
Self-Diffusion in Simple Liquids as a Random Walk Process


Abstract
1. Introduction
2. Results
2.1. Diffusion as Random Walk
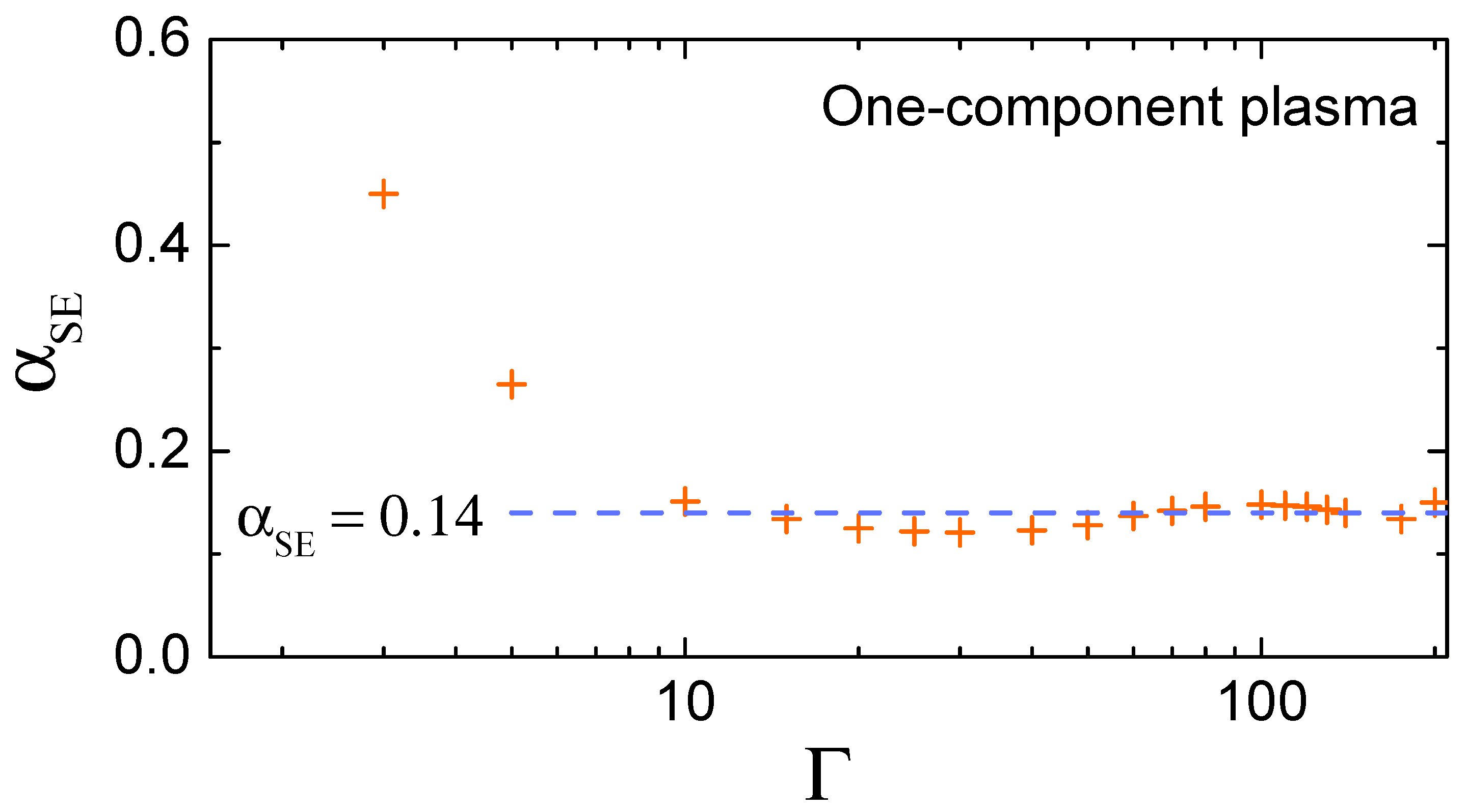
2.2. Relation to Collective Modes Properties
3. Discussion and Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| SE relation | Stokes–Einstein relation |
| OCP | one-component plasma |
| VDOS | vibrational density of states |
| QLCA | quasi-localized charge approximation |
Appendix A. Dispersion Relations of a Strongly Coupled OCP Fluid

| 0.514 | −0.8023 | 2.5856 | 9.7623 |
References
- Zwanzig, R. On the relation between self-diffusion and viscosity of liquids. J. Chem. Phys. 1983, 79, 4507–4508. [Google Scholar] [CrossRef]
- Frenkel, Y. Kinetic Theory of Liquids; Dover: New York, NY, USA, 1955. [Google Scholar]
- Khrapak, S.A. Vibrational model of thermal conduction for fluids with soft interactions. Phys. Rev. E 2021, 103, 013207. [Google Scholar] [CrossRef]
- Stillinger, F.H.; Weber, T.A. Hidden structure in liquids. Phys. Rev. A 1982, 25, 978–989. [Google Scholar] [CrossRef]
- Khrapak, S.A. Lindemann melting criterion in two dimensions. Phys. Rev. Res. 2020, 2, 012040. [Google Scholar] [CrossRef]
- Khrapak, S. Stokes–Einstein relation in simple fluids revisited. Mol. Phys. 2019, 118, e1643045. [Google Scholar] [CrossRef]
- Berezhkovskii, A.M.; Sutmann, G. Time and length scales for diffusion in liquids. Phys. Rev. E 2002, 65, 060201. [Google Scholar] [CrossRef] [PubMed]
- Khrapak, S.A.; Khrapak, A.G. Correlations between the Shear Viscosity and Thermal Conductivity Coefficients of Dense Simple Liquids. JETP Lett. 2021, 114, 540. [Google Scholar] [CrossRef]
- Brazhkin, V.V.; Fomin, Y.D.; Lyapin, A.G.; Ryzhov, V.N.; Trachenko, K. Two liquid states of matter: A dynamic line on a phase diagram. Phys. Rev. E 2012, 85, 031203. [Google Scholar] [CrossRef] [PubMed]
- Brazhkin, V.V.; Lyapin, A.; Ryzhov, V.N.; Trachenko, K.; Fomin, Y.D.; Tsiok, E.N. Where is the supercritical fluid on the phase diagram? Phys.-Uspekhi 2012, 182, 1137–1156. [Google Scholar] [CrossRef]
- Bryk, T.; Gorelli, F.A.; Mryglod, I.; Ruocco, G.; Santoro, M.; Scopigno, T. Reply to “Comment on ‘Behavior of Supercritical Fluids across the Frenkel Line’”. J. Phys. Chem. B 2018, 122, 6120–6123. [Google Scholar] [CrossRef]
- Lindemann, F. The calculation of molecular vibration frequencies. Z. Phys. 1910, 11, 609. [Google Scholar]
- Costigliola, L.; Heyes, D.M.; Schrøder, T.B.; Dyre, J.C. Revisiting the Stokes-Einstein relation without a hydrodynamic diameter. J. Chem. Phys. 2019, 150, 021101. [Google Scholar] [CrossRef] [PubMed]
- Khrapak, S.; Khrapak, A. Excess entropy and the Stokes–Einstein relation in simple fluids. Phys. Rev. E 2021, 104, 044110. [Google Scholar] [CrossRef] [PubMed]
- Mohanty, U. Inferences from a microscopic model for the Stokes-Einstein relation. Phys. Rev. A 1985, 32, 3055–3058. [Google Scholar] [CrossRef]
- Donko, Z.; Kalman, G.J.; Golden, K.I. Caging of Particles in One-Component Plasmas. Phys. Rev. Lett. 2002, 88, 225001. [Google Scholar] [CrossRef] [PubMed]
- Daligault, J. Universal Character of Atomic Motions at the Liquid-Solid Transition. arXiv 2020, arXiv:2009.14718. [Google Scholar]
- Brush, S.G.; Sahlin, H.L.; Teller, E. Monte Carlo Study of a One-Component Plasma. J. Chem. Phys. 1966, 45, 2102–2118. [Google Scholar] [CrossRef]
- Hansen, J.P. Statistical Mechanics of Dense Ionized Matter. I. Equilibrium Properties of the Classical One-Component Plasma. Phys. Rev. A 1973, 8, 3096–3109. [Google Scholar] [CrossRef]
- DeWitt, H.E. Statistical mechnics of dense plasmas: Numerical simulation and theory. J. Phys. Colloq. 1978, 39, C1-173–C1-180. [Google Scholar] [CrossRef][Green Version]
- Baus, M.; Hansen, J.P. Statistical mechanics of simple Coulomb systems. Phys. Rep. 1980, 59, 1–94. [Google Scholar] [CrossRef]
- Ichimaru, S. Strongly coupled plasmas: High-density classical plasmas and degenerate electron liquids. Rev. Mod. Phys. 1982, 54, 1017–1059. [Google Scholar] [CrossRef]
- Stringfellow, G.S.; DeWitt, H.E.; Slattery, W.L. Equation of state of the one-component plasma derived from precision Monte Carlo calculations. Phys. Rev. A 1990, 41, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Fortov, V.; Iakubov, I.; Khrapak, A. Physics of Strongly Coupled Plasma; OUP Oxford: New York, NY, USA; London, UK, 2006. [Google Scholar]
- Dubin, D.H.E.; O’Neil, T.M. Trapped nonneutral plasmas, liquids, and crystals (the thermal equilibrium states). Rev. Mod. Phys. 1999, 71, 87–172. [Google Scholar] [CrossRef]
- Khrapak, S.A.; Khrapak, A.G. Internal Energy of the Classical Two- and Three-Dimensional One-Component-Plasma. Contrib. Plasma Phys. 2016, 56, 270–280. [Google Scholar] [CrossRef]
- Khrapak, S.A.; Klumov, B.; Couedel, L.; Thomas, H.M. On the long-waves dispersion in Yukawa systems. Phys. Plasmas 2016, 23, 023702. [Google Scholar] [CrossRef]
- Daligault, J. Practical model for the self-diffusion coefficient in Yukawa one-component plasmas. Phys. Rev. E 2012, 86, 047401. [Google Scholar] [CrossRef]
- Daligault, J.; Rasmussen, K.; Baalrud, S.D. Determination of the shear viscosity of the one-component plasma. Phys. Rev. E 2014, 90, 033105. [Google Scholar] [CrossRef]
- Golden, K.I.; Kalman, G.J. Quasilocalized charge approximation in strongly coupled plasma physics. Phys. Plasmas 2000, 7, 14–32. [Google Scholar] [CrossRef]
- Rabani, E.; Gezelter, J.D.; Berne, B.J. Calculating the hopping rate for self-diffusion on rough potential energy surfaces: Cage correlations. J. Chem. Phys. 1997, 107, 6867–6876. [Google Scholar] [CrossRef]
- Hansen, J.P.; McDonald, I.R. Theory of Simple Liquids; Elsevier: Amsterdam, The Netherlands, 2006. [Google Scholar]
- Trachenko, K.; Brazhkin, V.V. Collective modes and thermodynamics of the liquid state. Rep. Progr. Phys. 2015, 79, 016502. [Google Scholar] [CrossRef]
- Bolmatov, D.; Zhernenkov, M.; Zav’yalov, D.; Stoupin, S.; Cunsolo, A.; Cai, Y.Q. Thermally triggered phononic gaps in liquids at THz scale. Sci. Rep. 2016, 6, 19469. [Google Scholar] [CrossRef] [PubMed]
- Kryuchkov, N.P.; Mistryukova, L.A.; Brazhkin, V.V.; Yurchenko, S.O. Excitation spectra in fluids: How to analyze them properly. Sci. Rep. 2019, 9, 10483. [Google Scholar] [CrossRef]
- Khrapak, S.A.; Khrapak, A.G.; Kryuchkov, N.P.; Yurchenko, S.O. Onset of transverse (shear) waves in strongly-coupled Yukawa fluids. J. Chem. Phys. 2019, 150, 104503. [Google Scholar] [CrossRef]
- Kryuchkov, N.P.; Mistryukova, L.A.; Sapelkin, A.V.; Brazhkin, V.V.; Yurchenko, S.O. Universal Effect of Excitation Dispersion on the Heat Capacity and Gapped States in Fluids. Phys. Rev. Lett. 2020, 125, 125501. [Google Scholar] [CrossRef]
- Khrapak, S.A. Practical dispersion relations for strongly coupled plasma fluids. AIP Adv. 2017, 7, 125026. [Google Scholar] [CrossRef]
- Khrapak, S.; Khrapak, A. Simple Dispersion Relations for Coulomb and Yukawa Fluids. IEEE Trans. Plasma Sci. 2018, 46, 737–742. [Google Scholar] [CrossRef]
- Fairushin, I.; Khrapak, S.; Mokshin, A. Direct evaluation of the physical characteristics of Yukawa fluids based on a simple approximation for the radial distribution function. Results Phys. 2020, 19, 103359. [Google Scholar] [CrossRef]
- Khrapak, S.A. Thermal conductivity of strongly coupled Yukawa fluids. Phys. Plasmas 2021, 28, 084501. [Google Scholar] [CrossRef]
- Khrapak, S.A.; Yurchenko, S.O. Entropy of simple fluids with repulsive interactions near freezing. J. Chem. Phys. 2021, 155, 134501. [Google Scholar] [CrossRef]

{kind=link}
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khrapak, S.A. Self-Diffusion in Simple Liquids as a Random Walk Process. Molecules 2021, 26, 7499. https://doi.org/10.3390/molecules26247499
Khrapak SA. Self-Diffusion in Simple Liquids as a Random Walk Process. Molecules. 2021; 26(24):7499. https://doi.org/10.3390/molecules26247499
Chicago/Turabian StyleKhrapak, Sergey A. 2021. "Self-Diffusion in Simple Liquids as a Random Walk Process" Molecules 26, no. 24: 7499. https://doi.org/10.3390/molecules26247499
APA StyleKhrapak, S. A. (2021). Self-Diffusion in Simple Liquids as a Random Walk Process. Molecules, 26(24), 7499. https://doi.org/10.3390/molecules26247499