
Reproducibility Evaluation of Urinary Peptide Detection Using CE-MS

 ,
,
Abstract
:1. Introduction
2. Results
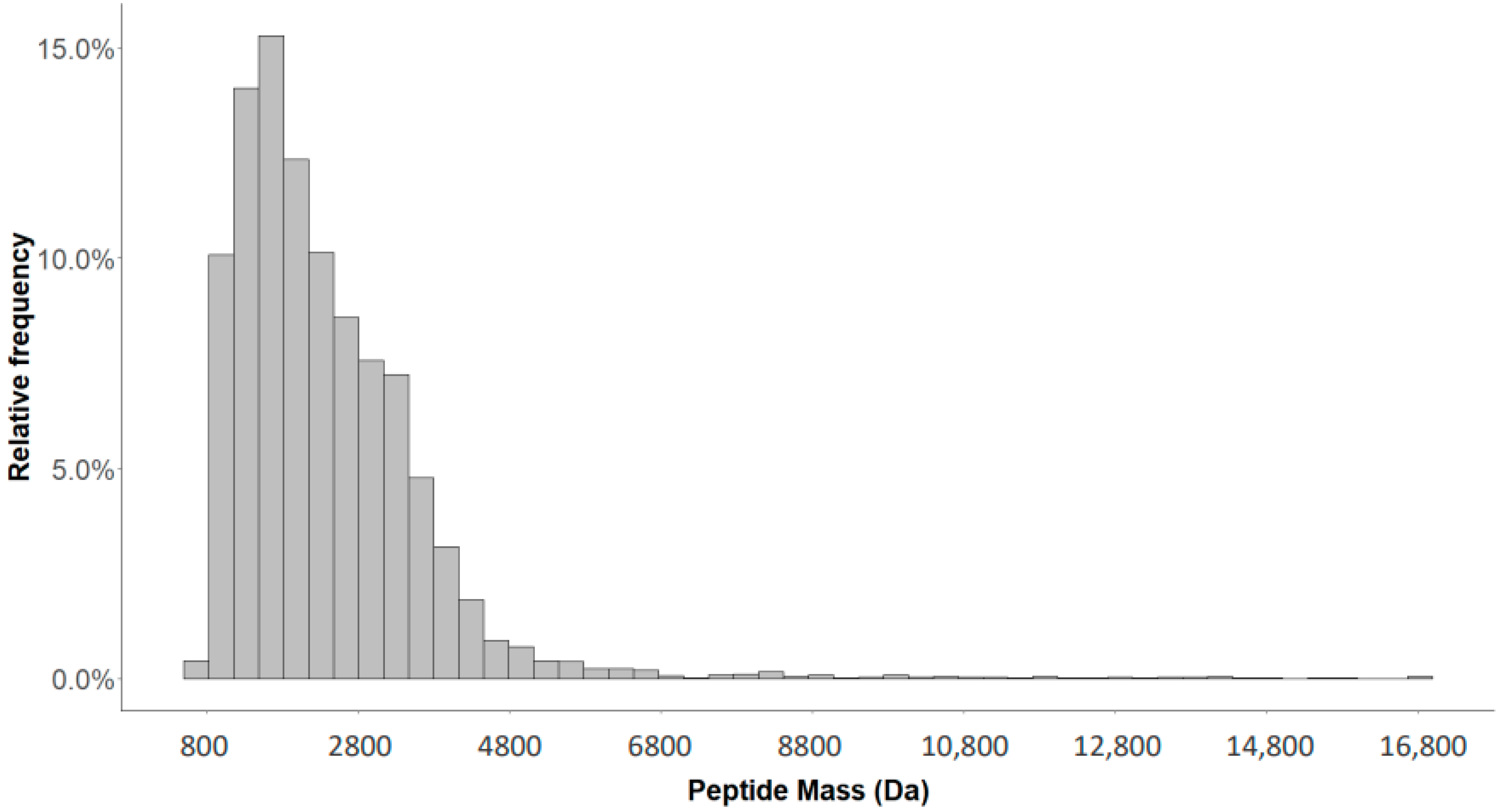
2.1. Detection of Naturally Occurring Peptides in the Standard Urine Sample
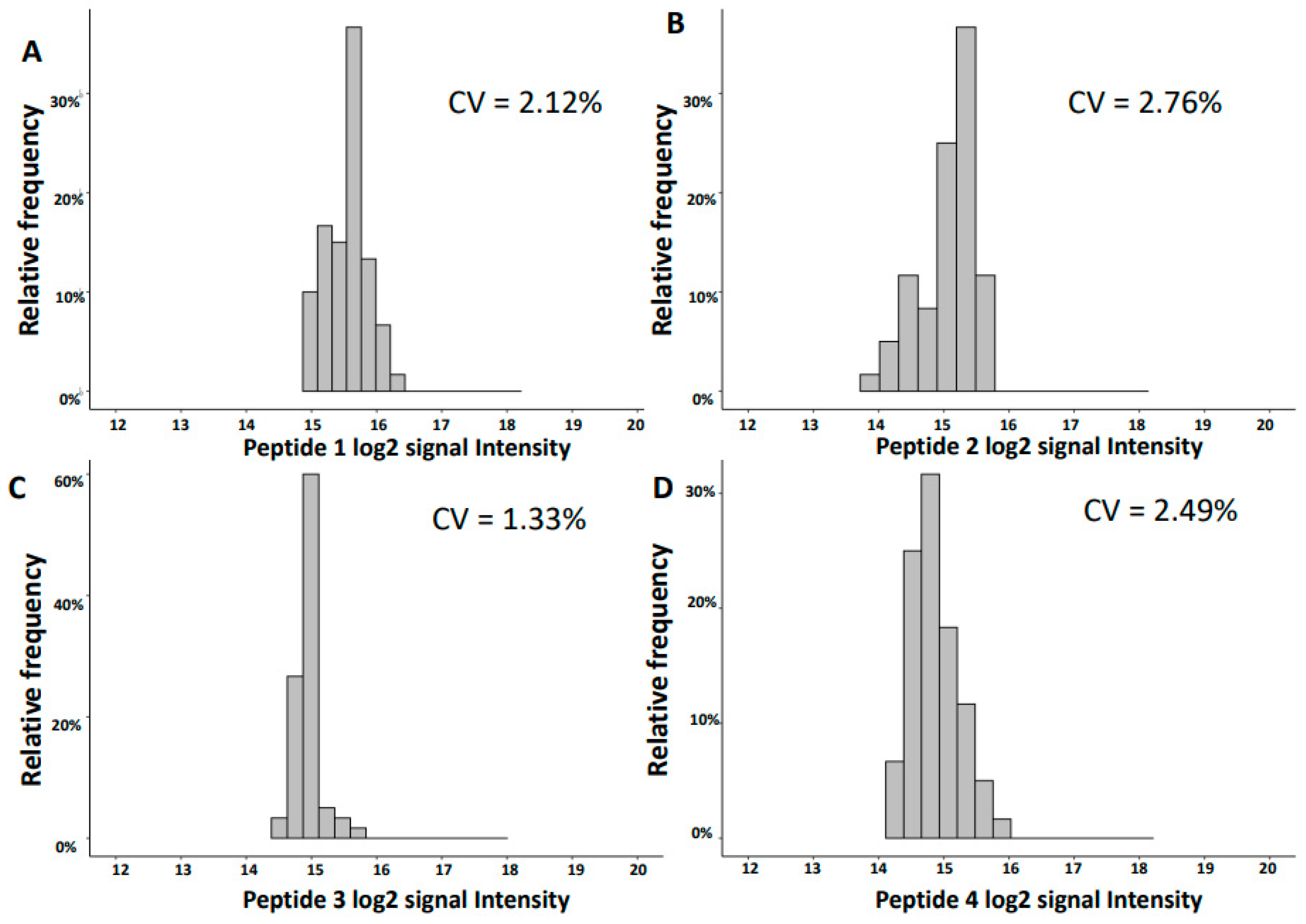
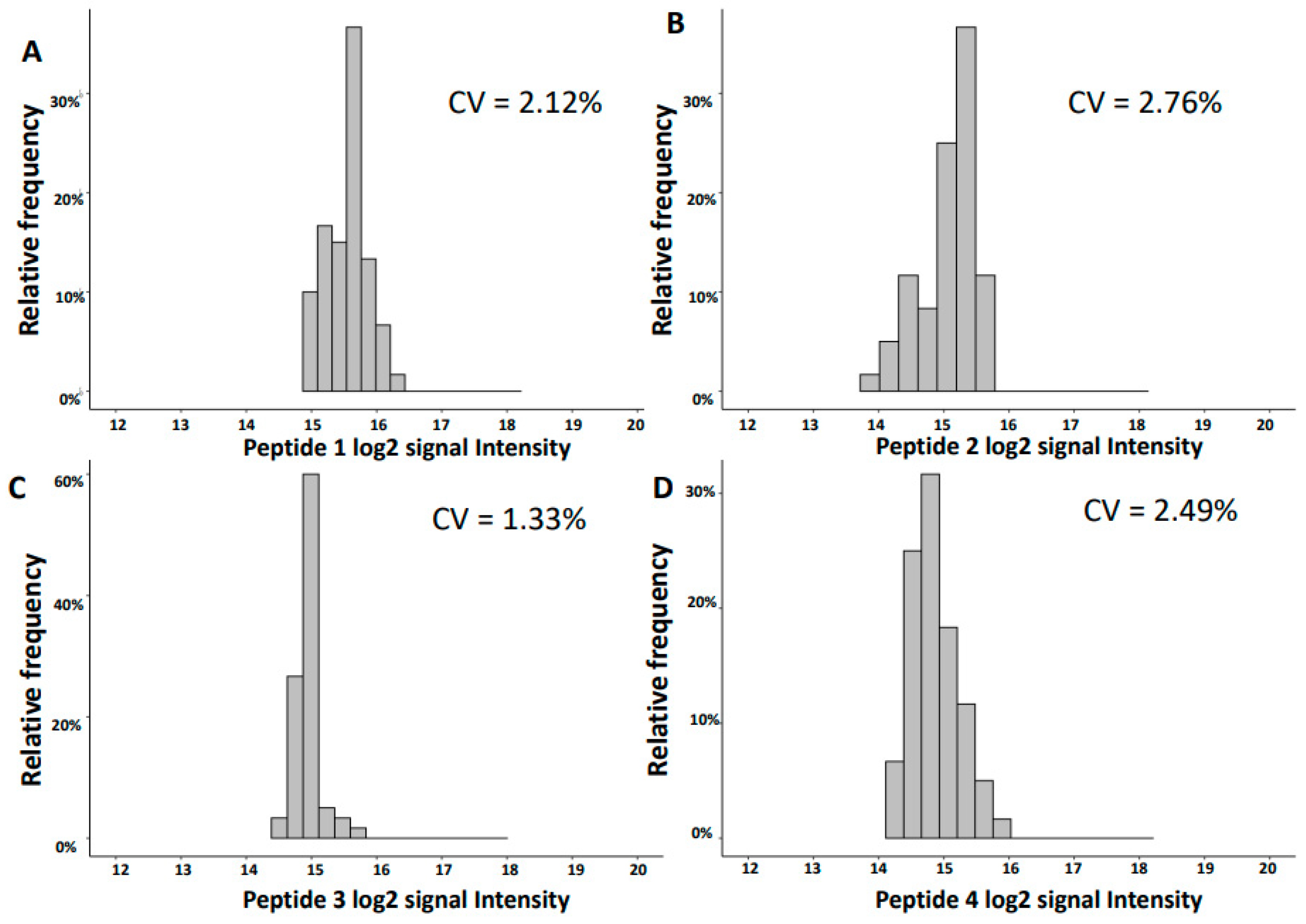
2.2. Variability of Signal Intensities of Individual Peptides
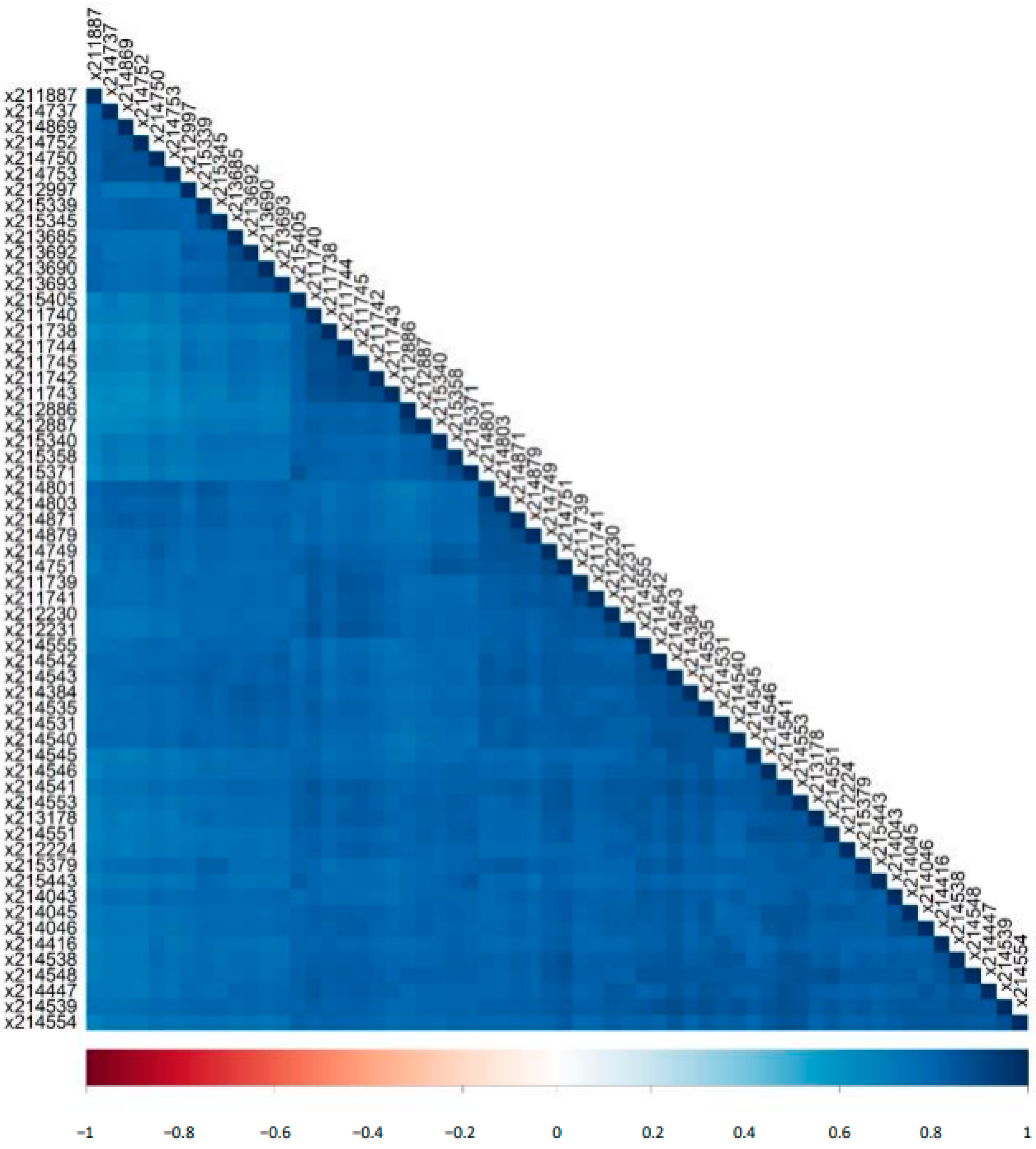
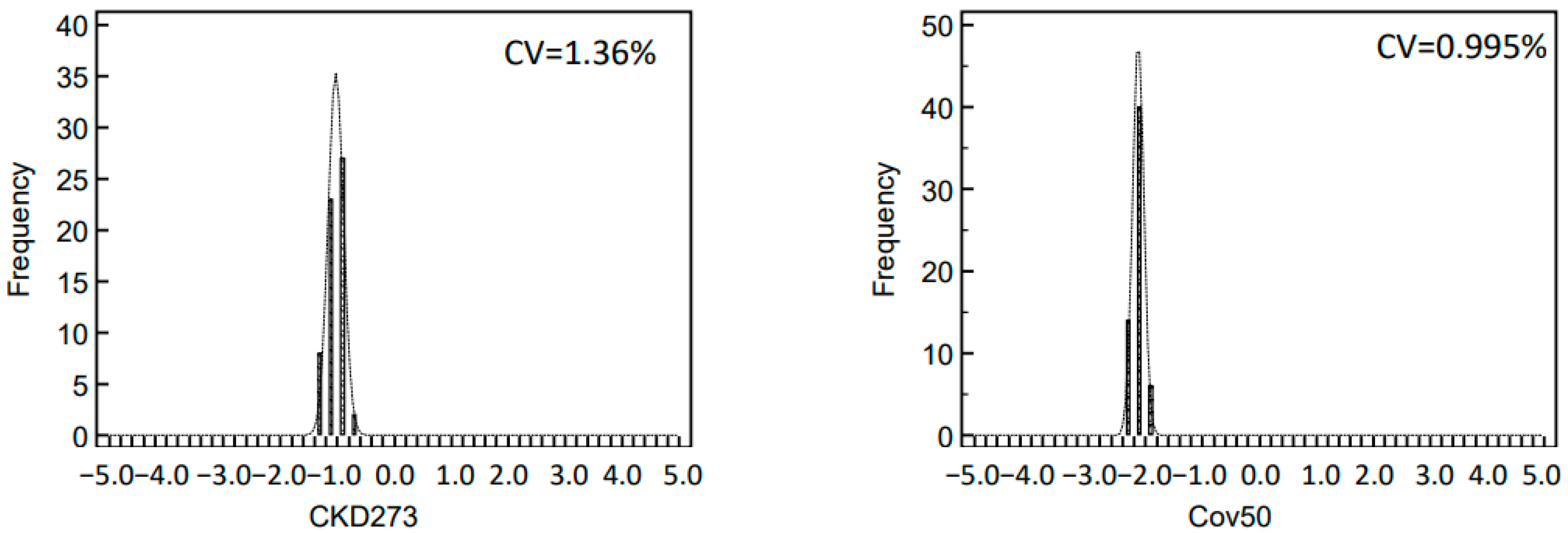
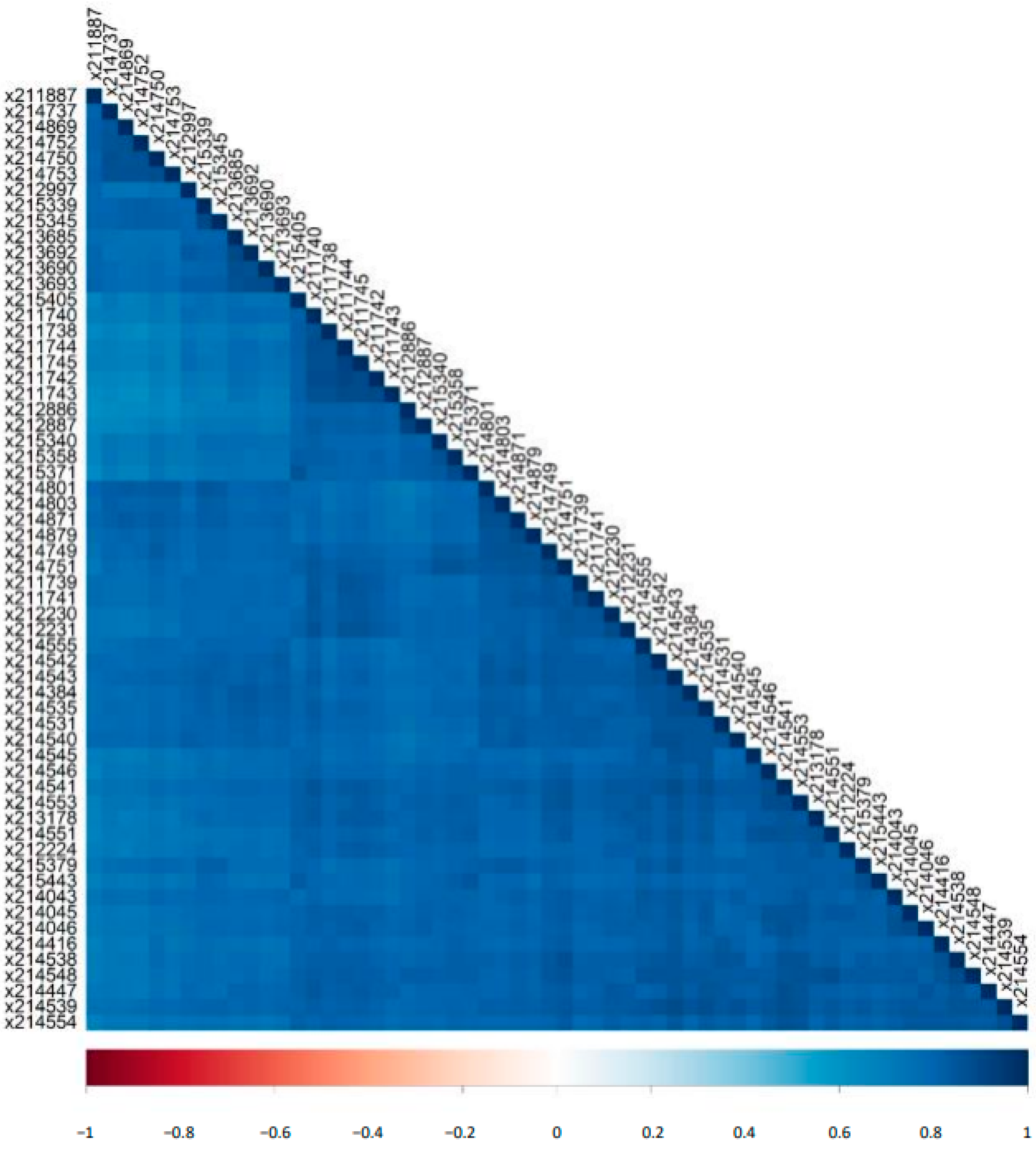
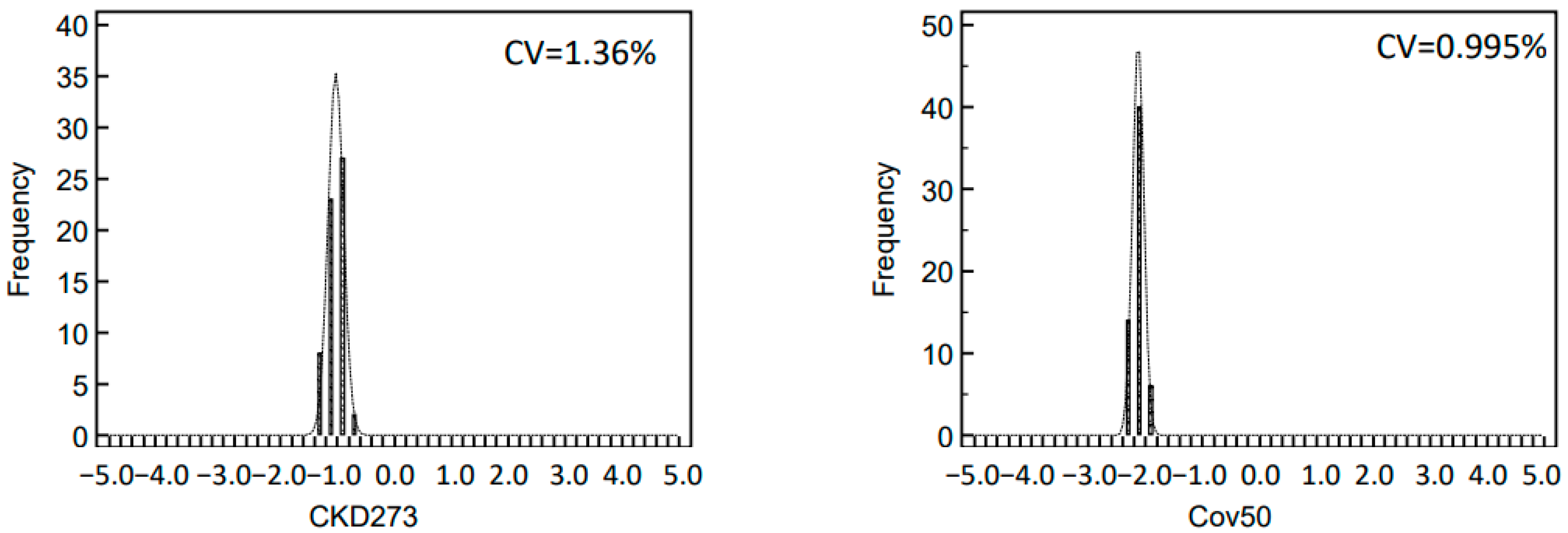
2.3. Variability of Biomarker Panels
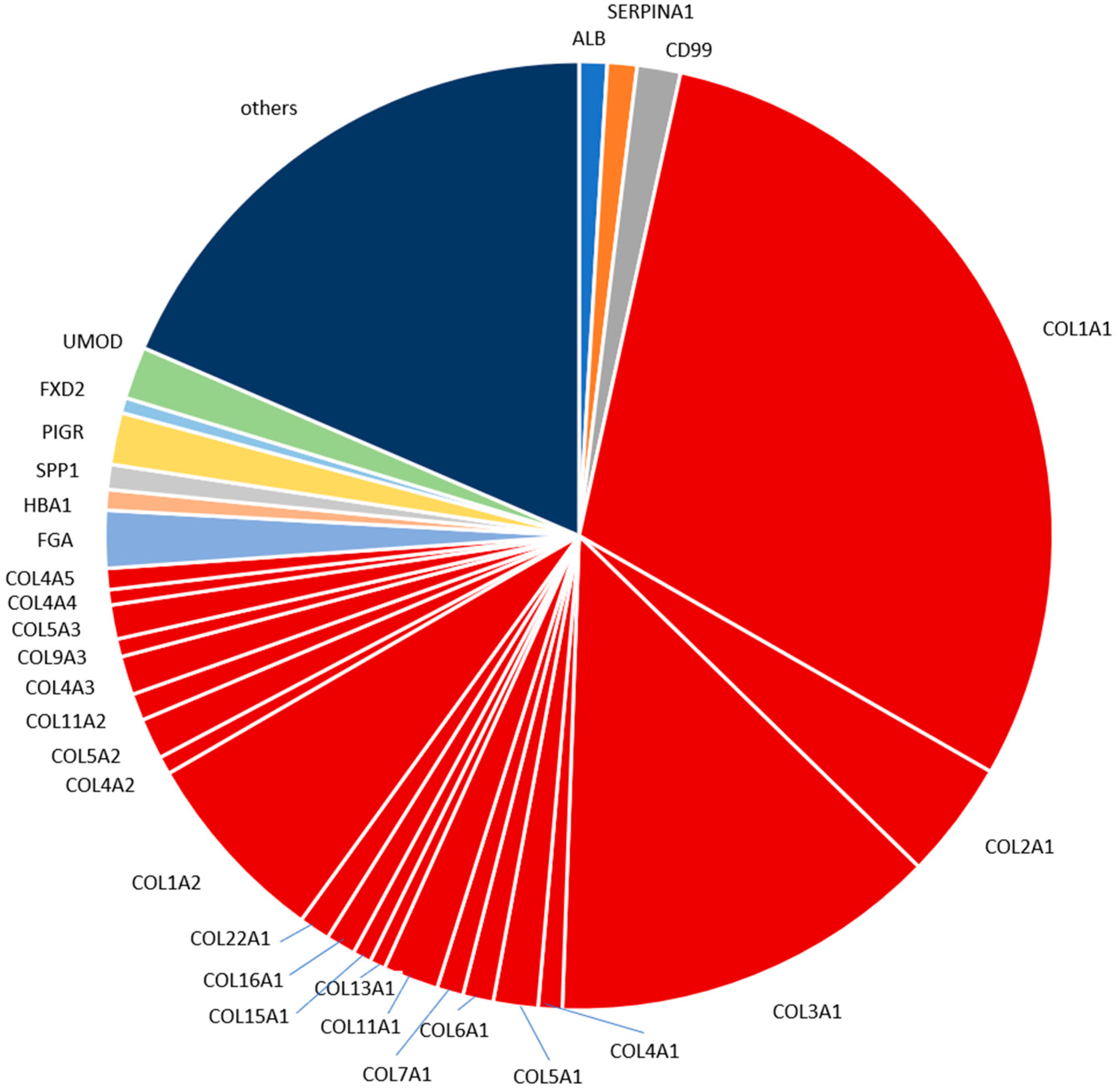
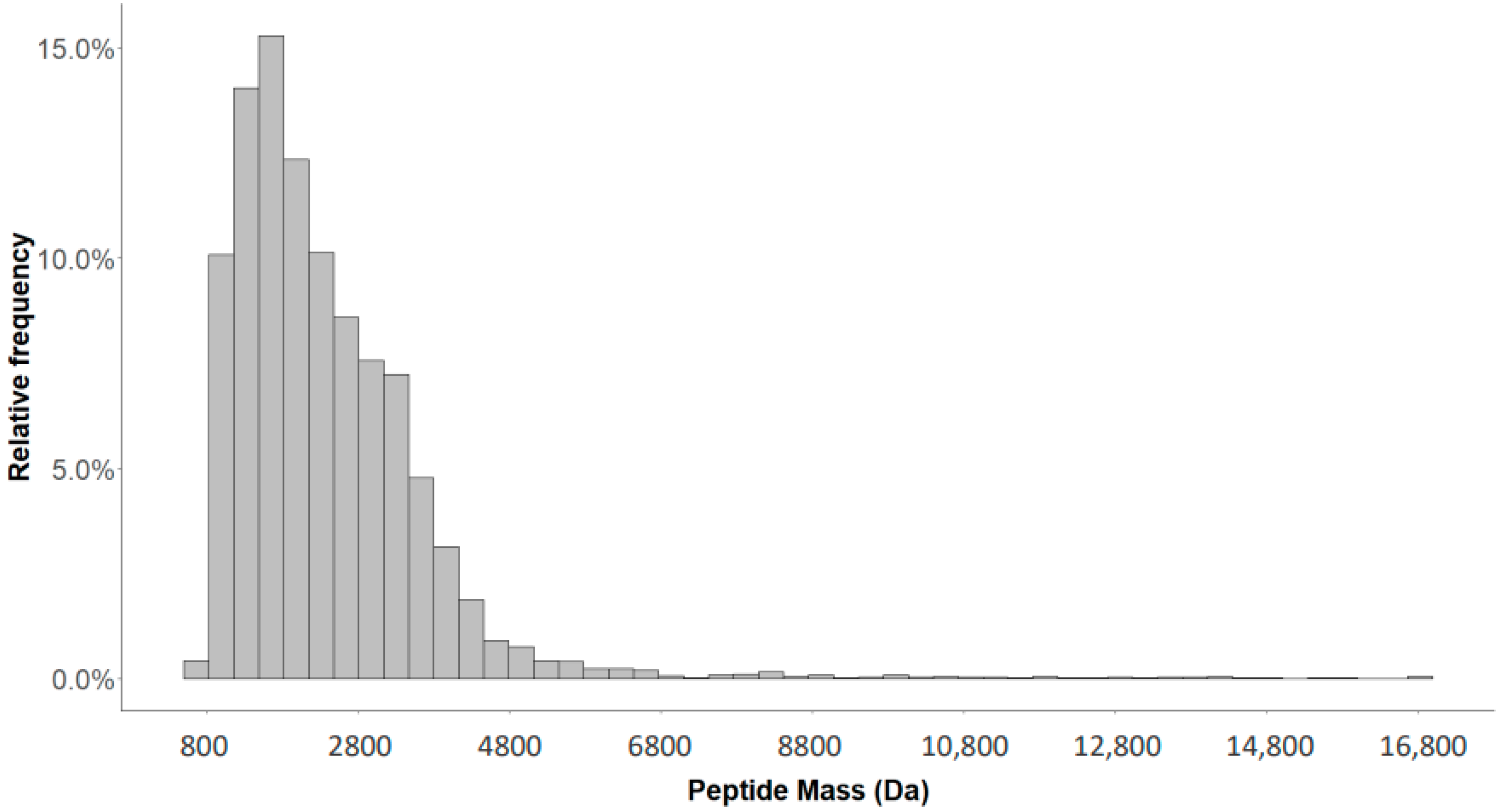
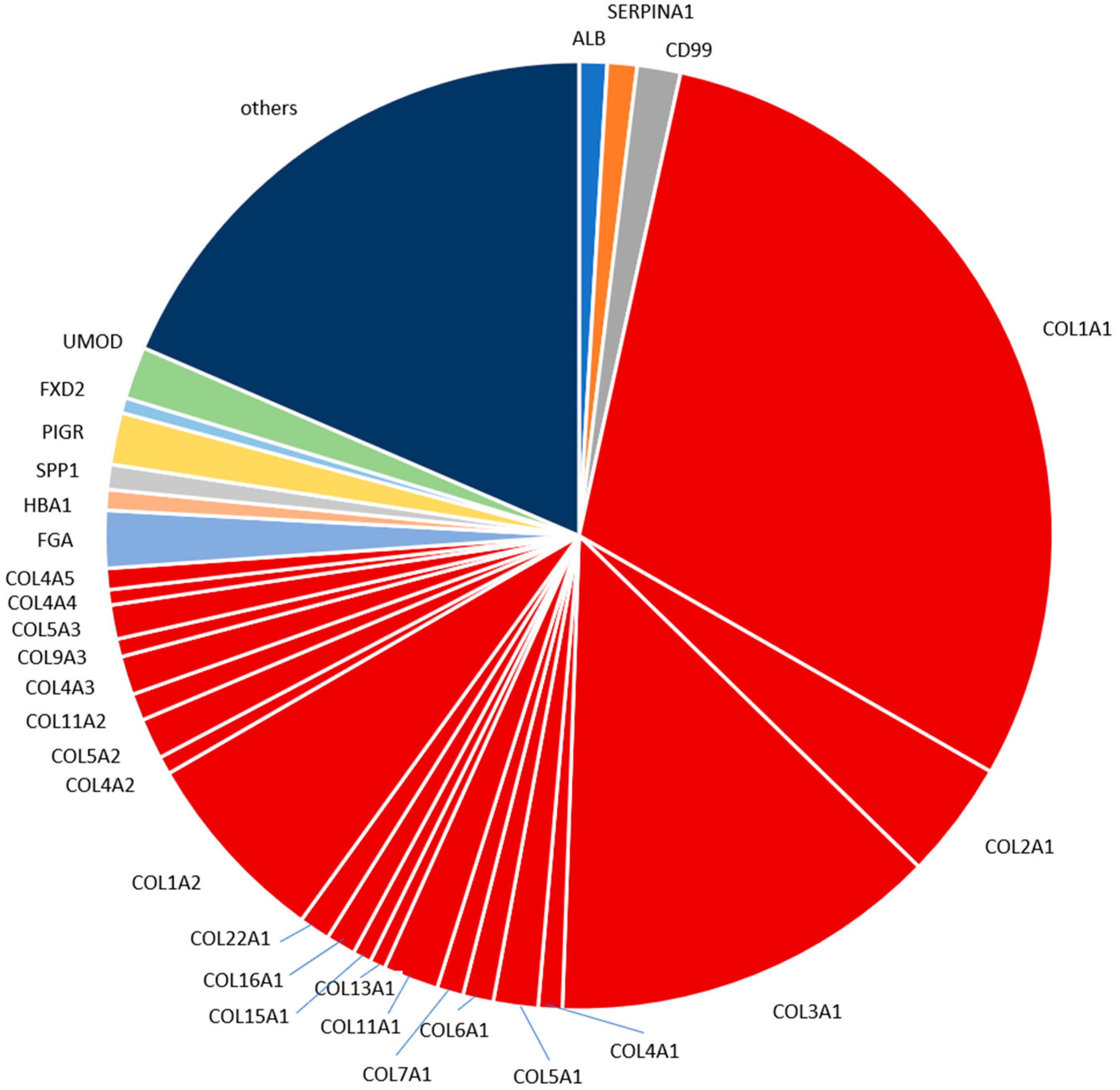
2.4. Characterization of the Urinary Peptidome Representative of a Healthy Individual
3. Discussion
4. Conclusions
5. Methods
5.1. Urine Sample
5.2. Capillary Electrophoresis Mass Spectrometry
5.3. MS Data Evaluation
5.4. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Latosinska, A.; Siwy, J.; Mischak, H.; Frantzi, M. Peptidomics and proteomics based on CE-MS as a robust tool in clinical application: The past, the present, and the future. Electrophoresis 2019, 40, 2294–2308. [Google Scholar] [CrossRef]
- Stalmach, A.; Albalat, A.; Mullen, W.; Mischak, H. Recent advances in capillary electrophoresis coupled to mass spectrometry for clinical proteomic applications. Electrophoresis 2013, 34, 1452–1464. [Google Scholar] [CrossRef]
- Latosinska, A.; Frantzi, M.; Vlahou, A.; Mischak, H. Clinical applications of capillary electrophoresis coupled to mass spectrometry in biomarker discovery: Focus on bladder cancer. Proteom. Clin. Appl. 2013, 7, 779–793. [Google Scholar] [CrossRef]
- Mischak, H.; Vlahou, A.; Ioannidis, J.P. Technical aspects and inter-laboratory variability in native peptide profiling: The CE–MS experience. Clin. Biochem. 2012, 46, 432–443. [Google Scholar] [CrossRef]
- Mischak, H.; Schanstra, J.P. CE-MS in biomarker discovery, validation, and clinical application. Proteom. Clin. Appl. 2010, 5, 9–23. [Google Scholar] [CrossRef]
- Huhn, C.; Ramautar, R.; Wuhrer, M.; Somsen, G.W. Relevance and use of capillary coatings in capillary electrophoresis–mass spectrometry. Anal. Bioanal. Chem. 2009, 396, 297–314. [Google Scholar] [CrossRef]
- Pontillo, C.; Filip, S.; Borràs, D.M.; Mullen, W.; Vlahou, A.; Mischak, H. CE-MS-based proteomics in biomarker discovery and clinical application. Proteom. Clin. Appl. 2015, 9, 322–334. [Google Scholar] [CrossRef]
- Kolch, W.; Neusüß, C.; Pelzing, M.; Mischak, H. Capillary electrophoresis-mass spectrometry as a powerful tool in clinical diagnosis and biomarker discovery. Mass Spectrom. Rev. 2005, 24, 959–977. [Google Scholar] [CrossRef]
- Klein, J.; Papadopoulos, T.; Mischak, H.; Mullen, W. Comparison of CE-MS/MS and LC-MS/MS sequencing demonstrates significant complementarity in natural peptide identification in human urine. Electrophoresis 2014, 35, 1060–1064. [Google Scholar] [CrossRef]
- Mischak, H.; Coon, J.J.; Novak, J.; Weissinger, E.M.; Schanstra, J.P.; Dominiczak, A. Capillary electrophoresis-mass spectrometry as a powerful tool in biomarker discovery and clinical diagnosis: An update of recent developments. Mass Spectrom. Rev. 2008, 28, 703–724. [Google Scholar] [CrossRef]
- Dakna, M.; Harris, K.; Kalousis, A.; Carpentier, S.; Kolch, W.; Schanstra, J.P.; Haubitz, M.; Vlahou, A.; Mischak, H.; Girolami, M. Addressing the Challenge of Defining Valid Proteomic Biomarkers and Classifiers. BMC Bioinform. 2010, 11, 594. [Google Scholar] [CrossRef] [Green Version]
- Stanley, E.; Delatola, E.I.; Nkuipou-Kenfack, E.; Spooner, W.; Kolch, W.; Schanstra, J.P.; Mischak, H.; Koeck, T. Comparison of different statistical approaches for urinary peptide biomarker detection in the context of coronary artery disease. BMC Bioinform. 2016, 17, 496. [Google Scholar] [CrossRef] [Green Version]
- Siwy, J.; Schiffer, E.; Brand, K.; Schumann, G.; Rossing, K.; Delles, C.; Mischak, H.; Metzger, J. Quantitative Urinary Proteome Analysis for Biomarker Evaluation in Chronic Kidney Disease. J. Proteome Res. 2008, 8, 268–281. [Google Scholar] [CrossRef]
- Rodríguez-Suárez, E.; Whetton, A.D. The application of quantification techniques in proteomics for biomedical research. Mass Spectrom. Rev. 2012, 32, 1–26. [Google Scholar] [CrossRef]
- Mavrogeorgis, E.; Mischak, H.; Beige, J.; Latosinska, A.; Siwy, J. Understanding glomerular diseases through proteomics. Expert Rev. Proteom. 2021, 18, 137–157. [Google Scholar] [CrossRef]
- Brown, C.E.; McCarthy, N.; Hughes, A.; Sever, P.; Stalmach, A.; Mullen, W.; Dominiczak, A.; Sattar, N.; Mischak, H.; Thom, S.; et al. Urinary proteomic biomarkers to predict cardiovascular events. Proteom. Clin. Appl. 2015, 9, 610–617. [Google Scholar] [CrossRef]
- He, T.; Mischak, M.; Clark, A.L.; Campbell, R.T.; Delles, C.; Díez, J.; Filippatos, G.; Mebazaa, A.; McMurray, J.J.; González, A.; et al. Urinary peptides in heart failure: A link to molecular pathophysiology. Eur. J. Heart Fail. 2021. [Google Scholar] [CrossRef]
- Wendt, R.; Thijs, L.; Kalbitz, S.; Mischak, H.; Siwy, J.; Raad, J.; Metzger, J.; Neuhaus, B.; von der Leyen, H.; Dudoignon, E.; et al. A urinary peptidomic profile predicts outcome in SARS-CoV-2-infected patients. EClinicalMedicine 2021, 36, 100883. [Google Scholar] [CrossRef]
- Wendt, R.; Kalbitz, S.; Lübbert, C.; Kellner, N.; Macholz, M.; Schroth, S.; Ermisch, J.; Latosisnka, A.; Arnold, B.; Mischak, H.; et al. Urinary Peptides Significantly Associate with COVID-19 Severity: Pilot Proof-of-Principle Data and Design of a Multicentric Diagnostic Study. Proteomics 2020, 20, 2000202. [Google Scholar] [CrossRef]
- Siwy, J.; Wendt, R.; Albalat, A.; He, T.; Mischak, H.; Mullen, W.; Latosinska, A.; Lübbert, C.; Kalbitz, S.; Mebazaa, A.; et al. CD99 and polymeric immunoglobulin receptor peptides deregulation in critical COVID-19: A potential link to molecular pathophysiology? Proteomics 2021, 21, 2100133. [Google Scholar] [CrossRef]
- Tofte, N.; Lindhardt, M.; Adamova, K.; Bakker, S.J.L.; Beige, J.; Beulens, J.W.J.; Birkenfeld, A.L.; Currie, G.; Delles, C.; Dimos, I.; et al. Early detection of diabetic kidney disease by urinary proteomics and subsequent intervention with spironolactone to delay progression (PRIORITY): A prospective observational study and embedded randomised placebo-controlled trial. Lancet Diabetes Endocrinol. 2020, 8, 301–312. [Google Scholar] [CrossRef]
- Pontillo, C.; Zhang, Z.-Y.; Schanstra, J.P.; Jacobs, L.; Zürbig, P.; Thijs, L.; Ramírez-Torres, A.; Heerspink, H.J.; Lindhardt, M.; Klein, R.; et al. Prediction of Chronic Kidney Disease Stage 3 by CKD273, a Urinary Proteomic Biomarker. Kidney Int. Rep. 2017, 2, 1066–1075. [Google Scholar] [CrossRef]
- Pontillo, C.; Jacobs, L.; Staessen, J.A.; Schanstra, J.P.; Rossing, P.; Heerspink, H.J.; Siwy, J.; Mullen, W.; Vlahou, A.; Mischak, H.; et al. A urinary proteome-based classifier for the early detection of decline in glomerular filtration. Nephrol. Dial. Transplant. 2016, 32, 1510–1516. [Google Scholar] [CrossRef] [Green Version]
- Lindhardt, M.; Persson, F.; Oxlund, C.; Jacobsen, I.A.; Zürbig, P.; Mischak, H.; Rossing, P.; Heerspink, H.J. Predicting albuminuria response to spironolactone treatment with urinary proteomics in patients with type 2 diabetes and hypertension. Nephrol. Dial. Transplant. 2017, 33, 296–303. [Google Scholar] [CrossRef] [Green Version]
- Latosinska, A.; Siwy, J.; Cherney, D.Z.; Perkins, B.A.; Mischak, H.; Beige, J. SGLT2-Inhibition reverts urinary peptide changes associated with severe COVID-19: An in-silico proof-of-principle of proteomics-based drug repurposing. Proteomics 2021, 21, 2100160. [Google Scholar] [CrossRef]
- Mischak, H.; Kolch, W.; Aivaliotis, M.; Bouyssié, D.; Court, M.; Dihazi, H.; Dihazi, G.H.; Franke, J.; Garin, J.; de Peredo, A.G.; et al. Comprehensive human urine standards for comparability and standardization in clinical proteome analysis. Proteom. Clin. Appl. 2010, 4, 464–478. [Google Scholar] [CrossRef] [Green Version]
- Kononikhin, A.S.; Zakharova, N.V.; Sergeeva, V.A.; Indeykina, M.I.; Starodubtseva, N.L.; Bugrova, A.E.; Muminova, K.T.; Khodzhaeva, Z.S.; Popov, I.A.; Shao, W.; et al. Differential Diagnosis of Preeclampsia Based on Urine Peptidome Features Revealed by High Resolution Mass Spectrometry. Diagnostics 2020, 10, 1039. [Google Scholar] [CrossRef]
- Van Huizen, N.A.; Van Rosmalen, J.; Dekker, L.J.M.; Braak, R.R.J.C.V.D.; Verhoef, C.; Ijzermans, J.N.M.; Luider, T.M. Identification of a Collagen Marker in Urine Improves the Detection of Colorectal Liver Metastases. J. Proteome Res. 2019, 19, 153–160. [Google Scholar] [CrossRef]
- Van, J.A.D.; Clotet-Freixas, S.; Zhou, J.; Batruch, I.; Sun, C.; Glogauer, M.; Rampoldi, L.; Elia, Y.; Mahmud, F.H.; Sochett, E.; et al. Peptidomic Analysis of Urine from Youths with Early Type 1 Diabetes Reveals Novel Bioactivity of Uromodulin Peptides In Vitro. Mol. Cell. Proteom. 2020, 19, 501–517. [Google Scholar] [CrossRef]
- Good, D.M.; Zürbig, P.; Argilés, A.; Bauer, H.W.; Behrens, G.; Coon, J.J.; Dakna, M.; Decramer, S.; Delles, C.; Dominiczak, A.; et al. Naturally Occurring Human Urinary Peptides for Use in Diagnosis of Chronic Kidney Disease. Mol. Cell. Proteom. 2010, 9, 2424–2437. [Google Scholar] [CrossRef] [Green Version]
- He, T.; Pejchinovski, M.; Mullen, W.; Beige, J.; Mischak, H.; Jankowski, V. Peptides in Plasma, Urine, and Dialysate: Toward Unravelling Renal Peptide Handling. Proteom. Clin. Appl. 2020, 15, e2000029. [Google Scholar] [CrossRef]
- Pontillo, C.; Mischak, H. Urinary peptide-based classifier CKD273: Towards clinical application in chronic kidney disease. Clin. Kidney J. 2017, 10, 192–201. [Google Scholar] [CrossRef]
- Thongboonkerd, V.; Mungdee, S.; Chiangjong, W. Should Urine pH Be Adjusted Prior to Gel-Based Proteome Analysis? J. Proteome Res. 2009, 8, 3206–3211. [Google Scholar] [CrossRef]
- Theodorescu, D.; Fliser, D.; Wittke, S.; Mischak, H.; Krebs, R.; Walden, M.; Ross, M.; Eltze, E.; Bettendorf, O.; Wülfing, C.; et al. Pilot study of capillary electrophoresis coupled to mass spectrometry as a tool to define potential prostate cancer biomarkers in urine. Electrophoresis 2005, 26, 2797–2808. [Google Scholar] [CrossRef]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016; ISBN 978-3-319-24277-4. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inter-Day | Intra-Day | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Mass [Da] | CE Time [Min] | Sequence | Gene Symbol | Mean log2 Int | SD | CV [%] | Mean log2 Int | SD | CV [%] |
| 1754.92 | 31.39 | SGSVIDQSRVLNLGPIT | UMOD | 15.56 | 0.33 | 2.12 | 15.48 | 0.17 | 1.07 |
| 3457.61 | 31.46 | NTGAPGSpGVSGpKGDAGQpGEKGSpGAQGppGAPGPLG | COL3A1 | 15.05 | 0.42 | 2.76 | 15.29 | 0.18 | 1.18 |
| 2248.99 | 26.16 | GGpGSDGKPGppGSQGESGRPGPpG | COL3A1 | 14.94 | 0.20 | 1.33 | 14.83 | 0.14 | 0.98 |
| 1882.80 | 20.24 | DEAGSEADHEGTHSTKRG | FGA | 14.87 | 0.37 | 2.49 | 14.64 | 0.13 | 0.91 |
| 2825.28 | 24.45 | ERGEAGIpGVpGAKGEDGKDGSpGEpGANG | COL3A1 | 14.76 | 0.28 | 1.92 | 14.72 | 0.13 | 0.88 |
| 1250.56 | 28.00 | ApGDRGEpGPPGp | COL1A1 | 14.65 | 0.18 | 1.26 | 14.34 | 0.11 | 0.75 |
| 2169.97 | 26.10 | NSGEpGApGSKGDTGAKGEPGpVG | COL1A1 | 14.61 | 0.21 | 1.46 | 14.45 | 0.16 | 1.10 |
| 2047.92 | 21.93 | NGDDGEAGKpGRpGERGPPGP | COL1A1 | 14.37 | 0.62 | 4.32 | 14.33 | 0.23 | 1.62 |
| 1911.05 | 25.23 | SGSVIDQSRVLNLGPITR | UMOD | 14.34 | 0.82 | 5.75 | 11.29 | 0.18 | 1.61 |
| 3441.61 | 31.36 | DGAPGQKGEMGPAGPTGPRGFpGppGPDGLPGSMGPP | COL4A1 | 14.04 | 0.33 | 2.36 | 14.49 | 0.23 | 1.57 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mavrogeorgis, E.; Mischak, H.; Latosinska, A.; Siwy, J.; Jankowski, V.; Jankowski, J. Reproducibility Evaluation of Urinary Peptide Detection Using CE-MS. Molecules 2021, 26, 7260. https://doi.org/10.3390/molecules26237260
Mavrogeorgis E, Mischak H, Latosinska A, Siwy J, Jankowski V, Jankowski J. Reproducibility Evaluation of Urinary Peptide Detection Using CE-MS. Molecules. 2021; 26(23):7260. https://doi.org/10.3390/molecules26237260
Chicago/Turabian StyleMavrogeorgis, Emmanouil, Harald Mischak, Agnieszka Latosinska, Justyna Siwy, Vera Jankowski, and Joachim Jankowski. 2021. "Reproducibility Evaluation of Urinary Peptide Detection Using CE-MS" Molecules 26, no. 23: 7260. https://doi.org/10.3390/molecules26237260
APA StyleMavrogeorgis, E., Mischak, H., Latosinska, A., Siwy, J., Jankowski, V., & Jankowski, J. (2021). Reproducibility Evaluation of Urinary Peptide Detection Using CE-MS. Molecules, 26(23), 7260. https://doi.org/10.3390/molecules26237260