
Synthesis of Rottlerone Analogues and Evaluation of Their α-Glucosidase and DPP-4 Dual Inhibitory and Glucose Consumption-Promoting Activity

 and
and
Abstract
1. Introduction
2. Results
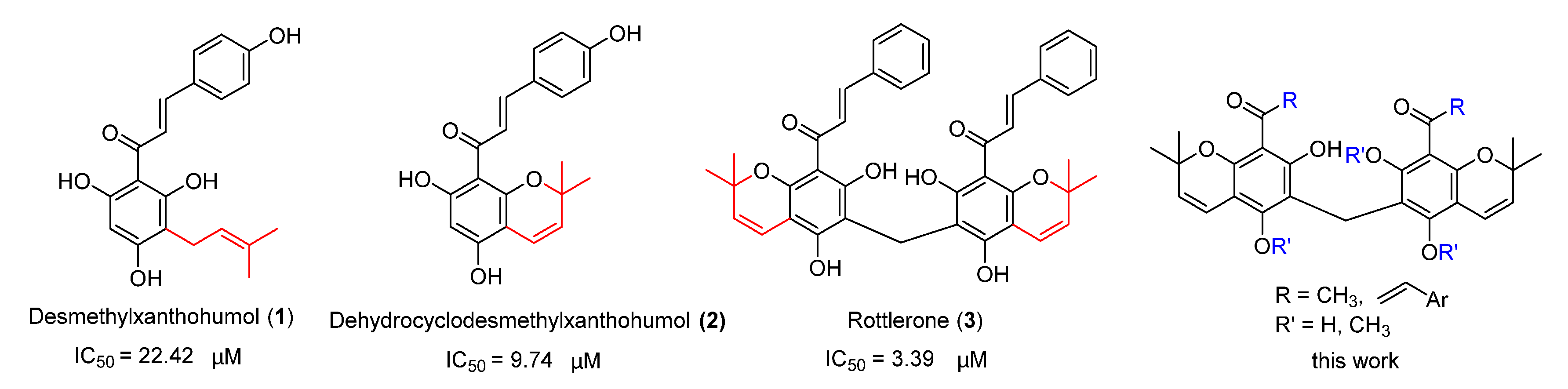
2.1. Synthesis and Characterisation of Rottlerone Analogues
2.2. Rottlerone Analogues Inhibited α-Glucosidase and DPP-4
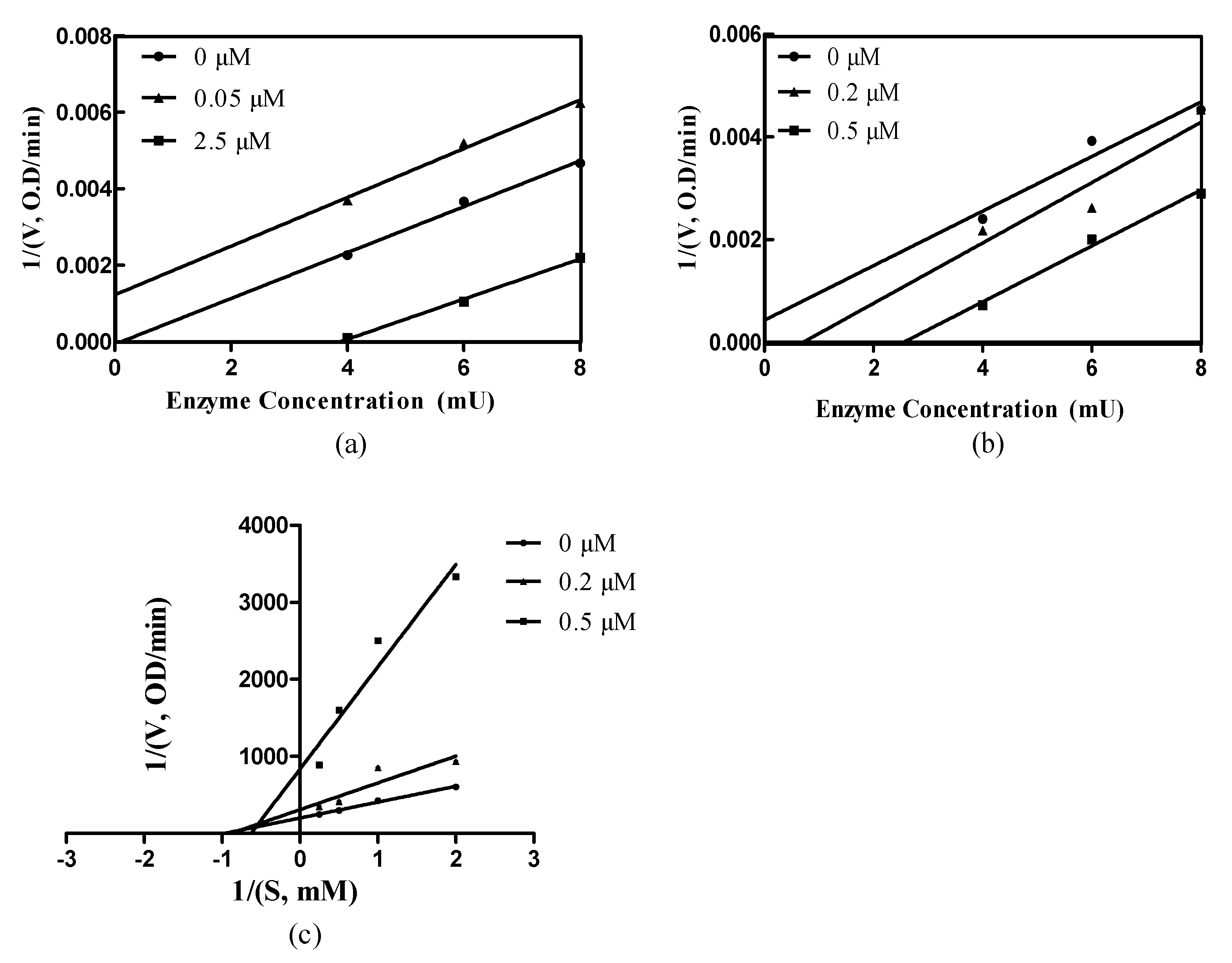
2.3. The Enzyme Kinetics of Compounds 4d and 5d
2.4. Glucose Consumption of Compounds 4d and 5d
3. Materials and Methods
3.1. Chemistry
3.1.1. 1-(2,6-Dihydroxy-4-(methoxymethoxy)phenyl)ethan-1-one (6)
3.1.2. 1-(7-Hydroxy-5-(methoxymethoxy)-2,2-dimethyl-2H-chromen-8-yl) ethan-1-one (7)
3.1.3. 1-(5,7-Dihydroxy-2,2-dimethyl-2H-chromen-8-yl)ethan-1-one (8)
3.1.4. 1,1’-(Methylenebis(5,7-dihydroxy-2,2-dimethyl-2H-chromene-6,8-diyl))bis (ethan-1-one) (9)
3.1.5. General Procedure for the Synthesis of Compounds 3 and 4a–4h
3.1.6. (2Z,2’Z)-1,1’-(Methylenebis(5,7-dihydroxy-2,2-dimethyl-2H-chromene-6,8-diyl))bis (3-phenylprop-2-en-1-one) (3)
3.1.7. (2Z,2’Z)-1,1’-(Methylenebis(5,7-dihydroxy-2,2-dimethyl-2H-chromene-6,8-diyl))bis (3-(4-methoxyphenyl)prop-2-en-1-one) (4a)
3.1.8. (2Z,2’Z)-1,1’-(Methylenebis(5,7-dihydroxy-2,2-dimethyl-2H-chromene-6,8-diyl))bis (3-(4-fluoro phenyl)prop-2-en-1-one) (4b)
3.1.9. (2Z,2’Z)-1,1’-(Methylenebis(5,7-dihydroxy-2,2-dimethyl-2H-chromene-6,8-diyl))bis (3-(furan-3-yl)prop-2-en-1-one) (4c)
3.1.10. (2Z,2’Z)-1,1’-(Methylenebis(5,7-dihydroxy-2,2-dimethyl-2H-chromene-6,8-diyl))bis (3-(thiophen-3-yl)prop-2-en-1-one) (4d)
3.1.11. (2Z,2’Z)-1,1’-(Methylenebis(5,7-dihydroxy-2,2-dimethyl-2H-chromene-6,8-diyl))bis (3-(4-(methoxymethoxy)phenyl)prop-2-en-1-one) (4e)
3.1.12. (2Z,2’Z)-1,1’-(Methylenebis(5,7-dihydroxy-2,2-dimethyl-2H-chromene-6,8-diyl))bis (3-(2,4-bis(methoxymethoxy)phenyl)prop-2-en-1-one) (4f)
3.1.13. (2Z,2’Z)-1,1’-(Methylenebis(5,7-dihydroxy-2,2-dimethyl-2H-chromene-6,8-diyl))bis (3-(2,3-bis(methoxymethoxy)phenyl)prop-2-en-1-one) (4g)
3.1.14. (2Z,2’Z)-1,1’-(Methylenebis(5,7-dihydroxy-2,2-dimethyl-2H-chromene-6,8-diyl))bis (3-(2,3,4-tris(methoxymethoxy)phenyl)prop-2-en-1-one) (4h)
3.1.15. General Procedure for the Synthesis of Compounds 5a–5d
3.1.16. (2Z,2’Z)-1,1’-(Methylenebis(5,7-dihydroxy-2,2-dimethyl-2H-chromene-6,8-diyl))bis (3-(4-hydroxyphenyl)prop-2-en-1-one) (5a)
3.1.17. (2Z,2’Z)-1,1’-(Methylenebis(5,7-dihydroxy-2,2-dimethyl-2H-chromene-6,8-diyl))bis (3-(2,4-dihydroxyphenyl)prop-2-en-1-one) (5b)
3.1.18. (2Z,2’Z)-1,1’-(Methylenebis(5,7-dihydroxy-2,2-dimethyl-2H-chromene-6,8-diyl))bis (3-(2,3-dihydroxyphenyl)prop-2-en-1-one) (5c)
3.1.19. (2Z,2’Z)-1,1’-(Methylenebis(5,7-dihydroxy-2,2-dimethyl-2H-chromene-6,8-diyl))bis (3-(2,3,4-trihydroxyphenyl)prop-2-en-1-one) (5d)
3.1.20. General Procedure for the Synthesis of Compounds 10–11
3.1.21. 1,1’-(Methylenebis(7-hydroxy-5-methoxy-2,2-dimethyl-2H-chromene-6,8-diyl))bis (ethan-1-one) (10)
3.1.22. 1-(6-((8-Acetyl-5,7-dimethoxy-2,2-dimethyl-2H-chromen-6-yl)methyl)-7-hydroxy-5-methoxy-2,2-dimethyl-2H-chromen-8-yl)ethan-1-one (11)
3.1.23. (2Z,2’Z)-1,1’-(Methylenebis(7-hydroxy-5-methoxy-2,2-dimethyl-2H-chromene-6,8-diyl))bis(3-phenylprop-2-en-1-one) (12)
3.1.24. (Z)-1-(6-((5,7-Dimethoxy-2,2-dimethyl-8-((Z)-3-phenylacryloyl)-2H-chromen-6-yl)methyl)-7-hydroxy-5-methoxy-2,2-dimethyl-2H-chromen-8-yl)-3-phenylprop-2-en-1-one (13)
3.2. α-Glucosidase Inhibition Assay
3.3. DPP-4 Inhibition Assay
3.4. Kinetics of Enzyme Inhibition
3.5. Glucose Consumption Assay
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Saeedi, P.; Petersohn, I.; Salpea, P.; Malanda, B.; Karuranga, S.; Unwin, N.; Colagiuri, S.; Guariguata, L.; Motala, A.A.; Ogurtsova, K.; et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas, 9th edition. Diabetes Res. Clin. Pract. 2019, 157, 107843. [Google Scholar] [CrossRef] [PubMed]
- Longo, M.; Bellastella, G.; Maiorino, M.I.; Meier, J.J.; Esposito, K.; Giugliano, D. Diabetes and Aging: From Treatment Goals to Pharmacologic Therapy. Front. Endocrinol. 2019, 10, 45. [Google Scholar] [CrossRef] [PubMed]
- Ghani, U. Re-exploring promising α-glucosidase inhibitors for potential development into oral anti-diabetic drugs: Finding needle in the haystack. Eur. J. Med. Chem. 2015, 103, 133–162. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Nakamura, J.; Koh, N.; Sakakibara, F.; Takeuchi, N.; Hotta, N. An importance of carbohydrate ingestion for the expression of the effect of alpha-glucosidase inhibitor in NIDDM. Diabetes Care 1996, 19, 642–647. [Google Scholar] [CrossRef] [PubMed]
- Min, S.H.; Yoon, J.H.; Hahn, S.; Cho, Y.M. Efficacy and safety of combination therapy with an α-glucosidase inhibitor and a dipeptidyl peptidase-4 inhibitor in patients with type 2 diabetes mellitus: A systematic review with meta-analysis. J. Diabetes Investig. 2018, 9, 893–902. [Google Scholar] [CrossRef] [PubMed]
- Delmulle, L.; Bellahcène, A.; Dhooge, W.; Comhaire, F.; Roelens, F.; Huvaere, K.; Heyerick, A.; Castronovo, V.; De Keukeleire, D. Anti-proliferative properties of prenylated flavonoids from hops (Humulus lupulus L.) in human prostate cancer cell lines. Phytomedicine 2006, 13, 732–734. [Google Scholar] [CrossRef] [PubMed]
- Vogel, S.; Ohmayer, S.; Brunner, G.; Heilmann, J. Natural and non-natural prenylated chalcones: Synthesis, cytotoxicity and anti-oxidative activity. Bioorg. Med. Chem. 2008, 16, 4286–4293. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Wang, D.; Song, X.; Zhang, Y.; Ding, W.; Peng, X.; Zhang, X.; Li, Y.; Ma, Y.; Wang, R.; et al. Natural Prenylchalconaringenins and Prenylnaringenins as Antidiabetic Agents: α-Glucosidase and α-Amylase Inhibition and in Vivo Antihyperglycemic and Antihyperlipidemic Effects. J. Agric. Food Chem. 2017, 65, 1574–1581. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.K.; Pathania, A.S.; Meena, S.; Sharma, R.; Sharma, A.; Singh, B.; Gupta, B.D.; Bhushan, S.; Bharate, S.B.; Vishwakarma, R.A. Semisynthesis of mallotus B from rottlerin: Evaluation of cytotoxicity and apoptosis-inducing activity. J. Nat. Prod. 2013, 76, 1724–1730. [Google Scholar] [CrossRef] [PubMed]
- Teng, Y.; Li, X.; Yang, K.; Li, X.; Zhang, Z.; Wang, L.; Deng, Z.; Song, B.; Yan, Z.; Zhang, Y.; et al. Synthesis and antioxidant evaluation of desmethylxanthohumol analogs and their dimers. Eur. J. Med. Chem. 2017, 125, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Akbaba, Y.H.T.; Gksu, S.; Ertan, A.; Menzek, A. Total Synthesis of the Biologically Active, Naturally Occurring 3,4-Dibromo-5-(2-bromo-3,4-dihydroxy-6-(methoxymethyl)benzyl)benzene-1,2-diol and Regioselective O-Demethylation of Aryl Methyl Ethers. Helv. Chim. Acta 2010, 93, 1127–1135. [Google Scholar] [CrossRef]
- Parmar, H.S.; Jain, P.; Chauhan, D.S.; Bhinchar, B.K.; Munjal, V.; Yusuf, M.; Choube, K.; Tawani, A.; Tiwari, V.; Manivannan, E.; et al. DPP-IV inhibitory potential of naringin: An in silico, in vitro and in vivo study. Diabetes Res. Clin. Pract. 2012, 97, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Yan, Y.; Chen, Q.; Luo, F.; Zhu, X.; Li, X.; Chen, K. Purification of naringin and neohesperidin from Huyou (Citrus changshanensis) fruit and their effects on glucose consumption in human HepG2 cells. Food Chem. 2012, 135, 1471–1478. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | IC50 (μM) a | |
|---|---|---|
| α-Glucosidase | DPP-4 | |
| 1 | 22.42 ± 3.10 | >50 |
| 2 | 9.74 ± 0.73 | >50 |
| 3 | 5.91 ± 0.19 | >50 |
| 4a | 6.41 ± 0.33 | >50 |
| 4b | 8.71 ± 0.54 | >50 |
| 4c | 1.84 ± 0.41 | 21.48 ± 0.09 |
| 4d | 0.22 ± 0.07 | 23.59 ± 0.14 |
| 5a | 0.51 ± 0.19 | >50 |
| 5b | 0.24 ± 0.02 | >50 |
| 5c | 1.07 ± 0.06 | >50 |
| 5d | 0.12 ± 0.02 | 26.19 ± 0.22 |
| 9 | 7.11 ± 0.05 | >50 |
| 12 | 3.39 ± 0.13 | >50 |
| 13 | 0.37 ± 0.04 | >50 |
| Sitagliptin | - | 0.08 ± 0.01 |
| Acarbose | 188.57 ± 5.52 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Wang, H.; Wu, Y.; Zhao, X.; Yan, Z.; Dodd, R.H.; Yu, P.; Lu, K.; Sun, H. Synthesis of Rottlerone Analogues and Evaluation of Their α-Glucosidase and DPP-4 Dual Inhibitory and Glucose Consumption-Promoting Activity. Molecules 2021, 26, 1024. https://doi.org/10.3390/molecules26041024
Zhang Y, Wang H, Wu Y, Zhao X, Yan Z, Dodd RH, Yu P, Lu K, Sun H. Synthesis of Rottlerone Analogues and Evaluation of Their α-Glucosidase and DPP-4 Dual Inhibitory and Glucose Consumption-Promoting Activity. Molecules. 2021; 26(4):1024. https://doi.org/10.3390/molecules26041024
Chicago/Turabian StyleZhang, Yinan, Haibo Wang, Yan Wu, Xue Zhao, Zhihong Yan, Robert H. Dodd, Peng Yu, Kui Lu, and Hua Sun. 2021. "Synthesis of Rottlerone Analogues and Evaluation of Their α-Glucosidase and DPP-4 Dual Inhibitory and Glucose Consumption-Promoting Activity" Molecules 26, no. 4: 1024. https://doi.org/10.3390/molecules26041024
APA StyleZhang, Y., Wang, H., Wu, Y., Zhao, X., Yan, Z., Dodd, R. H., Yu, P., Lu, K., & Sun, H. (2021). Synthesis of Rottlerone Analogues and Evaluation of Their α-Glucosidase and DPP-4 Dual Inhibitory and Glucose Consumption-Promoting Activity. Molecules, 26(4), 1024. https://doi.org/10.3390/molecules26041024