology 1982, 23, 135–147. [Google Scholar] [CrossRef]
- Jakli, G.; Van Hook, W.A. Excess Thermodynamic Properties of H2O and D2O Solutions of Tetramethylurea, an Azeotropic System. Vapor Pressures, Excess Vapor Pressures, and Vapor Pressure Isotope Effects. J. Chem. Eng. Data 2001, 46, 777–781. [Google Scholar] [CrossRef]
- Ribeiro da Silva, M.M.C.; Ribeiro da Silva, M.A.V.; Freitas, V.L.S.; Roux, M.V.; Jimenez, P.; Temprado, M.; Davalos, J.Z.; Cabildo, P.; Claramunt, R.M.; Elguero, J. Structural studies of cyclic ureas: 1. Enthalpies of formation of imidazolidin-2-one and N,N’-trimethyleneurea. J. Chem. Thermodyn. 2008, 40, 386–393. [Google Scholar] [CrossRef]
- Sawaya, T.; Mokbel, I.; Rauzy, E.; Saab, J.; Berro, C.; Jose, J. Experimental vapor pressures of alkyl and aryl sulfides Prediction by a group contribution method. Fluid Phase Equil. 2004, 226, 283–288. [Google Scholar] [CrossRef]
- Sassa, Y.; Konishi, R.; Katayama, T. Isothermal Vapor-Liquid Equilibrium Data of DMSO Solutions by Total Pressure Method. DMSO-Acetone, DMSO-Tetrahydrofuran, and DMSO-Ethyl Acetate Systems. J. Chem. Eng. Data 1974, 19, 44–48. [Google Scholar] [CrossRef]
- Markarian, S.A.; Zatikyan, A.L.; Grigoryan, V.V.; Grigoryan, G.S. Vapor Pressures of Pure Diethyl Sulfoxide from (298.15 to 318.15) K and Vapor-Liquid Equilibria of Binary Mixtures of Diethyl Sulfoxide with Water. J. Chem. Eng. Data 2005, 50, 23–25. [Google Scholar] [CrossRef]
- Grigoryan, G.S.; Markarian, S.A. Thermodynamics of Liquid–Gas Phase Equilibria in the Dipropylsulfoxide–Water System in the Range of 303.15 to 323.15 K. Russ. J. Phys. Chem. A 2013, 87, 191–193. [Google Scholar] [CrossRef]
- Grigoryan, G.S.; Markarian, S.A. Thermodynamics of Liquid–Gas Phase Equilibria of Diisopropyl and Dibutyl Sulfoxides. Russ. J. Phys. Chem. A 2014, 88, 1071–1072. [Google Scholar] [CrossRef]
- Edwards, D.R.; Prausnitz, J.M. Vapor Pressures of Some Sulfur-Containing, Coal-Related Compounds. J. Chem. Eng. Data 1981, 26, 121–124. [Google Scholar] [CrossRef]
- Anton, V.; Artigas, H.; Lomba, L.; Giner, B.; Lafuente, C. Thermophysical properties of the thiophene family. J. Therm. Anal. Calorim. 2016, 125, 509–518. [Google Scholar] [CrossRef]
- Anton, V.; Giner, B.; Artigas, H.; Gascon, I.; Lafuente, C. Comparative Study of the Thermophysical Properties of 2-Ethylthiophene and 2-Ethylfuran. J. Chem. Eng. Data 2018, 63, 3274–3284. [Google Scholar] [CrossRef]
- Burg, A.B.; Slota Jr., P.J. Dimethylaminodimethylphosphine. J. Am. Chem. Soc. 1958, 80, 1107–1109. [Google Scholar] [CrossRef]
- Burg, A.B.; Slota Jr., P.J. Chemistry of the C4H8P Ring: The Aminophosphine (CH3)2NPC4H8, the Cyclophosphine C4H8PH and the Tetracyclic Trimer (C4H8PBH2). J. Am. Chem. Soc. 1960, 82, 2148–2151. [Google Scholar] [CrossRef]
- Burg, A.B.; Griffiths, J.E. Trifluoromethyl-bromo-phosphines and Phosphoranes. J. Am. Chem. Soc. 1960, 82, 3514–3517. [Google Scholar] [CrossRef]
- Holmes, R.R.; Wagner, R.P. Phosphorus Nitrogen Chemistry. IV. The Reactions of Dimethylaminophosphines with Boron Trihalides and Trialkyls. J. Am. Chem. Soc. 1962, 84, 357–361. [Google Scholar] [CrossRef]
- Burg, A.B.; Gosling, K. Stable Thiotrifluoromethyl phosphines. J. Am. Chem. Soc. 1965, 87, 2113–2116. [Google Scholar] [CrossRef]
- Michou-Saucet, M.-A.; Jose, J.; Michou-Saucet, C.; Merlin, J.C. Pressions de Vapeur et Enthalpies Libre d’Exces de Systèmes Binaires: Hexamethylphosphorotriamide (HMPT) + n-Hexane; n-Heptane; n-Octane; A 288.15 K; 303.15 K; 313.15 K; 323.15 K; 333.15 K. Thermochim. Acta 1984, 75, 85–106. [Google Scholar] [CrossRef]
- Skene, W.G.; Krzymien, M.E. Vapor Pressure of Tri-n-butyl Phosphate. J. Chem. Eng. Data 1995, 40, 394–397. [Google Scholar] [CrossRef]
- Tsuzuki, M. Thermodynamic Estimation of Vapor Pressure for Organophosphorous Pesticides. Environ. Tox. Chem. 2000, 19, 1717–1726. [Google Scholar] [CrossRef]
- Butrow, A.B.; Buchanan, J.H.; Tevault, D.E. Vapor Pressure of Organophosphorus Nerve Agent Simulant Compounds. J. Chem. Eng. Data 2009, 54, 1876–1883. [Google Scholar] [CrossRef]
- Tevault, D.E.; Brozena, A.; Buchanan, J.H.; Abercrombie-Thomas, P.L.; Buettner, L.C. Thermophysical Properties of VX and RVX. J. Chem. Eng. Data 2012, 57, 1970–1977. [Google Scholar] [CrossRef]
- Brozena, A.; Buchanan, J.H.; Miles Jr., R.W. Vapor Pressure of Triethyl and Tri-n-Propyl Phosphates and Diethyl Malonate. J. Chem. Eng. Data 2014, 59, 2649–2659. [Google Scholar] [CrossRef]
- Althoff, M.A.; Grieger, K.; Härtel, M.A.C.; Karaghiosoff, K.L.; Klapötke, T.M.; Metzulat, M. Application of the Transpiration Method To Determine the Vapor Pressure and Related Physico-Chemical Data of Low Volatile, Thermolabile, and Toxic Organo(thio)phosphates. J Phys Chem. A 2017, 121, 2603-–2609. [Google Scholar] [CrossRef]
- Bikelyté, G.; Härtel, M.A.C.; Klapötke, T.M.; Krumm, B.; Sadaunykas, A. Experimental thermochemical data of CWA simulants: Triethyl phosphate, diethyl ethylphosphonate, malathion and methyl salicylate. J. Chem. Thermodyn. 2020, 143, 106043. [Google Scholar] [CrossRef]
- Schlesinger, H.I.; Ritter, D.M.; Burg, A.B. Hydrides of Boron. IX. The Preparation of Some Methyl Triborine Triamines. J. Am. Chem. Soc. 1938, 60, 1296–1300. [Google Scholar] [CrossRef]
- Burg, A.B.; Wagner, R.I. Chemistry of S-B Bonding: Polymeric Thioborines, Methanethiodiborane and Related Substances. J. Am. Chem. Soc. 1954, 76, 3307–3310. [Google Scholar] [CrossRef]
- Burg, A.B.; Good, C.D. Nitrogen bond strain effects in the chemistry of ring-amino boron hydrides. J. Inorg. Nucl. Chem. 1956, 2, 237–245. [Google Scholar] [CrossRef]
- Parsons, T.D.; Silverman, M.B.; Ritter, D.M. Alkenylboranes. I. Preparation and Properties of Some Vinyl- and Propenylboranes. J. Am. Chem. Soc. 1957, 79, 5091–5098. [Google Scholar] [CrossRef]
- Brotherton, R.J.; McCloskey, A.L. Tetra-(amino)-diborons. J. Am. Chem. Soc. 1960, 82, 6242–6245. [Google Scholar] [CrossRef]
- Brotherton, R.J.; McCloskey, A.L.; Boone, J.L.; Manasevit, H.M. The Preparation and Properties of Some Tetraalkoxydiborons. J. Am. Chem. Soc. 1960, 82, 6245–6248. [Google Scholar] [CrossRef]
- Christopher, P.M.; Shilman, A. Vapor pressures of trialkyl borates. J. Chem. Eng. Data 1967, 12, 333–335. [Google Scholar] [CrossRef]
- Burg, A.B.; Basi, J.S. Boron bis(trifluoromethyl)phosphinites. J. Am. Chem. Soc. 1969, 91, 1937–1940. [Google Scholar] [CrossRef]
- Emeléus, H.J.; Miller, N. 175. Derivatives of monosilane. Part, I. The reactions of chlorosilane with aliphatic amines. J. Chem. Soc. 1939, 819–823. [Google Scholar] [CrossRef]
- Aston, J.G.; Kennedy, R.M.; Messerly, G.H. The Heat Capacity and Entropy, Heats of Fusion and Vaporization and the Vapor Pressure of Silicon Tetramethyl. J. Am. Chem. Soc. 1941, 63, 2343–2348. [Google Scholar] [CrossRef]
- Emeléus, H.J.; Maddock, A.G. 251. The chemistry of the higher silanes. Part I. Tetrasilane. J. Chem. Soc. 1946, 1131–1134. [Google Scholar] [CrossRef]
- Tannenbaum, S.; Kaye, S.; Lewenz, G.F. Synthesis and Properties of Some Alkylsilanes. J. Am. Chem. Soc. 1953, 75, 3753–3757. [Google Scholar] [CrossRef]
- Opitz, H.E.; Peake, J.S.; Nebergall, W.H. The Preparation of Monobromosilane and Organic Silyl Derivatives. J. Am. Chem. Soc. 1956, 78, 292–294. [Google Scholar] [CrossRef]
- Emeléus, H.J.; Smythe, L.E. 118. Preparation and properties of dimethyliodosilane, 1:1′:2:2′-tetramethyl-disiloxane and -disilthiane. J. Chem. Soc. 1958, 609–611. [Google Scholar] [CrossRef]
- Takagi, S.; Ishikawa, M.; Kumada, M.; Kimura, T.; Fujishiro, R. Vapour Pressure and Thermodynamic Properties of Hexamethyldisilane at 305–387 K. Thermochim. Acta 1986, 109, 55–61. [Google Scholar] [CrossRef]
- Fulem, M.; Ruzicka, K.; Ruzicka, V.; Simecek, T.; Hulicius, E.; Pangrac, J.; Becker, J.; Koch, J.; Salzmann, A. Vapor Pressure of Di-tert-butylsilane. J. Chem. Eng. Data 2005, 50, 1613–1615. [Google Scholar] [CrossRef]
- Sternbach, B.; MacDiarmid, A.G. The Preparation of Methoxysilanes by the Interaction of Monosilane and Methanol. J. Am. Chem. Soc. 1959, 81, 5109–5110. [Google Scholar] [CrossRef]
- Marsh, K.N. Thermodynamics of octamethylcyclotetrasiloxane mixtures. Trans. Faraday Soc. 1968, 64, 883–893. [Google Scholar] [CrossRef]
- van der Vis, M.G.M.; Cordfunke, E.H.P. Tetraethoxysilane, Si(OC2H5)4: Vapour pressure measurements at temperatures from 323 to 442 K by means of a Bourdon spoon gauge. Thermochim. Acta 1995, 265, 129–134. [Google Scholar] [CrossRef]
- Kochetkov, A.; Smith, J.S.; Ravikrishna, R.; Valsaraj, K.T.; Thibodeaux, L.J. Air-Water Partition Constants for Volatile Methyl Siloxanes. Environ. Tox. Chem. 2009, 20, 2184–2188. [Google Scholar] [CrossRef]
- Abbas, R.; Schedemann, A.; Ihmels, C.; Enders, S.; Gmehling, J. Measurement of Thermophysical Pure Component Properties for a Few Siloxanes Used as Working Fluids for Organic Rankine Cycles. Ind. Eng. Chem. Res. 2011, 50, 9748–9757. [Google Scholar] [CrossRef]
- Emeléus, H.J.; MacDiarmid, A.G.; Maddock, A.G. Sulphur and Selenium Derivatives of Monosilanes. J. Inorg. Nucl. Chem. 1955, 1, 194–201. [Google Scholar] [CrossRef]
- Louis, E.; Urry, G. Preparation of hexamethyldisilthiane. Inorg. Chem. 1968, 7, 1253–1254. [Google Scholar] [CrossRef]
- Wisniewska, B.; Lencka, M.; Rogalski, M. Vapour pressures of 2,4-, 2,6-, and 3,5-dimethylpyridine at temperatures from 267 to 360 K. J. Chem. Thermodyn. 1986, 18, 703–708. [Google Scholar] [CrossRef]
- Verevkin, S.P.; Zaitseva, K.V.; Stanton, A.D.; Hindman, M.S.; Irvin, A.C.; Bara, J.E. Building Blocks for Ionic Liquids: Vapor Pressures and Vaporization Enthalpies of N-Functionalized Imidazoles with Branched and Cycloalkyl Substituents. Ind. Eng. Chem. Res. 2015, 54, 9850–9856. [Google Scholar] [CrossRef]
- Emel’yanenko, V.N.; Kaliner, M.; Strassner, T.; Verevkin, S.P. Thermochemical properties of different 1-(R-phenyl)-1H-imidazoles. Fluid Phase Equil. 2017, 433, 40–49. [Google Scholar] [CrossRef]
- Safronov, S.P.; Nagrimanov, R.N.; Samatov, A.A.; Emel’yanenko, V.N.; Zaitsau, D.H.; Pimerzin, A.A.; Skrzypczak, A.; Verevkin, S.P. Benchmark properties of pyrazole derivatives as a potential liquid organic hydrogen carrier: Evaluation of thermochemical data with complementary experimental and computational methods. J. Chem. Thermodyn. 2019, 128, 173–186. [Google Scholar] [CrossRef]
- Stiles, V.E.; Cady, G.H. Physical Properties of Perfluoro-n-hexane and Perfluoro-2-methylpentane. J. Am. Chem. Soc. 1952, 74, 3771–3773. [Google Scholar] [CrossRef]
- Barber, J.B.; Cady, G.H. Vapor Pressures of Perfluoropentanes. J. Phys. Chem. 1956, 60, 504–505. [Google Scholar] [CrossRef]
- Rowlinson, J.S.; Thacker, R. The physical properties of some fluorine compounds and their solutions. Part 3.—Perfluorocyclohexane and perfluoromethylcyclohexane. Trans. Faraday Soc. 1957, 53, 1–8. [Google Scholar] [CrossRef]
- Good, W.D.; Douslin, D.R.; Scott, D.W.; George, A.; Lacina, J.L.; dawson, J.P.; Waddington, G. Thermochemistry and Vapor Pressure of Aliphatic Fluorocarbons. A Comparison of the C–F and C–H Thermochemical Bond Energies. J. Phys. Chem. 1959, 63, 1133–1138. [Google Scholar] [CrossRef]
- Crowder, G.A.; Taylor, Z.L.; Reed III, T.M.; Young, J.A. Vapor pressures and triple point temperatures for several pure fluorocarbons. J. Chem. Eng. Data 1967, 12, 481–485. [Google Scholar] [CrossRef]
- Majer, V.; Svoboda, V.; Posta, A.; Pick, J. Determination of heats of vaporization and some other thermodynamic quantities for three fluorinated halogen ethanes. Collect. Czech. Chem. Commun. 1981, 46, 817–822. [Google Scholar] [CrossRef]
- Marrucho, I.M.; Oliveira, N.S.; Dohrn, R. Vapor-Phase Thermal Conductivity, Vapor Pressure, and Liquid Density of R365mfc. J. Chem. Eng. Data 2002, 47, 554–558. [Google Scholar] [CrossRef]
- Dias, A.M.A.; Goncalves, C.M.B.; Caco, A.I.; Santos, L.M.N.B.F.; Pineiro, M.M.; Vega, L.F.; Coutinho, J.A.P.; Marrucho, I.M. Densities and Vapor Pressures of Highly Fluorinated Compounds. J. Chem. Eng. Data 2005, 50, 1328–1333. [Google Scholar] [CrossRef]
- Sarraute, S.; Mokbel, I.; Costa Gomes, M.F.; Majer, V.; Jose, J. Atmosphere/water partition of halocyclohexanes from vapour pressure and solubility data. Atmos. Env. 2008, 42, 4724–4734. [Google Scholar] [CrossRef]
- Morgado, P.; Das, G.; McCabe, C.; Filipe, E.J.M. Vapor Pressure of Perfluoroalkylalkanes: The Role of the Dipole. J. Phys. Chem. B 2015, 119, 4–1623. [Google Scholar] [CrossRef]
- Nagrimanov, R.N.; Solomonov, B.N.; Emel’yanenko, V.N.; Verevkin, S.P. Six-membered ring aliphatic compounds: A search for regularities in phase transitions. Thermochim. Acta 2016, 638, 80–88. [Google Scholar] [CrossRef]
- Lister, M.W. Heats of Organic Reactions. X. Heats of Bromination of Cyclic Olefins. J. Am. Chem. Soc. 1941, 63, 143–149. [Google Scholar] [CrossRef]
- Bobbo, S.; Fedele, L.; Scattolini, M.; Camporese, M. Compressed Liquid Densities, Saturated Liquid Densities, and Vapor Pressures of Hexafluoro-1,3-butadiene (C4F6). J. Chem. Eng. Data 2002, 47, 179–182. [Google Scholar] [CrossRef]
- Subramoney, S.C.; Wayne, M.N.; Valtz, A.; Coquelet, C.; Richon, D.; Naidoo, P.; Ramjugernath, D. Pure Component and Binary Vapor-Liquid Equilibrium + Modeling for Hexafluoropropylene and Hexafluoropropylene Oxide with Toluene and Hexafluoroethane. J. Chem. Eng. Data 2010, 55, 411–418. [Google Scholar] [CrossRef]
- Fedele, L.; Di Nicola, G.; Brown, J.S.; Bobbo, S.; Zilio, C. Measurements and Correlations of cis-1,3,3,3-Tetrafluoroprop-1-ene (R1234ze(Z)) Saturation Pressure. Int. J. Thermophys. 2014, 35, 1–12. [Google Scholar] [CrossRef]
- Yang, Z.-q.; Kou, L.-g.; Mao, W.; Lu, J.; Zhang, W.; Lu, J. Isothermal Vapor–Liquid Equilibrium for the Binary System of 2,3,3,3-Tetrafluoropropene and 2-Chloro-1,1,1,2-tetrafluoropropane. J. Chem. Eng. Data 2015, 60, 1153–1156. [Google Scholar] [CrossRef]
- Sakoda, N.; Higashi, Y. Measurements of PvT Properties, Vapor Pressures, Saturated Densities, and Critical Parameters for cis-1-Chloro-2,3,3,3-tetrafluoropropene (R1224yd(Z)). J. Chem. Eng. Data 2019, 64, 3983–3987. [Google Scholar] [CrossRef]
- Li, S.; Yang, F.; Zhang, K.; Duan, Y.; Yang, Z. Vapor Pressure Measurements and Correlation for trans-1-Chloro-3,3,3-trifluoroprop-1-ene. J. Chem. Eng. Data 2019, 64, 2947–2954. [Google Scholar] [CrossRef]
- Sakoda, N.; Higashi, Y.; Akasaka, R. Measurements of Vapor Pressures for trans-1-Chloro-3,3,3-trifluoropropene (R1233zd(E)) and cis-1,1,1,4,4,4-Hexafluoro-2-butene (R1336mzz(Z)). J. Chem. Eng. Data 2020, 65, 4285–4289. [Google Scholar] [CrossRef]
- Bobbo, S.; Bet, A.; Scattolini, M.; Fedele, L. Saturated Pressure Measurements of cis-1-Chloro-2,3,3,3- tetrafluoropropene (R1224yd (Z)) Saturation Pressure. J. Chem. Eng. Data 2020, 65, 4263–4267. [Google Scholar] [CrossRef]
- Fang, Y.; Ye, G.; Ni, H.; Jiang, Q.; Bao, K.; Han, X.; Chen, G. Vapor–Liquid Equilibrium for the Binary Systems 1,1,2,3,3,3-Hexafluoro-1-propene (R1216) + 2,3,3,3-Tetrafluoroprop-1-ene (R1234yf) and 1,1,2,3,3,3-Hexafluoro-1-propene (R1216) + trans-1,3,3,3-Tetrafluoropropene (R1234ze(E)). J. Chem. Eng. Data 2020, 65, 4215–4222. [Google Scholar] [CrossRef]
- Yang, T.; Hu, X.; Meng, X.; Wu, J. Vapour-liquid equilibria for the binary systems of pentafluoroethane {(R125) + 2,3,3,3-tetrafluoroprop-1-ene (R1234yf)} and {trans-1,3,3,3-tetrafluoropropene R1234ze(E)}. J. Chem. Thermodyn. 2020, 150, 106222. [Google Scholar] [CrossRef]
- Yin, J.; Ke, J.; Zhao, G.; Ma, S. Experimental vapor pressures and gaseous pvT properties of trans-1-Chloro-3,3,3-trifluoropropene (R1233zd(E)). Int. J. Refrig. 2021, 121, 253–257. [Google Scholar] [CrossRef]
- Fischer, R.C.; Wittlinger, R.; Ballschmiter, K. Retention-index based vapor pressure estimation for polychlorobiphenyl (PCB) by gas chromatography. Fresenius, J. Anal. Chem. 1992, 342, 421–425. [Google Scholar] [CrossRef]
- Lei, Y.D.; Wania, F.; Shiu, W.Y. Vapor Pressures of the Polychlorinated Naphthalenes. J. Chem. Eng. Data 1999, 44, 577–582. [Google Scholar] [CrossRef]
- Verevkin, S.P.; Schick, C. Determination of vapor pressures, enthalpies of sublimation, enthalpies of vaporization, and enthalpies of fusion of a series of chloro-aminobenzenes and chloro-nitrobenzenes. Fluid Phase Equil. 2003, 211, 161–177. [Google Scholar] [CrossRef]
- Verevkin, S.P.; Emel’yanenko, V.N.; Klamt, A. Thermochemistry of Chlorobenzenes and Chlorophenols: Ambient Temperature Vapor Pressures and Enthalpies of Phase Transitions. J. Chem. Eng. Data 2007, 52, 499–510. [Google Scholar] [CrossRef]
- Nakajoh, K.; Grabda, M.; Oleszek-Kudlak, S.; Shibata, E.; Eckert, F.; Nakamura, T. Prediction of vapour pressures of chlorobenzenes and selected polychlorinated biphenyls using the COSMO-RS model. J. Mol. Struct. 2009, 895, 9–17. [Google Scholar] [CrossRef]
- Almeida, A.R.R.P.; Monte, M.J.S. Crystalline and liquid vapour pressures of the four p-monohalophenols: A thermodynamic study of their phase transitions. J. Chem. Thermodyn. 2013, 65, 150–158. [Google Scholar] [CrossRef]
- Verevkin, S.P.; Emel’yanenko, V.N.; Varfolomeev, M.A.; Solomonov, B.N.; Zherikova, K.V.; Melkhanova, S.V. Vaporization enthalpies of a series of the halogen-substituted fluorobenzenes. Fluid Phase Equil. 2015, 387, 160–168. [Google Scholar] [CrossRef]
- Oliveira, J.A.S.A.; Oliveira, T.S.M.; Gaspar, A.; Borges, F.; Ribeiro da Silva, M.D.M.C.; Monte, M.J.S. Study on the volatility of halogenated fluorenes. Chemosphere 2016, 157, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Almeida, A.R.R.P.; Pinheiro, B.D.A.; Lima, C.F.R.A.C.; Santos, A.F.L.O.M.; Ferreira, A.C.S.; Almeida Paz, F.A.; Monte, M.J.S. Thermodynamic Properties of Moldy-Musty Contaminants of Wine. J. Chem. Eng. Data 2019, 64, 4741–4753. [Google Scholar] [CrossRef]
- Rotariu, G.J.; Hanrahan, R.J.; Fruin, R.E. Liquid-Liquid Solubility and Vapor Pressure of Heptacosafluorotributylamine. J. Am. Chem. Soc. 1954, 76, 3752–3756. [Google Scholar] [CrossRef]
- Barr, D.A.; Haszeldine, R.N. 663. Perfluoroalkyl derivatives of nitrogen. Part III. Heptafluoronitrosopropane, perfluoro-2-n-propyl-1: 2-oxazetidine, perfluoro-(methylene-n-propylamine), and related compounds. J. Chem. Soc. 1956, 3416–3428. [Google Scholar] [CrossRef]
- Good, W.D.; Todd, S.S.; Messerly, J.F.; Lacina, J.L.; Dawson, J.P.; Scott, D.W.; McCullough, J.P. Perfluoropiperidine: Entropy, heat of formation, and vapor pressure.; N-F bond energy.; and solid-state transitions. J. Phys. Chem. 1963, 67, 1306–1311. [Google Scholar] [CrossRef]
- Nash, L.L.; Babb, D.P.; Couville, J.J.; Shreeve, J.M. Some bis(trifluoromethyl)nitroxides. J. Inorg. Nucl. Chem. 1968, 30, 3373–3375. [Google Scholar] [CrossRef]
- Takashima, M.; Shreeve, J.M. Preparation and Reactions of Heptafluoroazazcyclopentan-2-one. Inorg. Chem. 1979, 18, 3281–3283. [Google Scholar] [CrossRef]
- Varouchtchenko, R.M.; Droujinina, A.I. Thermodynamics of vaporization of some perfluorotrialkylamines. J. Chem. Thermodyn. 1995, 27, 355–368. [Google Scholar] [CrossRef]
- Aim, K.; Svoboda, V.; Majer, V. Vapour pressures, densities, and refractive indices of 2-chloro-1,1,2-trifluoroethyl ethyl ether, 2-chloro-1,1,2-trifluoroethyl propyl ether, 2-chloro-1,1,2-trifluoroethyl chloromethyl ether, and 2-chloro-1,1,2-trifluoroethyl difluoromethyl ether. J. Chem. Thermodyn. 1983, 15, 531–536. [Google Scholar] [CrossRef]
- Salvi-Narkhede, M.; Adcock, J.L.; Gakh, A.; Van Hook, W.A. Vapor pressures, liquid molar volumes, vapor non-ideality, and critical properties of CF3OCF2CF2CF3, c-CF2CF2CF2CF2O, CF3OCF2OCF3, and CF3OCF2CF2H. J. Chem. Thermodyn. 1993, 25, 643–647. [Google Scholar] [CrossRef]
- Satoh, K.; Nishiumi, H.; Kasatani, T. Vapor pressure of CH3OCHF2 synthesized from HCFC22. Fluid Phase Equil. 1998, 144, 211–216. [Google Scholar] [CrossRef]
- Murata, J.; Yamashita, S.; Akiyama, M.; Katayama, S.; Hiaki, T.; Sekiya, A. Vapor Pressures of Hydrofluoroethers. J. Chem. Eng. Data 2002, 47, 911–915. [Google Scholar] [CrossRef]
- Garate, M.P.; Bejarano, A.; de la Fuente, J.C. Vapour pressures for 1-(butoxymethoxy)butane (dibutoxymethane) and 1,1,1,2,2,3,3,4,4-nonafluoro-4-methoxybutane (methyl nonafluorobutyl ether) over the pressure range of (15–80) kPa. J. Chem. Thermodyn. 2016, 101, 351–355. [Google Scholar] [CrossRef]
- Black, J.E.; Silva, G.M.C.; Klein, C.; Iacovella, C.R.; Norgado, P.; Martins, L.F.G.; Filipe, E.J.M.; McCabe, C. Perfluoropolyethers: Development of an All-Atom Force Field for Molecular Simulations and Validation with New Experimental Vapor Pressures and Liquid Densities. J. Phys. Chem. B 2017, 121, 6588–6600. [Google Scholar] [CrossRef]
- Wong, A.; Lei, Y.D.; Alaee, M.; Wania, F. Vapor Pressures of the Polybrominated Diphenyl Ethers. J. Chem. Eng. Data 2001, 46, 239–242. [Google Scholar] [CrossRef]
- Brandt, G.A.R.; Emeléus, H.J.; Haszeldine, R.N. 410. Organometallic and organometalloidal fluorine compounds. Part III. Trifluoromethyl derivatives of sulphur. J. Chem. Soc. 1952, 2198–2205. [Google Scholar] [CrossRef]
- Shreeve, J.M.; Cady, G.H. Some Reactions of Peroxydisulfuryl Difluoride. J. Am. Chem. Soc. 1961, 83, 4521–4525. [Google Scholar] [CrossRef]
- Merrill, C.E.; Cady, G.H. Some Reactions of Bis(pentasulfur)peroxide. Defence Tech. Inf. Ctr. 1962, AD0277435. [Google Scholar]
- Delfing, J.J.; Shreeve, J.M. Enneafluoro-2-(fluorosulfato)butane and Trifluoroacetyl Fluorosulfate. Inorg. Chem. 1965, 5, 308–309. [Google Scholar] [CrossRef]
- Sauer, D.T.; Shreeve, J.M. Bis(perfluoroalkyl)sulfur difluorides and bis(perfluoroalkyl) sulfoxides. J. Fluor. Chem. 1971, 1, 1–11. [Google Scholar] [CrossRef]
- Abe, T.; Shreeve, J.M. Perfluorotetramethylenesulfur difluoride and its derivatives. Perfluoro-1,3-dithietane octafluoride and perfluoro-1,4-dithiane octafluoride. J. Fluor. Chem. 1973, 3, 17–26. [Google Scholar] [CrossRef]
- Kitazume, T.; Shreeve, J.M. Stable fluorinated sulfuranes and sulfurane oxides. Synthesis and reactions. Inorg. Chem. 1978, 17, 2173–2176. [Google Scholar] [CrossRef]
- Emeléus, H.J.; Wilkins, C.J. 122. Some new ethyl and phenyl silicon fluorides. J. Chem. Soc. 1944, 454–456. [Google Scholar] [CrossRef]
- Rugina, T.; Sacarescu, L. Isobaric vapor liquid equilibria for the binary systems dichloromethylsilane with chlorotrimethylsilane, dichlorodimethylsilane, trichloromethylsilane, or silicon tetrachloride. J. Chem. Eng. Data 1992, 37, 143–145. [Google Scholar] [CrossRef]
- Kopitzky, R.; Willner, H.; Hermann, A.; Oberhammer, H. Bis(trifluoroacetyl) Peroxide, CF3C(O)OOC(O)CF3. Inorg. Chem. 2001, 40, 2693–2698. [Google Scholar] [CrossRef]
- Schindler, B.J.; Buchanan, J.H.; Mahle, J.J.; Peterson, G.W.; Glover, T.G. Ambient Temperature Vapor Pressure and Adsorption Capacity for (Perfluorooctyl) Ethylene, 3-(Perfluorobutyl)propanol, Perfluorohexanoic Acid, Ethyl Perfluorooctanoate, and Perfluoro-3,6-dioxaheptanoic Acid. J. Chem. Eng. Data 2013, 58, 1806–1812. [Google Scholar] [CrossRef]
- Widegren, J.A.; Bruno, T.J. Vapor Pressure Measurements on Low-Volatility Terpenoid Compounds by the Concatenated Gas Saturation Method. Env. Sci. Tech. 2009, 44, 388–393. [Google Scholar] [CrossRef]
- Vilas-Boas, S.M.; Pokorny, V.; Stejfa, V.; Ferreira, O.; Pinho, S.P.; Ruzicka, K.; Fulem, M. Vapor pressure and thermophysical properties of eugenol and (+)-carvone. Fluid Phase Equil. 2019, 499, 112248. [Google Scholar] [CrossRef]
- Fonseca, L.A.A.P.; Sartoratto, A.; Cremasco, M.A. Experimental determination of thermodynamic properties of terpene and aromatic ketones by gas chromatography. J. Mol. Liquids 2020, 114531. [Google Scholar] [CrossRef]
- Goldfarb, J.L.; Suuberg, E.M. Vapor pressures and thermodynamics of oxygen-containing polycyclic aromatic hydrocarbons measured using Knudsen effusion. Environ. Tox. Chem. 2008, 27, 1244–1249. [Google Scholar] [CrossRef]
- Touhara, H.; Okazaki, S.; Okino, F.; Tanaka, H.; Nakanishi, K. Thermodynamic properties of aqueous mixtures of hydrophilic compounds 2. Aminoethanol and its methyl derivatives. J. Chem. Thermodyn. 1982, 14, 145–156. [Google Scholar] [CrossRef]
- Williams, B.R.; Hulet, M.S.; Brozena, A.; Miles Jr., R.W.; Tevault, D.E. Vapor Pressure of 2-Dialkyl Aminoethanethiols. J. Chem. Eng. Data 2013, 58, 1679–1684. [Google Scholar] [CrossRef]
- Belabbaci, A.; Ahmed, N.C.-B.; Mokbel, I.; Negadi, L. Investigation of the isothermal (vapour + liquid) equilibria of aqueous 2-amino-2-methyl-1-propanol (AMP), N-benzylethanolamine, or 3-dimethylamino-1-propanol solutions at several temperatures. J. Chem. Thermodyn. 2010, 42, 1158–1162. [Google Scholar] [CrossRef]
- Steele, W.V.; Chirico, R.D.; Knipmeyer, S.E.; Nguyen, A.; Smith, N.K.; Tasker, I.R. Thermodynamic Properties and Ideal-Gas Enthalpies of Formation for Cyclohexene, Phthalan (2,5-Dihydrobenzo-3,4-furan), Isoxazole, Octylamine, Dioctylamine, Trioctylamine, Phenyl Isocyanate, and 1,4,5,6-Tetrahydropyrimidine. J. Chem. Eng. Data 1996, 41, 1269–1284. [Google Scholar] [CrossRef]
- Espinosa Diaz, M.A.; Guetachew, T.; Landy, P.; Jose, J.; Voilley, A. Experimental and estimated saturated vapour pressures of aroma compounds. Fluid Phase Equil. 1999, 157, 257–270. [Google Scholar] [CrossRef]
- Petitjean, M.; Reyes-Perez, E.; Perez, D.; Mirabel, P.; Le Calvé, S. Vapor Pressure Measurements of Hydroxyacetaldehyde and Hydroxyacetone in the Temperature Range (273 to 356) K. J. Chem. Eng. Data 2010, 55, 852–855. [Google Scholar] [CrossRef]
- Gobble, C.; Vikman, J.; Chickos, J.S. Evaluation of the Vaporization Enthalpies and Liquid Vapor Pressures of (R)-Deprenyl, (S)-Benzphetamine, Alverine, and a Series of Aliphatic Tertiary Amines by Correlation Gas Chromatography at T/ K = 298.15. J. Chem. Eng. Data 2014, 59, 2551–2562. [Google Scholar] [CrossRef]
- Emel’yanenko, V.N.; Nagrimanov, R.N.; Solomonov, B.N.; Verevkin, S.P. Adamantanes: Benchmarking of thermochemical properties. J. Chem. Thermodyn. 2016, 101, 130–138. [Google Scholar] [CrossRef]
- Gobble, C.; Walker, B.; Chickos, J.S. The Vaporization Enthalpy and Vapor Pressure of Fenpropidin and Phencyclidine (PCP) at T/ K = 298.15 by Correlation Gas Chromatography. J. Chem. Eng. Data 2016, 61, 896–902. [Google Scholar] [CrossRef]
- Wootitunthipong, K.; Chickos, J. Vaporization enthalpy and vapor pressure of (−) Ambroxide and Galaxolide by correlation gas chromatography. J. Chem. Thermodyn. 2019, 129, 121–129. [Google Scholar] [CrossRef]
- Russo, A.Y.; Konnova, M.E.; Andreeva, I.V.; Verevkin, S.P. Vaporization thermodynamics of compounds modeling lignin structural units. Fluid Phase Equil. 2019, 491, 45–55. [Google Scholar] [CrossRef]
- Verevkin, S.P.; Emel’yanenko, V.N.; Siewert, R.; Pimerzin, A.A. Thermochemistry of the lignin broken bits. Fluid Phase Equil. 2020, 522, 112751. [Google Scholar] [CrossRef]
- Verevkin, S.P.; Konnova, M.E.; Emel’yanenko, V.N.; Pimerzin, A.A. Weaving a web of reliable thermochemistry around lignin building blocks: Vanillin and its isomers. J. Chem. Thermodyn. 2020, 106362. [Google Scholar] [CrossRef]
- Verevkin, S.P.; Emel’yanenko, V.N. Vapor pressures of methoxy substituted benzaldehydes. Fluid Phase Equil. 2020, 112912. [Google Scholar] [CrossRef]
- Ledo, J.M.; Flores, H.; Freitas, V.L.S.; Solano-Altamirano, J.M.; Hernandez-Perez, J.M.; Camarillo, E.A.; Ramos, F.; Ribeiro da Silva, M.D.M.C. Benzocaine: A comprehensive thermochemical study. J. Chem. Thermodyn. 2020, 147, 106119. [Google Scholar] [CrossRef]
- Umnahanant, P. Vaporization enthalpy and liquid vapor pressure of bicifadine using correlation gas chromatography. J. Chem. Thermodyn. 2020, 150, 106204. [Google Scholar] [CrossRef]
- Naef, R. Calculation of the Isobaric Heat Capacities of the Liquid and Solid Phase of Organic Compounds at and around 298.15 K Based on Their “True” Molecular Volume. Molecules 2019, 24, 1626. [Google Scholar] [CrossRef] [PubMed]
- Huelsekopf, M.; Ludwig, R. Temperature dependence of hydrogen bonding in alcohols. J. Mol. Liq. 2000, 85, 105–125. [Google Scholar] [CrossRef]
- Compernolle, S.; Ceulemans, K.; Müller, J.-F. EVAPORATION: A new vapour pressure estimation method for organic molecules including non-additivity and intramolecular interactions. Atmos. Chem. Phys. 2011, 11, 9431–9450. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Atom Type | Neighbours | Meaning |
|---|---|---|
| O(prim) | HC | Primary alcohol |
| O(sec) | HC | Secondary alcohol |
| O(tert) | HC | Tertiary alcohol |
| (COH)n | n > 1 | Molecule contains more than 1 saturated OH group |
| (COOH)n | n > 1 | Molecule contains more than 1 carboxylic acid group |
| Endocyclic bonds | No of single bonds | Number of single bonds in cyclic ring |
| Bridgehead atoms | No of bonds | Number of bridgehead C or N (e.g., camphor, DABCO) |
| Atom Type/Neighours | C sp3/H3C | C sp3/HC3 | C sp3/HC2O | C sp3/H2C2 | O(sec)/HC | Endocyclic Single bds | Const | Checksum |
|---|---|---|---|---|---|---|---|---|
| Contribution | 0.60 | −1.28 | −2.65 | −0.47 | 0.72 | 0.31 | 4.71 | |
| n | 1 | 1 | 1 | 4 | 1 | 6 | 1 | |
| n x Contrib. | 0.6 | −1.28 | −2.65 | −1.88 | 0.72 | 1.86 | 4.71 | 2.08 |
| Entry | Atom Type | Neighbours | Contribution | Occurrences | Molecules |
|---|---|---|---|---|---|
| 1 | Const | 4.71 | 2036 | 2036 | |
| 2 | B | HN2 | −1.17 | 6 | 2 |
| 3 | B | BN2 | −1.6 | 2 | 1 |
| 4 | B | BO2 | −1.71 | 4 | 2 |
| 5 | B | C2N | −0.35 | 1 | 1 |
| 6 | B | C2O | −0.44 | 1 | 1 |
| 7 | B | C2S | −0.44 | 1 | 1 |
| 8 | B | CO2 | −1.56 | 1 | 1 |
| 9 | B | O3 | −1.57 | 6 | 6 |
| 10 | B | S3 | −3.17 | 1 | 1 |
| 11 | C sp3 | H3B | 0 | 7 | 4 |
| 12 | C sp3 | H3C | 0.6 | 2211 | 1077 |
| 13 | C sp3 | H3N | −1.07 | 113 | 62 |
| 14 | C sp3 | H3N(+) | −1.64 | 1 | 1 |
| 15 | C sp3 | H3O | −0.95 | 152 | 116 |
| 16 | C sp3 | H3S | −0.52 | 23 | 17 |
| 17 | C sp3 | H3P | −1.22 | 8 | 7 |
| 18 | C sp3 | H3Si | −0.42 | 87 | 16 |
| 19 | C sp3 | H2C2 | −0.47 | 4196 | 831 |
| 20 | C sp3 | H2CN | −2.07 | 240 | 138 |
| 21 | C sp3 | H2CN(+) | −2.01 | 5 | 5 |
| 22 | C sp3 | H2CO | −1.82 | 460 | 314 |
| 23 | C sp3 | H2CP | −2.3 | 5 | 3 |
| 24 | C sp3 | H2CS | −1.6 | 79 | 54 |
| 25 | C sp3 | H2CF | 0.39 | 15 | 15 |
| 26 | C sp3 | H2CCl | −0.48 | 59 | 48 |
| 27 | C sp3 | H2CBr | −0.76 | 22 | 20 |
| 28 | C sp3 | H2CJ | −1.23 | 11 | 11 |
| 29 | C sp3 | H2CSi | −1.58 | 11 | 6 |
| 30 | C sp3 | H2N2 | −11.73 | 1 | 1 |
| 31 | C sp3 | H2NO | −3.82 | 2 | 2 |
| 32 | C sp3 | H2NS | −1.24 | 3 | 3 |
| 33 | C sp3 | H2O2 | −3.81 | 6 | 6 |
| 34 | C sp3 | H2OF | −1.22 | 3 | 3 |
| 35 | C sp3 | H2OCl | −2.1 | 2 | 2 |
| 36 | C sp3 | H2S2 | −2.59 | 3 | 3 |
| 37 | C sp3 | HC3 | −1.28 | 342 | 231 |
| 38 | C sp3 | HC2N | −2.87 | 35 | 28 |
| 39 | C sp3 | HC2N(+) | −2.87 | 3 | 3 |
| 40 | C sp3 | HC2O | −2.65 | 115 | 95 |
| 41 | C sp3 | HC2S | −2.36 | 11 | 8 |
| 42 | C sp3 | HC2F | −0.6 | 10 | 9 |
| 43 | C sp3 | HC2Cl | −1.22 | 31 | 15 |
| 44 | C sp3 | HC2Br | −1.59 | 16 | 12 |
| 45 | C sp3 | HC2J | −1.96 | 1 | 1 |
| 46 | C sp3 | HCN2 | −2.07 | 2 | 1 |
| 47 | C sp3 | HCNO | −5.99 | 1 | 1 |
| 48 | C sp3 | HCNS | −2.48 | 1 | 1 |
| 49 | C sp3 | HCO2 | −3.65 | 7 | 7 |
| 50 | C sp3 | HCOBr | −4.78 | 1 | 1 |
| 51 | C sp3 | HCF2 | 0.37 | 31 | 27 |
| 52 | C sp3 | HCFCl | −0.01 | 7 | 7 |
| 53 | C sp3 | HCCl2 | −0.94 | 12 | 11 |
| 54 | C sp3 | HCClBr | −0.76 | 1 | 1 |
| 55 | C sp3 | HCBr2 | −1.93 | 3 | 2 |
| 56 | C sp3 | HOF2 | −1.09 | 6 | 6 |
| 57 | C sp3 | C4 | −2.19 | 98 | 87 |
| 58 | C sp3 | C3N | −3.6 | 11 | 11 |
| 59 | C sp3 | C3N(+) | −3.57 | 2 | 2 |
| 60 | C sp3 | C3O | −3.46 | 36 | 35 |
| 61 | C sp3 | C3S | −3.21 | 6 | 6 |
| 62 | C sp3 | C3Si | −3.37 | 3 | 2 |
| 63 | C sp3 | C3Cl | −2.84 | 6 | 3 |
| 64 | C sp3 | C3Br | −2.2 | 2 | 2 |
| 65 | C sp3 | C3F | −1.39 | 13 | 10 |
| 66 | C sp3 | C2O2 | −5.46 | 4 | 2 |
| 67 | C sp3 | C2OF | −2.8 | 5 | 5 |
| 68 | C sp3 | C2F2 | −0.37 | 184 | 71 |
| 69 | C sp3 | C2FCl | −0.8 | 1 | 1 |
| 70 | C sp3 | C2Cl2 | 0 | 3 | 3 |
| 71 | C sp3 | CNF2 | −2.03 | 12 | 5 |
| 72 | C sp3 | CNF2(+) | −0.37 | 1 | 1 |
| 73 | C sp3 | CNCl2 | −0.4 | 1 | 1 |
| 74 | C sp3 | COF2 | −1.69 | 49 | 39 |
| 75 | C sp3 | CSF2 | −1.15 | 24 | 12 |
| 76 | C sp3 | CF3 | 0.67 | 152 | 107 |
| 77 | C sp3 | CF2Cl | 0.3 | 8 | 7 |
| 78 | C sp3 | CF2Br | −0.07 | 5 | 4 |
| 79 | C sp3 | CFCl2 | −0.37 | 5 | 4 |
| 80 | C sp3 | CFClBr | −0.73 | 1 | 1 |
| 81 | C sp3 | CCl3 | −0.98 | 15 | 14 |
| 82 | C sp3 | CCl2Br | 0 | 1 | 1 |
| 83 | C sp3 | NF3 | −1.09 | 5 | 3 |
| 84 | C sp3 | OF3 | −0.36 | 13 | 10 |
| 85 | C sp3 | O2F2 | −2.67 | 1 | 1 |
| 86 | C sp3 | S2F2 | −1.83 | 4 | 2 |
| 87 | C sp3 | SF3 | −0.01 | 10 | 7 |
| 88 | C sp3 | SCl3 | −7.92 | 1 | 1 |
| 89 | C sp3 | PF3 | −0.08 | 20 | 8 |
| 90 | C sp2 | H2=C | 0.67 | 127 | 113 |
| 91 | C sp2 | HC=C | −0.38 | 272 | 175 |
| 92 | C sp2 | HC=N | −1.49 | 7 | 7 |
| 93 | C sp2 | HC=O | −0.47 | 27 | 27 |
| 94 | C sp2 | H=CN | −1.84 | 19 | 12 |
| 95 | C sp2 | H=CO | −0.79 | 5 | 5 |
| 96 | C sp2 | H=CS | −0.79 | 8 | 6 |
| 97 | C sp2 | H=CP | −1.03 | 3 | 1 |
| 98 | C sp2 | H=CF | 0.68 | 3 | 3 |
| 99 | C sp2 | H=CCl | −0.15 | 13 | 11 |
| 100 | C sp2 | H=CBr | −0.56 | 5 | 3 |
| 101 | C sp2 | H=CJ | −1.2 | 2 | 1 |
| 102 | C sp2 | HN=N | −1.89 | 11 | 9 |
| 103 | C sp2 | HN=O | −2.47 | 9 | 8 |
| 104 | C sp2 | HO=O | −1.25 | 8 | 8 |
| 105 | C sp2 | C2=C | −1.25 | 79 | 67 |
| 106 | C sp2 | C2=N | −3.09 | 2 | 2 |
| 107 | C sp2 | C=CN | −2.26 | 2 | 2 |
| 108 | C sp2 | C2=O | −1.27 | 56 | 53 |
| 109 | C sp2 | C=CO | −1.5 | 6 | 6 |
| 110 | C sp2 | C=CP | −3.09 | 1 | 1 |
| 111 | C sp2 | C=CS | −1.78 | 6 | 5 |
| 112 | C sp2 | C=CF | −0.25 | 3 | 3 |
| 113 | C sp2 | C=CCl | −1.24 | 18 | 13 |
| 114 | C sp2 | CN=N | −4.13 | 2 | 2 |
| 115 | C sp2 | CN=O | −3.17 | 35 | 32 |
| 116 | C sp2 | C=NS | −1.47 | 2 | 1 |
| 117 | C sp2 | CO=O | −2.33 | 222 | 184 |
| 118 | C sp2 | C=OCl | −0.54 | 4 | 4 |
| 119 | C sp2 | C=OBr | −1.1 | 1 | 1 |
| 120 | C sp2 | C=OJ | −1.67 | 1 | 1 |
| 121 | C sp2 | =CF2 | 0.95 | 7 | 6 |
| 122 | C sp2 | =CFCl | 0.14 | 1 | 1 |
| 123 | C sp2 | =CFBr | −0.25 | 1 | 1 |
| 124 | C sp2 | =CCl2 | −0.53 | 10 | 8 |
| 125 | C sp2 | =CBr2 | 0.65 | 1 | 1 |
| 126 | C sp2 | N2=N | −4.7 | 1 | 1 |
| 127 | C sp2 | N2=O | −5.19 | 5 | 5 |
| 128 | C sp2 | N=NS | −1.58 | 1 | 1 |
| 129 | C sp2 | N2=S | 0.14 | 2 | 1 |
| 130 | C sp2 | NO=O | −4.55 | 15 | 13 |
| 131 | C sp2 | N=OS | −0.64 | 7 | 7 |
| 132 | C sp2 | =NOS | −0.26 | 1 | 1 |
| 133 | C sp2 | NS=S | 1.24 | 1 | 1 |
| 134 | C sp2 | O2=O | −3.59 | 4 | 4 |
| 135 | C aromatic | H:C2 | −0.2 | 3662 | 751 |
| 136 | C aromatic | H:C:N | −0.41 | 34 | 21 |
| 137 | C aromatic | H:N2 | 0.48 | 2 | 2 |
| 138 | C aromatic | :C3 | −1.06 | 260 | 85 |
| 139 | C aromatic | C:C2 | −1.06 | 929 | 508 |
| 140 | C aromatic | C:C:N | −1.21 | 15 | 13 |
| 141 | C aromatic | :C2N | −2.3 | 40 | 38 |
| 142 | C aromatic | :C2N(+) | −2.59 | 33 | 29 |
| 143 | C aromatic | :C2:N | −1.42 | 4 | 3 |
| 144 | C aromatic | :C2O | −2 | 381 | 195 |
| 145 | C aromatic | :C2P | −4.07 | 1 | 1 |
| 146 | C aromatic | :C2S | −1.69 | 8 | 6 |
| 147 | C aromatic | :C2F | −0.1 | 63 | 26 |
| 148 | C aromatic | :C2Cl | −0.84 | 1630 | 386 |
| 149 | C aromatic | :C2Br | −1.13 | 166 | 58 |
| 150 | C aromatic | :C2J | −1.57 | 10 | 9 |
| 151 | C aromatic | :C2Si | 0.89 | 1 | 1 |
| 152 | C aromatic | C:N2 | −1.39 | 2 | 2 |
| 153 | C aromatic | :C:NO | −1.85 | 6 | 6 |
| 154 | C aromatic | :C:NCl | −1.33 | 5 | 5 |
| 155 | C aromatic | N:N2 | −2.72 | 17 | 10 |
| 156 | C aromatic | :N2O | −0.96 | 2 | 2 |
| 157 | C aromatic | :N2S | 1.89 | 3 | 3 |
| 158 | C aromatic | :N2Cl | −1.38 | 3 | 3 |
| 159 | C sp | H#C | 0.81 | 14 | 13 |
| 160 | C sp | C#C | −0.49 | 22 | 17 |
| 161 | C sp | =C2 | −0.51 | 3 | 3 |
| 162 | C sp | C#N | −0.61 | 34 | 27 |
| 163 | C sp | =N=O | 0.75 | 3 | 3 |
| 164 | C sp | =N=S | 1.19 | 1 | 1 |
| 165 | N sp3 | HB2 | 0.45 | 3 | 2 |
| 166 | N sp3 | H2C | 1.45 | 62 | 47 |
| 167 | N sp3 | H2C(pi) | 0.15 | 18 | 18 |
| 168 | N sp3 | H2N | −0.52 | 3 | 3 |
| 169 | N sp3 | HC2 | 2.36 | 26 | 26 |
| 170 | N sp3 | HC2(pi) | 0.56 | 35 | 26 |
| 171 | N sp3 | HC2(2pi) | 0.44 | 14 | 10 |
| 172 | N sp3 | HCN | 0.7 | 3 | 2 |
| 173 | N sp3 | HCN(pi) | −0.36 | 1 | 1 |
| 174 | N sp3 | HCN(2pi) | 0.38 | 1 | 1 |
| 175 | N sp3 | HCP(pi) | −4.25 | 1 | 1 |
| 176 | N sp3 | HCS(pi) | 5.56 | 1 | 1 |
| 177 | N sp3 | B2C | 1.1 | 3 | 2 |
| 178 | N sp3 | BC2 | 2.13 | 5 | 2 |
| 179 | N sp3 | C3 | 3.52 | 49 | 45 |
| 180 | N sp3 | C3(pi) | 2.95 | 27 | 26 |
| 181 | N sp3 | C3(2pi) | 3.52 | 11 | 11 |
| 182 | N sp3 | C3(3pi) | 3.4 | 3 | 3 |
| 183 | N sp3 | C2N(pi) | 0.11 | 4 | 4 |
| 184 | N sp3 | C2N(2pi) | 3.37 | 8 | 8 |
| 185 | N sp3 | C2N(3pi) | 2.89 | 1 | 1 |
| 186 | N sp3 | C2O | 3.47 | 1 | 1 |
| 187 | N sp3 | C2S | 2.57 | 3 | 3 |
| 188 | N sp3 | C2S(pi) | 3.96 | 3 | 2 |
| 189 | N sp3 | C2S(2pi) | 7.1 | 1 | 1 |
| 190 | N sp3 | C2P | 2.07 | 7 | 4 |
| 191 | N sp3 | C2F(pi) | 4.38 | 1 | 1 |
| 192 | N sp3 | CF2 | 0.61 | 1 | 1 |
| 193 | N sp3 | CSi2 | 1.18 | 2 | 2 |
| 194 | N sp3 | SF2 | 0.07 | 1 | 1 |
| 195 | N sp2 | C=C | 0.39 | 16 | 15 |
| 196 | N sp2 | C=N | −3.19 | 1 | 1 |
| 197 | N sp2 | C=N(+) | 0.96 | 7 | 7 |
| 198 | N sp2 | =CN | −0.04 | 10 | 9 |
| 199 | N sp2 | =CO | 0.68 | 4 | 3 |
| 200 | N sp2 | =CS | −0.39 | 1 | 1 |
| 201 | N sp2 | N=N | 0 | 1 | 1 |
| 202 | N sp2 | N=O | 0 | 4 | 4 |
| 203 | N sp2 | =NP(+) | −0.39 | 1 | 1 |
| 204 | N sp2 | O=O | 1.58 | 6 | 6 |
| 205 | N aromatic | :C2 | −0.06 | 61 | 39 |
| 206 | N(+) sp2 | CO=O(−) | 0.34 | 45 | 41 |
| 207 | N(+) sp2 | O2=O(−) | 0.54 | 50 | 26 |
| 208 | N(+) sp | =N2(−) | 0 | 8 | 8 |
| 209 | O(prim) | HC | 0.44 | 95 | 78 |
| 210 | O(sec) | HC | 0.72 | 48 | 47 |
| 211 | O(tert) | HC | 0.74 | 11 | 11 |
| 212 | O | HC(pi) | 0.04 | 102 | 90 |
| 213 | O | HN(pi) | −1.29 | 1 | 1 |
| 214 | O | HO | −1.16 | 4 | 3 |
| 215 | O | BC | 1.39 | 26 | 8 |
| 216 | O | BP | 0.16 | 3 | 2 |
| 217 | O | C2 | 2.38 | 150 | 132 |
| 218 | O | C2(pi) | 2.3 | 228 | 191 |
| 219 | O | C2(2pi) | 1.49 | 151 | 130 |
| 220 | O | CN | 0 | 1 | 1 |
| 221 | O | CN(pi) | 0 | 6 | 6 |
| 222 | O | CN(2pi) | 0.26 | 3 | 2 |
| 223 | O | CN(+)(pi) | 0 | 50 | 26 |
| 224 | O | CO | 1.03 | 8 | 3 |
| 225 | O | CO(pi) | 1.59 | 3 | 2 |
| 226 | O | CS | 1.25 | 6 | 4 |
| 227 | O | CS(pi) | 1.44 | 2 | 2 |
| 228 | O | CP | 0.06 | 95 | 44 |
| 229 | O | CP(pi) | −0.29 | 14 | 12 |
| 230 | O | CSi | 0.65 | 7 | 2 |
| 231 | O | OS | −0.67 | 3 | 2 |
| 232 | O | S2 | −1.14 | 5 | 3 |
| 233 | O | Si2 | −0.3 | 22 | 7 |
| 234 | P3 | C3 | 0 | 1 | 1 |
| 235 | P3 | HC2 | 2.57 | 1 | 1 |
| 236 | P3 | C2N | 1.59 | 2 | 2 |
| 237 | P3 | C2O | 0 | 3 | 2 |
| 238 | P3 | C2S | −0.09 | 5 | 4 |
| 239 | P3 | CN2 | −0.35 | 1 | 1 |
| 240 | P3 | CS2 | −0.94 | 1 | 1 |
| 241 | P4 | HO2=O | −0.55 | 1 | 1 |
| 242 | P4 | C3=S | 0.19 | 1 | 1 |
| 243 | P4 | CO2=O | 0.62 | 4 | 4 |
| 244 | P4 | CO2=S | 3.03 | 1 | 1 |
| 245 | P4 | CO=OS | 0.38 | 2 | 2 |
| 246 | P4 | COS=S | −0.5 | 1 | 1 |
| 247 | P4 | N3=O | −0.83 | 1 | 1 |
| 248 | P4 | NO=OS | −0.06 | 1 | 1 |
| 249 | P4 | N=OF2 | 0 | 1 | 1 |
| 250 | P4 | O3=O | 0.23 | 9 | 9 |
| 251 | P4 | O3=S | 0.22 | 13 | 13 |
| 252 | P4 | O2=OS | −0.36 | 1 | 1 |
| 253 | P4 | O=OS2 | −1.76 | 1 | 1 |
| 254 | P4 | O2S=S | −0.58 | 12 | 11 |
| 255 | S2 | HC | 0.83 | 29 | 23 |
| 256 | S2 | HC(pi) | 0.28 | 1 | 1 |
| 257 | S2 | HS | −0.26 | 2 | 1 |
| 258 | S2 | HP | 0.06 | 1 | 1 |
| 259 | S2 | BC | 0.52 | 4 | 2 |
| 260 | S2 | C2 | 1.07 | 30 | 28 |
| 261 | S2 | C2(pi) | −1.97 | 14 | 13 |
| 262 | S2 | C2(2pi) | 1.52 | 9 | 9 |
| 263 | S2 | CN | 0 | 1 | 1 |
| 264 | S2 | CN(2pi) | −2.3 | 1 | 1 |
| 265 | S2 | CS | 0.05 | 8 | 4 |
| 266 | S2 | CP | −0.07 | 22 | 19 |
| 267 | S2 | CP(pi) | 0 | 1 | 1 |
| 268 | S2 | N2 | −1.45 | 2 | 2 |
| 269 | S2 | NCl | −0.43 | 1 | 1 |
| 270 | S2 | P2 | −0.7 | 1 | 1 |
| 271 | S2 | Si2 | 0.33 | 3 | 3 |
| 272 | S4 | C2=O | −0.96 | 4 | 4 |
| 273 | S4 | C2=O2 | 1.6 | 2 | 2 |
| 274 | S4 | C2O2 | −2.15 | 1 | 1 |
| 275 | S4 | C2F2 | 0.41 | 5 | 5 |
| 276 | S4 | CO=O2 | 2.16 | 1 | 1 |
| 277 | S4 | CN=O2 | −2.13 | 1 | 1 |
| 278 | S4 | NO=O2 | −2.51 | 1 | 1 |
| 279 | S4 | N=O2Cl | 0 | 1 | 1 |
| 280 | S4 | O2=O | −0.56 | 1 | 1 |
| 281 | S4 | O2=O2 | −0.94 | 1 | 1 |
| 282 | S4 | O=O2F | 0.12 | 4 | 4 |
| 283 | S6 | C2F4 | 0.72 | 5 | 3 |
| 284 | S6 | O2F4 | −0.78 | 1 | 1 |
| 285 | S6 | OF5 | 1.08 | 7 | 5 |
| 286 | Si | H3C | 1.72 | 4 | 4 |
| 287 | Si | H3N | 0 | 4 | 2 |
| 288 | Si | H3S | −0.3 | 2 | 1 |
| 289 | Si | H3Si | −0.53 | 2 | 1 |
| 290 | Si | H2C2 | 1.78 | 2 | 2 |
| 291 | Si | H2Si2 | 0 | 2 | 1 |
| 292 | Si | HC2O | 0.78 | 2 | 1 |
| 293 | Si | HC2S | 0.11 | 2 | 1 |
| 294 | Si | HC2J | 0.23 | 1 | 1 |
| 295 | Si | HCCl2 | 0.47 | 1 | 1 |
| 296 | Si | HO3 | 0.21 | 1 | 1 |
| 297 | Si | C4 | 1.97 | 2 | 2 |
| 298 | Si | C3O | 1.08 | 6 | 3 |
| 299 | Si | C3S | 0.19 | 2 | 1 |
| 300 | Si | C3Cl | 1.05 | 1 | 1 |
| 301 | Si | C3Si | −0.79 | 2 | 1 |
| 302 | Si | C2O2 | −0.18 | 18 | 5 |
| 303 | Si | C2F2 | 1.69 | 1 | 1 |
| 304 | Si | C2Cl2 | 0.41 | 1 | 1 |
| 305 | Si | CF3 | 0 | 1 | 1 |
| 306 | Si | CCl3 | 0.06 | 1 | 1 |
| 307 | Si | O4 | −0.16 | 1 | 1 |
| 308 | (COH)n | n>1 | −0.74 | 23 | 22 |
| 309 | (COOH)n | n>1 | −1.73 | 12 | 12 |
| 310 | Endocyclic bonds | No of single bds | 0.31 | 1072 | 193 |
| 311 | Bridgehead atoms | No of atoms | 0.23 | 80 | 27 |
| 312 | Angle60 | 0.19 | 42 | 14 | |
| 313 | Angle90 | 0.17 | 72 | 21 | |
| 314 | Angle102 | 0.11 | 323 | 110 | |
| A | Based on | Valid groups | 171 | 2036 | |
| B | Goodness of fit | R2 | 0.9946 | 1908 | |
| C | Deviation | Average | 0.18 | 1908 | |
| D | Deviation | Standard | 0.24 | 1908 | |
| E | K-fold cv | K | 10 | 1842 | |
| F | Goodness of fit | Q2 | 0.9938 | 1842 | |
| G | Deviation | Average (cv) | 0.2 | 1842 | |
| H | Deviation | Standard (cv) | 0.26 | 1842 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Naef, R.; Acree, W.E., Jr. Calculation of the Vapour Pressure of Organic Molecules by Means of a Group-Additivity Method and Their Resultant Gibbs Free Energy and Entropy of Vaporization at 298.15 K. Molecules 2021, 26, 1045. https://doi.org/10.3390/molecules26041045
Naef R, Acree WE Jr. Calculation of the Vapour Pressure of Organic Molecules by Means of a Group-Additivity Method and Their Resultant Gibbs Free Energy and Entropy of Vaporization at 298.15 K. Molecules. 2021; 26(4):1045. https://doi.org/10.3390/molecules26041045
Chicago/Turabian StyleNaef, Rudolf, and William E. Acree, Jr. 2021. "Calculation of the Vapour Pressure of Organic Molecules by Means of a Group-Additivity Method and Their Resultant Gibbs Free Energy and Entropy of Vaporization at 298.15 K" Molecules 26, no. 4: 1045. https://doi.org/10.3390/molecules26041045
APA StyleNaef, R., & Acree, W. E., Jr. (2021). Calculation of the Vapour Pressure of Organic Molecules by Means of a Group-Additivity Method and Their Resultant Gibbs Free Energy and Entropy of Vaporization at 298.15 K. Molecules, 26(4), 1045. https://doi.org/10.3390/molecules26041045