
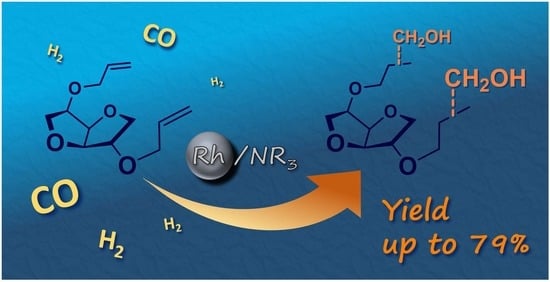
Reductive Hydroformylation of Isosorbide Diallyl Ether

 , , and
, , and
Abstract
:
1. Introduction
2. Results
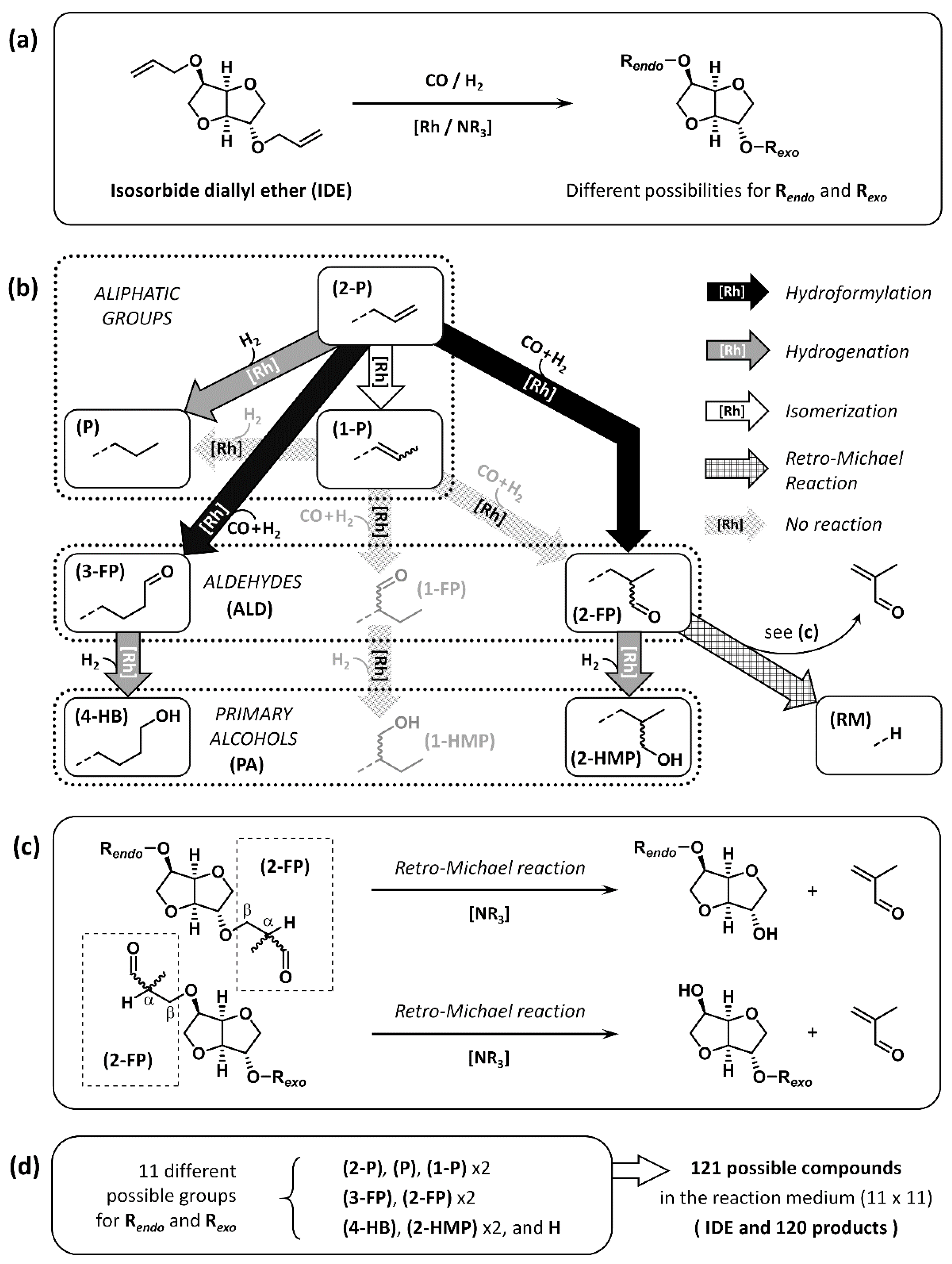
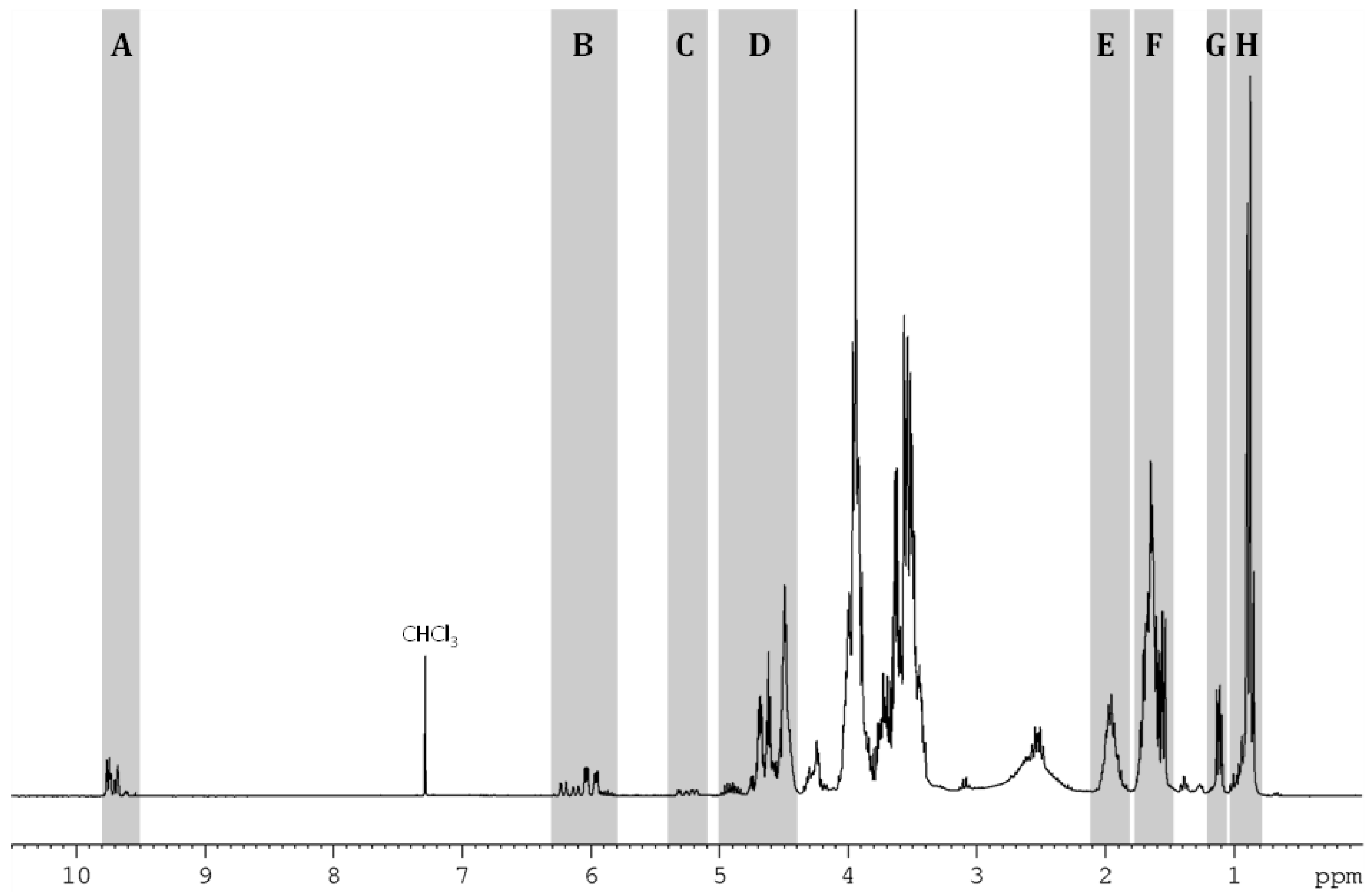
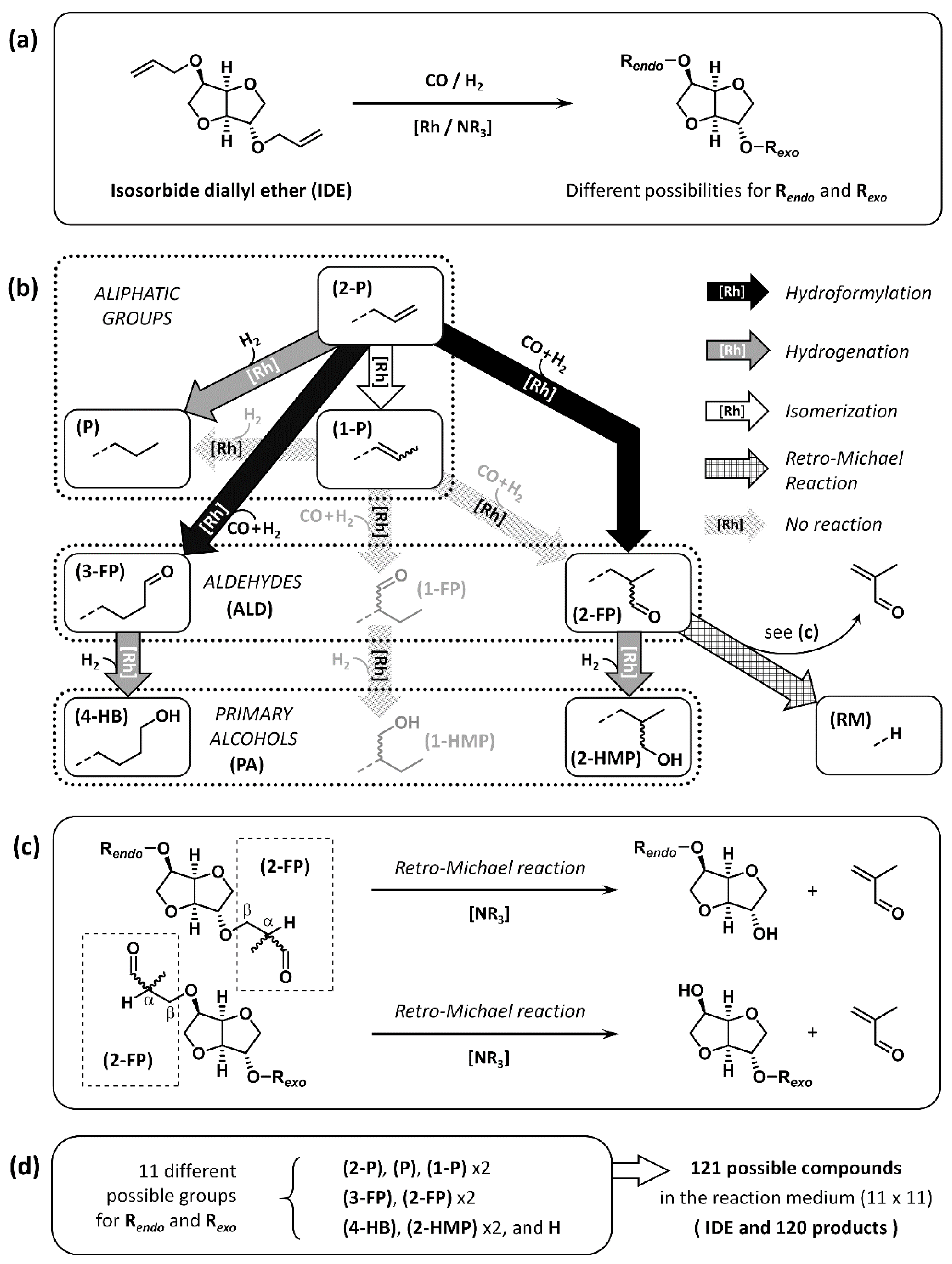
2.1. Presentation of the Compounds Resulting from HHM of IDE
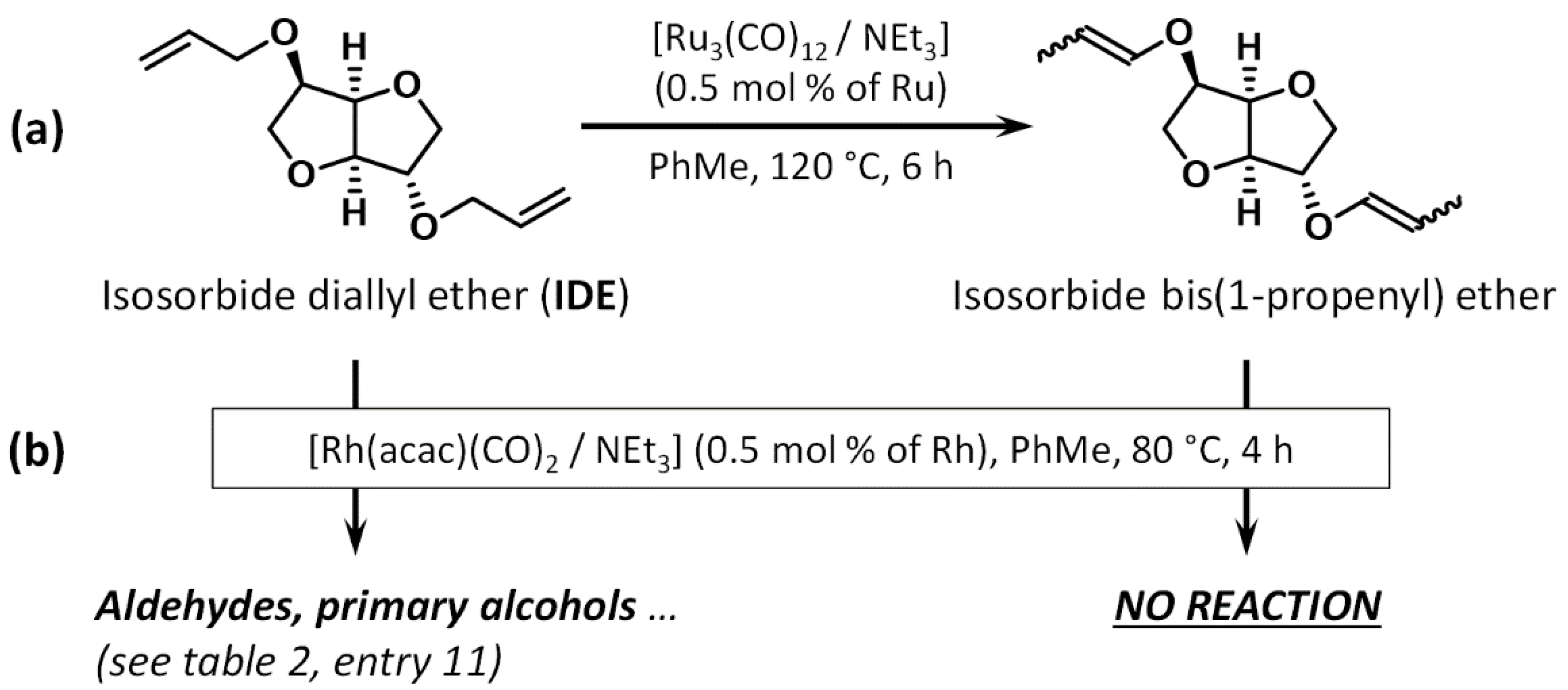
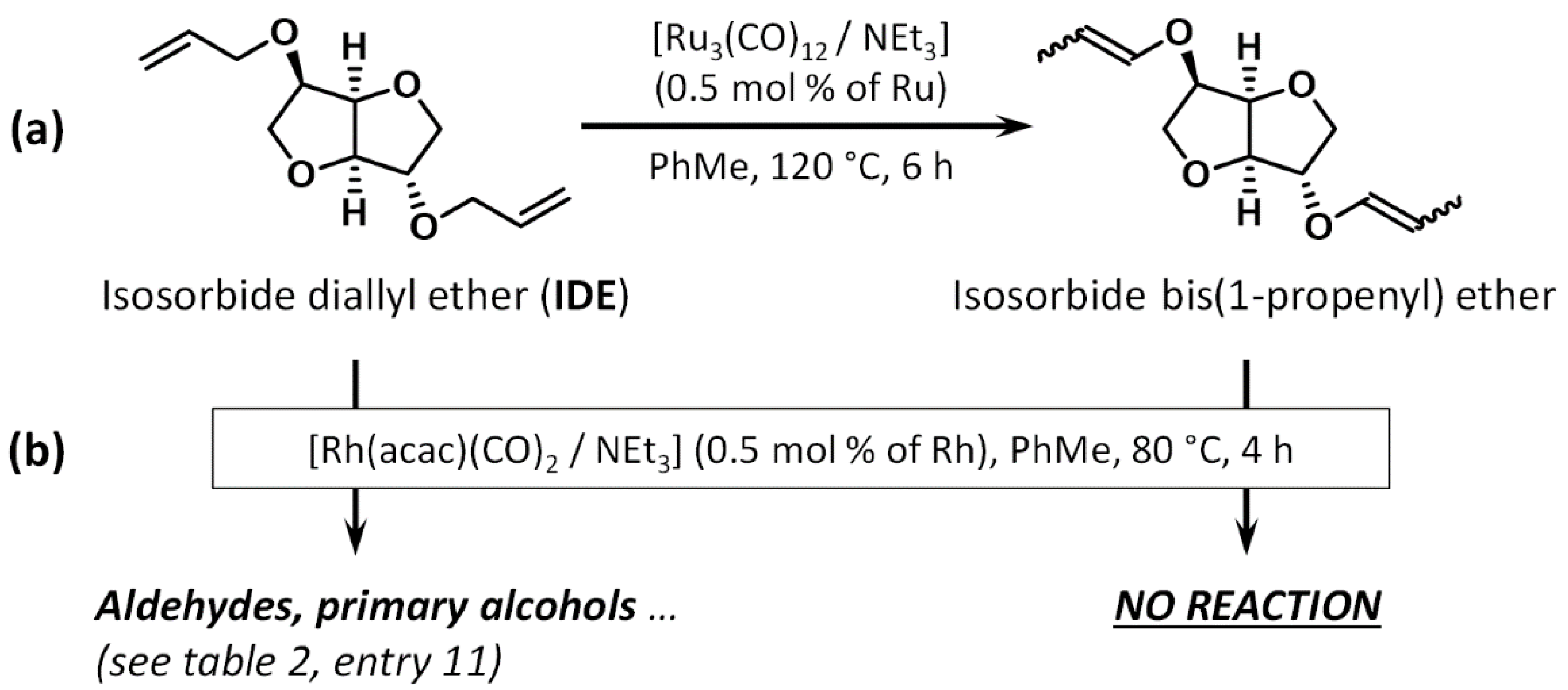
2.2. Preliminary Reactions
2.3. HHM of IDE: Effect of the TEA Amount and Reaction Time
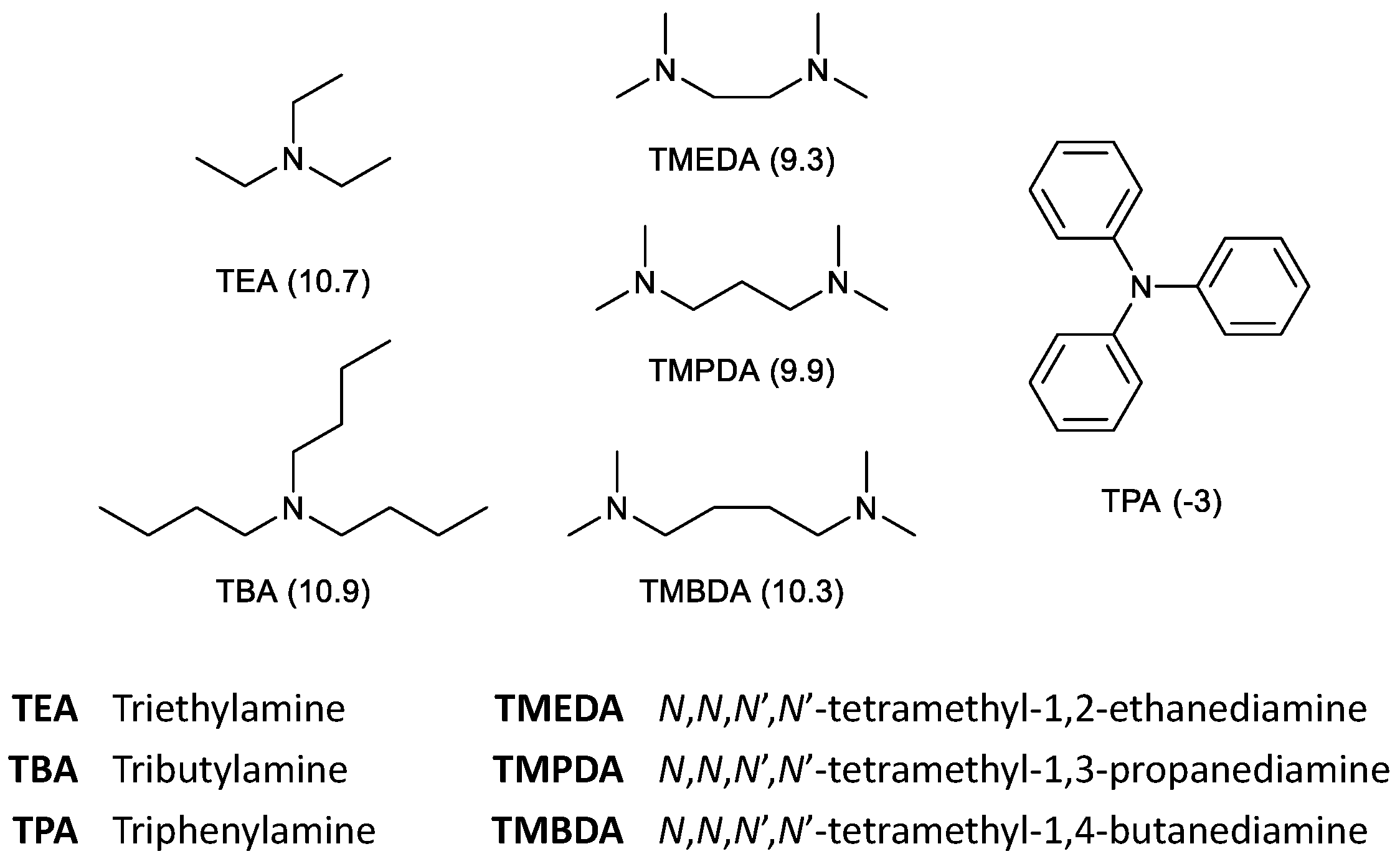
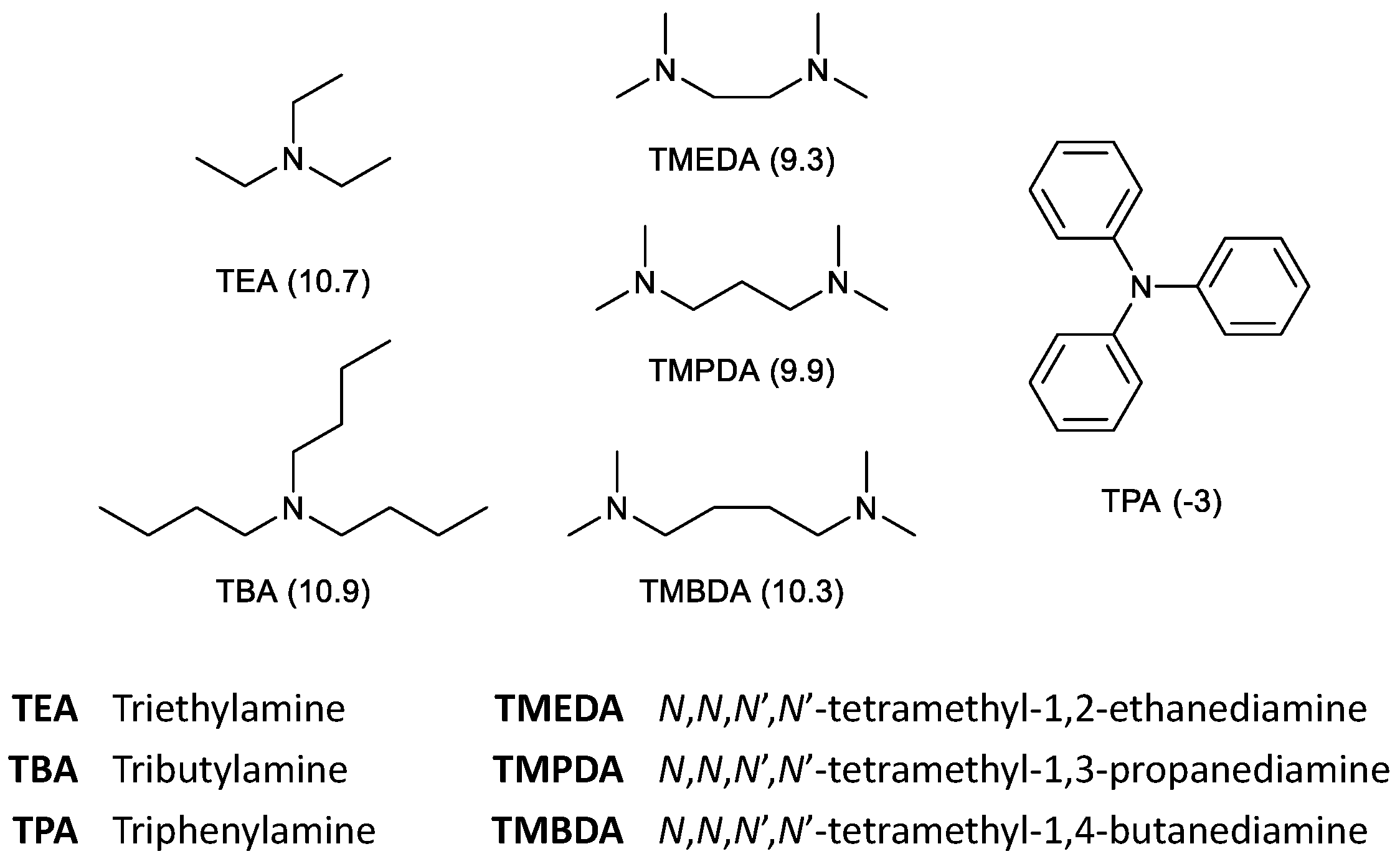
2.4. HHM of IDE: Effect of the Nitrogen Ligand
2.5. HHM of IDE: Effect of the Reaction Pressure
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Bozell, J.J.; Petersen, G.R. Technology development for the production of biobased products from biorefinery carbohydrates—the US Department of Energy’s “Top 10” revisited. Green Chem. 2010, 12, 539–554. [Google Scholar] [CrossRef]
- Cole, R.T.; Kalogeropoulos, A.P.; Georgiopoulou, V.V.; Gheorghiade, M.; Quyyumi, A.; Yancy, C.; Butler, J. Hydralazine and Isosorbide Dinitrate in Heart Failure Historical Perspective, Mechanisms, and Future Directions. Circulation 2011, 123, 2414–2422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delbecq, F.; Khodadadi, M.R.; Rodriguez Padron, D.; Varma, R.; Len, C. Isosorbide: Recent advances in catalytic production. Mol. Catal. 2020, 482, 110648. [Google Scholar] [CrossRef]
- Tundo, P.; Aricò, F.; Gauthier, G.; Rossi, L.; Rosamilia, A.E.; Bevinakatti, H.S.; Sievert, R.L.; Newman, C.P. Green Synthesis of Dimethyl Isosorbide. ChemSusChem 2010, 3, 566–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rose, M.; Palkovits, R. Isosorbide as a Renewable Platform chemical for Versatile Applications—Quo Vadis? ChemSusChem 2012, 5, 167–176. [Google Scholar] [CrossRef]
- Feng, X.; East, A.J.; Hammond, W.B.; Zhang, Y.; Jaffe, M. Overview of advances in sugar-based polymers. Polym. Adv. Technol. 2011, 22, 139–150. [Google Scholar] [CrossRef]
- Fenouillot, F.; Rousseau, A.; Colomines, G.; Saint-Loup, R.; Pascault, J.P. Polymers from renewable 1,4:3,6-dianhydrohexitols (isosorbide, isomannide and isoidide): A review. Prog. Polym. Sci. 2010, 35, 578–622. [Google Scholar] [CrossRef]
- Saxon, D.J.; Luke, A.M.; Sajjad, H.; Tolman, W.B.; Reineke, T.M. Next-generation polymers: Isosorbide as a renewable alternative. Prog. Polym. Sci. 2020, 101, 101196. [Google Scholar] [CrossRef]
- Kricheldorf, H.R. “Sugar Diols” as Building Blocks of Polycondensates. J. Macromol. Sci. 1997, 37, 599–631. [Google Scholar] [CrossRef]
- Cognet-Georjon, E.; Mechin, F.; Pascault, J.P. New polyurethanes based on 4,4′-diphenylmethane diisocyanate and 1,4:3,6 dianhydrosorbitol, 2. Synthesis and properties of segmented polyurethane elastomers. Macromol. Chem. Phys. 1996, 197, 3593–3612. [Google Scholar] [CrossRef]
- Majdoub, M.; Loupy, A.; Fleche, G. Nouveaux polyéthers et polyesters à base d’isosorbide: Synthèse et caractérisation. Eur. Polym. J. 1994, 30, 1431–1437. [Google Scholar] [CrossRef]
- Noordover, B.A.J.; van Staalduinen, V.G.; Duchateau, R.; Koning, C.E.; van, B.; Mak, M.; Heise, A.; Frissen, A.E.; van Haveren, J. Co- and Terpolyesters Based on Isosorbide and Succinic Acid for Coating Applications: Synthesis and Characterization. Biomacromolecules 2006, 7, 3406–3416. [Google Scholar] [CrossRef] [PubMed]
- Thiyagarajan, S.; Gootjes, L.; Vogelzang, W.; van Haveren, J.; Lutz, M.; van Es, D.S. Renewable Rigid Diamines: Efficient, Stereospecific Synthesis of High Purity Isohexide Diamines. ChemSusChem 2011, 4, 1823–1829. [Google Scholar] [CrossRef]
- Thiyagarajan, S.; Gootjes, L.; Vogelzang, W.; Wu, J.; van Haveren, J.; van Es, D.S. Chiral building blocks from biomass: 2,5-diamino-2,5-dideoxy-1,4-3,6-dianhydroiditol. Tetrahedron 2011, 67, 383–389. [Google Scholar] [CrossRef]
- Thiyagarajan, S.; Wu, J.; Knoop, R.; Haveren, J.; Lutz, M.; Van Es, D. Isohexide hydroxy esters: Synthesis and application of a new class of biobased AB-type building blocks. Rsc Adv. 2014, 4, 7937–47950. [Google Scholar] [CrossRef]
- Stensrud, K. Monoallyl, Monoglycidyl Ethers and Bisglycidyl Ethers of Isohexides. WO/2013/188253, 19 December 2013. [Google Scholar]
- Torres, G.M.; Frauenlob, R.; Franke, R.; Börner, A. Production of alcohols via hydroformylation. Catal. Sci. Technol. 2015, 5, 34–54. [Google Scholar] [CrossRef]
- Diebolt, O.; Müller, C.; Vogt, D. “On-water” rhodium-catalysed hydroformylation for the production of linear alcohols. Catal. Sci. Technol. 2012, 2, 773–777. [Google Scholar] [CrossRef]
- Rodrigues, F.M.S.; Kucmierczyk, P.K.; Pineiro, M.; Jackstell, R.; Franke, R.; Pereira, M.M.; Beller, M. Dual Rh−Ru Catalysts for Reductive Hydroformylation of Olefins to Alcohols. ChemSusChem 2018, 11, 2310–2314. [Google Scholar] [CrossRef]
- Furst, M.R.L.; Korkmaz, V.; Gaide, T.; Seidensticker, T.; Behr, A.; Vorholt, A.J. Tandem Reductive Hydroformylation of Castor Oil Derived Substrates and Catalyst Recycling by Selective Product Crystallization. ChemCatChem 2017, 9, 4319–4323. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamashita, M.; Nozaki, K. Tandem Hydroformylation/Hydrogenation of Alkenes to Normal Alcohols Using Rh/Ru Dual Catalyst or Ru Single Component Catalyst. J. Am. Chem. Soc. 2012, 134, 18746–18757. [Google Scholar] [CrossRef]
- Fell, B.; Shanshool, J.; Asinger, F. Einstufige oxoalkoholsynthese mit einem Co2(CO)8/Fe(CO)5/N-methylpyrrolidin-katalysatorsystem. J. Organomet. Chem. 1971, 33, 69–72. [Google Scholar] [CrossRef]
- Fell, B.; Geurts, A. Hydroformylierung mit Rhodiumcarbonyl-tert.-Amin-Komplexkatalysatoren. Chem. Ing. Tech. 1972, 44, 708–712. [Google Scholar] [CrossRef]
- Cheung, L.L.W.; Vasapollo, G.; Alper, H. Synthesis of Alcohols via a Rhodium-Catalyzed Hydroformylation–Reduction Sequence using Tertiary Bidentate Amine Ligands. Adv. Synth. Catal. 2012, 354, 2019–2022. [Google Scholar] [CrossRef]
- Mizoroki, T.; Kioka, M.; Suzuki, M.; Sakatani, S.; Okumura, A.; Maruya, K.-i. Behavior of Amine in Rhodium Complex–Tertiary Amine Catalyst System Active for Hydrogenation of Aldehyde under Oxo Reaction Conditions. Bull. Chem. Soc. Jpn. 1984, 57, 577–578. [Google Scholar] [CrossRef]
- Hunter, D.L.; Moore, S.E.; Garrou, P.E.; Dubois, R.A. Dicyclopentadiene hydroformylation catalyzed by RhxCo4-x(CO)12 (x = 4, 2-0)/tertiary amine catalysts. Appl. Catal. 1985, 19, 259–273. [Google Scholar] [CrossRef]
- Hunter, D.L.; Moore, S.E.; Dubois, R.A.; Garrou, P.E. Deactivation of rhodium hydroformylation catalysts on amine functionalized organic supports. Appl. Catal. 1985, 19, 275–285. [Google Scholar] [CrossRef]
- Alvila, L.; Pakkanen, T.A.; Pakkanen, T.T.; Krause, O. Hydroformylation of olefins catalysed by rhodium and cobalt clusters supported on organic (Dowex) resins. J. Mol. Catal. 1992, 71, 281–290. [Google Scholar] [CrossRef]
- Jurewicz, A.T.; Rollmann, L.D.; Whitehurst, D.D. Hydroformylation with Rhodium-Amine Complexes. Homogeneous Catalysis—II. Adv. Chem. 1974, 132, 240–251. [Google Scholar] [CrossRef]
- Fuchs, S.; Lichte, D.; Dittmar, M.; Meier, G.; Strutz, H.; Behr, A.; Vorholt, A.J. Tertiary Amines as Ligands in a Four-Step Tandem Reaction of Hydroformylation and Hydrogenation: An Alternative Route to Industrial Diol Monomers. ChemCatChem 2017, 9, 1436–1441. [Google Scholar] [CrossRef]
- Vanbésien, T.; Monflier, E.; Hapiot, F. Rhodium-catalyzed one pot synthesis of hydroxymethylated triglycerides. Green Chem. 2016, 18, 6687–6694. [Google Scholar] [CrossRef]
- Rösler, T.; Ehmann, K.R.; Köhnke, K.; Leutzsch, M.; Wessel, N.; Vorholt, A.J.; Leitner, W. Reductive hydroformylation with a selective and highly active rhodium amine system. J. Catal. 2021, 400, 234–243. [Google Scholar] [CrossRef]
- Gorbunov, D.; Nenasheva, M.; Naranov, E.; Maximov, A.; Rosenberg, E.; Karakhanov, E. Tandem hydroformylation/hydrogenation over novel immobilized Rh-containing catalysts based on tertiary amine-functionalized hybrid inorganic-organic materials. Appl. Catal. A Gen. 2021, 623, 118266. [Google Scholar] [CrossRef]
- Hong, J.; Radojčić, D.; Ionescu, M.; Petrović, Z.S.; Eastwood, R. Advanced materials from corn: Isosorbide-based epoxy resins. Polym. Chem. 2014, 5, 5360. [Google Scholar] [CrossRef]
- ÇAkmakÇI, E.; ŞEn, F.; Kahraman, M.V. Isosorbide Diallyl Based Antibacterial Thiol–Ene Photocured Coatings Containing Polymerizable Fluorous Quaternary Phosphonium Salt. ACS Sustain. Chem. Eng. 2019, 7, 10605–10615. [Google Scholar] [CrossRef]
- Montassier, C.; Menezo, J.C.; Moukolo, J.; Naja, J.; Barbier, J.; Boitiaux, J.P. Furanic Derivatives Synthesis from Polyols by Heterogeneous Catalysis Over Metals. In Studies in Surface Science and Catalysis; Guisnet, M., Barrault, J., Bouchoule, C., Duprez, D., Pérot, G., Maurel, R., Montassier, C., Eds.; Elsevier: Amsterdam, The Netherlands, 1991; Volume 59, pp. 223–230. [Google Scholar]
- Jiao, Y.; Torne, M.S.; Gracia, J.; Niemantsverdriet, J.W.; van Leeuwen, P.W.N.M. Ligand effects in rhodium-catalyzed hydroformylation with bisphosphines: Steric or electronic? Catal. Sci. Technol. 2017, 7, 1404–1414. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
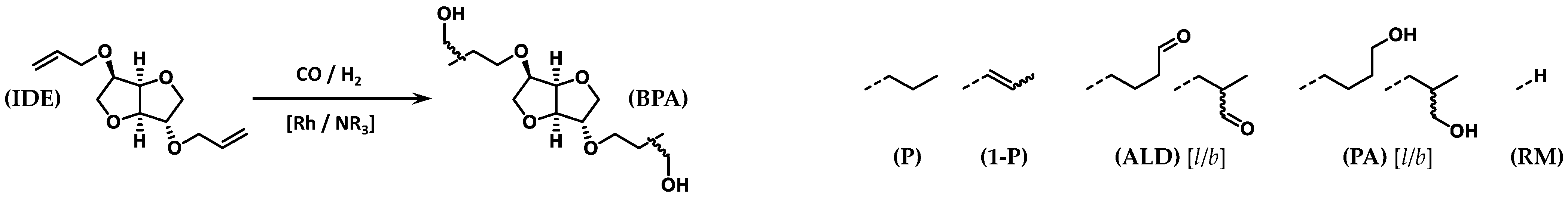 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | TEA/Rh | t (h) | Conv. b (%) | Y(P) c (%) | Y(1-P) c (%) | Y(ALD) c (%) [l/b] d | Y(PA) c (%) [l/b] e | Y(RM) c (%) | Y’(BPA) f (%) [ll/lb/bb] g |
| 1h | 0 | 18 | 5 | 0 | 5 | 0 | 0 | 0 | 0 |
| 2 | 0 | 18 | 100 | 9 | 3 | 82 [50/50] | 0 | 6 | 0 |
| 3 | 10 | 6 | 95 | 2 | 12 | 21 [45/55] | 54 [38/62] | 6 | 38 [12/40/48] |
| 4 | 20 | 6 | 100 | 2 | 4 | 22 [45/55] | 67 [40/60] | 5 | 44 [12/46/42] |
| 5 | 50 | 6 | 100 | 2 | 7 | 6 [49/51] | 80 [45/55] | 5 | 63 [17/49/34] |
| 6 | 100 | 6 | 100 | 2 | 5 | 0 | 87 [45/55] | 6 | 67 [17/50/33] |
| 7 | 200 | 6 | 100 | 2 | 5 | 0 | 87 [44/56] | 6 | 69 [17/49/34] |
| 8 i | 1200 | 18 | 100 | 3 | 6 | 0 | 85 [43/57] | 6 | 62 [16/49/35] |
| 9 | 20 | 4 | 79 | 2 | 3 | 38 [50/50] | 30 [40/60] | 5 | 22 [12/43/45] |
| 10 | 20 | 18 | 100 | 3 | 4 | 0 | 89 [46/54] | 4 | 67 [17/50/33] |
| 11 | 200 | 4 | 95 | 1 | 4 | 16 [48/52] | 70 [46/54] | 4 | 49 [17/48/35] |
| 12 | 200 | 18 | 100 | 3 | 5 | 0 | 86 [45/55] | 6 | 67 [17/79/34] |
| | ||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Ligand (equiv/Rh) | Conv. b (%) | Y(P) c (%) | Y(1-P) c (%) | Y(ALD) c (%) [l/b] d | Y(PA) c (%) [l/b] e | Y(RM) c (%) | Y’(BPA) f (%) [ll/lb/bb] g |
| 1 | TEA (200) | 100 | 2 | 5 | 0 | 87 [44/56] | 6 | 69 [17/49/34] |
| 2 | TPA (200) | 100 | 9 | 2 | 80 [50/50] | 0 | 9 | 0 |
| 3 | TBA (200) | 100 | 5 | 5 | 0 | 84 [42/58] | 6 | 60 [16/48/36] |
| 4 | TMEDA (100) | 100 | 1 | 3 | 0 | 90 [46/54] | 6 | 74 [16/49/35] |
| 5 | TMPDA (100) | 100 | 2 | 4 | 0 | 89 [47/53] | 5 | 75 [15/48/37] |
| 6 | TMBDA (100) | 100 | 1 | 1 | 0 | 91 [46/54] | 7 | 79 [16/49/37] |
| 7 | TMEDA (200) | 100 | 2 | 3 | 0 | 90 [46/54] | 5 | 75 [15/48/37] |
| 8 | TMPDA (200) | 100 | 1 | 3 | 0 | 90 [47/53] | 6 | 76 [16/48/36] |
| 9 | TMBDA (200) | 100 | 2 | 2 | 0 | 91 [46/54] | 5 | 79 [16/49/37] |
| | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Ligand (equiv/Rh) | P (bar) (CO:H2) | Conv. b (%) | Y(P) c (%) | Y(1-P) c (%) | Y(ALD) c (%) [l/b] d | Y(PA) c (%) [l/b] e | Y(RM) c (%) | Y’(BPA) f (%) [ll/lb/bb] g |
| 1 | TEA (200) | 80 (1:1) | 100 | 2 | 5 | 0 | 87 [44/56] | 6 | 69 [17/49/34] |
| 2 | TEA (200) | 80 (1:2) | 100 | 3 | 4 | 0 | 87 [45/55] | 6 | 69 [17/48/35] |
| 3 | TEA (200) | 60 (1:1) | 100 | 2 | 13 | 0 | 80 [48/52] | 5 | 60 [20/51/29] |
| 4 | TEA (200) | 40 (1:1) | 85 | 2 | 18 | 13 [48/52] | 48 [45/55] | 4 | 22 [17/48/35] |
| 5 | TMPDA (100) | 80 (1:1) | 100 | 2 | 4 | 0 | 89 [47/53] | 5 | 75 [15/48/37] |
| 6 | TMPDA (100) | 80 (1:2) | 100 | 3 | 2 | 0 | 88 [48/52] | 7 | 74 [16/49/37] |
| 7 | TMBDA (100) | 80 (1:1) | 100 | 1 | 1 | 0 | 91 [46/54] | 7 | 79 [16/49/37] |
| 8 | TMBDA (100) | 80 (1:2) | 100 | 3 | 3 | 0 | 87 [46/54] | 7 | 78 [15/49/36] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ternel, J.; Lopes, A.; Sauthier, M.; Buffe, C.; Wiatz, V.; Bricout, H.; Tilloy, S.; Monflier, E. Reductive Hydroformylation of Isosorbide Diallyl Ether. Molecules 2021, 26, 7322. https://doi.org/10.3390/molecules26237322
Ternel J, Lopes A, Sauthier M, Buffe C, Wiatz V, Bricout H, Tilloy S, Monflier E. Reductive Hydroformylation of Isosorbide Diallyl Ether. Molecules. 2021; 26(23):7322. https://doi.org/10.3390/molecules26237322
Chicago/Turabian StyleTernel, Jérémy, Adrien Lopes, Mathieu Sauthier, Clothilde Buffe, Vincent Wiatz, Hervé Bricout, Sébastien Tilloy, and Eric Monflier. 2021. "Reductive Hydroformylation of Isosorbide Diallyl Ether" Molecules 26, no. 23: 7322. https://doi.org/10.3390/molecules26237322
APA StyleTernel, J., Lopes, A., Sauthier, M., Buffe, C., Wiatz, V., Bricout, H., Tilloy, S., & Monflier, E. (2021). Reductive Hydroformylation of Isosorbide Diallyl Ether. Molecules, 26(23), 7322. https://doi.org/10.3390/molecules26237322