
Tetraalkyl Hydroxymethylene-bisphosphonate and Dialkyl 1-Diphenylphosphinoyl-1-hydroxy-ethylphosphonate Derivatives by the Pudovik Reaction and Their Rearranged Products



Abstract
:1. Introduction
2. Results and Discussion
3. Experimental
3.1. General
3.2. Preparation of the α-Oxoethylphosphonates (1 and 6)
3.3. Synthesis of the Target Compounds
3.3.1. Tetraethyl α-Hydroxy-ethylidenebisphosphonate (2)
3.3.2. Diethyl 1-(Diethylphosphonoylethyl)phosphate (3)
3.3.3. Diethyl-Dimethyl α-Hydroxy-ethylidenebisphosphonate (4)
3.3.4. Dimethyl 1-(Diethylphosphonoylethyl)phosphate (5-1) and Diethyl 1-(Dimethylphosphonoylethyl)phosphate (5-2)
3.3.5. Diethyl (Diethylphosphonoylbenzyl)phosphate (8)
3.3.6. Dimethyl (Diethylphosphonoylbenzyl)phosphate (10-1) and Dimthylphosphonoylbenzyl)phosphate (10-2)
3.3.7. Diethyl 1-Diphenylphosphinoyl-1-hydroxy-ethylphosphonate (11)
3.3.8. Diethyl (Diphenylphosphinoyloxybenzyl)phosphonate and Diethyl (Diphenylphosphinoylbenzyl)phosphate (13-1 and 13-2)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Breuer, E. The development of bisphosphonates as drugs. In Analogue-Based Drug Discovery; Fischer, J., Ganellin, C.R., Eds.; Wiley-VCH: Weinheim, Germany, 2006. [Google Scholar]
- Mucha, A.; Kafarski, P.; Berlicki, L. Remarkable potential of the α-aminophosphonate/phosphinate structural motif in medicinal chemistry. J. Med. Chem. 2011, 54, 5955–5980. [Google Scholar] [CrossRef] [PubMed]
- Grembecka, J.; Mucha, A.; Cierpicki, T.; Kafarski, P. The most potent organophosphorus inhibitors of leucine aminopeptidase. Structure-based design, chemistry, and activity. J. Med. Chem. 2003, 46, 2641–2655. [Google Scholar] [CrossRef] [PubMed]
- Hudson, H.R.; Wardle, N.J.; Bligh, S.W.A.; Greiner, I.; Grün, A.; Keglevich, G. N-Heterocyclic Dronic acids: Applications and synthesis. Mini-Rev. Med. Chem. 2012, 12, 313–325. [Google Scholar] [CrossRef] [PubMed]
- Nagy, D.I.; Grün, A.; Greiner, I.; Keglevich, G. The role of phosphorus trichloride and phoshorous acid in the formation of α-hydroxymethylenebisphosphonic acids from the corresponding carboxylic acids—A mechanistic overview. Curr. Org. Chem. 2017, 21, 1567–1578. [Google Scholar] [CrossRef]
- Nagy, D.I.; Grün, A.; Garadnay, S.; Greiner, I.; Keglevich, G. Synthesis of hydroxymethylenebisphosphonic acid derivatives in different solvents. Molecules 2016, 21, 1046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quin, L.D. A Guide to Organophosphorus Chemistry; Wiley & Sons: New York, NY, USA, 2000. [Google Scholar]
- Rádai, Z.; Keglevich, G. Synthesis and reactions of α-hydroxyphosphonates. Molecules 2018, 23, 1493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McConnell, R.L.; Coover, H.W., Jr. Preparation of 1-hydroxyalkylidenediphosphonates. J. Am. Chem. Soc. 1956, 8, 4450–4452. [Google Scholar] [CrossRef]
- Fitch, S.J.; Moedritzer, K. NMR study of the P-C(OH)-P to P-CO-P rearrangement: Tetraethyl 1-hydroxyalkylidenediphosphonates. J. Am. Chem. Soc. 1962, 84, 1876–1880. [Google Scholar] [CrossRef]
- Nicholson, D.A.; Vaughn, H. A general method of preparation of tetramethyl alkyl-1-hydroxy-1,1-diphosphonates. J. Org. Chem. 1971, 36, 3843–3845. [Google Scholar] [CrossRef]
- Turhanen, P.A.; Ahlgren, M.J.; Jarvinen, T.; Vepsalainen, J.J. Bisphosphonate prodrugs. Synthesis and identification of (1-hydroxyetrylidene)-1,1-bisphosphonic acid tetraesters by mass spectrometry, NMR spectroscopy and X-ray crystallography. Phosphorus Sulfur Silicon 2001, 170, 115–133. [Google Scholar] [CrossRef]
- Turhanen, P.A.; Ahlgren, M.J.; Jarvinen, T.; Vepsalainen, J.J. Bisphosphonate prodrugs, selective synthesis of (1-hydroxyethylidene)-1,1-bisphosphonate partial esters. Synthesis 2001, 4, 633–637. [Google Scholar] [CrossRef]
- Grün, A.; Molnár, I.G.; Bertók, B.; Greiner, I.; Keglevich, G. Synthesis of α-hydroxy-methylenebisphosphonates by the microwave-assisted reaction of α-oxophosphonates and dialkyl phosphites under solventless conditions. Heteroat. Chem. 2009, 20, 350–354. [Google Scholar] [CrossRef]
- Keglevich, G.; Grün, A.; Molnár, I.G.; Greiner, I. Phenyl-, benzyl- and unsymmetrical hydroxy-methylenebisphosphonates as dronic acid ester analogues from α-oxophosphonates by microwave-assisted synthesis. Heteroat. Chem. 2011, 22, 640–648. [Google Scholar] [CrossRef]
- Pallitsch, K.; Roller, A.; Hammerschmidt, F. The stereochemical course of the α-hydroxyphosphonate-phosphate rearrangement. Chem. Eur. J. 2015, 21, 10200–10206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prechelmacher, S.; Mereiter, K.; Hammerschmidt, F. The α-hydroxyphosphonate-phosphate rearrangement of a noncyclic substrate—Some new observations. Org. Biomol. Chem. 2018, 16, 3672–3680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arbuzova, S.N.; Gusarova, N.K.; Bogdanova, M.V.; Ushakov, I.A.; Mal’kina, A.G.; Trofimov, B.A. Nucleophilic addition of secondary phosphine chalcogenides to α,β-acetylenic γ-hydroxy acid nitriles and a rearrangement of the adducts. Mendeleev Commun. 2007, 17, 325–326. [Google Scholar] [CrossRef]
- Hartley, S.B.; Holmes, W.S.; Jacques, J.K.; Mole, M.F.; McCoubrey, J.C. Thermochemical properties of phosphorus compounds. Q. Rev. Chem. Soc. 1963, 17, 204–223. [Google Scholar] [CrossRef]

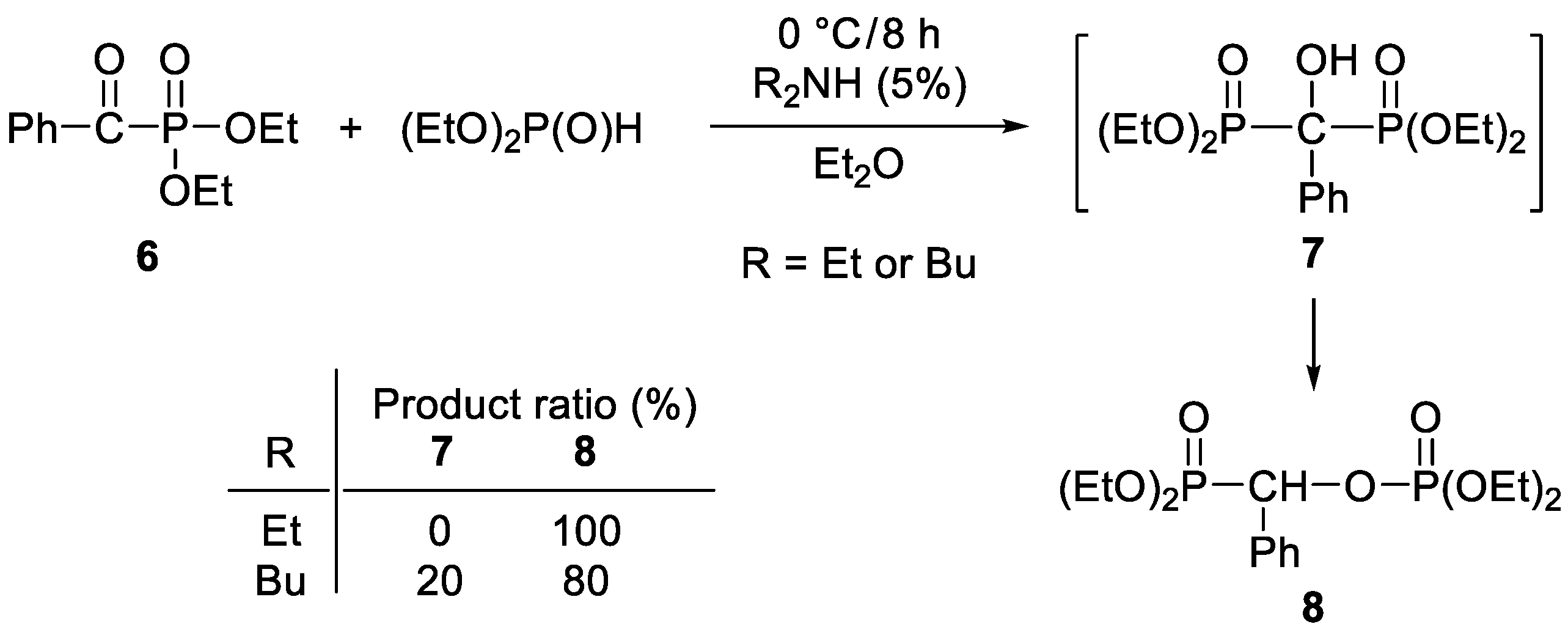
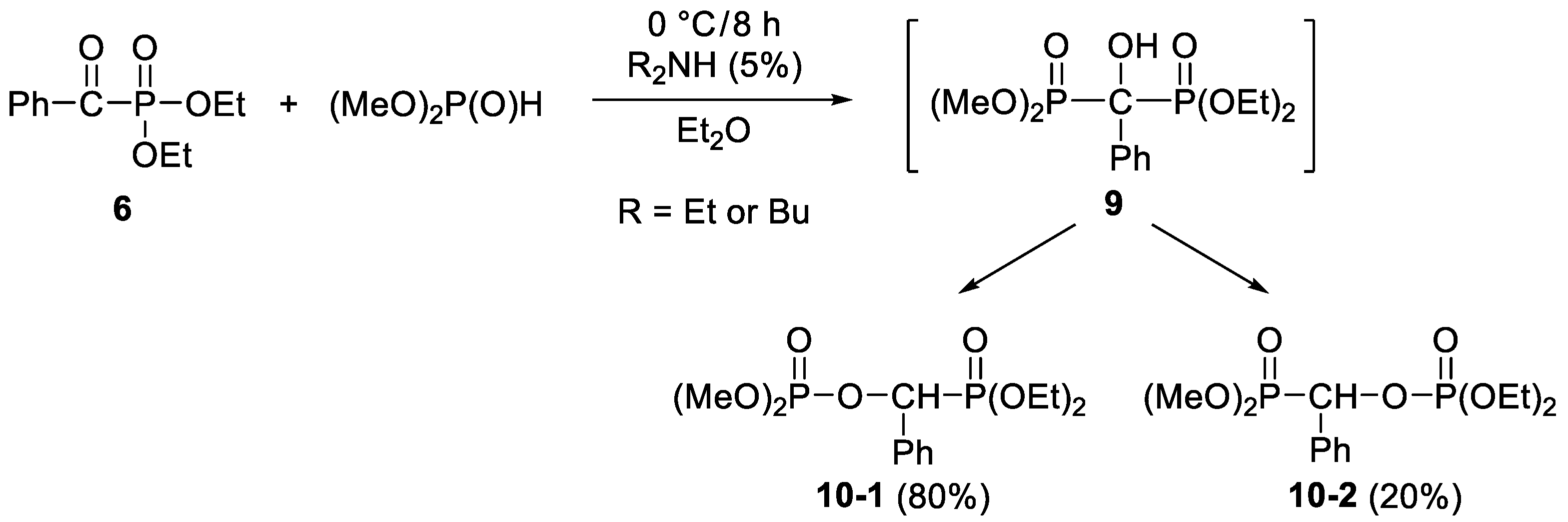

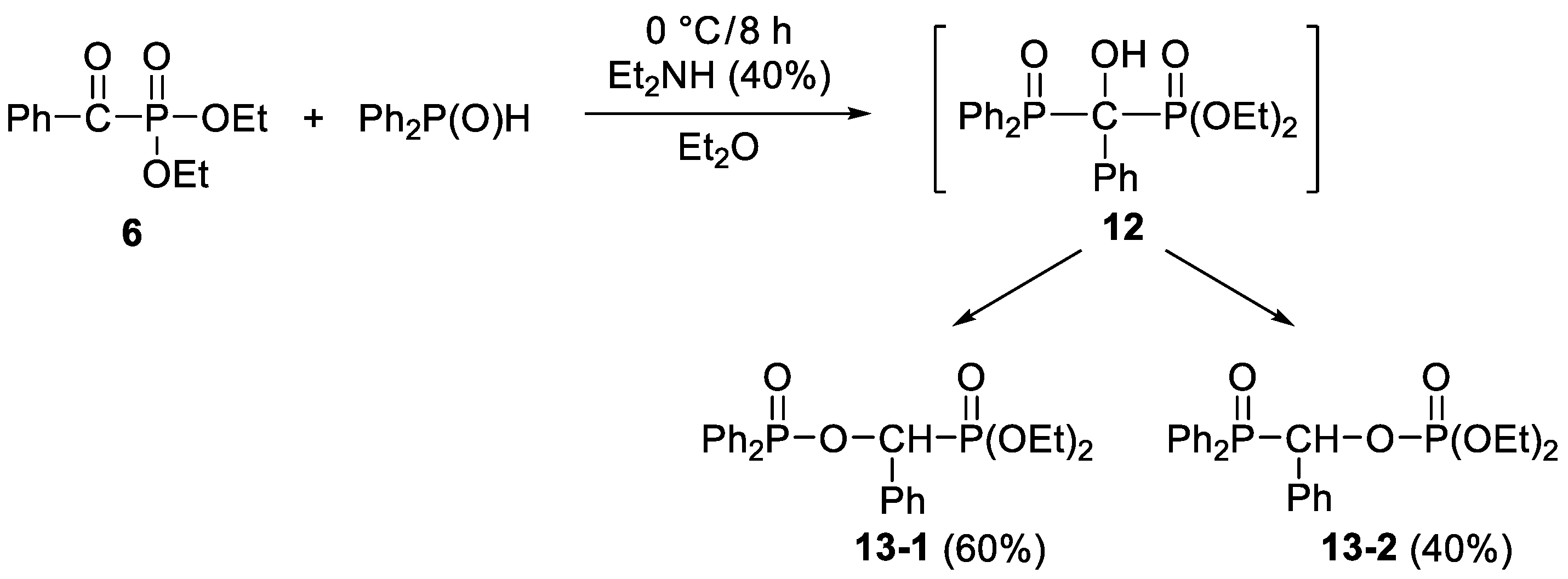

{kind=link}
{kind=link}
{kind=link}
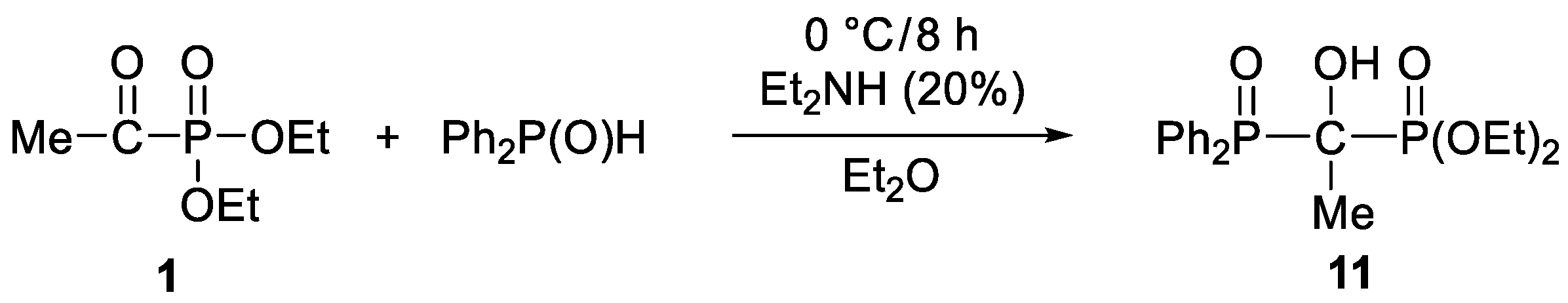{kind=link}
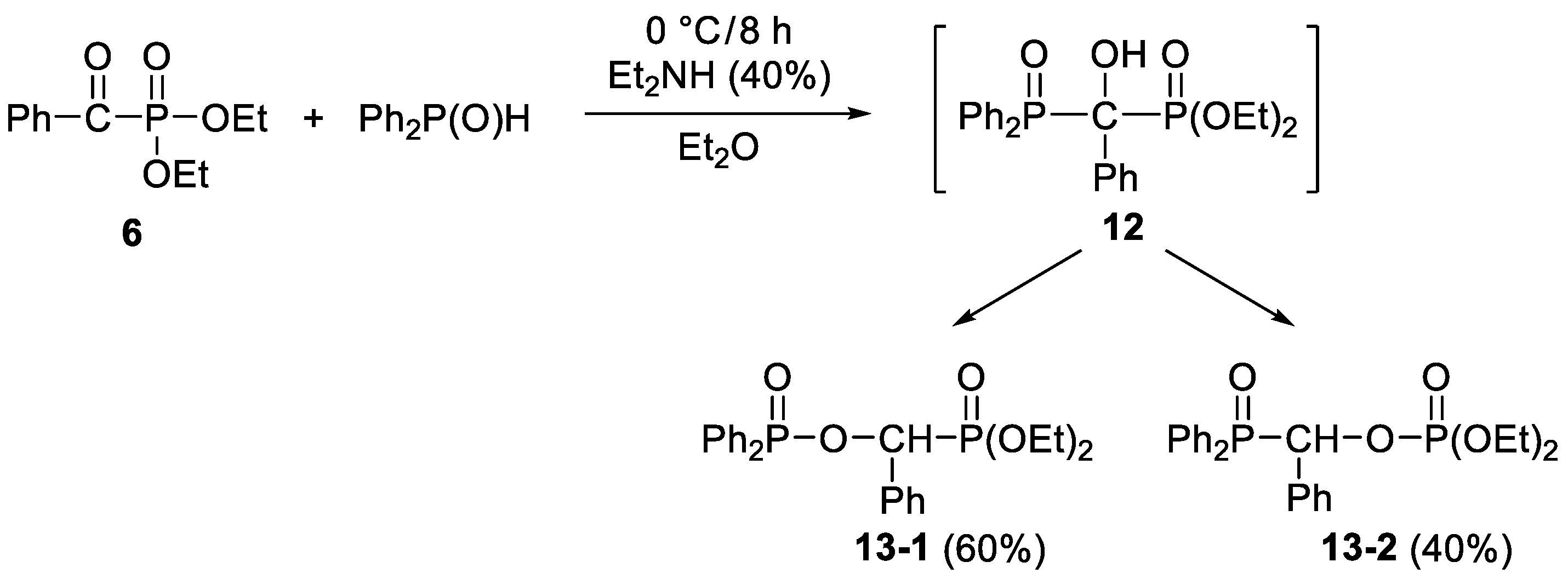{kind=link}
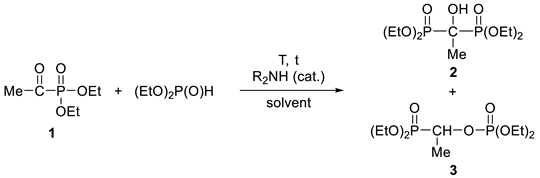 | |||||||
|---|---|---|---|---|---|---|---|
| Exp. | Catalyst (%) | Solvent | T (°C) | t | Product Composition (%) a | Yield (%) | |
| 2 | 3 | ||||||
| 1 | Et2NH (5) | Et2O | 0 | 8 h | 100 | 0 | 86% of 2 |
| 2 | Et2NH (20) | Et2O | 0 | 8 h | 98 | 2 | |
| 3 | Et2NH (40) | Et2O | 0 | 8 h | 87 | 13 | |
| 4 | Bu2NH (5) | Et2O | 0 | 8 h | 99 | 1 | 82% of 2 |
| 5 | Et2NH (40) | – | 120 | 20 min b | 66 | 34 | |
| 6 | Et2NH (40) | – | 135 | 20 min | 51 | 49 | |
| 7 | Bu2NH (5) | – | 120 | 20 min | 94 | 6 | |
| 8 | Et2NH (40) | PhMe | 110 | 7 h c | 5 | 95 | 75% of 3 |
| 9 | Et2NH (20) | PhMe | 110 | 5 h | 88 | 12 | |
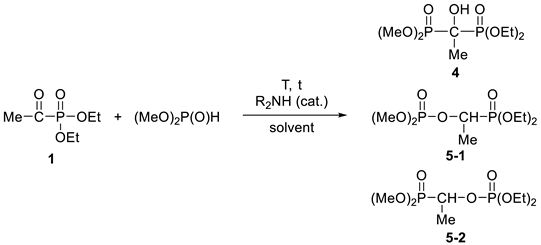 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Exp. | Catalyst (%) | Solvent | T (°C) | t | Product Composition (%) a | Yield (%) | ||
| 4 | 5-1 | 5-2 | ||||||
| 1 | Et2NH (5) | Et2O | 0 | 8 h | 94 | 4 | 2 | |
| 2 | Et2NH (20) | Et2O | 0 | 8 h | 12 | 71 | 17 | |
| 3 | Bu2NH (5) | Et2O | 0 | 8 h | 100 | 0 | 0 | 87% (4) |
| 4 | Et2NH (40) | Et2O | 0 | 8 h | 3 | 76 | 21 | 76% (5) |
| 5 | Bu2NH (5) | – | 120 | 20 min | 79 | 17 | 4 | |
| 6 | Et2NH (20) | PhMe | 110 | 5 h | 15 | 64 | 21 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szalai, Z.; Keglevich, G. Tetraalkyl Hydroxymethylene-bisphosphonate and Dialkyl 1-Diphenylphosphinoyl-1-hydroxy-ethylphosphonate Derivatives by the Pudovik Reaction and Their Rearranged Products. Molecules 2021, 26, 7575. https://doi.org/10.3390/molecules26247575
Szalai Z, Keglevich G. Tetraalkyl Hydroxymethylene-bisphosphonate and Dialkyl 1-Diphenylphosphinoyl-1-hydroxy-ethylphosphonate Derivatives by the Pudovik Reaction and Their Rearranged Products. Molecules. 2021; 26(24):7575. https://doi.org/10.3390/molecules26247575
Chicago/Turabian StyleSzalai, Zsuzsanna, and György Keglevich. 2021. "Tetraalkyl Hydroxymethylene-bisphosphonate and Dialkyl 1-Diphenylphosphinoyl-1-hydroxy-ethylphosphonate Derivatives by the Pudovik Reaction and Their Rearranged Products" Molecules 26, no. 24: 7575. https://doi.org/10.3390/molecules26247575
APA StyleSzalai, Z., & Keglevich, G. (2021). Tetraalkyl Hydroxymethylene-bisphosphonate and Dialkyl 1-Diphenylphosphinoyl-1-hydroxy-ethylphosphonate Derivatives by the Pudovik Reaction and Their Rearranged Products. Molecules, 26(24), 7575. https://doi.org/10.3390/molecules26247575