
Theoretical Prediction and Explanation of Reaction Site Selectivity in the Addition of a Phenoxy Group to Perfluoropyrimidine, Perfluoropyridazine, and Perfluoropyrazine


Abstract
:1. Introduction
2. Results
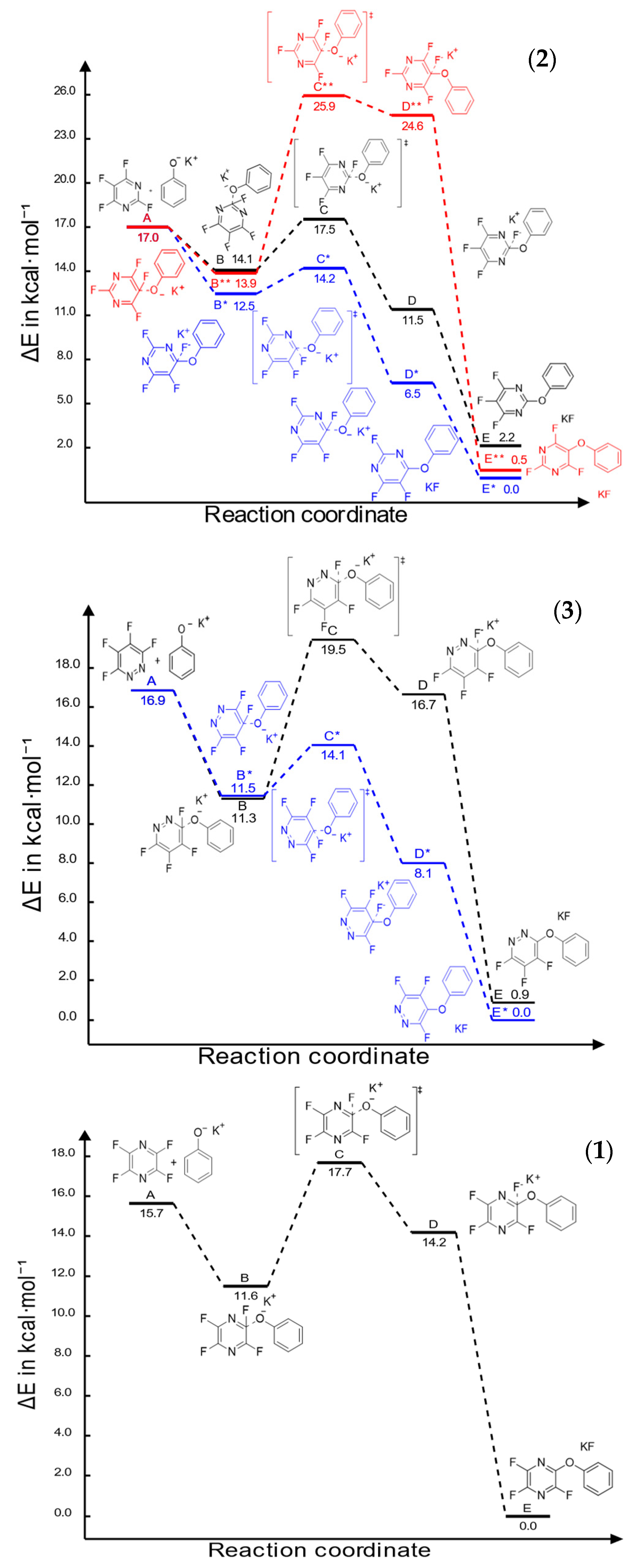
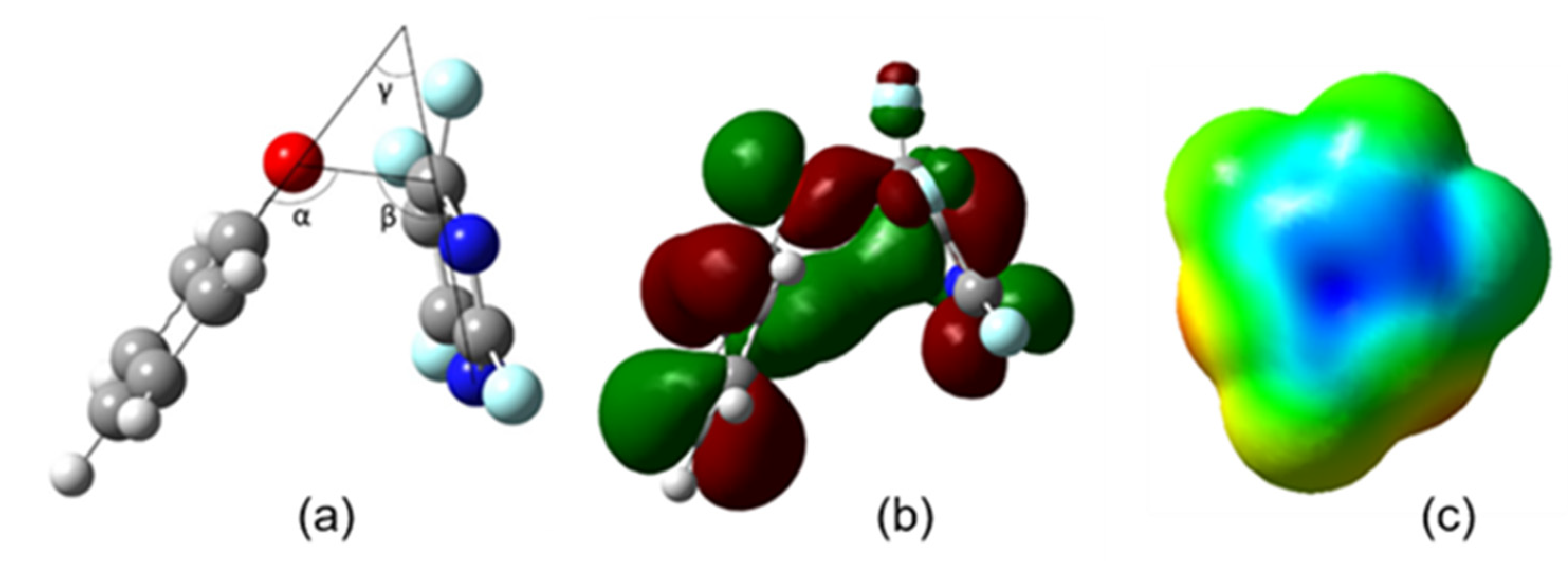
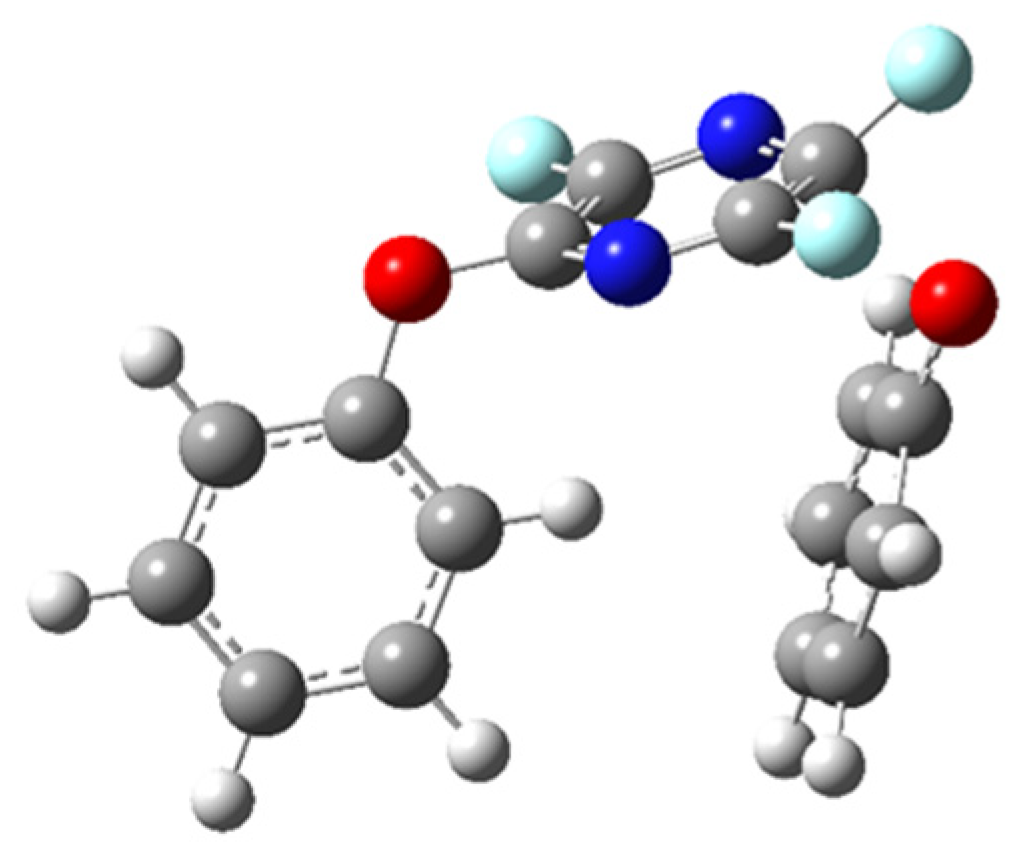
2.1. First Phenoxide Substitution
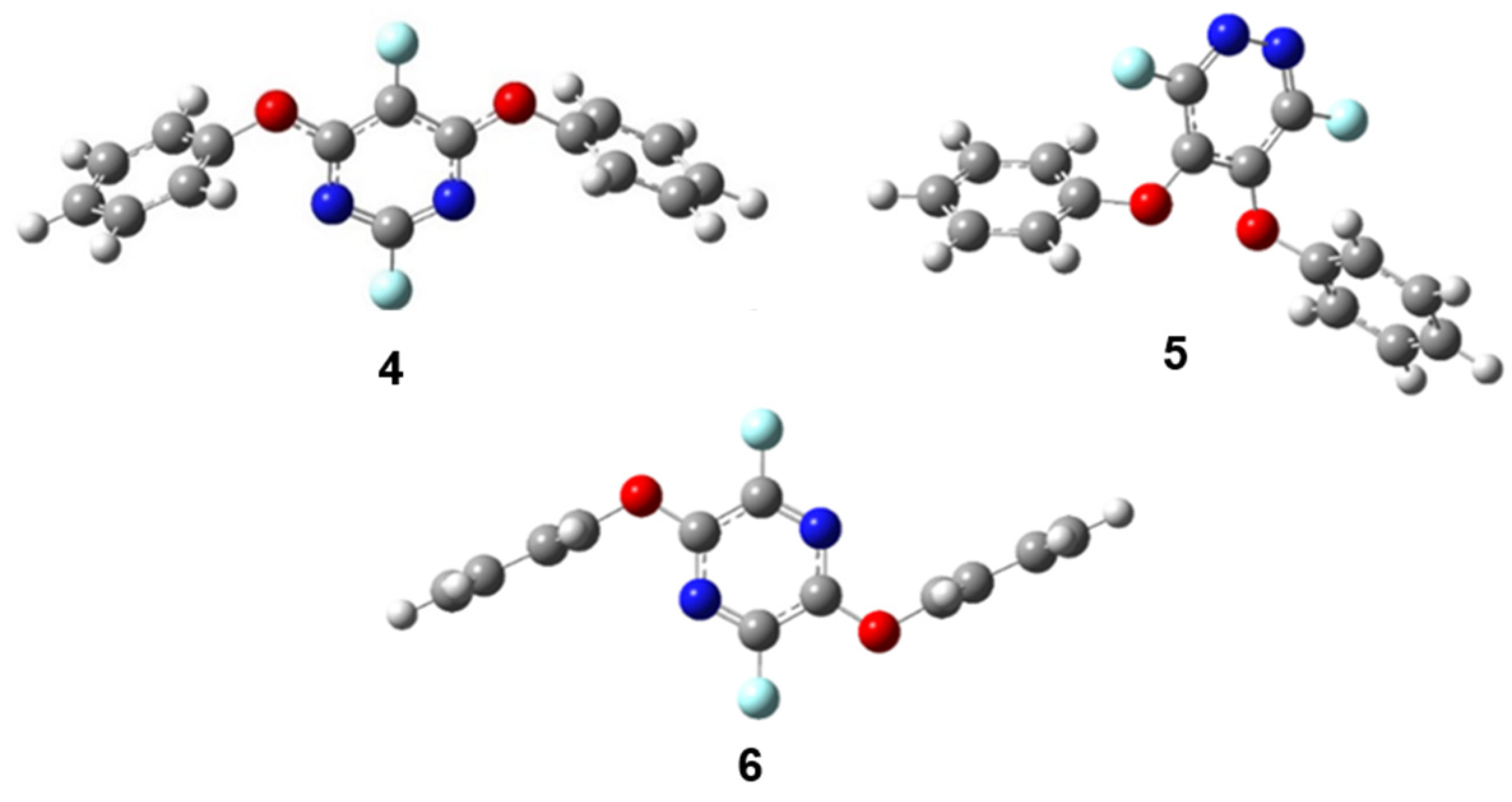
2.2. Second Phenoxide Substitution
3. Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Corley, C.A.; Kobra, K.; Peloquin, A.J.; Salmon, K.; Gumireddy, L.; Knoerzer, T.A.; McMillen, C.D.; Pennington, W.T.; Schoffstall, A.M.; Iacono, S.T. Utilizing the Regioselectivity of Perfluoropyridine towards the Preparation of Phenyoxyacetylene Precursors for Partially Fluorinated Polymers of Diverse Architecture. J. Fluor. Chem. 2019, 228, 109409. [Google Scholar] [CrossRef]
- Seyb, C.; Kerres, J. Novel partially fluorinated sulfonated poly(arylenethioether)s and poly(aryleneether)s prepared from octafluorotoluene and pentafluoropyridine, and their blends with PBI-Celazol. Eur. Polym. J. 2013, 49, 518–531. [Google Scholar] [CrossRef]
- Moore, L.M.J.; Greeson, K.T.; Stewart, K.A.; Kure, D.A.; Corley, C.A.; Jennings, A.R.; Iacono, S.T.; Ghiassi, K.B. Perfluoropyridine as a Scaffold for Semifluorinated Thiol-ene Networks with Readily Tunable Thermal Properties. Macromol. Chem. Phys. 2020, 221, 2000100. [Google Scholar] [CrossRef]
- Iacono, S.T.; Budy, S.M.; Jin, J.; Smith, J.; Dennis, W. Science and technology of perfluorocyclobutyl aryl ether polymers. J. Polym. Sci. Part A Polym. Chem. 2007, 45, 5705–5721. [Google Scholar] [CrossRef]
- Bhebhe, M.N.; De Eulate, E.A.; Pei, Y.; Arrigan, D.W.M.; Roth, P.J.; Lowe, A.B. Reactive Conjugated Polymers: Synthesis, Modification, and Electrochemical Properties of Polypentafluorophenylacetylene (Co)Polymers. Macromol. Rapid Commun. 2017, 38, 1600450. [Google Scholar] [CrossRef] [PubMed]
- Houck, M.B.; Fuhrer, T.J.; Phelps, C.R.; Brown, L.C.; Iacono, S.T. Toward Taming the Chemical Reversibility of Perfluoropyridine through Molecular Design with Applications to Pre- and Postmodifiable Polymer Architectures. Macromolecules 2021, 54, 5586–5594. [Google Scholar] [CrossRef]
- Gimenez, D.; Dose, A.; Robson, N.L.; Sandford, G.; Cobb, S.L.; Coxon, C.R. 2,2,2-Trifluoroethanol as a solvent to control nucleophilic peptide arylation. Org. Biomol. Chem. 2017, 15, 4081–4085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gimenez, D.; Mooney, C.A.; Dose, A.; Sandford, G.; Coxon, C.R.; Cobb, S.L. The application of perfluoroheteroaromatic reagents in the preparation of modified peptide systems. Org. Biomol. Chem. 2017, 15, 4086–4095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lühmann, T.; Mong, S.K.; Simon, M.D.; Meinel, L.; Pentelute, B.L. A perfluoroaromatic abiotic analog of H2 relaxin enabled by rapid flow-based peptide synthesis. Org. Biomol. Chem. 2016, 14, 3345–3349. [Google Scholar] [CrossRef]
- Dai, P.; Williams, J.K.; Zhang, C.; Welborn, M.; Shepherd, J.J.; Zhu, T.; Van Voorhis, T.; Hong, M.; Pentelute, B.L. A structural and mechanistic study of π-clamp-mediated cysteine perfluoroarylation. Sci. Rep. 2017, 7, 7954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fadzen, C.M.; Wolfe, J.M.; Cho, C.-F.; Chiocca, E.A.; Lawler, S.E.; Pentelute, B.L. Perfluoroarene–Based Peptide Macrocycles to Enhance Penetration Across the Blood–Brain Barrier. J. Am. Chem. Soc. 2017, 139, 15628–15631. [Google Scholar] [CrossRef] [Green Version]
- Fadzen, C.M.; Wolfe, J.M.; Zhou, W.; Cho, C.-F.; von Spreckelsen, N.; Hutchinson, K.T.; Lee, Y.-C.; Chiocca, E.A.; Lawler, S.E.; Yilmaz, O.H.; et al. A Platinum(IV) Prodrug—Perfluoroaryl Macrocyclic Peptide Conjugate Enhances Platinum Uptake in the Brain. J. Med. Chem. 2020, 63, 6741–6747. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, J.M.; Fadzen, C.M.; Holden, R.L.; Yao, M.; Hanson, G.J.; Pentelute, B.L. Perfluoroaryl Bicyclic Cell-Penetrating Peptides for Delivery of Antisense Oligonucleotides. Angew. Chem. Int. Ed. 2018, 57, 4756–4759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandford, G. Perfluoroheteroaromatic Chemistry: Multifunctional Systems from Perfluorinated Heterocycles by Nucleophilic Aromatic Substitution Processes. In Halogenated Heterocycles: Synthesis, Application and Environment; Iskra, J., Ed.; Springer: Berlin/Heidelberg, Germany, 2012; pp. 1–31. [Google Scholar]
- Cockcroft, J.K.; Ghosh, R.E.; Shephard, J.J.; Singh, A.; Williams, J.H. Investigation of the phase behaviour of the 1 : 1 adduct of mesitylene and hexafluorobenzene. CrystEngComm 2017, 19, 1019–1023. [Google Scholar] [CrossRef] [Green Version]
- Calabrese, C.; Gou, Q.; Maris, A.; Caminati, W.; Melandri, S. Probing the Lone Pair···π-Hole Interaction in Perfluorinated Heteroaromatic Rings: The Rotational Spectrum of Pentafluoropyridine·Water. J. Phys. Chem. Lett. 2016, 7, 1513–1517. [Google Scholar] [CrossRef]
- Senaweera, S.; Weaver, J.D. SNAr catalysis enhanced by an aromatic donor–acceptor interaction; facile access to chlorinated polyfluoroarenes. Chem. Commun. 2017, 53, 7545–7548. [Google Scholar] [CrossRef] [PubMed]
- Rozzell, J.D.; Hartwig, J.F. Fluoroalkyl-Substituted Derivatives of Pyridine, Pyrimidine, and Pyrazine. U.S. Patent. No. 20150284341A1, 8 October 2015. [Google Scholar]
- Darehkordi, A.; Ramezani, M.; Rahmani, F.; Ramezani, M. Design, synthesis and evaluation of antibacterial effects of a new class of piperazinylquinolone derivatives. J. Heterocycl. Chem. 2016, 53, 89–94. [Google Scholar] [CrossRef]
- Clark, D.T.; Abu-shbak, M.M. Plasma polymerization. XI. A comparison of the plasma polymerization of the isomeric perfluorinated diazines. J. Polym. Sci. Polym. Chem. Ed. 1984, 22, 17–28. [Google Scholar] [CrossRef]
- Clark, D.T.; Johnson, S.A.; Brennan, W.J. An MNDO SCF MO investigation of some structural isomers of the perfluoro diazines (pyrimidine, pyrazine, and pyridazine) of relevance to their plasma polymerization. J. Polym. Sci. Polym. Chem. Ed. 1984, 22, 2145–2158. [Google Scholar] [CrossRef]
- Volochnyuk, D.M.; Grygorenko, O.O.; Gorlova, A.O. Fluorine Containing Diazines. Synthesis and Properties. In Fluorine in Heterocyclic Chemistry Volume 2: 6-Membered Heterocycles; Nenajdenko, V., Ed.; Springer International Publishing: Cham, Switzerland, 2014; pp. 291–575. [Google Scholar]
- Fuhrer, T.J.; Houck, M.; Corley, C.A.; Iacono, S.T. Theoretical Explanation of Reaction Site Selectivity in the Addition of a Phenoxy Group to Perfluoropyridine. J. Phys. Chem. A 2019, 123, 9450–9455. [Google Scholar] [CrossRef] [PubMed]
- Neumann, C.N.; Hooker, J.M.; Ritter, T. Concerted nucleophilic aromatic substitution with 19F− and 18F−. Nature 2016, 534, 369–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neumann, C.N.; Ritter, T. Facile C–F Bond Formation through a Concerted Nucleophilic Aromatic Substitution Mediated by the PhenoFluor Reagent. Acc. Chem. Res. 2017, 50, 2822–2833. [Google Scholar] [CrossRef] [PubMed]
- Kwan, E.E.; Zeng, Y.; Besser, H.A.; Jacobsen, E.N. Concerted nucleophilic aromatic substitutions. Nat. Chem. 2018, 10, 917–923. [Google Scholar] [CrossRef] [PubMed]
- Kenwright, A.M.; Sandford, G.; Tadeusiak, A.J.; Yufit, D.S.; Howard, J.A.K.; Kilickiran, P.; Nelles, G. Strategies for the synthesis of fluorinated liquid crystal derivatives from perbromofluoroaromatic systems. Tetrahedron 2010, 66, 9819–9827. [Google Scholar] [CrossRef]
- Pattison, G.; Sandford, G.; Wilson, I.; Yufit, D.S.; Howard, J.A.K.; Christopher, J.A.; Miller, D.D. Polysubstituted and ring-fused pyridazine systems from tetrafluoropyridazine. Tetrahedron 2017, 73, 437–454. [Google Scholar] [CrossRef] [Green Version]
- Banks, R.E.; Field, D.S.; Haszeldine, R.N. Heterocyclic polyfluoro-compounds. Part X. Nucleophilic substitution in tetrafluoropyrimidine. J. Chem. Soc. C Org. 1967, 1822–1826. [Google Scholar] [CrossRef]
- Sinnokrot, M.O.; Sherrill, C.D. Substituent Effects in π−π Interactions: Sandwich and T-Shaped Configurations. J. Am. Chem. Soc. 2004, 126, 7690–7697. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.-P.; Yang, X.; Zhou, D.-G.; Liu, S. T-shaped phenol–benzene complexation driven by π-involved noncovalent interactions. Theor. Chem. Acc. 2016, 135, 100. [Google Scholar] [CrossRef]
- Banerjee, A.; Saha, A.; Saha, B.K. Understanding the Behavior of π–π Interactions in Crystal Structures in Light of Geometry Corrected Statistical Analysis: Similarities and Differences with the Theoretical Models. Cryst. Growth Des. 2019, 19, 2245–2252. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision B.01; Gaussian. Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Ii, R.; Keith, T.; Millam, J.; Eppinnett, K.; Hovell, L.; Gilliland, R. GaussView, Version 5; Semichem Inc.: Shawnee Mission, KS, USA, 2016. [Google Scholar]
- Thompson, M.A. Molecular docking using ArgusLab, an efficient shape-based search algorithm and AScore scoring function. In Proceedings of the ACS Meeting, Philadelphia, PA, USA, 22–26 August 2004; p. 172. [Google Scholar]
- Miertuš, S.; Scrocco, E.; Tomasi, J. Electrostatic interaction of a solute with a continuum. A direct utilizaion of AB initio molecular potentials for the prevision of solvent effects. Chem. Phys. 1981, 55, 117–129. [Google Scholar] [CrossRef]
- Miertus, S.; Tomasi, J. Approximate evaluations of the electrostatic free energy and internal energy changes in solution processes. Chem. Phys. 1982, 65, 239–245. [Google Scholar] [CrossRef]
- Pascual-ahuir, J.L.; Silla, E.; Tunon, I. GEPOL: An improved description of molecular surfaces. III. A new algorithm for the computation of a solvent-excluding surface. J. Comput. Chem. 1994, 15, 1127–1138. [Google Scholar] [CrossRef]
- Peng, C.; Bernhard Schlegel, H. Combining Synchronous Transit and Quasi-Newton Methods to Find Transition States. Isr. J. Chem. 1993, 33, 449–454. [Google Scholar] [CrossRef]
- Fukui, K. The path of chemical reactions—the IRC approach. Acc. Chem. Res. 1981, 14, 363–368. [Google Scholar] [CrossRef]

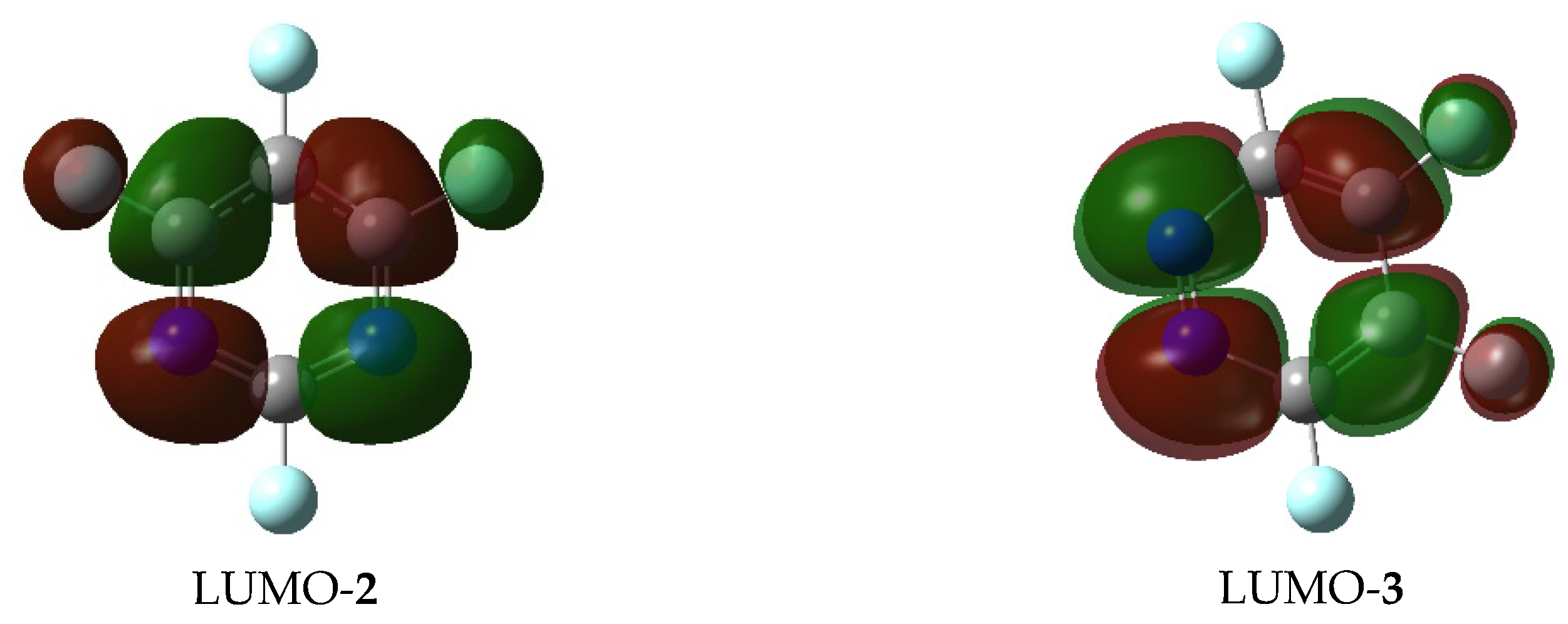

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Transition State | Angle α | Angle β | Angle γ | Ea (kcal/mol) |
|---|---|---|---|---|
| 2-phenoxyperfluoropyrimidine | 116.385 | 111.131 | 47.516 | 3.437 |
| 4-phenoxyperfluoropyrimidine | 114.961 | 112.551 | 47.512 | 1.699 |
| 5-phenoxyperfluoropyrimidine | 115.486 | 126.353 | 61.839 | 12.017 |
| 3-phenoxyperfluoropyridazine | 116.108 | 118.859 | 54.967 | 8.137 |
| 4-phenoxyperfluoropyridazine | 115.628 | 115.345 | 50.973 | 2.572 |
| Transition State | Angle α | Angle β | Angle γ | Ea (kcal/mol) |
|---|---|---|---|---|
| 2,4-phenoxyperfluoropyrimidine | 117.838 | 113.505 | 51.343 | 5.945 |
| 4,5-phenoxyperfluoropyrimidine | 115.658 | 126.230 | 61.888 | 15.945 |
| 4,6-phenoxyperfluoropyrimidine | 115.536 | 115.234 | 50.770 | 3.937 |
| 3,4-phenoxyperfluoropyridazine | 116.588 | 117.413 | 54.002 | 8.260 |
| 4,5-phenoxyperfluoropyridazine | 115.849 | 114.699 | 50.549 | 2.600 |
| 4,6-phenoxyperfluoropyridazine | 117.044 | 120.732 | 57.776 | 8.241 |
| 2,3-phenoxyperfluoropyrazine | 116.616 | 116.291 | 52.907 | 6.270 |
| 2,5-phenoxyperfluoropyrazine | 117.493 | 116.755 | 54.248 | 6.189 |
| 2,6-phenoxyperfluoropyrazine | 115.942 | 119.439 | 55.381 | 8.310 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fuhrer, T.J.; Houck, M.; Chapman, R.M.; Iacono, S.T. Theoretical Prediction and Explanation of Reaction Site Selectivity in the Addition of a Phenoxy Group to Perfluoropyrimidine, Perfluoropyridazine, and Perfluoropyrazine. Molecules 2021, 26, 7637. https://doi.org/10.3390/molecules26247637
Fuhrer TJ, Houck M, Chapman RM, Iacono ST. Theoretical Prediction and Explanation of Reaction Site Selectivity in the Addition of a Phenoxy Group to Perfluoropyrimidine, Perfluoropyridazine, and Perfluoropyrazine. Molecules. 2021; 26(24):7637. https://doi.org/10.3390/molecules26247637
Chicago/Turabian StyleFuhrer, Timothy J., Matthew Houck, Rachel M. Chapman, and Scott T. Iacono. 2021. "Theoretical Prediction and Explanation of Reaction Site Selectivity in the Addition of a Phenoxy Group to Perfluoropyrimidine, Perfluoropyridazine, and Perfluoropyrazine" Molecules 26, no. 24: 7637. https://doi.org/10.3390/molecules26247637
APA StyleFuhrer, T. J., Houck, M., Chapman, R. M., & Iacono, S. T. (2021). Theoretical Prediction and Explanation of Reaction Site Selectivity in the Addition of a Phenoxy Group to Perfluoropyrimidine, Perfluoropyridazine, and Perfluoropyrazine. Molecules, 26(24), 7637. https://doi.org/10.3390/molecules26247637