
Experimental and Computational Analysis of Para-Hydroxy Methylcinnamate following Photoexcitation

 , ,
, ,
Abstract
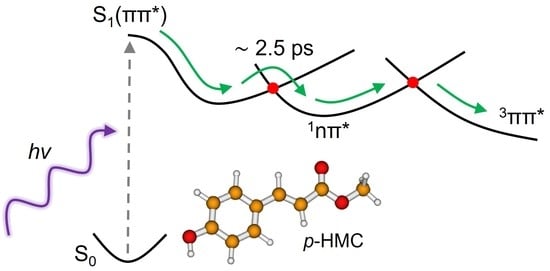
:
1. Introduction
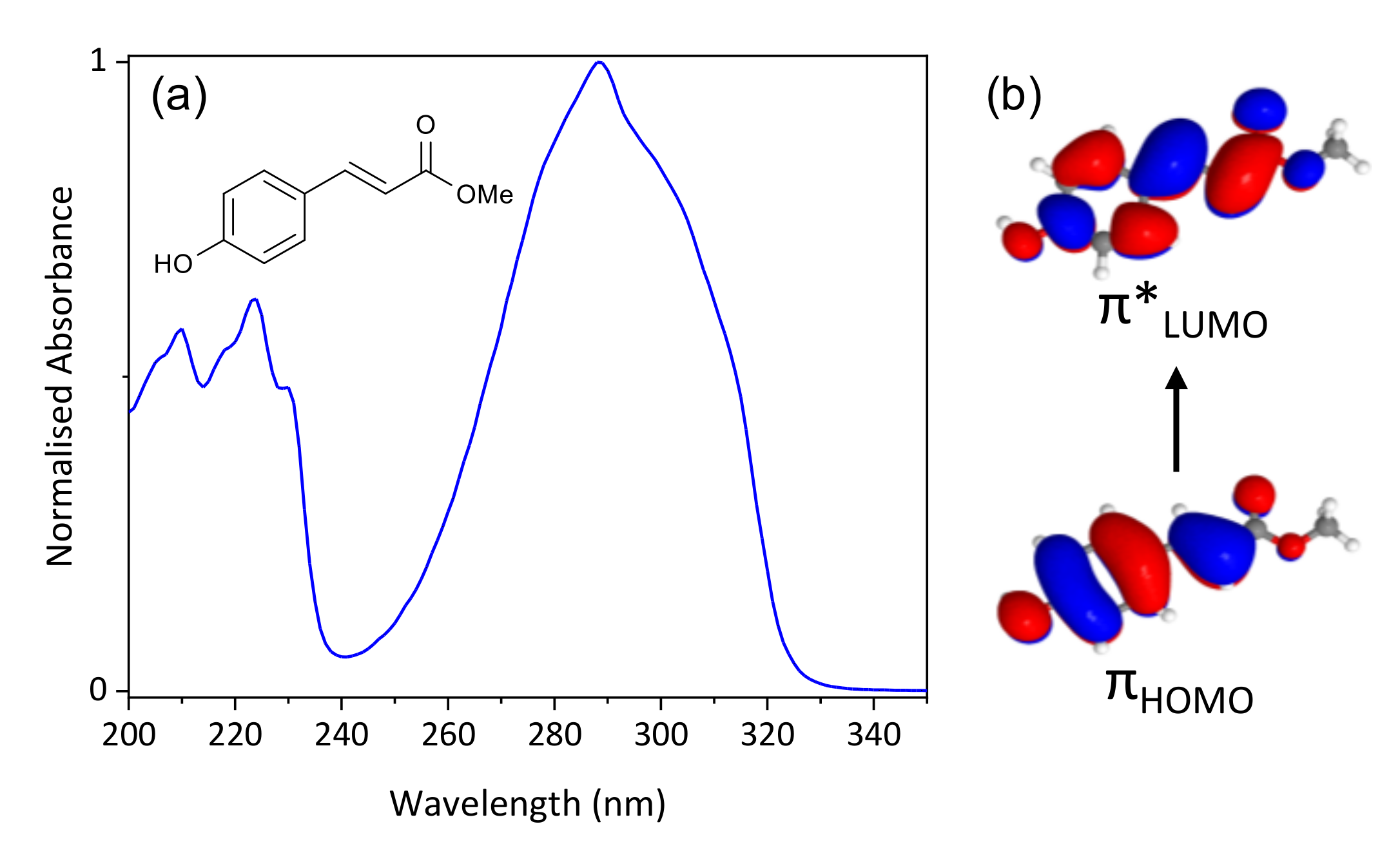
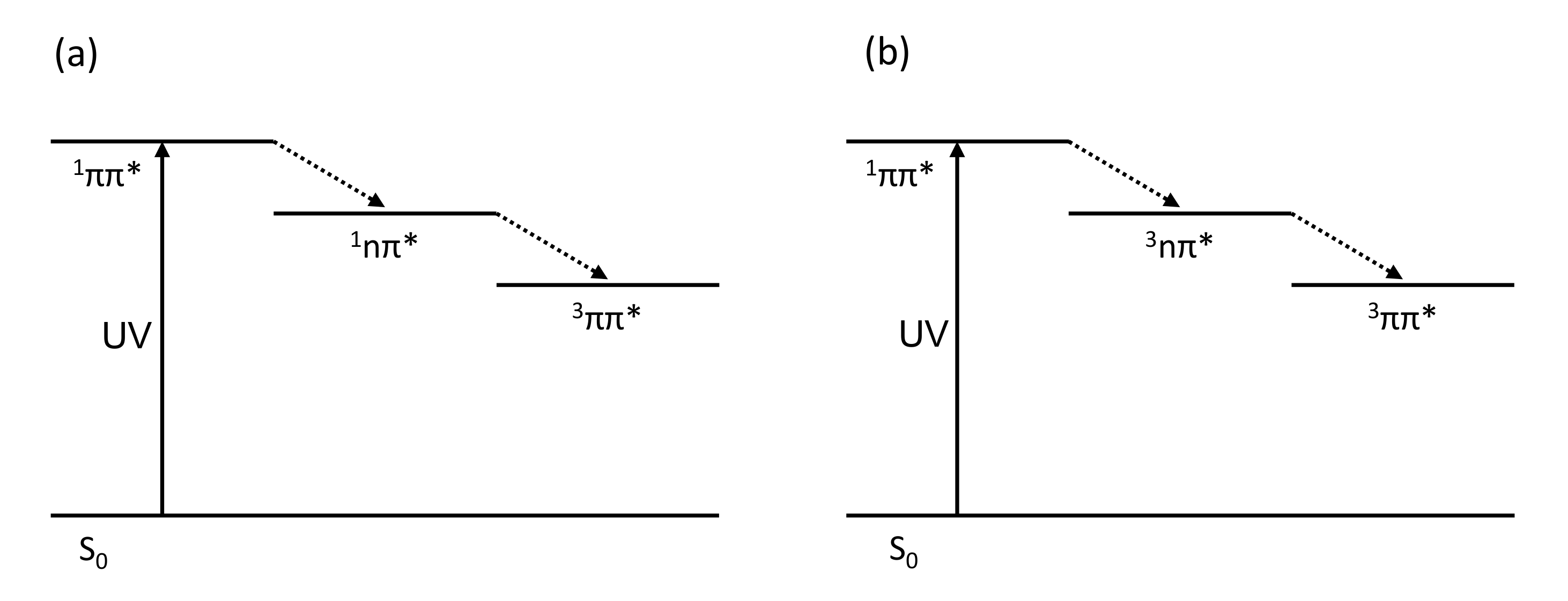
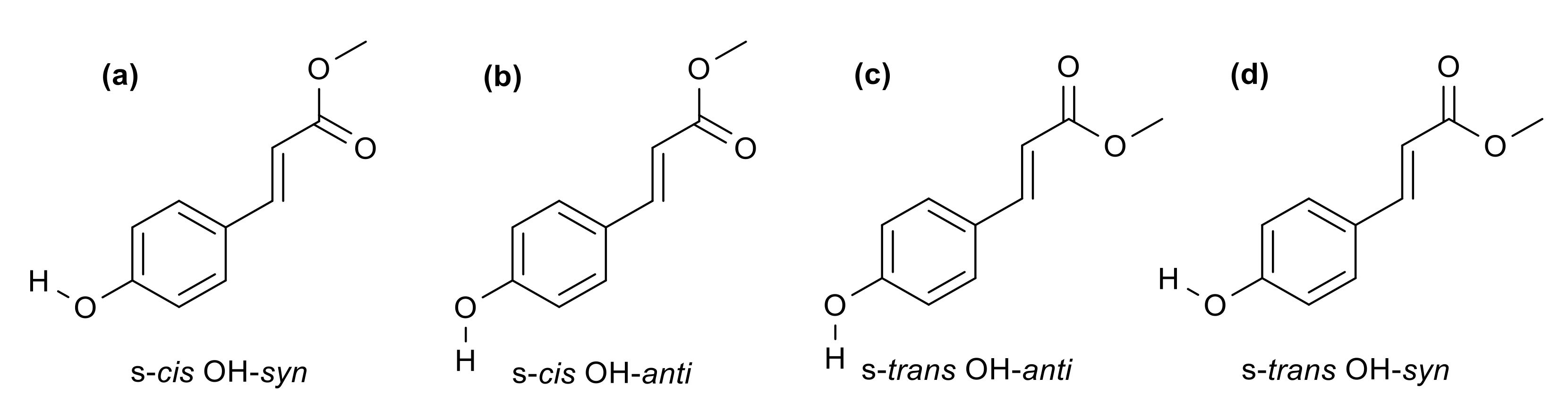
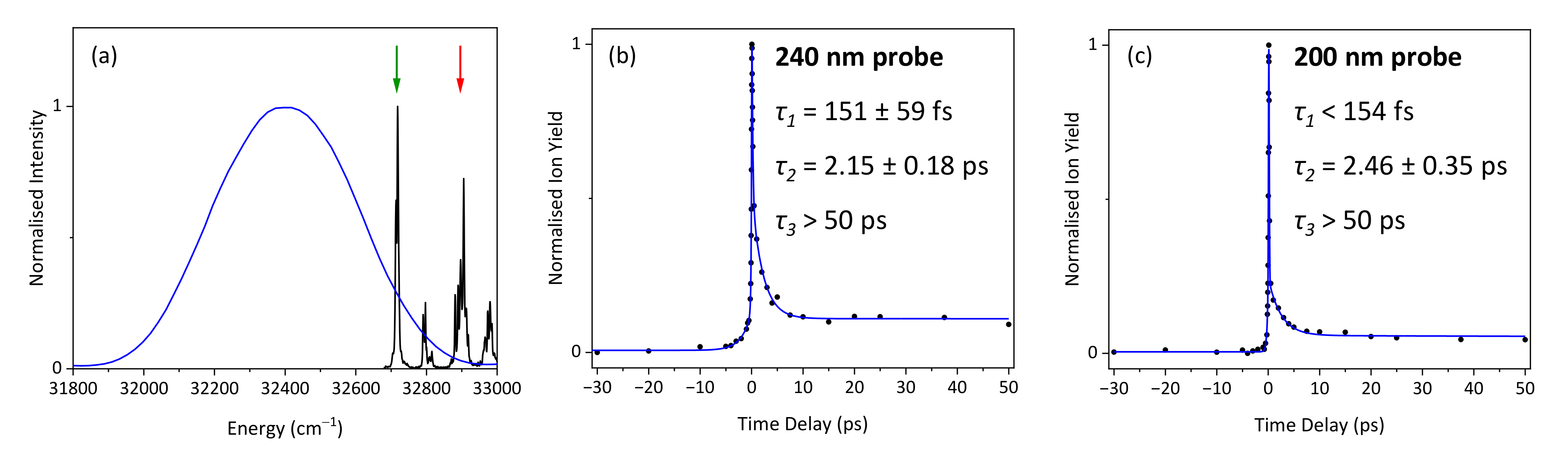
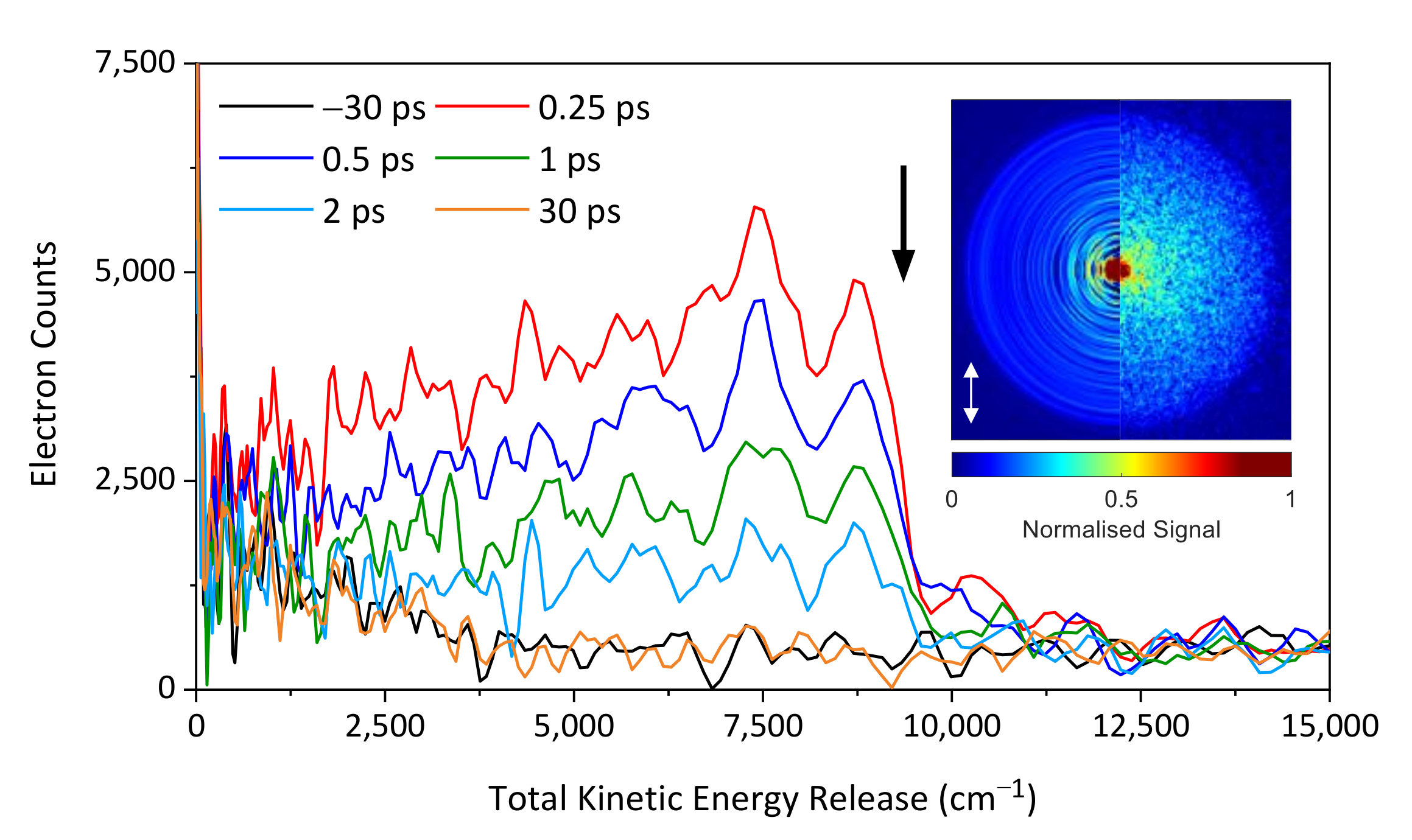
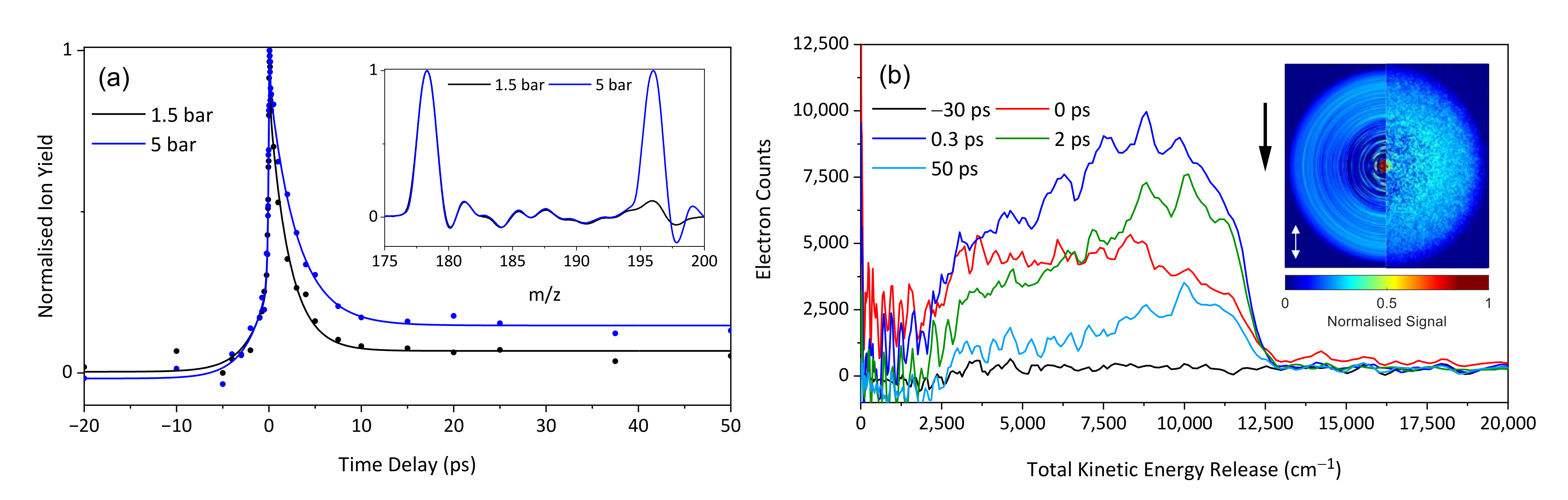
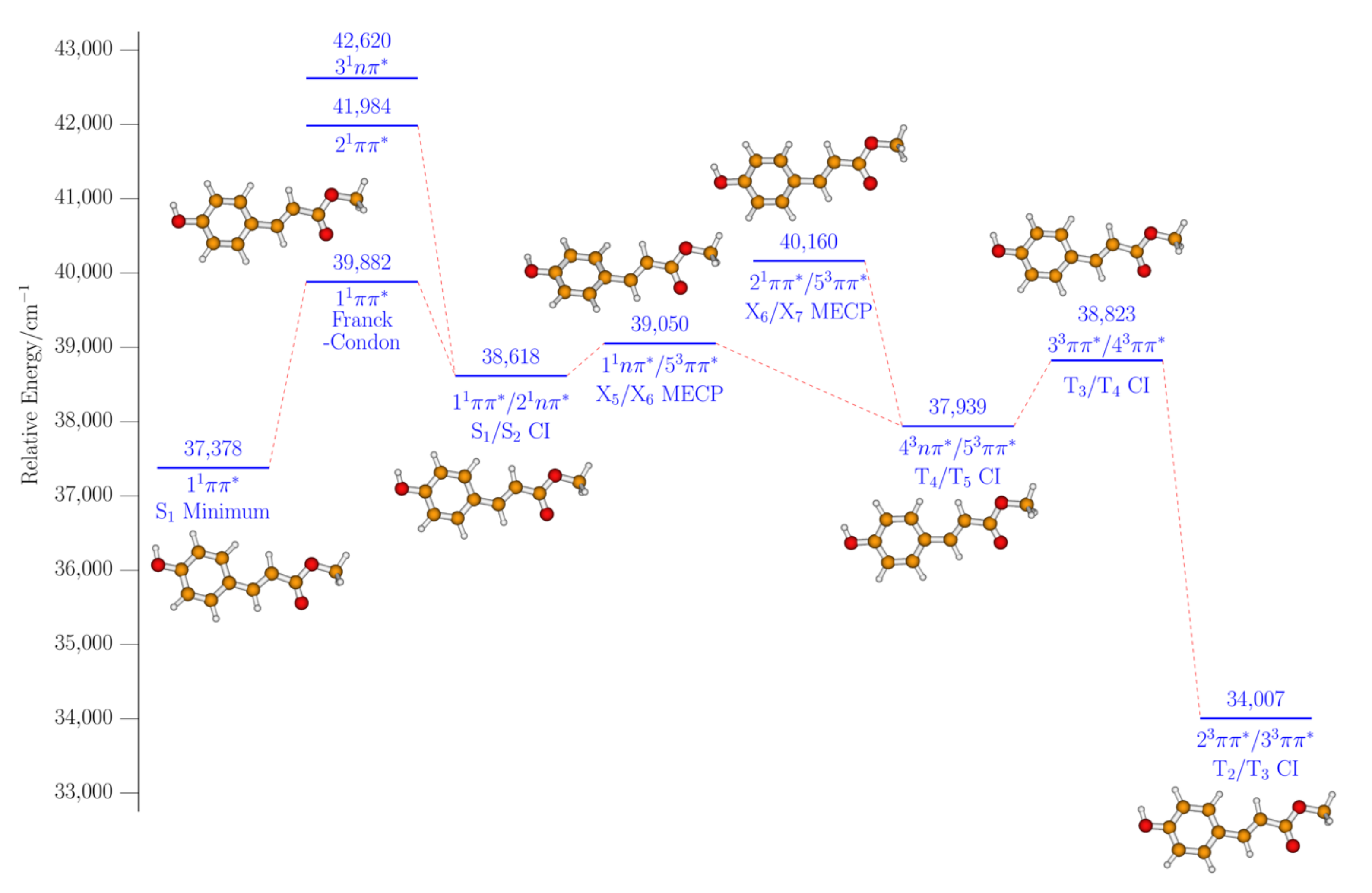
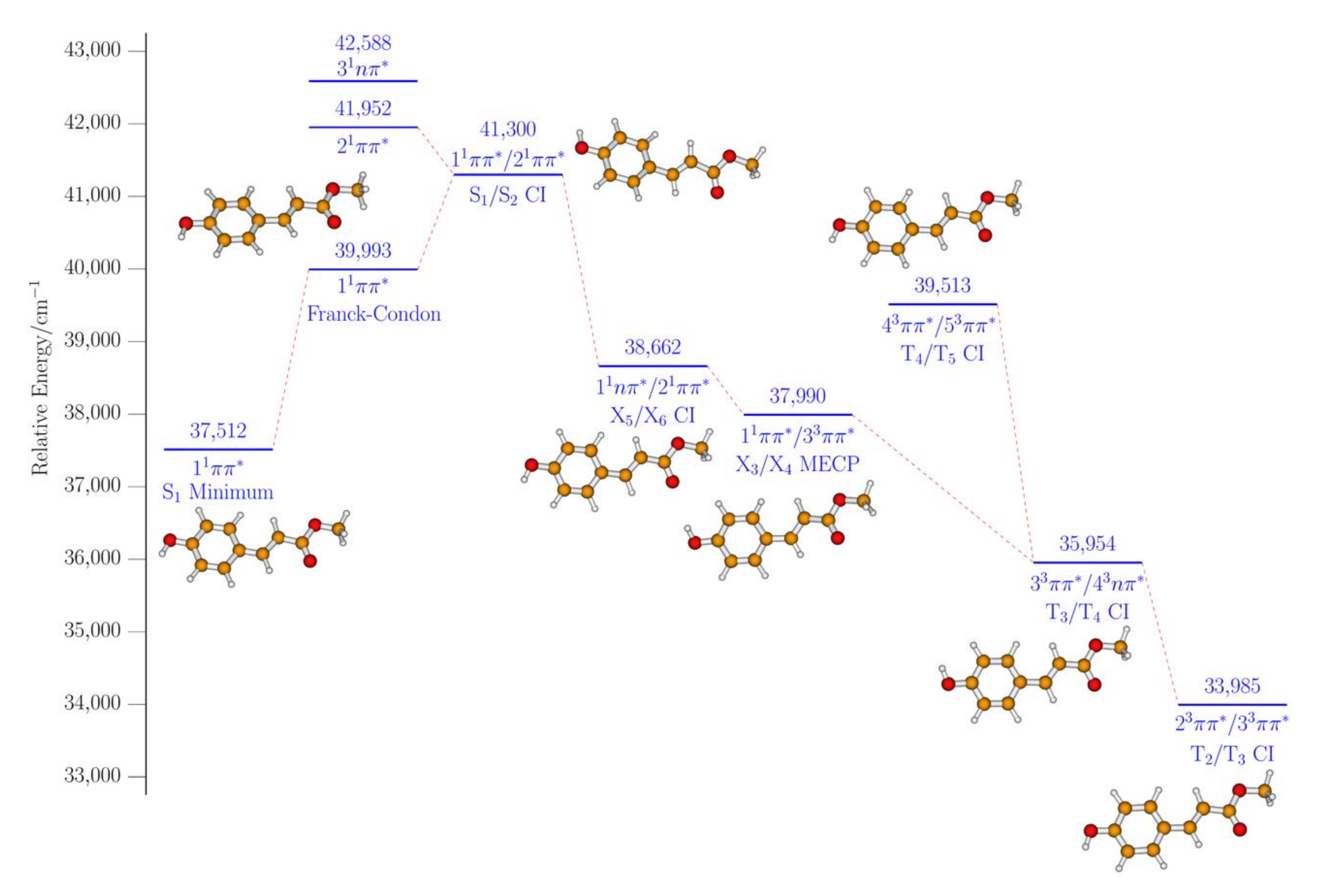
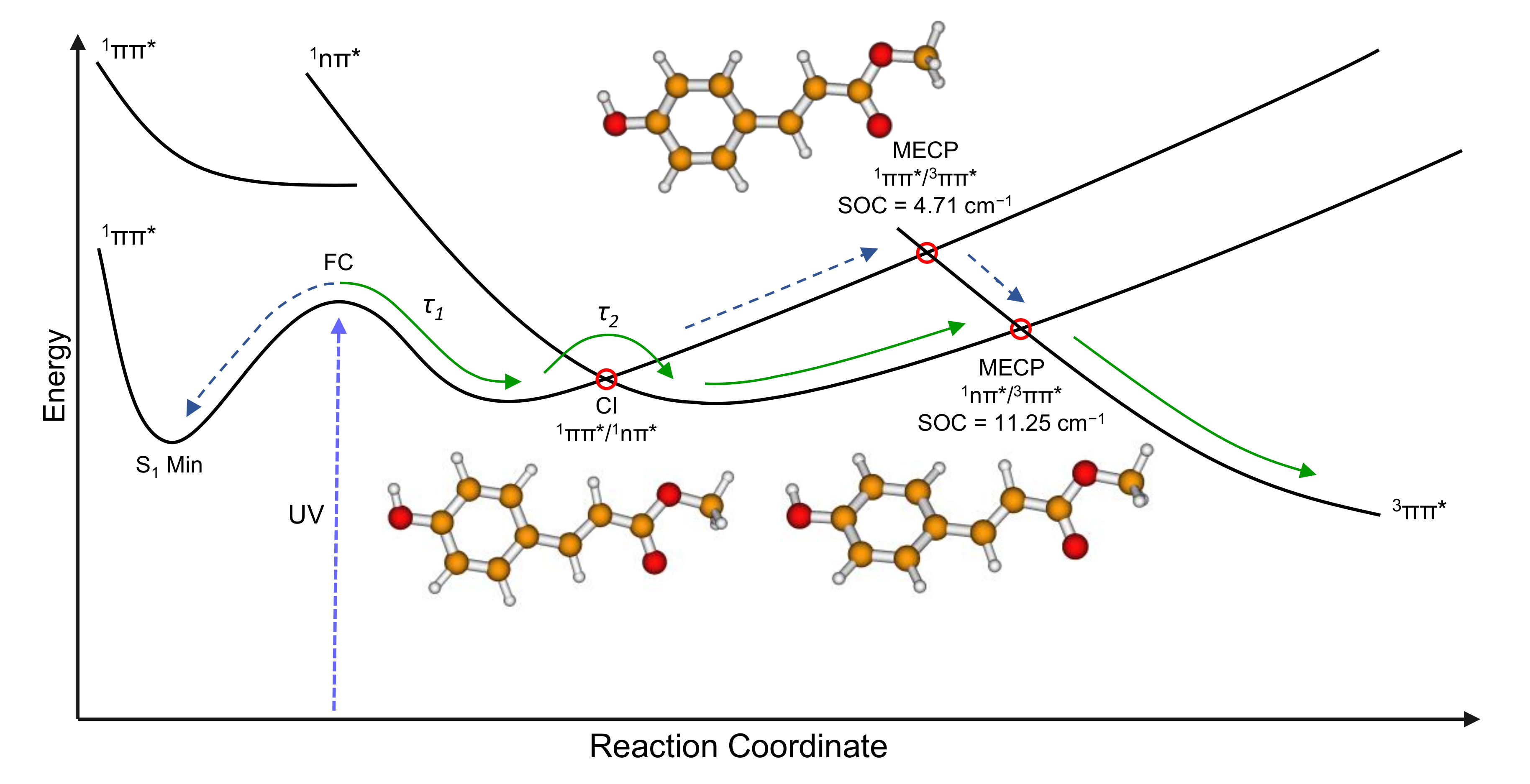
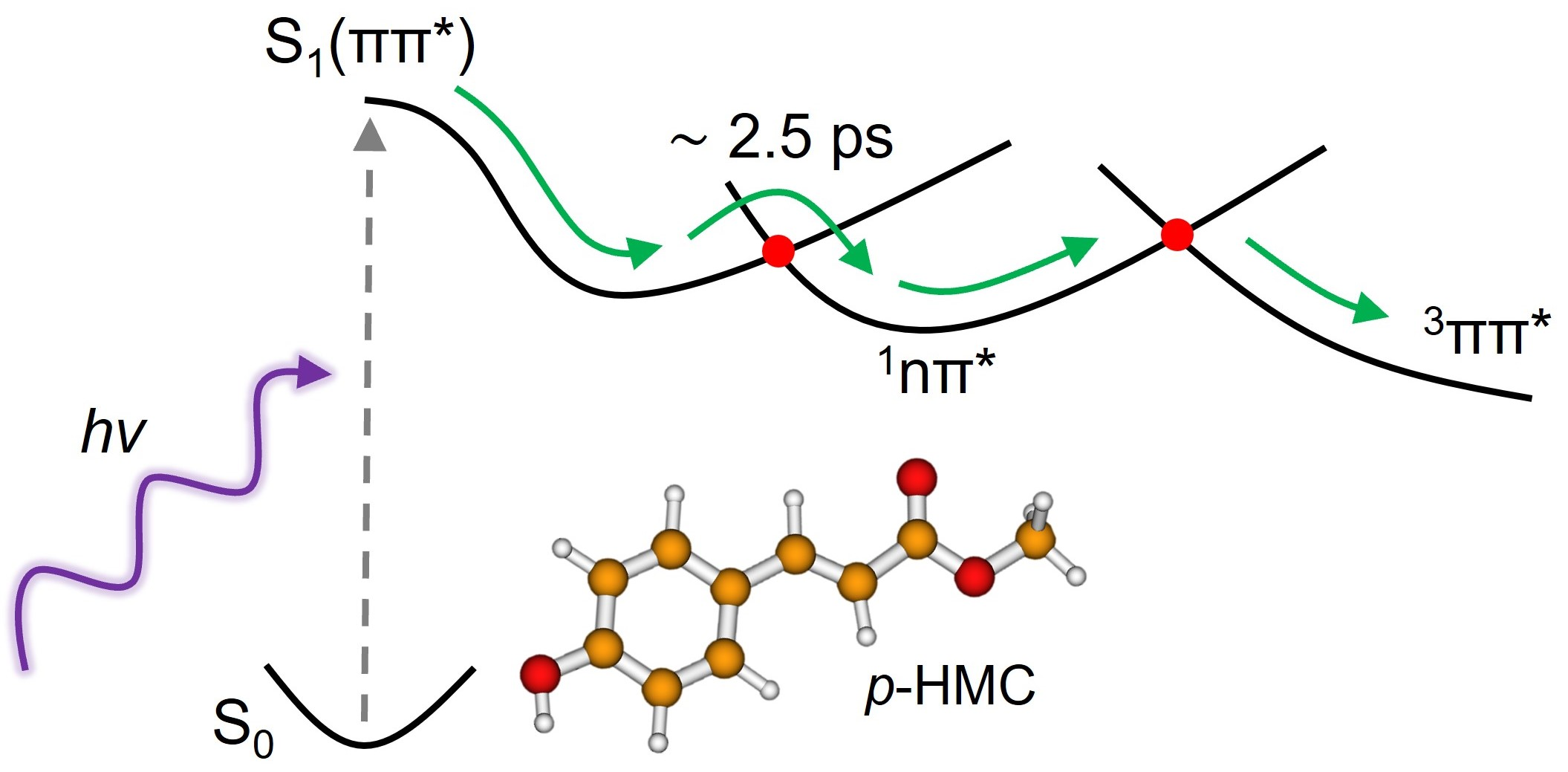
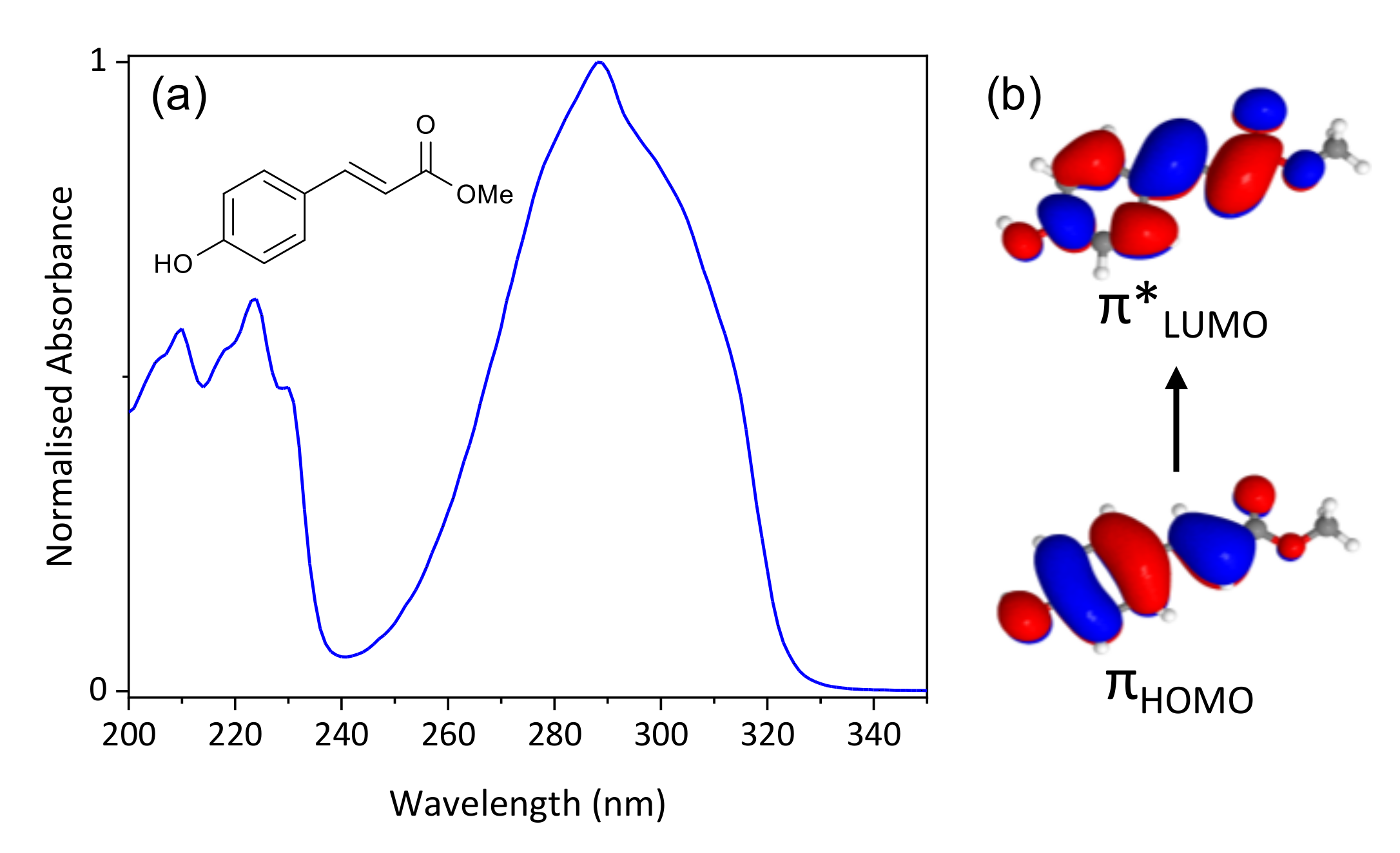
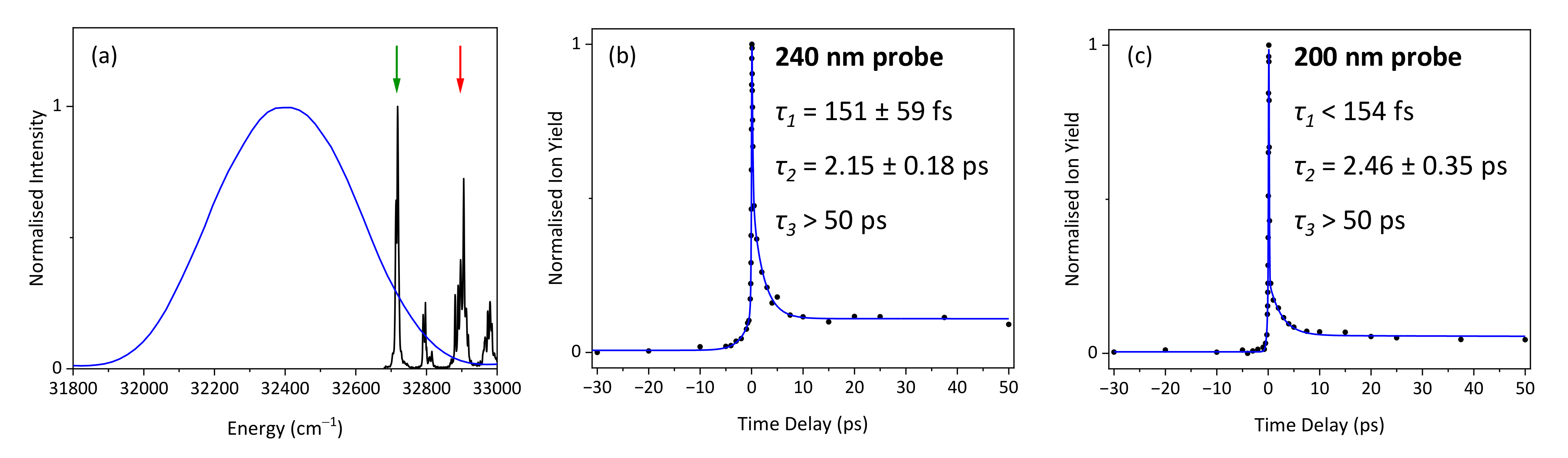
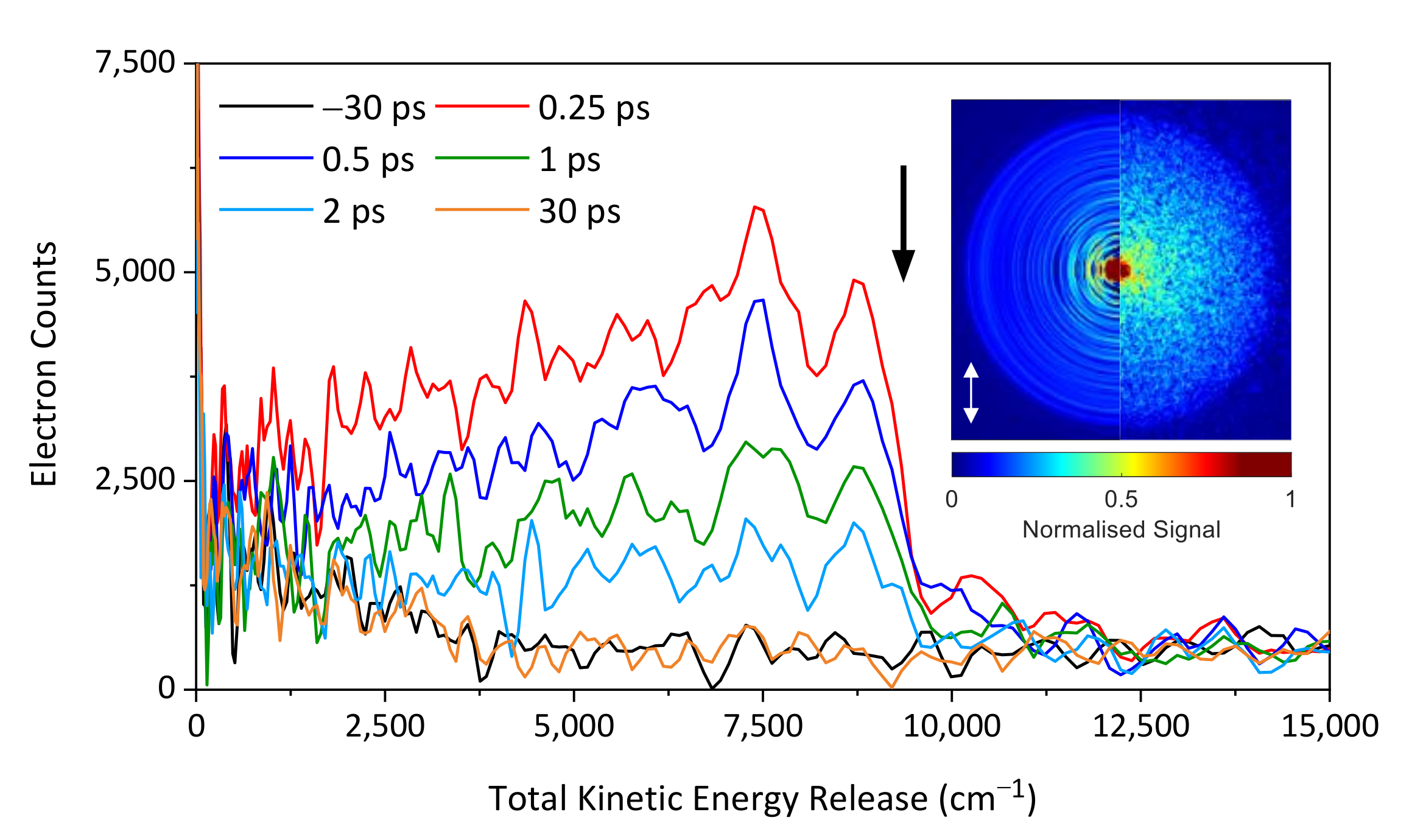
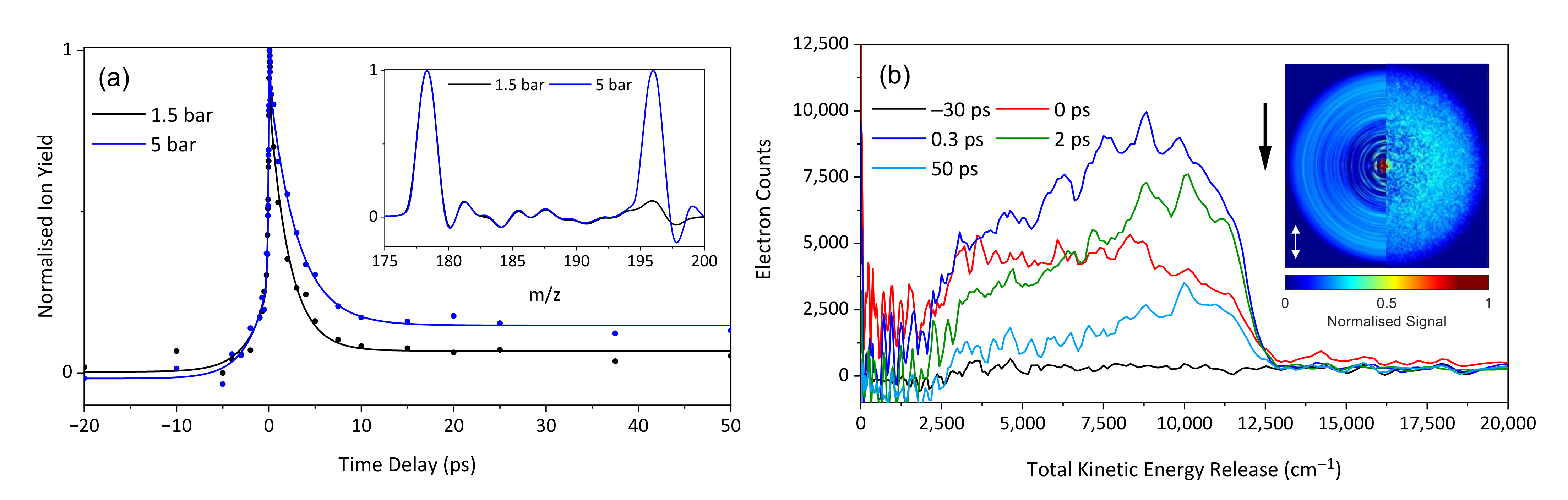
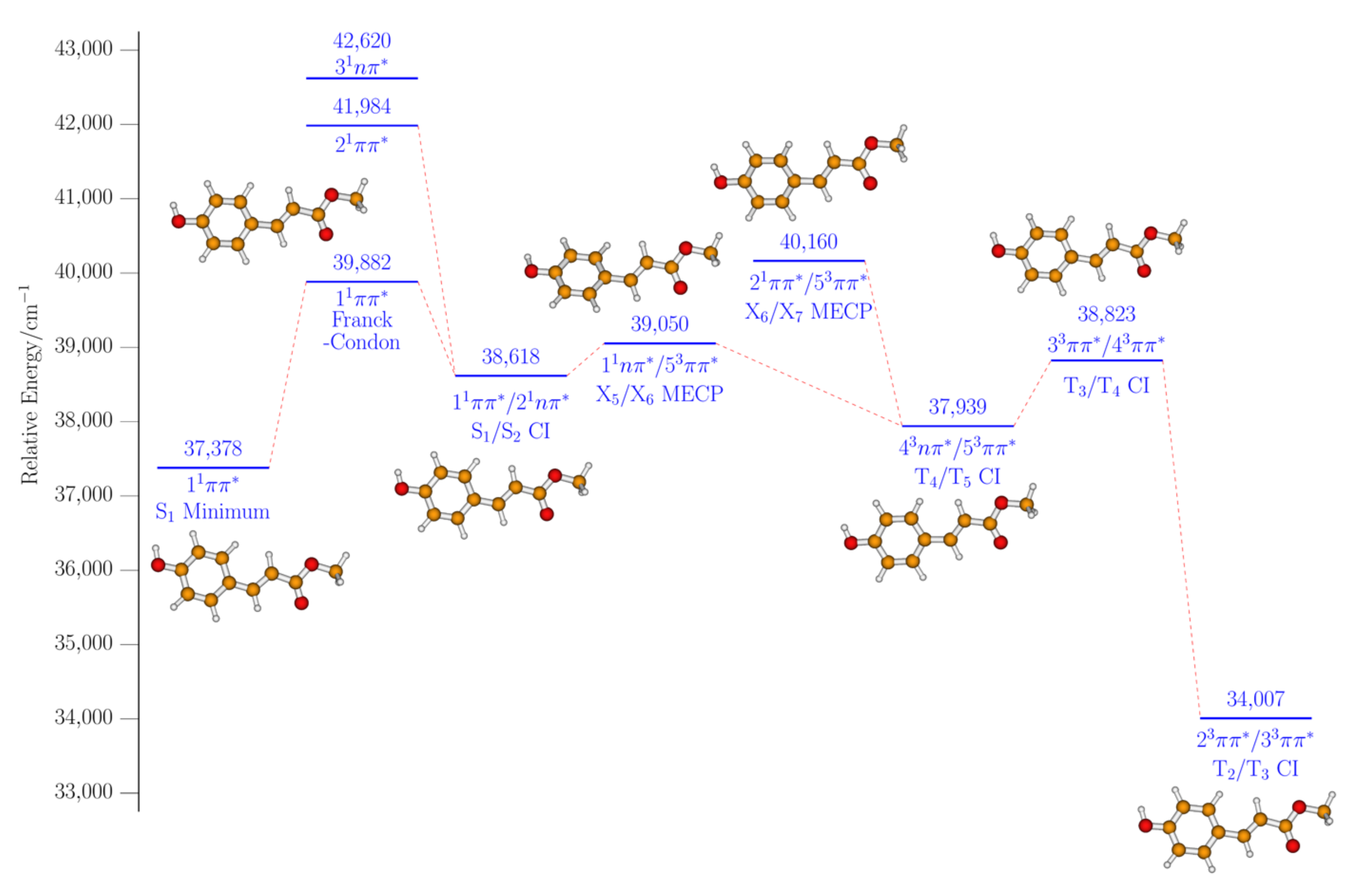
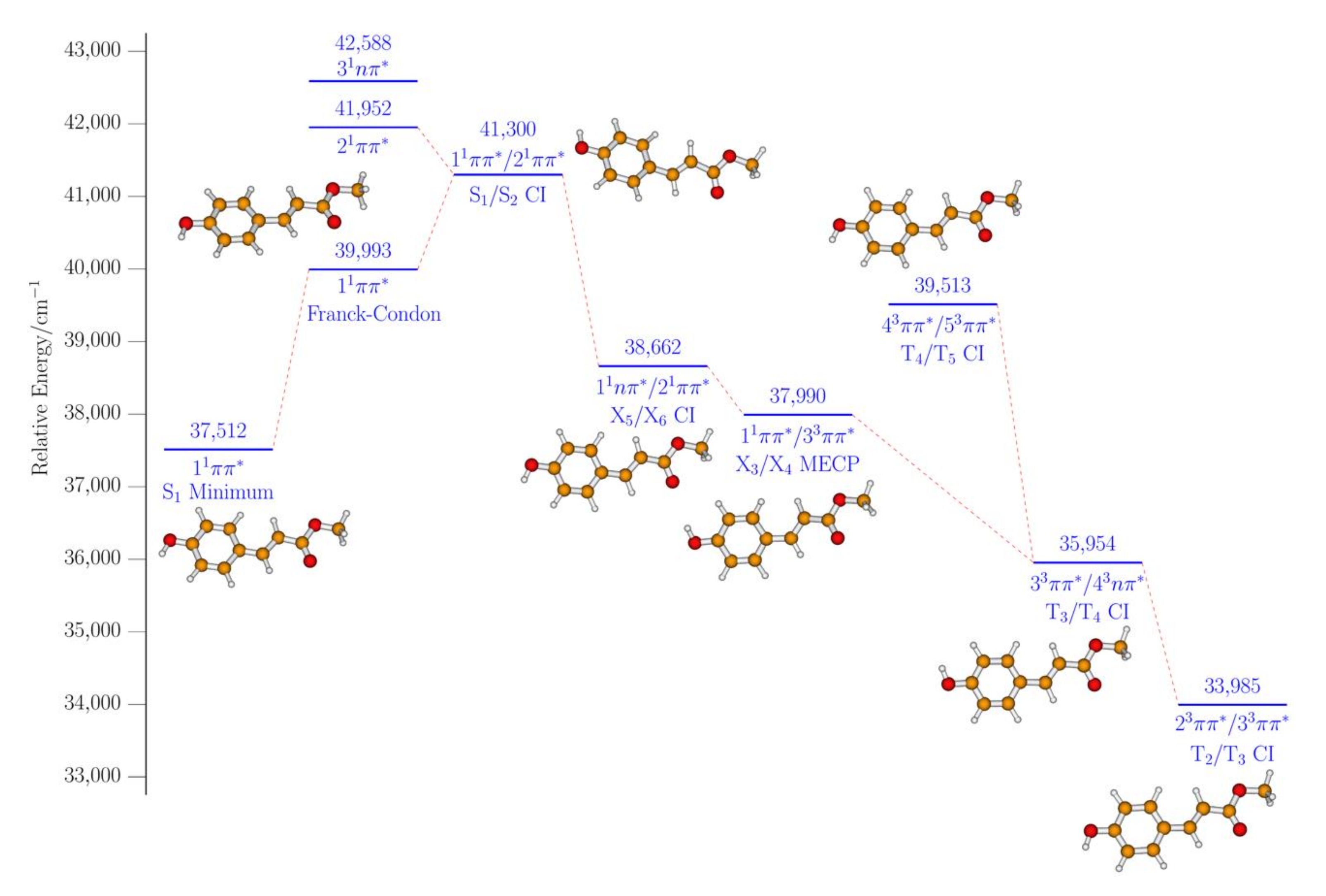
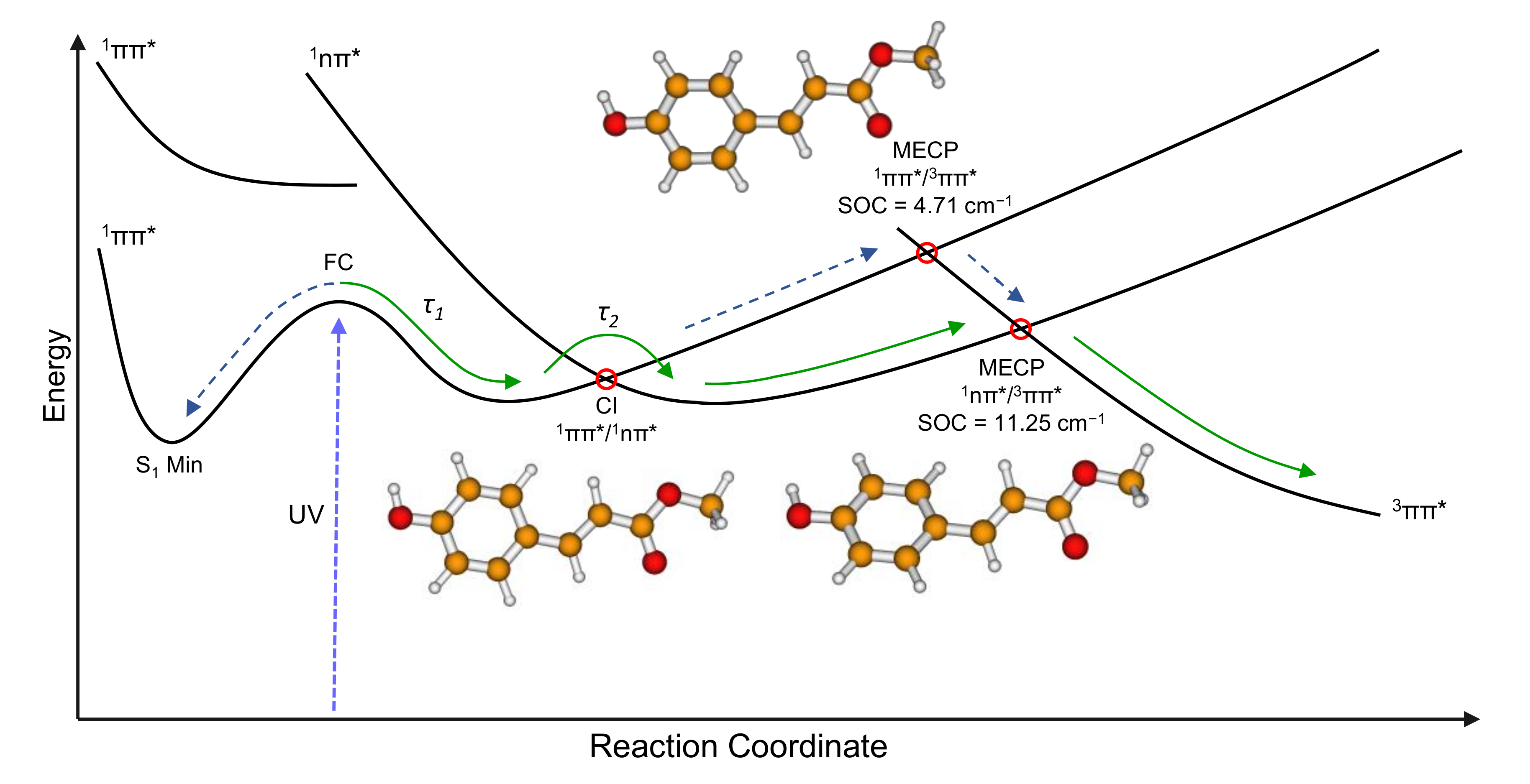
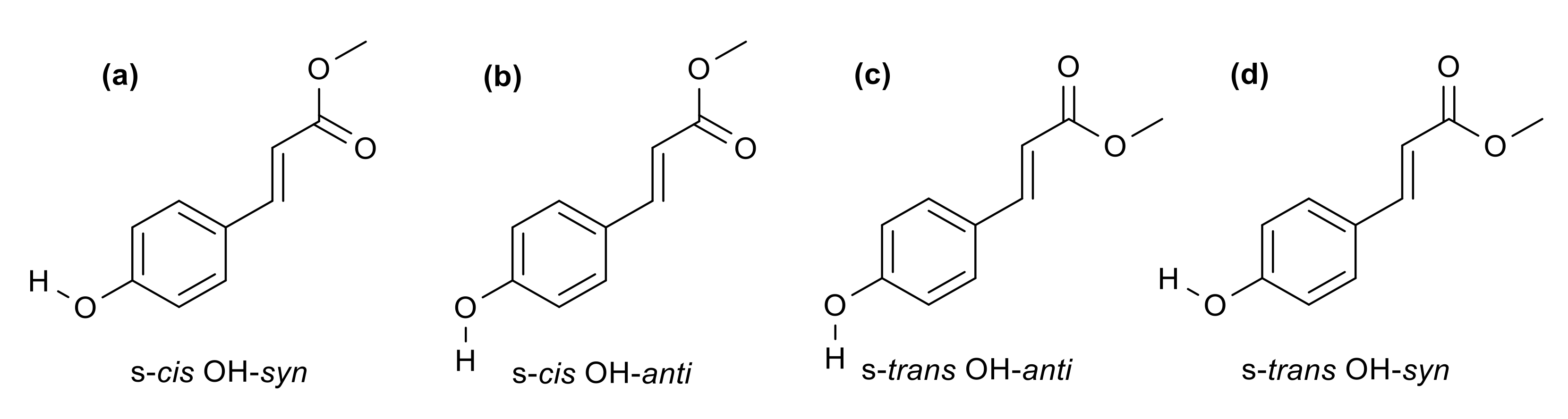
2. Results and Discussion
2.1. Experimental
2.2. Computational
3. Materials and Methods
3.1. Experimental Setup
3.2. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kinoshita, S.N.; Harabuchi, Y.; Inokuchi, Y.; Maeda, S.; Ehara, M.; Yamazaki, K.; Ebata, T. Substitution Effect on the Nonradiative Decay and Trans Cis Photoisomerization Route: A Guideline to Develop Efficient Cinnamate-Based Sunscreens. Phys. Chem. Chem. Phys. 2021, 23, 834–845. [Google Scholar] [CrossRef] [PubMed]
- Promkatkaew, M.; Suramitr, S.; Karpkird, T.; Wanichwecharungruang, S.; Ehara, M.; Hannongbua, S. Photophysical Properties and Photochemistry of Substituted Cinnamates and Cinnamic Acids for UVB Blocking: Effect of Hydroxy, Nitro, and Fluoro Substitutions at Ortho, Meta, and Para Positions. Photochem. Photobiol. Sci. 2014, 13, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Peperstraete, Y.; Staniforth, M.; Baker, L.A.; Rodrigues, N.D.N.; Cole-Filipiak, N.C.; Quan, W.D.; Stavros, V.G. Bottom-up Excited State Dynamics of Two Cinnamate-Based Sunscreen Filter Molecules. Phys. Chem. Chem. Phys. 2016, 18, 28140–28149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karpkird, T.M.; Wanichweacharungruang, S.; Albinsson, B. Photophysical Characterization of Cinnamates. Photochem. Photobiol. Sci. 2009, 8, 1455–1460. [Google Scholar] [CrossRef] [PubMed]
- Iida, Y.; Kinoshita, S.N.; Kenjo, S.; Muramatsu, S.; Inokuchi, Y.; Zhu, C.; Ebata, T. Electronic States and Nonradiative Decay of Cold Gas-Phase Cinnamic Acid Derivatives Studied by Laser Spectroscopy with a Laser-Ablation Technique. J. Phys. Chem. A 2020, 124, 5580–5589. [Google Scholar] [CrossRef] [PubMed]
- Meyer, T.E. Isolation and Characterization of Soluble Cytochromes, Ferredoxins and Other Chromophoric Proteins from the Halophilic Phototrophic Bacterium Ectothiorhodospira Halophila. Biochim. Biophys. Acta 1985, 806, 175–183. [Google Scholar] [CrossRef]
- Meyer, T.E.; Fitch, J.C.; Bartsch, R.G.; Tollin, G.; Cusanovich, M.A. Soluble Cytochromes and a Photoactive Yellow Protein Isolated from the Moderately Halophilic Purple Phototrophic Bacterium, Rhodospirillum Salexigens. Biochim. Biophys. Acta 1990, 1016, 364–370. [Google Scholar] [CrossRef]
- Meyer, T.E.; Yakali, E.; Cusanovich, M.A.; Tollin, G. Properties of a Water-Soluble, Yellow Protein Isolated from a Halophilic Phototrophic Bacterium That Has Photochemical Activity Analogous to Sensory Rhodopsin. Biochemistry 1987, 26, 418–423. [Google Scholar] [CrossRef] [PubMed]
- Imamoto, Y.; Kataoka, M. Structure and Photoreaction of Photoactive Yellow Protein, a Structural Prototype of the PAS Domain Superfamily. Photochem. Photobiol. 2007, 83, 40–49. [Google Scholar] [CrossRef]
- Carroll, E.C.; Hospes, M.; Valladares, C.; Hellingwerf, K.J.; Larsen, D.S. Is the Photoactive Yellow Protein a UV-B/Blue Light Photoreceptor? Photochem. Photobiol. Sci. 2011, 10, 464–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlichting, I.; Berendzen, J. Out of the Blue: The Photocycle of the Photoactive Yellow Protein. Structure 1997, 5, 735–739. [Google Scholar] [CrossRef] [Green Version]
- Sprenger, W.W.; Hoff, W.D.; Armitage, J.P.; Hellingwerf, K.J. The Eubacterium Ectothiorhodospira Halophila Is Negatively Phototactic, with a Wavelength Dependence That Fits the Absorption Spectrum of the Photoactive Yellow Protein. J. Bacteriol. 1993, 175, 3096–3104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burnett, M.E.; Hu, J.Y.; Wang, S.Q. Sunscreens: Obtaining Adequate Photoprotection. Dermatol. Ther. 2012, 25, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, J.B.; Maruthi, R.; Wang, S.Q.; Lim, H.W. Sunscreens: An Update. Am. J. Clin. Dermatol. 2017, 18, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Palm, M.D.; O’Donoghue, M.N. Update on Photoprotection. Dermatol. Ther. 2007, 20, 360–376. [Google Scholar] [CrossRef] [PubMed]
- De Groot, M.; Gromov, E.V.; Köppel, H.; Buma, W.J. High-Resolution Spectroscopy of Methyl 4-Hydroxycinnamate and Its Hydrogen-Bonded Water Complex. J. Phys. Chem. B 2008, 112, 4427–4434. [Google Scholar] [CrossRef] [PubMed]
- Roberts, G.M.; Stavros, V.G. The Role of Πσ* States in the Photochemistry of Heteroaromatic Biomolecules and Their Subunits: Insights from Gas-Phase Femtosecond Spectroscopy. Chem. Sci. 2014, 5, 1698–1722. [Google Scholar] [CrossRef]
- Sobolewski, A.L.; Domcke, W.; Dedonder-Lardeux, C.; Jouvet, C. Excited-State Hydrogen Detachment and Hydrogen Transfer Driven by Repulsive 1πσ* States: A New Paradigm for Nonradiative Decay in Aromatic Biomolecules. Phys. Chem. Chem. Phys. 2002, 4, 1093–1100. [Google Scholar] [CrossRef]
- Ratzer, C.; Küpper, J.; Spangenberg, D.; Schmitt, M. The Structure of Phenol in the S1-State Determined by High Resolution UV-Spectroscopy. Chem. Phys. 2002, 283, 153–169. [Google Scholar] [CrossRef]
- Dixon, R.N.; Oliver, T.A.A.; Ashfold, M.N.R. Tunnelling under a Conical Intersection: Application to the Product Vibrational State Distributions in the UV Photodissociation of Phenols. J. Chem. Phys. 2011, 134, 194303. [Google Scholar] [CrossRef] [PubMed]
- Smolarek, S.; Vdovin, A.; Tan, E.M.M.; De Groot, M.; Buma, W.J. Spectroscopy and Dynamics of Methyl-4-Hydroxycinnamate: The Influence of Isotopic Substitution and Water Complexation. Phys. Chem. Chem. Phys. 2011, 13, 4393–4399. [Google Scholar] [CrossRef] [PubMed]
- Smolarek, S.; Vdovin, A.; Perrier, D.L.; Smit, J.P.; Drabbels, M.; Buma, W.J. High-Resolution Excitation and Absorption Spectroscopy of Gas-Phase p-Coumaric Acid: Unveiling an Elusive Chromophore. J. Am. Chem. Soc. 2010, 132, 6315–6317. [Google Scholar] [CrossRef] [PubMed]
- Krokidi, K.M.; Turner, M.A.P.; Pearcy, P.A.J.; Stavros, V.G. A Systematic Approach to Methyl Cinnamate Photodynamics. Mol. Phys. 2021, 119, e1811910. [Google Scholar] [CrossRef]
- Kinoshita, S.N.; Inokuchi, Y.; Onitsuka, Y.; Kohguchi, H.; Akai, N.; Shiraogawa, T.; Ehara, M.; Yamazaki, K.; Harabuchi, Y.; Maeda, S.; et al. The Direct Observation of the Doorway 1nπ∗ State of Methylcinnamate and Hydrogen-Bonding Effects on the Photochemistry of Cinnamate-Based Sunscreens. Phys. Chem. Chem. Phys. 2019, 21, 19755–19763. [Google Scholar] [CrossRef]
- Yamazaki, K.; Miyazaki, Y.; Harabuchi, Y.; Taketsugu, T.; Maeda, S.; Inokuchi, Y.; Kinoshita, S.N.; Sumida, M.; Onitsuka, Y.; Kohguchi, H.; et al. Multistep Intersystem Crossing Pathways in Cinnamate-Based UV-B Sunscreens. J. Phys. Chem. Lett. 2016, 7, 4001–4007. [Google Scholar] [CrossRef]
- Tan, E.M.M.; Amirjalayer, S.; Bakker, B.H.; Buma, W.J. Excited State Dynamics of Photoactive Yellow Protein Chromophores Elucidated by High-Resolution Spectroscopy and Ab Initio Calculations. Faraday Discuss. 2013, 163, 321–340. [Google Scholar] [CrossRef] [Green Version]
- Shimada, D.; Kusaka, R.; Inokuchi, Y.; Ehara, M.; Ebata, T. Nonradiative Decay Dynamics of Methyl-4-Hydroxycinnamate and Its Hydrated Complex Revealed by Picosecond Pump-Probe Spectroscopy. Phys. Chem. Chem. Phys. 2012, 14, 8999–9005. [Google Scholar] [CrossRef] [Green Version]
- Kinoshita, S.N.; Miyazaki, Y.; Sumida, M.; Onitsuka, Y.; Kohguchi, H.; Inokuchi, Y.; Akai, N.; Shiraogawa, T.; Ehara, M.; Yamazaki, K.; et al. Different Photoisomerization Routes Found in the Structural Isomers of Hydroxy Methylcinnamate. Phys. Chem. Chem. Phys. 2018, 20, 17583–17598. [Google Scholar] [CrossRef]
- Kotsina, N.; Candelaresi, M.; Saalbach, L.; Zawadzki, M.M.; Crane, S.W.; Sparling, C.; Townsend, D. Short-Wavelength Probes in Time-Resolved Photoelectron Spectroscopy: An Extended View of the Excited State Dynamics in Acetylacetone. Phys. Chem. Chem. Phys. 2020, 22, 4647–4658. [Google Scholar] [CrossRef] [PubMed]
- De Camillis, S.; Miles, J.; Alexander, G.; Ghafur, O.; Williams, I.D.; Townsend, D.; Greenwood, J.B. Ultrafast Non-Radiative Decay of Gas-Phase Nucleosides. Phys. Chem. Chem. Phys. 2015, 17, 23643–23650. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.-Z.; Qin, C.-C.; Zhang, S.; Wang, Y.-M.; Zhang, B. Ultrafast Dynamics of the First Excited State of Chlorobenzene. Acta Phys. Chim. Sin. 2011, 27, 965–970. [Google Scholar]
- El-Sayed, M.A. Spin—Orbit Coupling and the Radiationless Processes in Nitrogen Heterocyclics. J. Chem. Phys. 1963, 38, 2834–2838. [Google Scholar] [CrossRef]
- Iqbal, A.; Pegg, L.J.; Stavros, V.G. Direct versus Indirect H Atom Elimination from Photoexcited Phenol Molecules. J. Phys. Chem. A 2008, 112, 9531–9534. [Google Scholar] [CrossRef] [PubMed]
- Even, U. The Even-Lavie Valve as a Source for High Intensity Supersonic Beam. EPJ Tech. Instrum. 2015, 2, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Even, U.; Jortner, J.; Noy, D.; Lavie, N.; Cossart-Magos, C. Cooling of Large Molecules below 1 K and He Clusters Formation. J. Chem. Phys. 2000, 112, 8068–8071. [Google Scholar] [CrossRef] [Green Version]
- Eppink, A.T.J.B.; Parker, D.H. Velocity Map Imaging of Ions and Electrons Using Electrostatic Lenses: Application in Photoelectron and Photofragment Ion Imaging of Molecular Oxygen. Rev. Sci. Instrum. 1997, 68, 3477–3484. [Google Scholar] [CrossRef]
- Roberts, G.M.; Nixon, J.L.; Lecointre, J.; Wrede, E.; Verlet, J.R.R. Toward Real-Time Charged-Particle Image Reconstruction Using Polar Onion-Peeling. Rev. Sci. Instrum. 2009, 80, 053104. [Google Scholar] [CrossRef] [Green Version]
- Compton, R.N.; Miller, J.C.; Carter, A.E.; Kruit, P. Resonantly Enhanced Multiphoton Ionization of Xenon: Photoelectron Energy Analysis. Chem. Phys. Lett. 1980, 71, 87–90. [Google Scholar] [CrossRef]
- Neese, F. The ORCA Program System. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Ekström, U.; Visscher, L.; Bast, R.; Thorvaldsen, A.J.; Ruud, K. Arbitrary-Order Density Functional Response Theory from Automatic Differentiation. J. Chem. Theory Comput. 2010, 6, 1971–1980. [Google Scholar] [CrossRef]
- Weigend, F. Accurate Coulomb-Fitting Basis Sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-Consistent Molecular Orbital Methods. XX. A Basis Set for Correlated Wave Functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Frisch, M.J.; Pople, J.A.; Binkley, J.S. Self-Consistent Molecular Orbital Methods 25. Supplementary Functions for Gaussian Basis Sets. J. Chem. Phys. 1984, 80, 3265–3269. [Google Scholar] [CrossRef]
- De Souza, B.; Farias, G.; Neese, F.; Izsák, R. Predicting Phosphorescence Rates of Light Organic Molecules Using Time-Dependent Density Functional Theory and the Path Integral Approach to Dynamics. J. Chem. Theory Comput. 2019, 15, 1896–1904. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Initial Geometry | MECP | Spin Orbit Coupling Values (cm−1) | |||
|---|---|---|---|---|---|
| S→T (x) | S→T (y) | S→T (z) | Magnitude | ||
| Anti | 11ππ*/33ππ* (S1/T3) | 0.66i | 0.52i | −0.95i | 1.27 |
| 11ππ*/63ππ* (S1/T6) | 0.01i | 0.00 | −0.02i | 0.02 | |
| 21ππ*2/63ππ* (S2/T6) | 0.00 | 0.00 | 0.01i | 0.01 | |
| 31ππ*/73ππ* (S3/T7) | 0.00 | 0.00 | −0.01i | 0.01 | |
| Syn | 11ππ*/33ππ* (S1/T3) | 0.02i | 0.71i | −0.06i | 0.71 |
| 11ππ*/43ππ* (S1/T4) | 0.19i | −0.53i | −0.02i | 0.56 | |
| 11nπ*/53ππ* (S1/T5) | 4.29i | 10.28i | 1.58i | 11.25 | |
| 21ππ*/53ππ* (S2/T5) | −0.59i | 4.57i | 0.99i | 4.71 | |
| 21ππ*/63ππ* (S2/T6) | 0.01i | 0.00 | 0.00 | 0.01 | |
| 31ππ*/73ππ* (S3/T7) | −0.01i | 0.00 | 0.00 | 0.01 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dalton, J.; Richings, G.W.; Woolley, J.M.; Abiola, T.T.; Habershon, S.; Stavros, V.G. Experimental and Computational Analysis of Para-Hydroxy Methylcinnamate following Photoexcitation. Molecules 2021, 26, 7621. https://doi.org/10.3390/molecules26247621
Dalton J, Richings GW, Woolley JM, Abiola TT, Habershon S, Stavros VG. Experimental and Computational Analysis of Para-Hydroxy Methylcinnamate following Photoexcitation. Molecules. 2021; 26(24):7621. https://doi.org/10.3390/molecules26247621
Chicago/Turabian StyleDalton, Jack, Gareth W. Richings, Jack M. Woolley, Temitope T. Abiola, Scott Habershon, and Vasilios G. Stavros. 2021. "Experimental and Computational Analysis of Para-Hydroxy Methylcinnamate following Photoexcitation" Molecules 26, no. 24: 7621. https://doi.org/10.3390/molecules26247621
APA StyleDalton, J., Richings, G. W., Woolley, J. M., Abiola, T. T., Habershon, S., & Stavros, V. G. (2021). Experimental and Computational Analysis of Para-Hydroxy Methylcinnamate following Photoexcitation. Molecules, 26(24), 7621. https://doi.org/10.3390/molecules26247621