
Synthesis and Exon-Skipping Properties of a 3′-Ursodeoxycholic Acid-Conjugated Oligonucleotide Targeting DMD Pre-mRNA: Pre-Synthetic versus Post-Synthetic Approach

 ,
,  ,
,  , and
, and 
Abstract
:
1. Introduction
2. Results
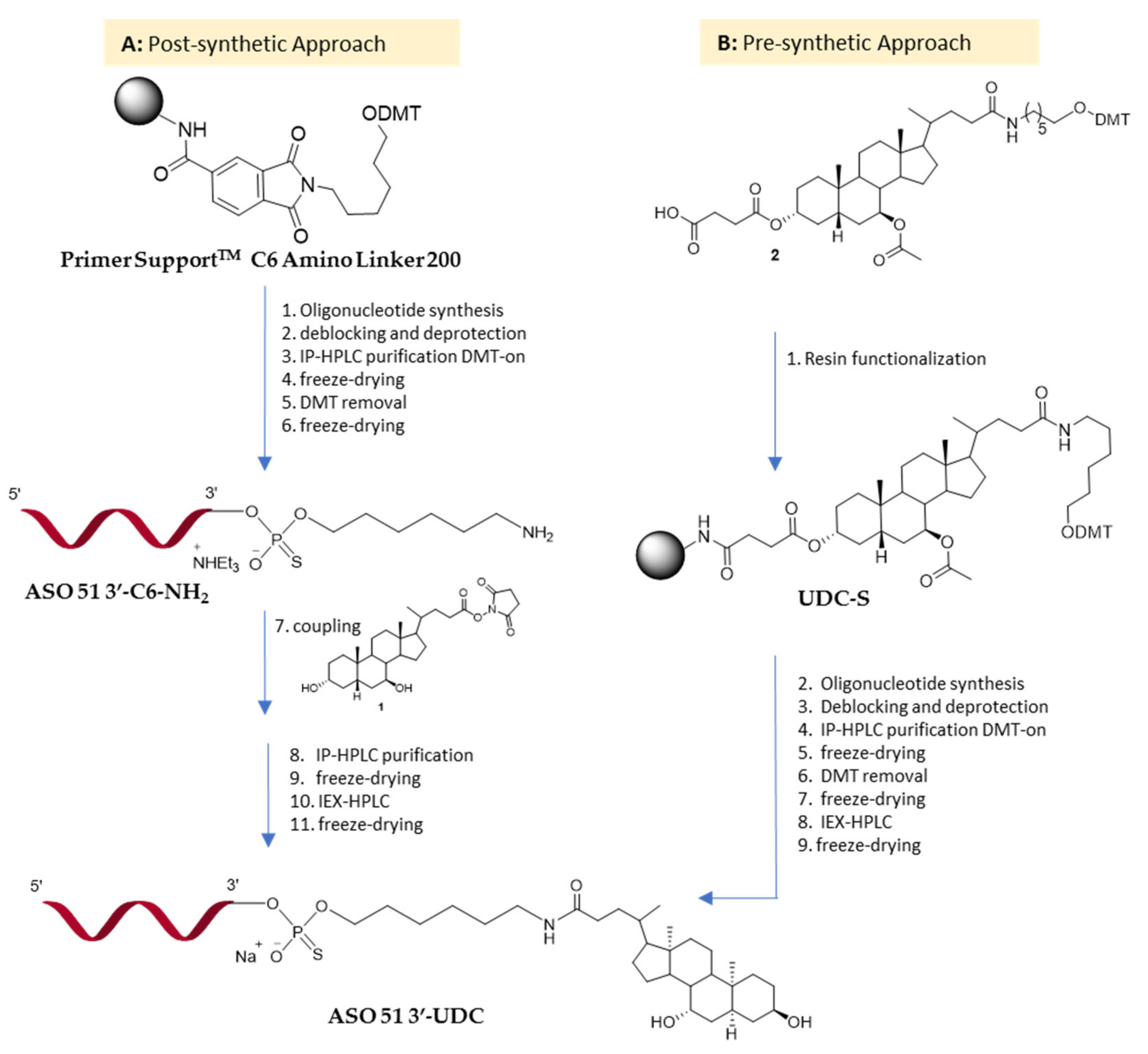
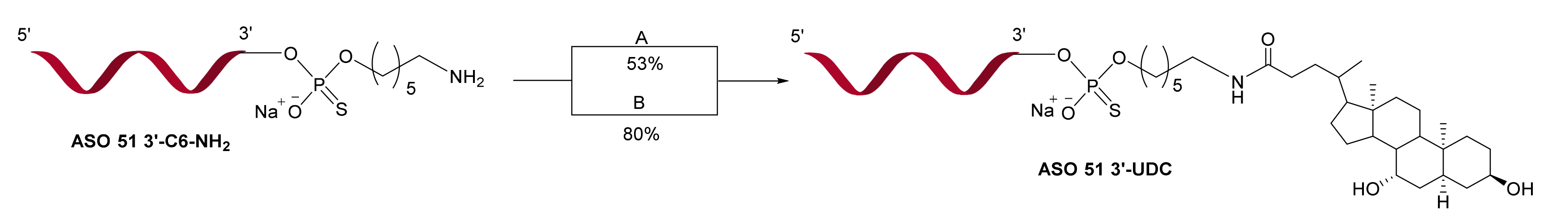
2.1. Synthesis of ASO 51 3′-UDC via Post-Synthetic Approach
2.2. ASO 51 3′-UDC Is Stable in Cleavage/Deprotection Conditions
2.3. Synthesis of ASO 51 3′-UDC via Pre-Synthetic Approach
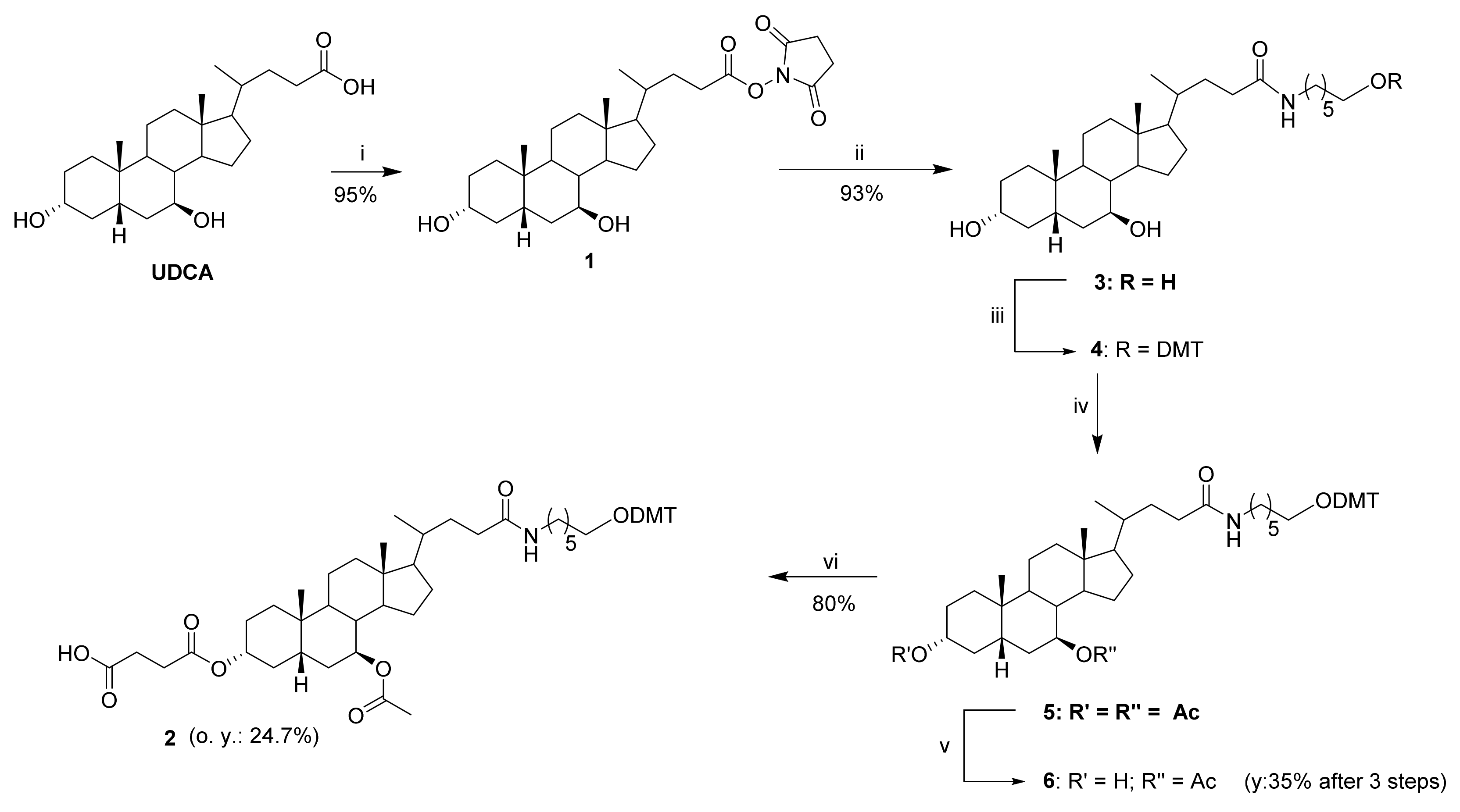
2.3.1. Ursodeoxycholic Acid Derivatization
2.3.2. Solid Support Functionalization: Synthesis of UDC-S Support
2.3.3. Synthesis of ASO 51 3′UDC on UDC-S Support
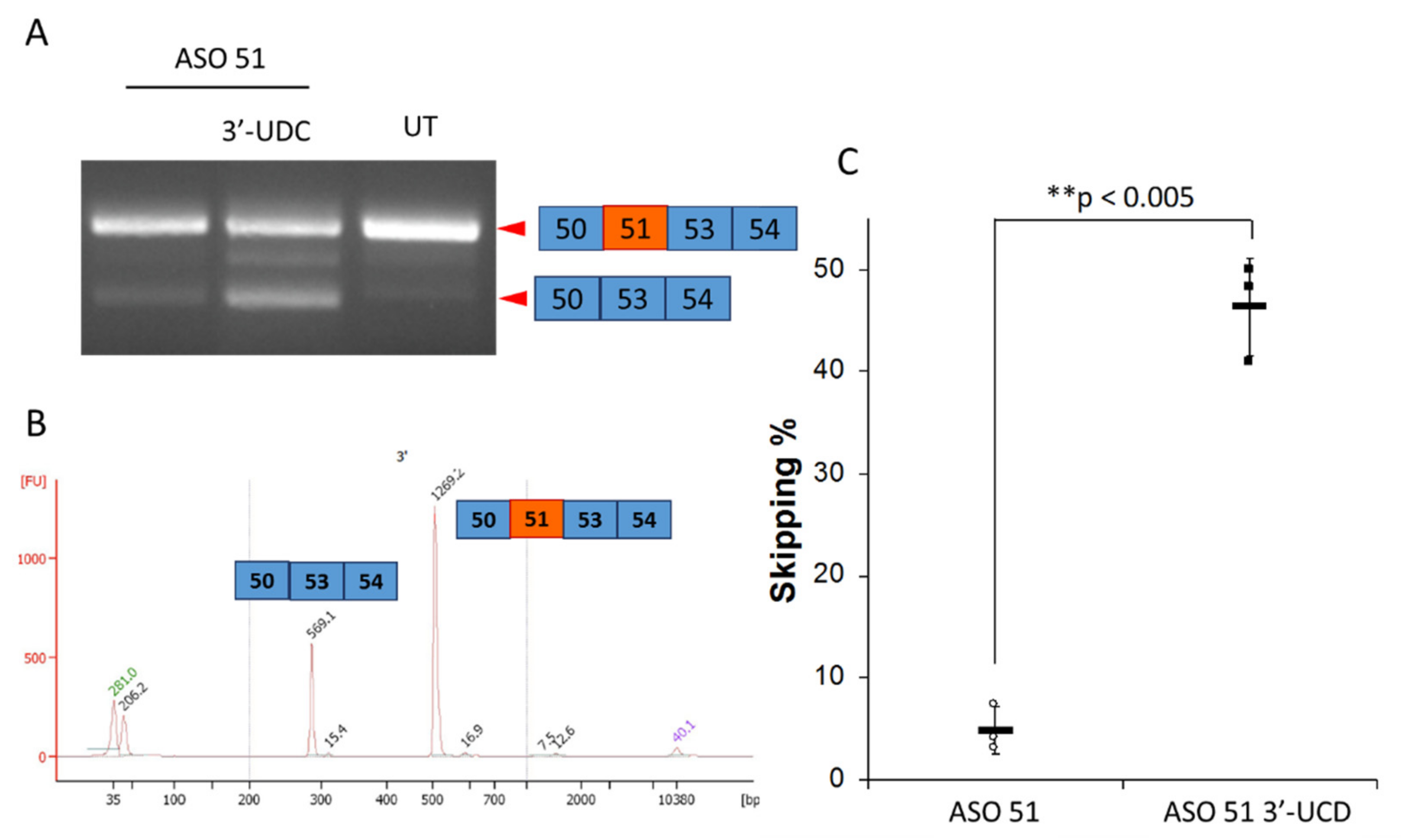
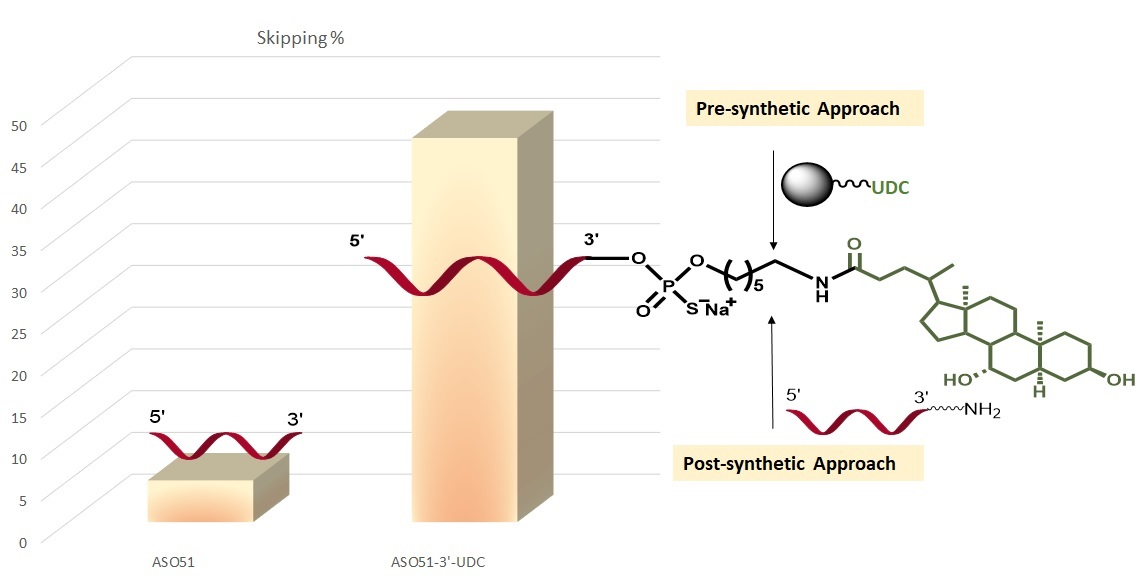
2.4. ASO 51 3′UDC Has Improved Efficiency in Inducing Exon-Skipping without Toxicity
2.5. 3′-UDC Conjugation Is Not a Source of Duplex Instability
2.6. ASO 51 and ASO 51 3′-UDC Aggregation Studies by Dynamic Light Scattering
3. Discussion
4. Materials and Methods
4.1. General Procedure for Solid Phase Synthesis
4.2. General Procedure for Oligonucleotide Purification
4.3. Synthesis of ASO 51 3′-UDC by Coupling of UDC-NHS Ester 1 in DMSO
4.4. Synthesis of UDC-NHS Ester 1
4.5. Synthesis of UDC-Aminohexyl Alcohol 3
4.6. Synthesis of UDC-Amino-DMT-Hexanol 4
4.7. Synthesis of 3,7-Diacetyl-UDC-Amino-DMT-Hexanol 5
4.8. Synthesis of 7-Acetyl-UDC-Amino-DMT-Hexanol 6
4.9. Synthesis of 3-Hemisuccinyl-7-Acetyl-UDC-Amino-DMT-Hexanol 2
4.10. Synthesis of UDC-S Support
4.11. Loading Determination
4.12. Synthesis of ASO 51 3′-UDC on UDC-S Support
4.13. ASO 51 3′-UDC Stability in Cleavage/Deprotection Conditions
4.14. Melting Studies
4.15. Dynamic Light Scattering (DLS) Characterization
4.16. Exon-Skipping and Toxicity Studies
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, M.; Wu, B.; Shah, S.N.; Lu, P.; Lu, Q. Saponins enhance exon skipping of 2’-O-methyl phosphorothioate oligonucleotide in vitro and in vivo. Drug Des. Devel. Ther. 2018, 12, 3705–3715. [Google Scholar] [CrossRef] [Green Version]
- Echevarría, L.; Aupy, P.; Relizani, K.; Bestetti, T.; Griffith, G.; Blandel, F.; Komisarski, M.; Haeberli, A.; Svinartchouk, F.; Garcia, L.; et al. Evaluating the Impact of Variable Phosphorothioate Content in Tricyclo-DNA Antisense Oligonucleotides in a Duchenne Muscular Dystrophy Mouse Model. Nucleic Acid Ther. 2019, 29, 148–160. [Google Scholar] [CrossRef]
- Tang, Z.; Zhao, J.; Pearson, Z.J.; Boskovic, Z.V.; Wang, J. RNA-Targeting Splicing Modifiers: Drug Development and Screening Assays. Molecules 2021, 26, 2263. [Google Scholar] [CrossRef]
- Hammond, S.M.; Aartsma-Rus, A.; Alves, S.; Borgos, S.E.; Buijsen, R.A.M.; Collin, R.W.J.; Covello, G.; Denti, M.A.; Desviat, L.R.; Echevarría, L.; et al. Delivery of oligonucleotide-based therapeutics: Challenges and opportunities. EMBO Mol. Med. 2021, 13, e13243. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Hu, C.; Moufawad El Achkar, C.; Black, L.E.; Douville, J.; Larson, A.; Pendergast, M.K.; Goldkind, S.F.; Lee, E.A.; Kuniholm, A.; et al. Patient-Customized Oligonucleotide Therapy for a Rare Genetic Disease. N. Engl. J. Med. 2019, 381, 1644. [Google Scholar] [CrossRef] [PubMed]
- Hanson, B.; Wood, M.J.A.; Roberts, T.C. Molecular correction of Duchenne muscular dystrophy by splice modulation and gene editing. RNA Biol. 2021, 18, 1048–1062. [Google Scholar] [CrossRef] [PubMed]
- Fortunato, F.; Rossi, R.; Falzarano, M.S.; Ferlini, A. Innovative therapeutic approaches for duchenne muscular dystrophy. J. Clin. Med. 2021, 10, 820. [Google Scholar] [CrossRef] [PubMed]
- Godfrey, C.; Desviat, L.R.; Smedsrød, B.; Piétri-Rouxel, F.; Denti, M.A.; Disterer, P.; Lorain, S.; Nogales-Gadea, G.; Sardone, V.; Anwar, R.; et al. Delivery is key: Lessons learnt from developing splice-switching antisense therapies. EMBO Mol. Med. 2017, 9, 545–557. [Google Scholar] [CrossRef]
- Lorenzer, C.; Dirin, M.; Winkler, A.M.; Baumann, V.; Winkler, J. Going beyond the liver: Progress and challenges of targeted delivery of siRNA therapeutics. J. Control. Release 2015, 203, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juliano, R.L. The delivery of therapeutic oligonucleotides. Nucleic Acids Res. 2016, 44, 6518–6548. [Google Scholar] [CrossRef] [PubMed]
- Falzarano, M.S.; Passarelli, C.; Bassi, E.; Fabris, M.; Perrone, D.; Sabatelli, P.; Maraldi, N.M.; Donà, S.; Selvatici, R.; Bonaldo, P.; et al. Biodistribution and molecular studies on orally administered nanoparticle-AON complexes encapsulated with alginate aiming at inducing dystrophin rescue in mdx mice. Biomed. Res. Int. 2013, 2013, 13. [Google Scholar] [CrossRef] [Green Version]
- Roberts, T.C.; Langer, R.; Wood, M.J.A. Advances in oligonucleotide drug delivery. Nat. Rev. Drug Discov. 2020, 19, 673–694. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yu, R.Z.; Henry, S.; Geary, R.S. Pharmacokinetics and Clinical Pharmacology Considerations of GalNAc3-Conjugated Antisense Oligonucleotides. Expert Opin. Drug Metab. Toxicol. 2019, 15, 475–485. [Google Scholar] [CrossRef]
- Nair, J.K.; Willoughby, J.L.S.; Chan, A.; Charisse, K.; Alam, M.R.; Wang, Q.; Hoekstra, M.; Kandasamy, P.; Kel’in, A.V.; Milstein, S.; et al. Multivalent N-Acetylgalactosamine-Conjugated siRNA Localizes in Hepatocytes and Elicits Robust RNAi-Mediated Gene Silencing. J. Am. Chem. Soc. 2014, 136, 16958–16961. [Google Scholar] [CrossRef] [Green Version]
- Springer, A.D.; Dowdy, S.F. GalNAc-siRNA Conjugates: Leading the Way for Delivery of RNAi Therapeutics. Nucleic Acid Ther. 2018, 28, 109–118. [Google Scholar] [CrossRef]
- Thiel, K.W.; Giangrande, P.H. Intracellular delivery of RNA-based therapeutics using aptamers. Ther. Deliv. 2010, 1, 849–861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dassie, J.P.; Giangrande, P.H. Current progress on aptamer-targeted oligonucleotide therapeutics. Ther. Deliv. 2013, 4, 1527–1546. [Google Scholar] [CrossRef] [Green Version]
- Soutschek, J.; Akinc, A.; Bramlage, B.; Charisse, K.; Constien, R.; Donoghue, M.; Elbashir, S.; Gelck, A.; Hadwiger, P.; Harborth, J.; et al. Therapeutic silencing of an endogenous gene by systemic administration of modified siRNAs. Nature 2004, 432, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Nishina, K.; Unno, T.; Uno, Y.; Kubodera, T.; Kanouchi, T.; Mizusawa, H.; Yokota, T. Efficient in vivo delivery of siRNA to the liver by conjugation of α-tocopherol. Mol. Ther. 2008, 16, 734–740. [Google Scholar] [CrossRef]
- Wolfrum, C.; Shi, S.; Jayaprakash, K.N.; Jayaraman, M.; Wang, G.; Pandey, R.K.; Rajeev, K.G.; Nakayama, T.; Charrise, K.; Ndungo, E.M.; et al. Mechanisms and optimization of in vivo delivery of lipophilic siRNAs. Nat. Biotechnol. 2007, 25, 1149–1157. [Google Scholar] [CrossRef]
- Kurrikoff, K.; Vunk, B.; Langel, Ü. Status update in the use of cell-penetrating peptides for the delivery of macromolecular therapeutics. Expert Opin. Biol. Ther. 2021, 21, 361–370. [Google Scholar] [CrossRef]
- Wang, S.; Allen, N.; Prakash, T.P.; Liang, X.H.; Crooke, S.T. Lipid Conjugates Enhance Endosomal Release of Antisense Oligonucleotides into Cells. Nucleic Acid Ther. 2019, 29, 245–255. [Google Scholar] [CrossRef]
- Prakash, T.P.; Mullick, A.E.; Lee, R.G.; Yu, J.; Yeh, S.T.; Low, A.; Chappell, A.E.; Østergaard, M.E.; Murray, S.; Gaus, H.J.; et al. Fatty acid conjugation enhances potency of antisense oligonucleotides in muscle. Nucleic Acids Res. 2019, 47, 6029–6044. [Google Scholar] [CrossRef]
- Lönnberg, H. Solid-Phase Synthesis of Oligonucleotide Conjugates Useful for Delivery and Targeting of Potential Nucleic Acid Therapeutics. Bioconjug. Chem. 2009, 20, 1065–1094. [Google Scholar] [CrossRef]
- Hawner, M.; Ducho, C. Cellular Targeting of Oligonucleotides by Conjugation with Small Molecules. Molecules 2020, 25, 5963. [Google Scholar] [CrossRef]
- Forget, D.; Renaudet, O.; Boturyn, D.; Defrancq, E.; Dumy, P. 3′-Oligonucleotides conjugation via chemoselective oxime bond formation. Tetrahedron Lett. 2001, 42, 9171–9174. [Google Scholar] [CrossRef]
- Hovinen, J.; Guzaev, A.; Azhayev, A.; Lönnberg, H. Novel solid supports for the preparation of 3′-derivatized oligonucleotides: Introduction of 3′-alkylphosphate tether groups bearing amino, carboxy, carboxamido, and mercapto functionalities. Tetrahedron 1994, 50, 7203–7218. [Google Scholar] [CrossRef]
- Osborn, M.F.; Khvorova, A. Improving siRNA Delivery in Vivo through Lipid Conjugation. Nucleic Acid Ther. 2018, 28, 128–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stetsenko, D.A.; Gait, M.J. A convenient solid-phase method for synthesis of 3′-conjugates of oligonucleotides. Bioconjug. Chem. 2001, 12, 576–586. [Google Scholar] [CrossRef] [PubMed]
- Spinelli, N.; Edupuganti, O.P.; Defrancq, E.; Dumy, P. New solid support for the synthesis of 3′-oligonucleotide conjugates through glyoxylic oxime bond formation. Org. Lett. 2007, 9, 219–222. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, T.J.; Engels, J.W. Synthesis and Properties of Bile Acid Phosphoramidites 5′-Tethered to Antisense Oligodeoxynucleotides against HCV. Bioorg. Med. Chem. 2001, 9, 1827–1835. [Google Scholar] [CrossRef]
- Tuma, J.; Richert, C. Solution structure of a steroid-DNA complex with cholic acid residues sealing the termini of a Watson-Crick duplex. Biochemistry 2003, 42, 8957–8965. [Google Scholar] [CrossRef] [PubMed]
- Bleczinski, C.F.; Richert, C. Steroid-DNA interactions increasing stability, sequence-selectivity, DNA/RNA discrimination, and hypochromicity of oligonucleotide duplexes. J. Am. Chem. Soc. 1999, 121, 10889–10894. [Google Scholar] [CrossRef]
- Palmieri, B.; Bovolenta, M.; Braghetta, P.; Capobianco, M.L.; Marchesi, E.; Medici, A.; Molon, S.; Perrone, D.; Rimessi, P. Conjugates of Oligonucleotides and Bile Acids and Their Derivatives for Pharmaceutical Active Molecules Delivery. WO 2020084488, 30 April.
- Pavlović, N.; Goločorbin-Kon, S.; Ðanić, M.; Stanimirov, B.; Al-Salami, H.; Stankov, K.; Mikov, M. Bile Acids and Their Derivatives as Potential Modifiers of Drug Release and Pharmacokinetic Profiles. Front. Pharmacol. 2018, 9, 1283. [Google Scholar] [CrossRef] [PubMed]
- Bosgra, S.; Sipkens, J.; De Kimpe, S.; Den Besten, C.; Datson, N.; Van Deutekom, J. The Pharmacokinetics of 2′-O-Methyl Phosphorothioate Antisense Oligonucleotides: Experiences from Developing Exon Skipping Therapies for Duchenne Muscular Dystrophy. Nucleic Acid Ther. 2019, 29, 305–322. [Google Scholar] [CrossRef] [PubMed]
- Pascual-Morena, C.; Cavero-Redondo, I.; Álvarez-Bueno, C.; Mesas, A.E.; Pozuelo-Carrascosa, D.; Martínez-Vizcaíno, V. Restorative treatments of dystrophin expression in Duchenne muscular dystrophy: A systematic review. Ann. Clin. Transl. Neurol. 2020, 7, 1738–1752. [Google Scholar] [CrossRef]
- Goemans, N.M.; Tulinius, M.; Van Den Hauwe, M.; Kroksmark, A.K.; Buyse, G.; Wilson, R.J.; Van Deutekom, J.C.; De Kimpe, S.J.; Lourbakos, A.; Campion, G. Long-term efficacy, safety, and pharmacokinetics of drisapersen in duchenne muscular dystrophy: Results from an open-label extension study. PLoS ONE 2016, 11, e0161955. [Google Scholar] [CrossRef]
- Van Deutekom, J.C.; Janson, A.A.; Ginjaar, I.B.; Frankhuizen, W.S.; Aartsma-Rus, A.; Bremmer-Bout, M.; den Dunnen, J.T.; Koop, K.; van der Kooi, A.J.; Goemans, N.M.; et al. Local Dystrophin Restoration with Antisense Oligonucleotide PRO051. N. Engl. J. Med. 2009, 357, 2677–2686. [Google Scholar] [CrossRef] [Green Version]
- Goossens, J.F.; Bailly, C. Ursodeoxycholic acid and cancer: From chemoprevention to chemotherapy. Pharmacol. Ther. 2019, 203, 107396. [Google Scholar] [CrossRef]
- Catani, M.; De Luca, C.; Medeiros Garcia Alcântara, J.; Manfredini, N.; Perrone, D.; Marchesi, E.; Weldon, R.; Müller-Späth, T.; Cavazzini, A.; Morbidelli, M.; et al. Oligonucleotides: Current Trends and Innovative Applications in the Synthesis, Characterization, and Purification. Biotechnol. J. 2020, 15, 1900226. [Google Scholar] [CrossRef]
- Dalpiaz, A.; Paganetto, G.; Pavan, B.; Fogagnolo, M.; Medici, A.; Beggiato, S.; Perrone, D. Zidovudine and Ursodeoxycholic Acid Conjugation: Design of a New Prodrug Potentially Able to Bypass the Active Efflux Transport Systems of the Central Nervous System. Mol. Pharm. 2012, 9, 957–968. [Google Scholar] [CrossRef] [PubMed]
- Faustino, C.; Serafim, C.; Rijo, P.; Reis, C.P. Bile acids and bile acid derivatives: Use in drug delivery systems and as therapeutic agents. Expert Opin. Drug Deliv. 2016, 13, 1133–1148. [Google Scholar] [CrossRef]
- Yuen, L.H.; Franzini, R.M. Stability of Oligonucleotide–Small Molecule Conjugates to DNA-Deprotection Conditions. Bioconjug. Chem. 2017, 28, 1076–1083. [Google Scholar] [CrossRef]
- Massarenti, C.; Bortolini, O.; Fantin, G.; Cristofaro, D.; Ragno, D.; Perrone, D.; Marchesi, E.; Toniolo, G.; Massi, A. Fluorous-tag assisted synthesis of bile acid-bisphosphonate conjugates: Via orthogonal click reactions: An access to potential anti-resorption bone drugs. Org. Biomol. Chem. 2017, 15, 4907–4920. [Google Scholar] [CrossRef] [PubMed]
- Marchesi, E.; Chinaglia, N.; Capobianco, M.L.; Marchetti, P.; Huang, T.E.; Weng, H.C.; Guh, J.H.; Hsu, L.C.; Perrone, D.; Navacchia, M.L. Dihydroartemisinin–Bile Acid Hybridization as an Effective Approach to Enhance Dihydroartemisinin Anticancer Activity. ChemMedChem 2019, 14, 779–787. [Google Scholar] [CrossRef]
- Derzhalova, A.; Markov, O.; Fokina, A.; Shiohama, Y.; Zatsepin, T.; Fujii, M.; Zenkova, M.; Stetsenko, D. Novel Lipid-Oligonucleotide Conjugates Containing Long-Chain Sulfonyl Phosphoramidate Groups: Synthesis and Biological Properties. Appl. Sci. 2021, 11, 1174. [Google Scholar] [CrossRef]
- Ezzat, K.; Aoki, Y.; Koo, T.; McClorey, G.; Benner, L.; Coenen-Stass, A.; O’Donovan, L.; Lehto, T.; Garcia-Guerra, A.; Nordin, J.; et al. Self-Assembly into Nanoparticles Is Essential for Receptor Mediated Uptake of Therapeutic Antisense Oligonucleotides. Nano Lett. 2015, 15, 4364–4373. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry a | Support (eq.) | Coupling Reagent (eq.) | DIPEA (eq.) | Efficiency b (%) |
|---|---|---|---|---|
| 1 | 1.00 | HBTU (1.00) | 3.00 | <1 |
| 2 | 0.50 | HBTU (1.00) | 3.00 | <1 |
| 3 c | 0.90 | / | 2.00 | <1 |
| 4 | 0.90 | HCTU (1.00) | 2.00 | 70 |
| 5 | 0.90 | HCTU (1.00) | 3.00 | 65 |
| 6 | 0.90 | HCTU (1.05) | 2.00 | 68 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marchesi, E.; Bovolenta, M.; Preti, L.; Capobianco, M.L.; Mamchaoui, K.; Bertoldo, M.; Perrone, D. Synthesis and Exon-Skipping Properties of a 3′-Ursodeoxycholic Acid-Conjugated Oligonucleotide Targeting DMD Pre-mRNA: Pre-Synthetic versus Post-Synthetic Approach. Molecules 2021, 26, 7662. https://doi.org/10.3390/molecules26247662
Marchesi E, Bovolenta M, Preti L, Capobianco ML, Mamchaoui K, Bertoldo M, Perrone D. Synthesis and Exon-Skipping Properties of a 3′-Ursodeoxycholic Acid-Conjugated Oligonucleotide Targeting DMD Pre-mRNA: Pre-Synthetic versus Post-Synthetic Approach. Molecules. 2021; 26(24):7662. https://doi.org/10.3390/molecules26247662
Chicago/Turabian StyleMarchesi, Elena, Matteo Bovolenta, Lorenzo Preti, Massimo L. Capobianco, Kamel Mamchaoui, Monica Bertoldo, and Daniela Perrone. 2021. "Synthesis and Exon-Skipping Properties of a 3′-Ursodeoxycholic Acid-Conjugated Oligonucleotide Targeting DMD Pre-mRNA: Pre-Synthetic versus Post-Synthetic Approach" Molecules 26, no. 24: 7662. https://doi.org/10.3390/molecules26247662
APA StyleMarchesi, E., Bovolenta, M., Preti, L., Capobianco, M. L., Mamchaoui, K., Bertoldo, M., & Perrone, D. (2021). Synthesis and Exon-Skipping Properties of a 3′-Ursodeoxycholic Acid-Conjugated Oligonucleotide Targeting DMD Pre-mRNA: Pre-Synthetic versus Post-Synthetic Approach. Molecules, 26(24), 7662. https://doi.org/10.3390/molecules26247662