
A Validated HPLC-PDA-HRMS Method to Investigate the Biological Stability and Metabolism of Antiparasitic Triterpenic Esters

 ,
,  ,
,
Abstract
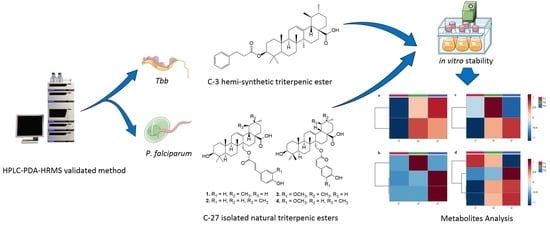
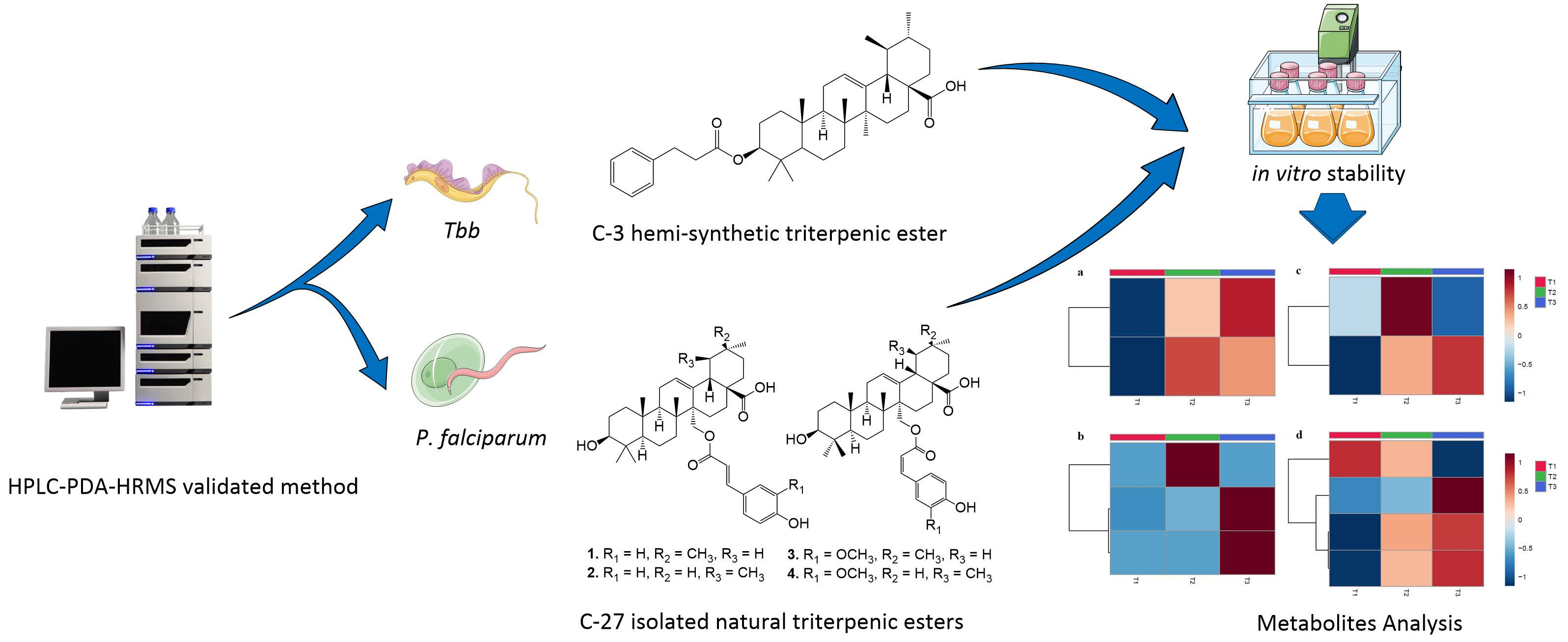
:
1. Introduction
2. Results and Discussion
2.1. Method Validation
2.1.1. Selectivity
2.1.2. Response Function
2.1.3. Trueness, Precision and Accuracy
2.1.4. Detection and Quantification Limits
2.1.5. Uncertainty of Measurement
2.1.6. Linearity
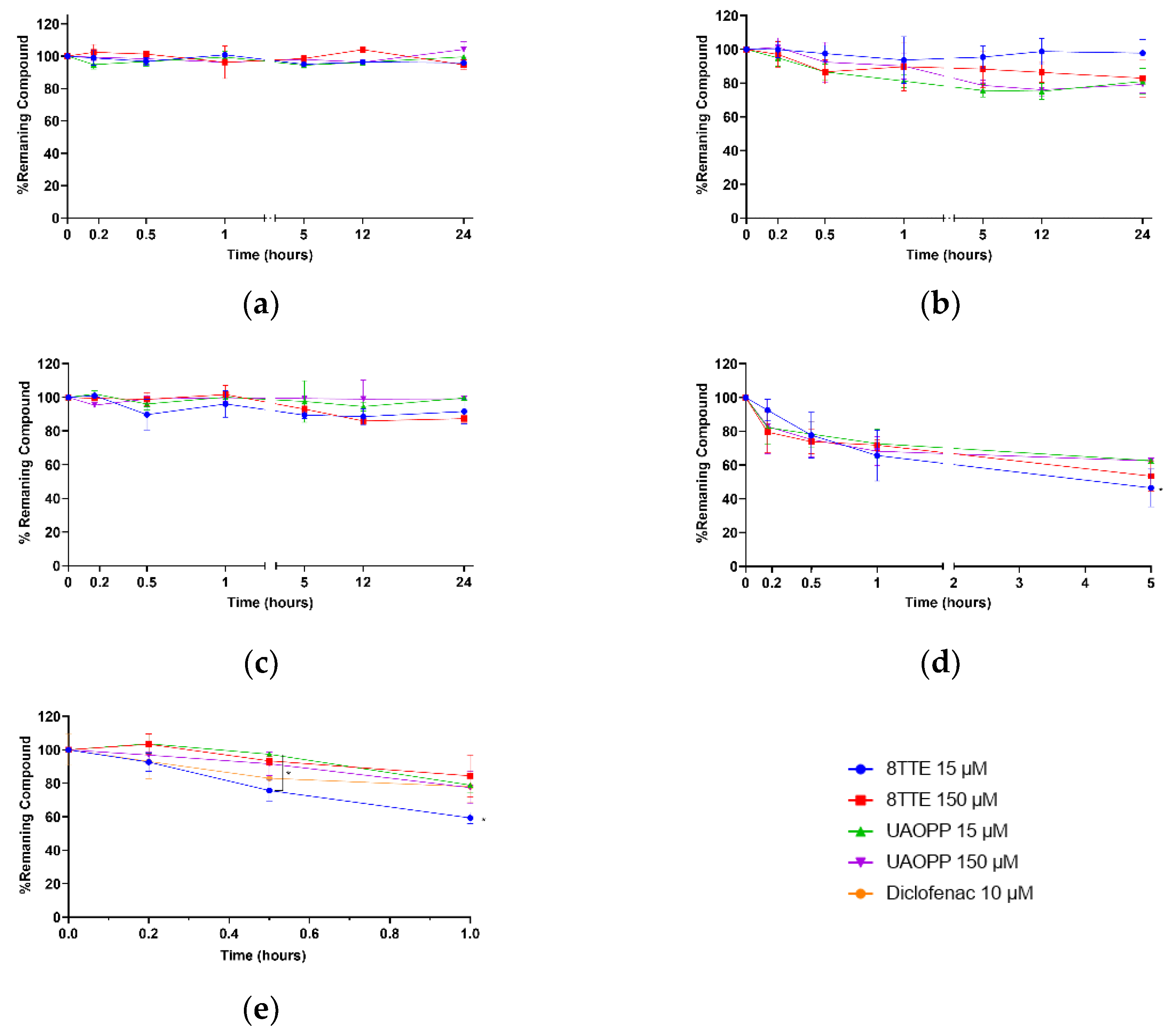
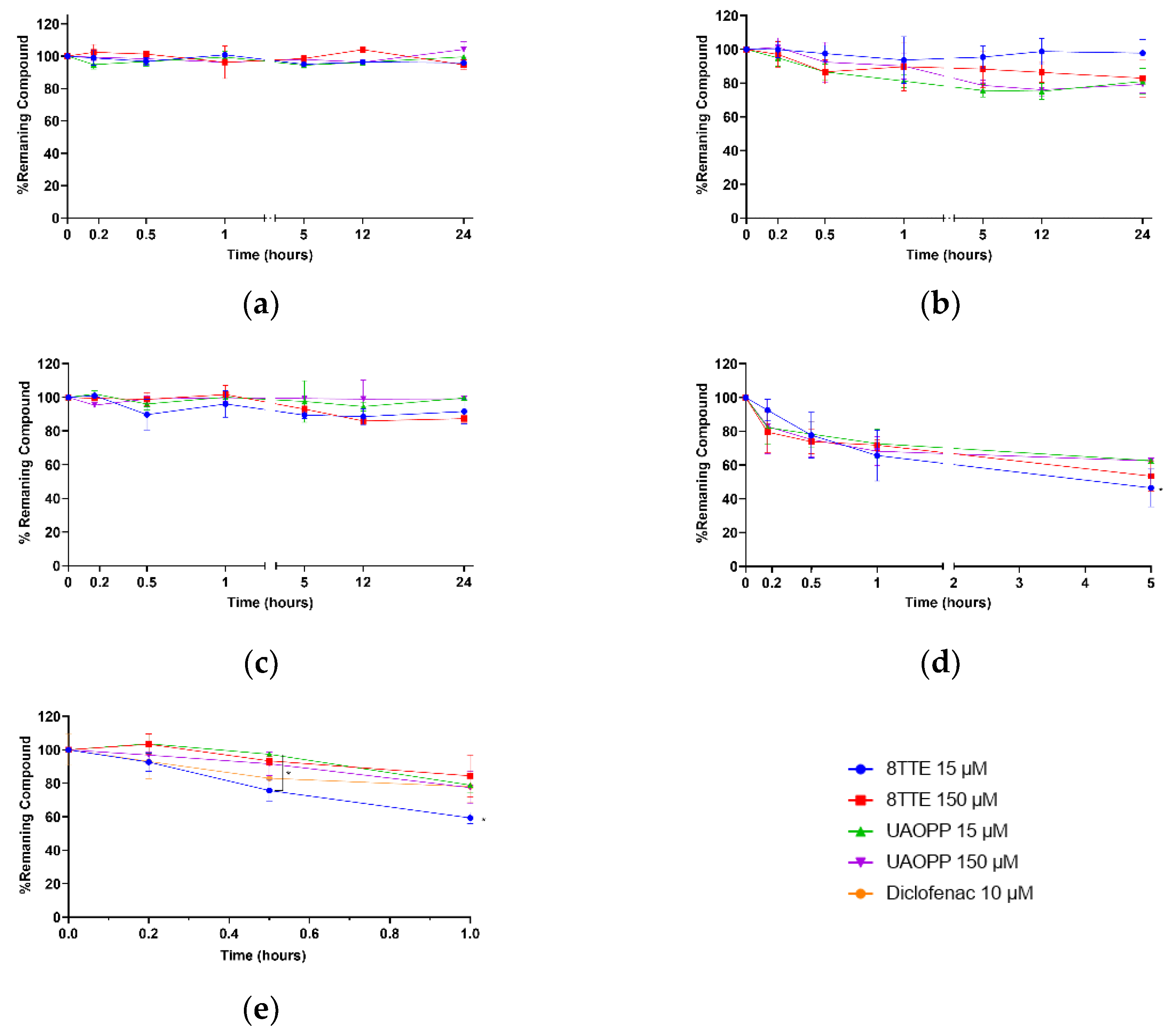
2.2. Stability Profiles Investigation
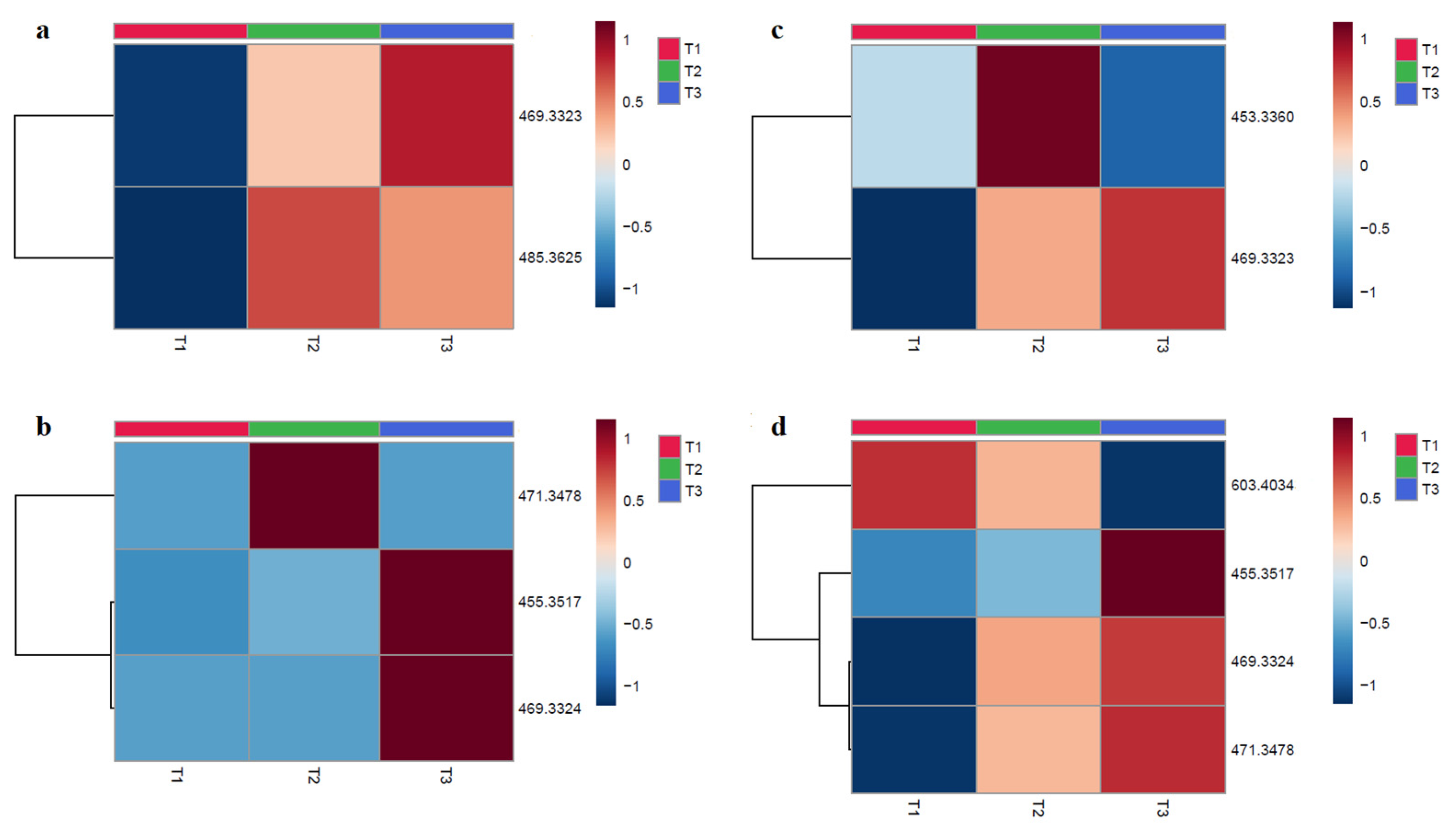
2.3. Metabolites Analysis
3. Materials and Methods
3.1. Compound Isolation and Synthesis
3.2. Standard solutions
3.3. Plasma Samples
3.4. HPLC-PDA- and HPLC-PDA-HRMS Analysis
3.5. Method Validation
3.6. Stability Tests
3.7. Sampling and Data Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Control of Neglected Tropical Diseases. Available online: https://www.who.int/teams/control-of-neglected-tropical-diseases (accessed on 10 February 2021).
- Büscher, P.; Cecchi, G.; Jamonneau, V.; Priotto, G. Human African trypanosomiasis. Lancet 2017, 390, 2397–2409. [Google Scholar] [CrossRef]
- Prevention, C.-C. For D.C. and CDC—Malaria—Malaria Worldwide—Impact of Malaria. 2021. Available online: https://www.cdc.gov/malaria/malaria_worldwide/impact.html (accessed on 20 October 2021).
- Conteh, L.; Engels, T.; Molyneux, D.H. Socioeconomic aspects of neglected tropical diseases. Lancet 2010, 375, 239–247. [Google Scholar] [CrossRef]
- Ashley, E.A.; Dhorda, M.; Fairhurst, R.M.; Amaratunga, C.; Lim, P.; Suon, S.; Sreng, S.; Anderson, J.M.; Mao, S.; Sam, B.; et al. Spread of artemisinin resistance in Plasmodium falciparum malaria. N. Engl. J. Med. 2014, 371, 411–423. [Google Scholar] [CrossRef] [Green Version]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paduch, R.; Kandefer-Szerszen, M. Antitumor and Antiviral Activity of Pentacyclic Triterpenes. Mini. Rev. Org. Chem. 2014, 11, 262–268. [Google Scholar] [CrossRef]
- Isah, M.B.; Ibrahim, M.A.; Mohammed, A.; Aliyu, A.B.; Masola, B.; Coetzer, T.H.T. A systematic review of pentacyclic triterpenes and their derivatives as chemotherapeutic agents against tropical parasitic diseases. Parasitology 2016, 143, 1219–1231. [Google Scholar] [CrossRef]
- Bero, J.; Hérent, M.F.; Schmeda-Hirschmann, G.; Frédérich, M.; Quetin-Leclercq, J. In Vivo antimalarial activity of Keetia leucantha twigs extracts and in vitro antiplasmodial effect of their constituents. J. Ethnopharmacol. 2013, 149, 176–183. [Google Scholar] [CrossRef]
- Beaufay, C.; Hérent, M.F.; Quetin-Leclercq, J.; Bero, J. In Vivo anti-malarial activity and toxicity studies of triterpenic esters isolated form Keetia leucantha and crude extracts. Malar. J. 2017, 16, 406. [Google Scholar] [CrossRef] [Green Version]
- Schioppa, L.; Beaufay, D.C.; Bonneau, D.N.; Sanchez, D.M.; Girardi, D.C.; Leverrier, D.A.; Ortiz, D.S.; Palermo, P.J.; Poupaert, P.J.H.; Quetin-Leclercq, P.J. Design, Synthesis and Biological Activity of C3 Hemisynthetic Triterpenic Esters as Novel Antitrypanosomal Hits. ChemistryOpen 2021, 10, 896. [Google Scholar] [CrossRef]
- Rozet, E.; Hubert, C.; Ceccato, A.; Dewé, W.; Ziemons, E.; Moonen, F.; Michail, K.; Wintersteiger, R.; Streel, B.; Boulanger, B.; et al. Using tolerance intervals in pre-study validation of analytical methods to predict in-study results. The fit-for-future-purpose concept. J. Chromatogr. A 2007, 1158, 126–137. [Google Scholar] [CrossRef]
- Hubert, P.; Nguyen-Huu, J.J.; Boulanger, B.; Chapuzet, E.; Chiap, P.; Cohen, N.; Compagnon, P.A.; Dewé, W.; Feinberg, M.; Lallier, M.; et al. Harmonization of strategies for the validation of quantitative analytical procedures. A SFSTP proposal—Part II. J. Pharm. Biomed. Anal. 2007, 45, 70–81. [Google Scholar] [CrossRef] [PubMed]
- Hubert, P.; Nguyen-Huu, J.J.; Boulanger, B.; Chapuzet, E.; Chiap, P.; Cohen, N.; Compagnon, P.A.; Dewé, W.; Feinberg, M.; Lallier, M.; et al. Harmonization of strategies for the validation of quantitative analytical procedures: A SFSTP proposal—Part I. J. Pharm. Biomed. Anal. 2004, 36, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Rozet, E.; Ceccato, A.; Hubert, C.; Ziemons, E.; Oprean, R.; Rudaz, S.; Boulanger, B.; Hubert, P. Analysis of recent pharmaceutical regulatory documents on analytical method validation. J. Chromatogr. A 2007, 1158, 111–125. [Google Scholar] [CrossRef]
- Q2 (R1) Validation of Analytical Procedures: Text and Methodology|FDA. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/q2-r1-validation-analytical-procedures-text-and-methodology (accessed on 15 March 2021).
- Medicines Agency, E. 2 ** Committee for Medicinal Products for Human Use (CHMP) Guideline on bioanalytical method validation; 1922. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/draft-guideline-validation-bionanalytical-methods_en.pdf (accessed on 20 October 2021).
- European Pharmacopoeia (Ph. Eur.) 10th Edition|EDQM—European Directorate for the Quality of Medicines. Available online: https://www.edqm.eu/en/european-pharmacopoeia-ph-eur-10th-edition (accessed on 15 March 2021).
- Lautié, E.; Rozet, E.; Hubert, P.; Quetin Leclercq, J. Quantification of rotenone in seeds of different species of yam bean (Pachyrhizus sp.) by a SPE HPLC-UV method. Food Chem. 2012, 131, 1531–1538. [Google Scholar] [CrossRef]
- Rozet, E.; Marini, R.D.; Ziemons, E.; Boulanger, B.; Hubert, P. Advances in validation, risk and uncertainty assessment of bioanalytical methods. J. Pharm. Biomed. Anal. 2011, 55, 848–858. [Google Scholar] [CrossRef]
- Hubert, P.; Nguyen-Huu, J.J.; Boulanger, B.; Chapuzet, E.; Cohen, N.; Compagnon, P.A.; Dewé, W.; Feinberg, M.; Laurentie, M.; Mercier, N.; et al. Harmonization of strategies for the validation of quantitative analytical procedures: A SFSTP proposal. Part IV. Examples of application. J. Pharm. Biomed. Anal. 2008, 48, 760–771. [Google Scholar] [CrossRef] [PubMed]
- Di, L.; Kerns, E.H. Stability Challenges in Drug Discovery. Chem. Biodivers. 2009, 6, 1875–1886. [Google Scholar] [CrossRef] [PubMed]
- Catteau, L.; Schioppa, L.; Beaufay, C.; Girardi, C.; Hérent, M.F.; Frédérich, M.; Quetin-Leclercq, J. Antiprotozoal activities of Triterpenic Acids and Ester Derivatives Isolated from the Leaves of Vitellaria paradoxa. Planta Med. 2021, 87, 860–867. [Google Scholar] [CrossRef]
- Yu, Z.; Sun, W.; Peng, W.; Yu, R.; Li, G.; Jiang, T. Pharmacokinetics in vitro and in Vivo of Two Novel Prodrugs of Oleanolic Acid in Rats and Its Hepatoprotective Effects against Liver Injury Induced by CCl4. Mol. Pharm. 2016, 13, 1699–1710. [Google Scholar] [CrossRef]
- Chand Gupta, A.; Bawankule, D.U.; Verma, A.K.; Shanker, K. Nanoemulsion preconcentrate of a pentacyclic triterpene for improved oral efficacy: Formulation design and in-vivo antimalarial activity. J. Drug Deliv. Sci. Technol. 2020, 57, 101734. [Google Scholar] [CrossRef]
- Soica, C.; Trandafirescu, C.; Danciu, C.; Muntean, D.; Dehelean, C.; Simu, G. New improved drug delivery technologies for pentacyclic triterpenes: A review. Protein Pept. Lett. 2014, 21, 1137–1145. [Google Scholar] [CrossRef]
- Furtado, N.A.J.C.; Pirson, L.; Edelberg, H.; Miranda, L.M.; Loira-Pastoriza, C.; Preat, V.; Larondelle, Y.; André, C.M. Pentacyclic triterpene bioavailability: An overview of in vitro and in vivo studies. Molecules 2017, 22, 400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, D.W.; Kim, Y.H.; Kim, H.H.; Ji, H.Y.; Yoo, S.D.; Choi, W.R.; Lee, S.M.; Han, C.K.; Lee, H.S. Dose-linear pharmacokinetics of oleanolic acid after intravenous and oral administration in rats. Biopharm. Drug Dispos. 2007, 28, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Kartasasmita, R.; Musfiroh, I.; Muhtadi, A.; Ibrahim, S. Binding affinity of asiatic acid derivatives design against Inducible Nitric Oxide Synthase and ADMET Prediction ARTICLE INFO ABSTRACT. J. Appl. Pharm. Sci. 2014, 4, 75–80. [Google Scholar] [CrossRef]
- Chen, D.; Huang, X.; Zhou, H.; Luo, H.; Wang, P.; Chang, Y.; He, X.; Ni, S.; Shen, Q.; Cao, G.; et al. Discovery of pentacyclic triterpene 3β-ester derivatives as a new class of cholesterol ester transfer protein inhibitors. Eur. J. Med. Chem. 2017, 139, 201–213. [Google Scholar] [CrossRef]
- Mukkavilli, R.; Pinjari, J.; Patel, B.; Sengottuvelan, S.; Mondal, S.; Gadekar, A.; Verma, M.; Patel, J.; Pothuri, L.; Chandrashekar, G.; et al. In Vitro metabolism, disposition, preclinical pharmacokinetics and prediction of human pharmacokinetics of DNDI-VL-2098, a potential oral treatment for Visceral Leishmaniasis. Eur. J. Pharm. Sci. 2014, 65, 147–155. [Google Scholar] [CrossRef] [Green Version]
- Viant, M.R.; Kurland, I.J.; Jones, M.R.; Dunn, W.B. How close are we to complete annotation of metabolomes? Curr. Opin. Chem. Biol. 2017, 36, 64–69. [Google Scholar] [CrossRef]
- Sánchez-González, M.; Lozano-Mena, G.; Parra, A.; Juan, M.E.; Planas, J.M. Identification in rat plasma and urine by linear trap quadrupole-orbitrap mass spectrometry of the metabolites of maslinic acid, a triterpene from olives. J. Agric. Food Chem. 2015, 63, 1126–1132. [Google Scholar] [CrossRef]
- Xia, B.; Bai, L.; Li, X.; Xiong, J.; Xu, P.; Xue, M. Structural Analysis of Metabolites of Asiatic Acid and Its Analogue Madecassic Acid in Zebrafish Using LC/IT-MSn. Molecules 2015, 20, 3001–3019. [Google Scholar] [CrossRef] [Green Version]
- Moneriz, C.; Mestres, J.; Bautista, J.M.; Diez, A.; Puyet, A. Multi-targeted activity of maslinic acid as an antimalarial natural compound. FEBS J. 2011, 278, 2951–2961. [Google Scholar] [CrossRef]
- Mullié, C.; Jonet, A.; Dassonville-Klimpt, A.; Gosmann, G.; Sonnet, P. Inhibitory effect of ursolic acid derivatives on hydrogen peroxide- and glutathione-mediated degradation of hemin: A possible additional mechanism of action for antimalarial activity. Exp. Parasitol. 2010, 125, 202–207. [Google Scholar] [CrossRef] [PubMed]
- Maya, J.D.; Cassels, B.K.; Iturriaga-Vásquez, P.; Ferreira, J.; Faúndez, M.; Galanti, N.; Ferreira, A.; Morello, A. Mode of action of natural and synthetic drugs against Trypanosoma cruzi and their interaction with the mammalian host. Comp. Biochem. Physiol. Part A Mol. Integr. Physiol. 2007, 146, 601–620. [Google Scholar] [CrossRef]
- Agency, E.M. Guideline on Bioanalytical Method Validation. 2011. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-bioanalytical-method-validation_en.pdf (accessed on 20 October 2021).
- Kessner, D.; Chambers, M.; Burke, R.; Agus, D.; Mallick, P. ProteoWizard: Open source software for rapid proteomics tools development. Bioinformatics 2008, 24, 2534–2536. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.A.; Want, E.J.; O’Maille, G.; Abagyan, R.; Siuzdak, G. XCMS: Processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification. Anal. Chem. 2006, 78, 779–787. [Google Scholar] [CrossRef]
- Giacomoni, F.; Le Corguille, G.; Monsoor, M.; Landi, M.; Pericard, P.; Pétéra, M.; Duperier, C.; Tremblay-Franco, M.; Martin, J.-F.; Jacob, D. Workflow4Metabolomics: A collaborative research infrastructure for computational metabolomics. Bioinformatics 2015, 31, 1493–1495. [Google Scholar] [CrossRef] [Green Version]
- Kuhl, C.; Tautenhahn, R.; Böttcher, C.; Larson, T.R.; Neumann, S. CAMERA: An integrated strategy for compound spectra extraction and annotation of liquid chromatography/mass spectrometry data sets. Anal. Chem. 2012, 84, 283–289. [Google Scholar] [CrossRef] [Green Version]
- Rev 1 * EU Individual Case Safety Report (ICSR) 1 Implementation Guide Start of Public Consultation of First Version Individual Case Safety Reports (ICSRs) in Pharmacovigilance—Part 2: Human Pharmaceutical Reporting Requirements for ICSR (ISO 27953-2:2011) and the ICH E2B(R3) Implementation Guide. 2017. Available online: https://ich.org/page/e2br3-individual-case-safety-report-icsr-specification-and-related-files (accessed on 20 October 2021).
- Hubert, P.; Nguyen-Huu, J.J.; Boulanger, B.; Chapuzet, E.; Cohen, N.; Compagnon, P.A.; Dewé, W.; Feinberg, M.; Laurentie, M.; Mercier, N.; et al. Harmonization of strategies for the validation of quantitative analytical procedures. A SFSTP proposal-Part III. J. Pharm. Biomed. Anal. 2007, 45, 82–96. [Google Scholar] [CrossRef] [PubMed]
- Rafamantanana, M.H.; Rozet, E.; Raoelison, G.E.; Cheuk, K.; Ratsimamanga, S.U.; Hubert, P.; Quetin-Leclercq, J. An improved HPLC-UV method for the simultaneous quantification of triterpenic glycosides and aglycones in leaves of Centella asiatica (L.) Urb (APIACEAE). J. Chromatogr. B 2009, 877, 2396–2402. [Google Scholar] [CrossRef]
- Corning® GentestTM Mouse (B6C3F1) Pooled Liver Microsomes, Male, 0.5 mL|B6C3F1, Male|Microsome Type|Life Sciences EU Other Consumer Site|Corning. Available online: https://ecatalog.corning.com/life-sciences/b2c/EUOther/en/ADME-Tox-Research/Tissue-Fractions/Animal-Liver-Fractions/Animal-Liver-Tissue-Fraction-Products---Corning®-GentestTM-Animal-Pooled-Liver-Microsomes-Mouse-Liver-Microsomes/p/452220 (accessed on 4 March 2021).
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [CrossRef]
- Mohutsky, M.A.; Chien, J.Y.; Ring, B.J.; Wrighton, S.A. Predictions of the in vivo clearance of drugs from rate of loss using human liver microsomes for phase I and phase II biotransformations. Pharm. Res. 2006, 23, 654–662. [Google Scholar] [CrossRef]
- Greenblatt, D.J. Elimination half-life of drugs: Value and limitations. Annu. Rev. Med. 1985, 36, 421–427. [Google Scholar] [CrossRef] [PubMed]
- Bohnert, T.; Gan, L.L. In Vitro Experimental Models for Studying Drug Biotransformation. In Handbook of Metabolic Pathways of Xenobiotics; John Wiley & Sons, Ltd.: Chichester, UK, 2013; pp. 1–61. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
| Valid. Criteria | Concentration Levels (µg/mL) for Methanolic Samples | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| UA-3-O-PP | 8TTE | |||||||||
| 2.5 | 10 | 37.5 | 100 | 1.25 | 2.5 | 37.5 | 75 | 100 | ||
| Response function | Linear regression | |||||||||
| Calib. range | 4 points | 5 points | ||||||||
| µg/mL | 2.5–100 | 1.25–100 | ||||||||
| Trueness | Relative Bias (%) | −1.87 | 5.05 | 0.06 | 0.51 | 7.24 | −2.80 | −4.14 | −2.38 | −1.58 |
| Precision | Repeatability (RSD 1 %) | 2.18 | 1.66 | 1.72 | 1.19 | 4.78 | 4.72 | 4.55 | 2.17 | 4.08 |
| Intermediate precision (RSD 1 %) | 3.84 | 2.88 | 2.71 | 2.46 | 4.41 | 4.16 | 3.81 | 2.99 | 4.23 | |
| Accuracy | (lower and uppertol. Limits %) | −15.59 | −5.26 | −8.34 | −8.35 | −3.27 | −12.63 | −13.23 | −11.54 | −12.18 |
| 11.84 | 15.35 | 8.45 | 9.37 | 17.76 | 7.02 | 4.95 | 6.79 | 9.03 | ||
| Linearity | Slope | 1.003 | 0.982 | |||||||
| Int.pt | 0.122 | −0.201 | ||||||||
| R2 | 0.999 | 0.997 | ||||||||
| Valid. Criteria | Concentration Levels 1 (µg/mL) for Plasma Samples | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| UA-3-O-PP | 8TTE | ||||||||
| 5 | 25 | 50 | 100 | 5 | 25 | 50 | 100 | ||
| Response function | Linear regression | ||||||||
| Calib. range | 5 points | 6 points | |||||||
| µg/mL | 5–125 | 2.5–125 | |||||||
| Trueness | Relative Bias (%) | −13.03 | −0.57 | −2.25 | −0.57 | 15.47 | 7.18 | −0.94 | 1.61 |
| Precision | Repeatability (RSD 2 %) | 2.89 | 2.70 | 1.57 | 1.79 | 1.81 | 2.66 | 1.05 | 1.52 |
| Intermediate precision (RSD 2 %) | 2.65 | 3.25 | 3.19 | 3.72 | 1.48 | 2.47 | 2.49 | 1.83 | |
| Accuracy | (lower and uppertol. limits %) | −19.35 | −9.67 | −13.74 | −13.97 | 11.85 | 1.28 | −9.97 | −3.26 |
| −6.71 | 8.52 | 9.23 | 12.83 | 19.09 | 13.09 | 8.07 | 6.48 | ||
| Linearity | Slope | 0.998 | 1.004 | ||||||
| Int.pt | −0.551 | 0.755 | |||||||
| R2 | 0.998 | 0.999 | |||||||
| Concentration Level | Uncertainty (µg/mL) | Expanded Uncertainty (µg/mL) | Relative Expanded Uncertainty (%) | |||
|---|---|---|---|---|---|---|
| (µg/mL) | UA-3-O-PP | 8TTE | UA-3-O-PP | 8TTE | UA-3-O-PP | 8TTE |
| Methanolic sample method | ||||||
| 1.25 | - | 0.061 | - | 0.122 | - | 9.780 |
| 2.5 | 0.106 | 0.104 | 0.211 | 0.207 | 8.460 | 8.285 |
| 10 | 0.340 | - | 0.680 | - | 6.801 | - |
| 37.5 | 1.135 | 1.382 | 0.586 | 2.764 | 6.052 | 7.371 |
| 75 | - | 2.417 | - | 4.834 | - | 6.445 |
| 100 | 2.979 | 4.414 | 5.595 | 8.828 | 5.595 | 8.828 |
| Plasma sample method | ||||||
| 5 1 | 0.119 | 0.085 | 0.239 | 0.171 | 4.765 | 3.419 |
| 25 1 | 0.879 | 0.686 | 1.759 | 1.373 | 7.034 | 5.491 |
| 50 1 | 1.763 | 1.402 | 3.527 | 2.805 | 7.054 | 5.610 |
| 100 1 | 4.186 | 2.022 | 8.371 | 4.045 | 8.371 | 4.045 |
| Kloss (min−1) | t1/2 (min) | Cl Int (µg/min/mg) | |
|---|---|---|---|
| 8TTE | 0.009 | 76.82 | 18.04 |
| UA-3-O-PP | 0.003 | 264.30 | 5.24 |
| Diclofenac | 0.004 | 153.25 | 9.04 |
| 8TTE | Conc. | RT (sec) | Mol. Ion | Formula (M) | m/zexp | m/zthr | Δ ppm | ID vl | Reaction |
| 150 µM | 1005.73 | [M − 1]− | C30H46O3 | 453.3361 | 453.3374 | −2.8 | 2 | Hydrolysis + dehydration | |
| 884.76 | [M − 1]− | C30H46O4 | 469.3323 | 469.3323 | 0 | 2 | Hydrolysis + Reduction | ||
| 15 µM | 884.76 | [M − 1]− | C30H46O4 | 469.3323 | 469.3323 | 0 | 2 | Hydrolysis + Reduction | |
| 860.19 | [M − 1]− | C31H50O4 | 485.3625 | 485.3631 | −1.2 | 2 | Hydrolysis + 0-Methylation | ||
| UA-3-O-PP | 150 µM | 951.46 | [M − 1]− | C30H48O3 | 455.3518 | 455.3530 | 2.6 | 2 | Hydrolysis |
| 869.94 | [M − 1]− | C30H46O4 | 469.3324 | 469.3323 | 0.2 | 2 | Hydrolysis + Hydroxylation + Reduction | ||
| 717.12 | [M − 1]− | C30H48O4 | 471.3479 | 471.3479 | 0 | 2 | Hydrolysis + Hydroxylation | ||
| 983.24 | [M − 1]− | C39H56O5 | 603.4034 | 603.4055 | −3.5 | 2 | Hydroxylation | ||
| 15 µM | 951.46 | [M − 1]− | C30H48O3 | 455.3518 | 455.3530 | 2.6 | 2 | Hydrolysis | |
| 869.94 | [M − 1]− | C30H46O4 | 469.3324 | 469.3323 | 0.2 | 2 | Hydrolysis + Hydroxylation + Reduction | ||
| 717.12 | [M − 1]− | C30H48O4 | 471.3479 | 471.3479 | 0 | 2 | Hydrolysis + Hydroxylation |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schioppa, L.; Fall, F.; Ortiz, S.; Poupaert, J.H.; Quetin-Leclercq, J. A Validated HPLC-PDA-HRMS Method to Investigate the Biological Stability and Metabolism of Antiparasitic Triterpenic Esters. Molecules 2021, 26, 7154. https://doi.org/10.3390/molecules26237154
Schioppa L, Fall F, Ortiz S, Poupaert JH, Quetin-Leclercq J. A Validated HPLC-PDA-HRMS Method to Investigate the Biological Stability and Metabolism of Antiparasitic Triterpenic Esters. Molecules. 2021; 26(23):7154. https://doi.org/10.3390/molecules26237154
Chicago/Turabian StyleSchioppa, Laura, Fanta Fall, Sergio Ortiz, Jacques H. Poupaert, and Joelle Quetin-Leclercq. 2021. "A Validated HPLC-PDA-HRMS Method to Investigate the Biological Stability and Metabolism of Antiparasitic Triterpenic Esters" Molecules 26, no. 23: 7154. https://doi.org/10.3390/molecules26237154
APA StyleSchioppa, L., Fall, F., Ortiz, S., Poupaert, J. H., & Quetin-Leclercq, J. (2021). A Validated HPLC-PDA-HRMS Method to Investigate the Biological Stability and Metabolism of Antiparasitic Triterpenic Esters. Molecules, 26(23), 7154. https://doi.org/10.3390/molecules26237154