
A Comprehensive Analysis of the Metal–Nitrile Bonding in an Organo-Diiron System

 , , ,
, , ,  and
and 
Abstract
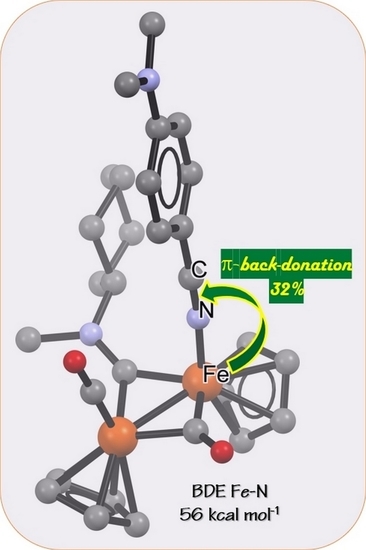
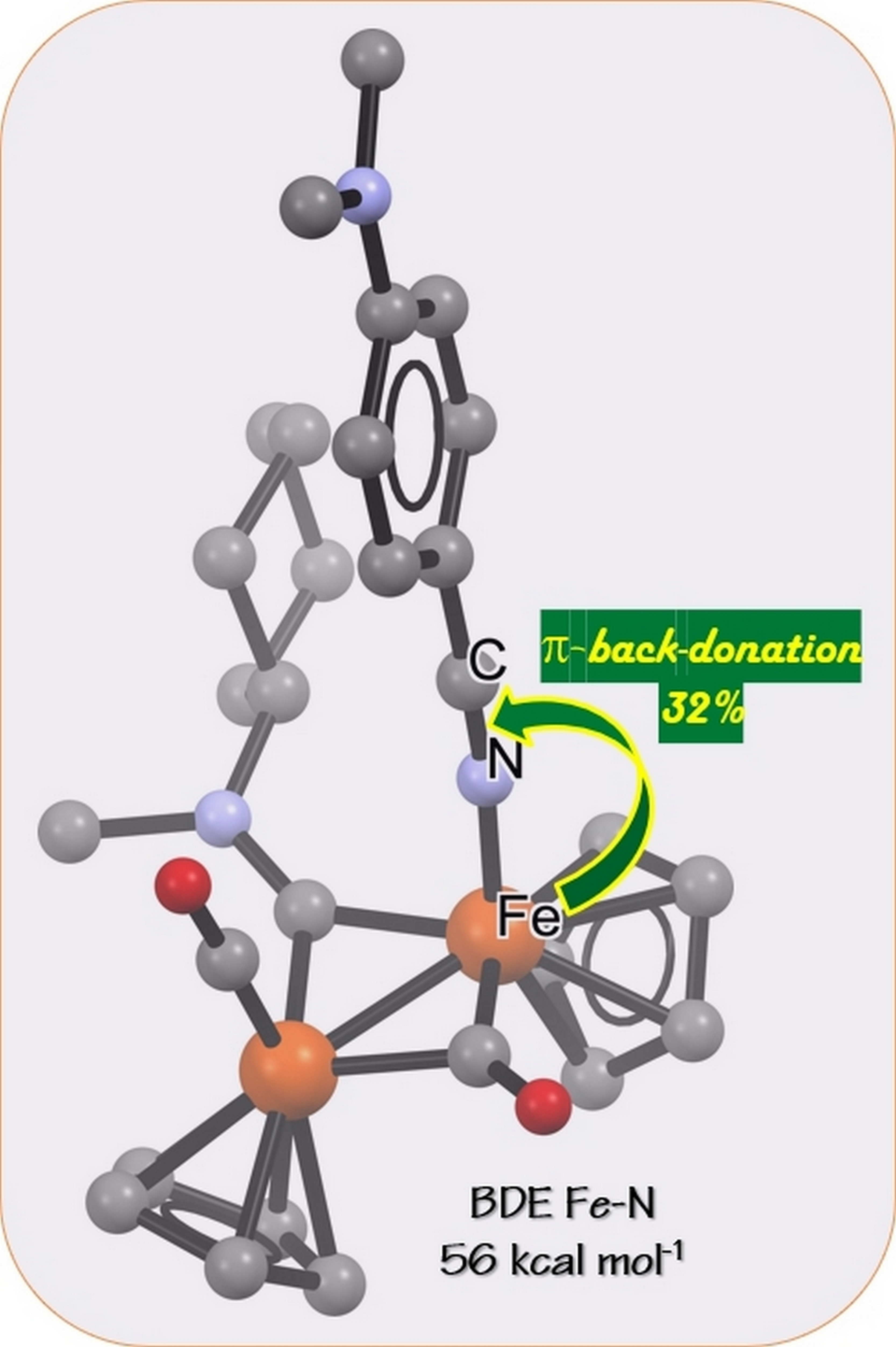
1. Introduction
2. Results and Discussion
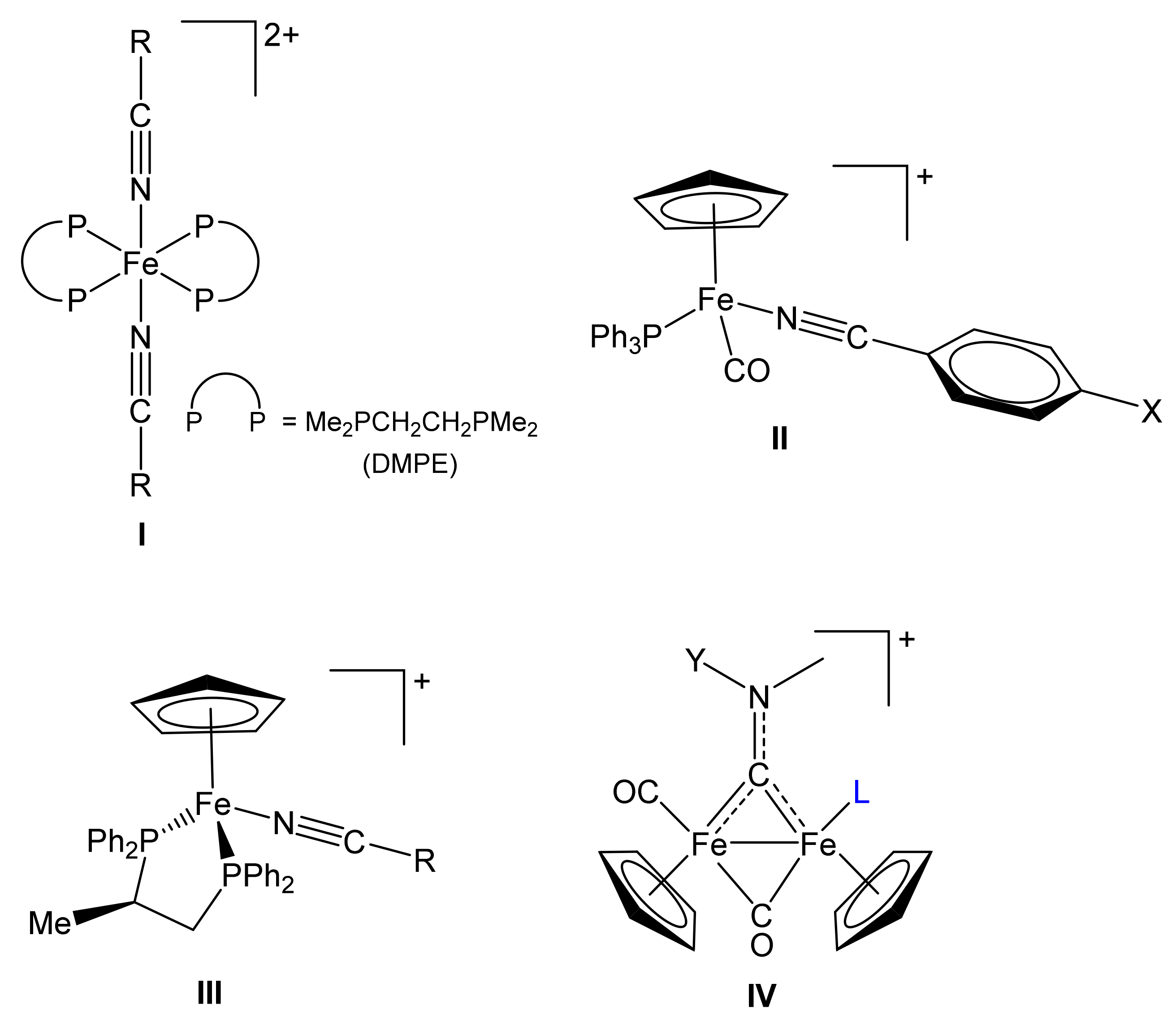
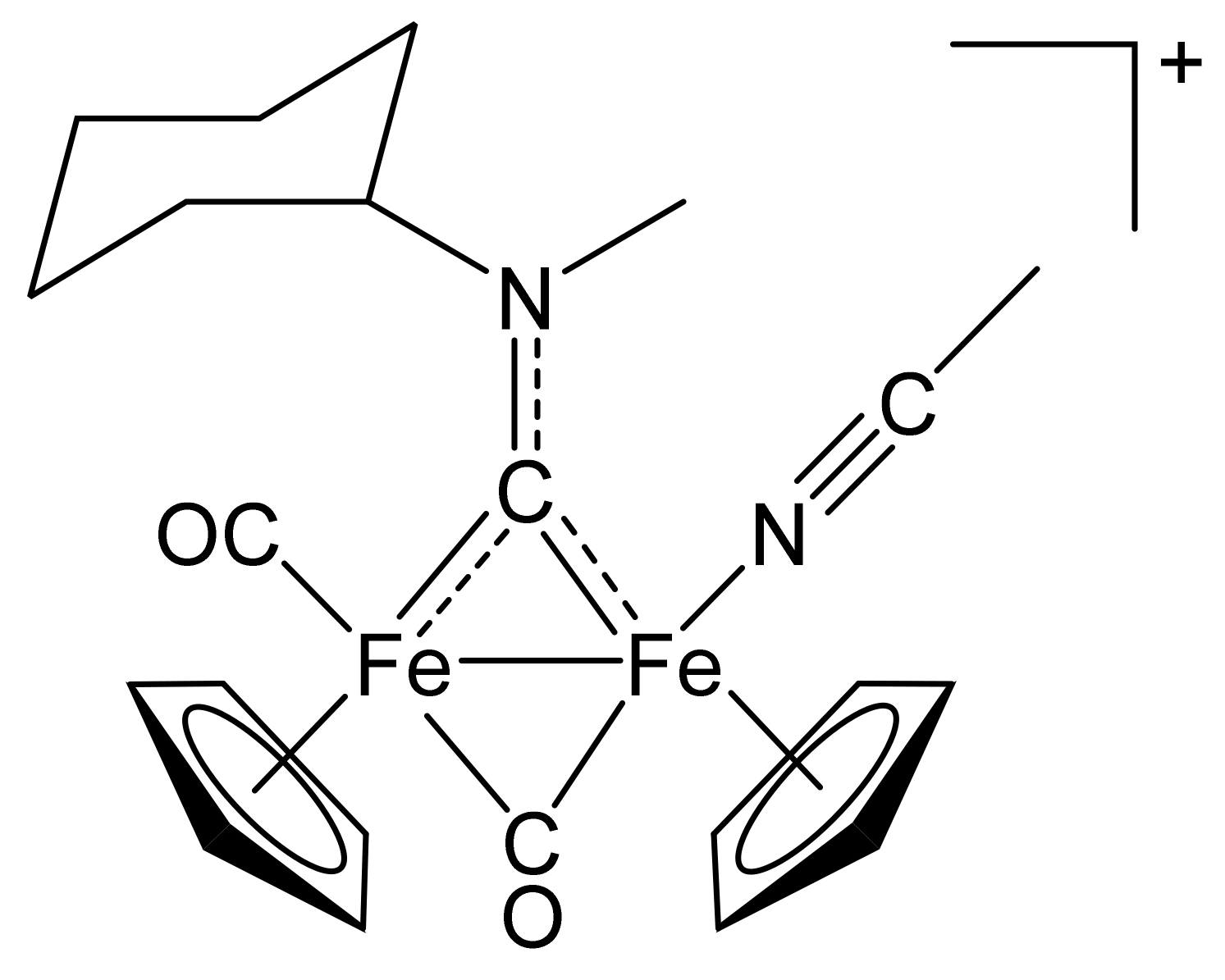
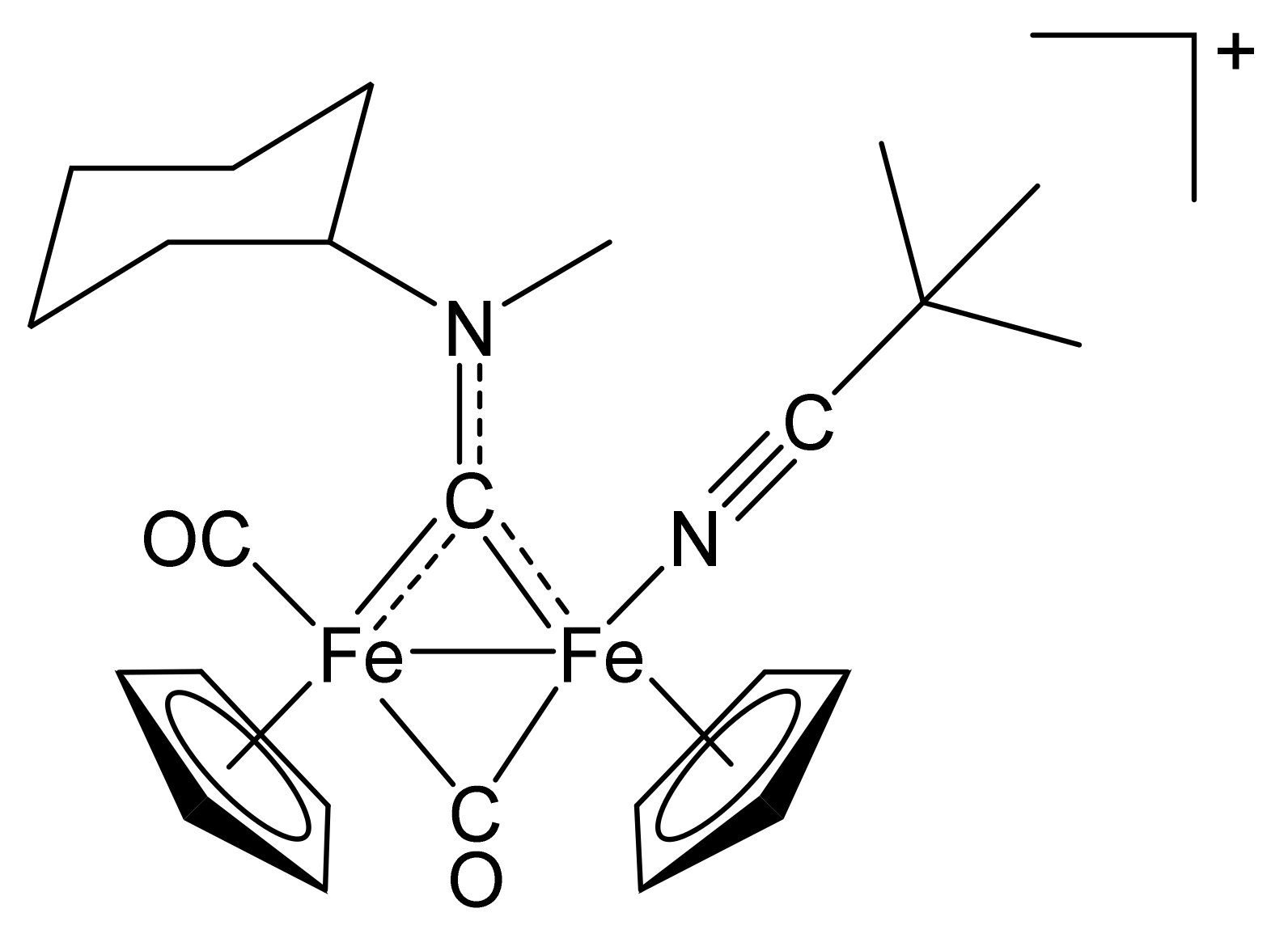
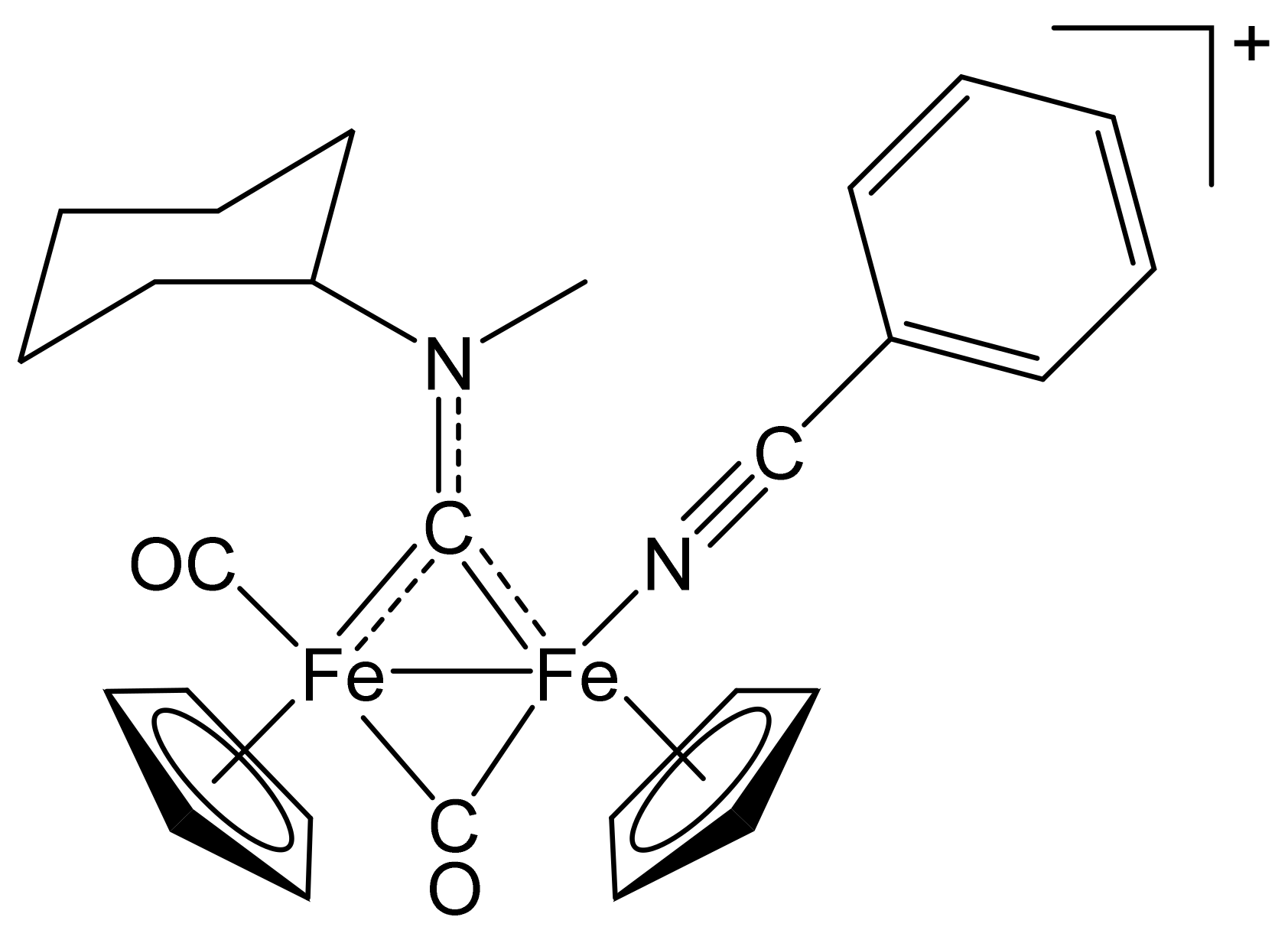
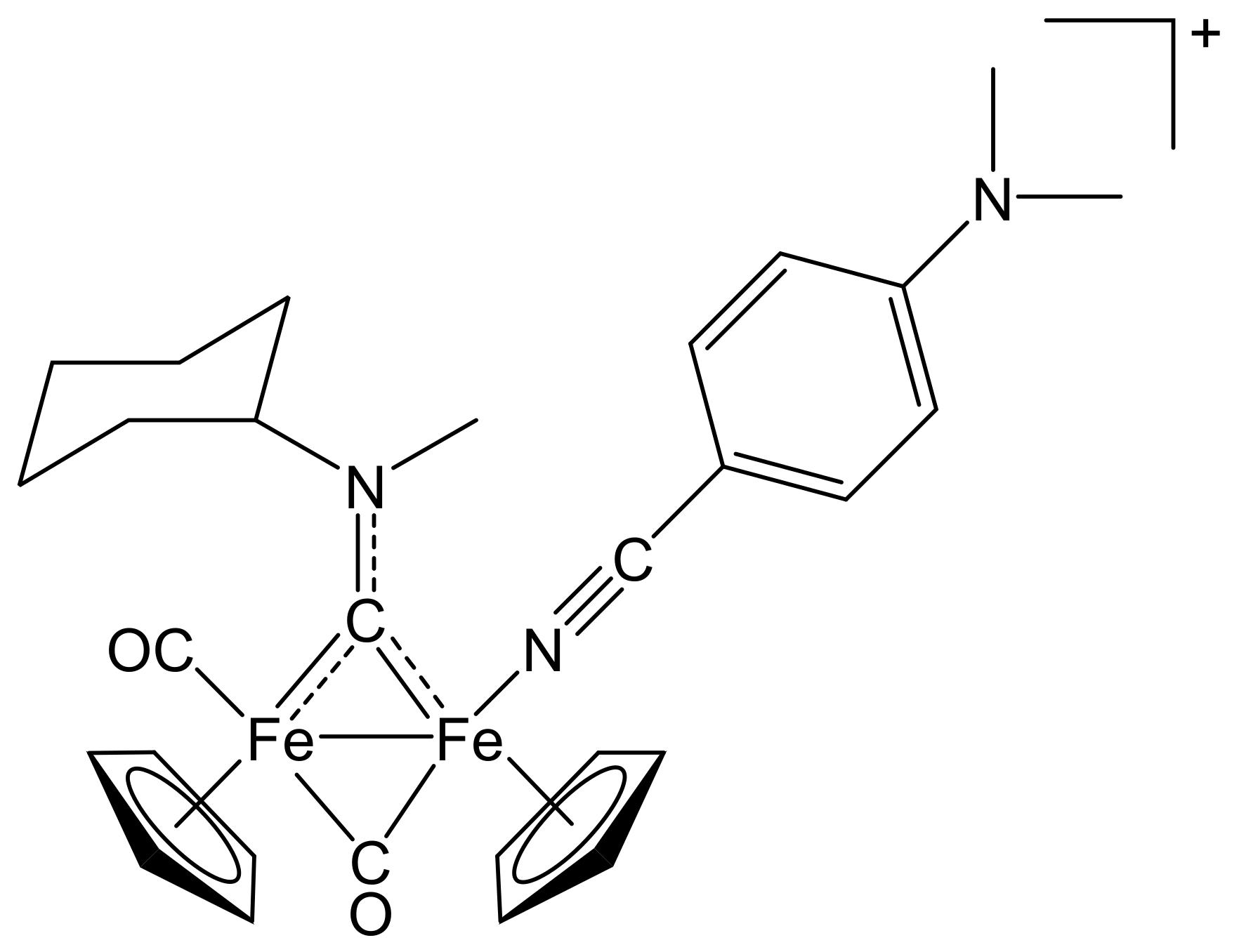
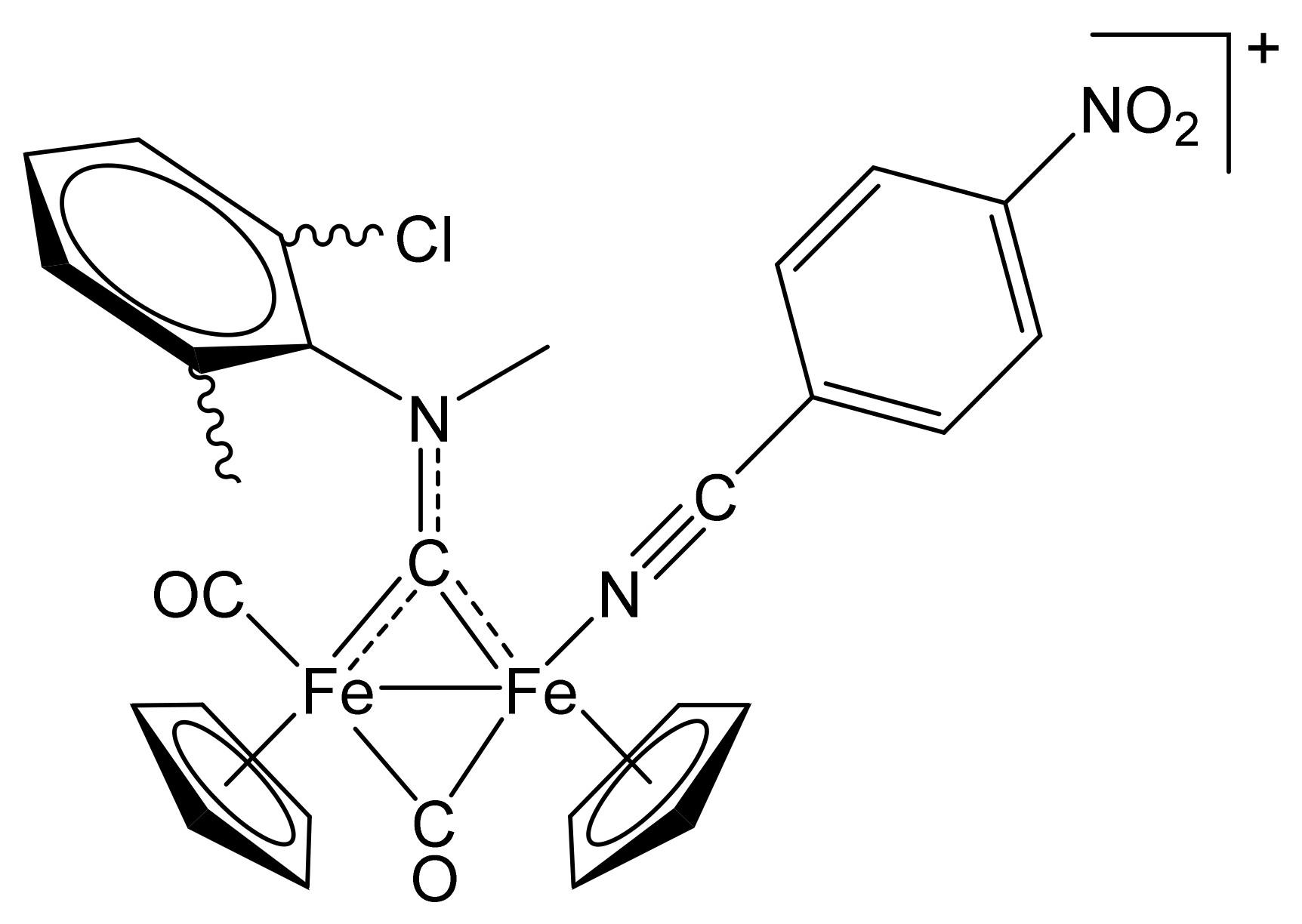
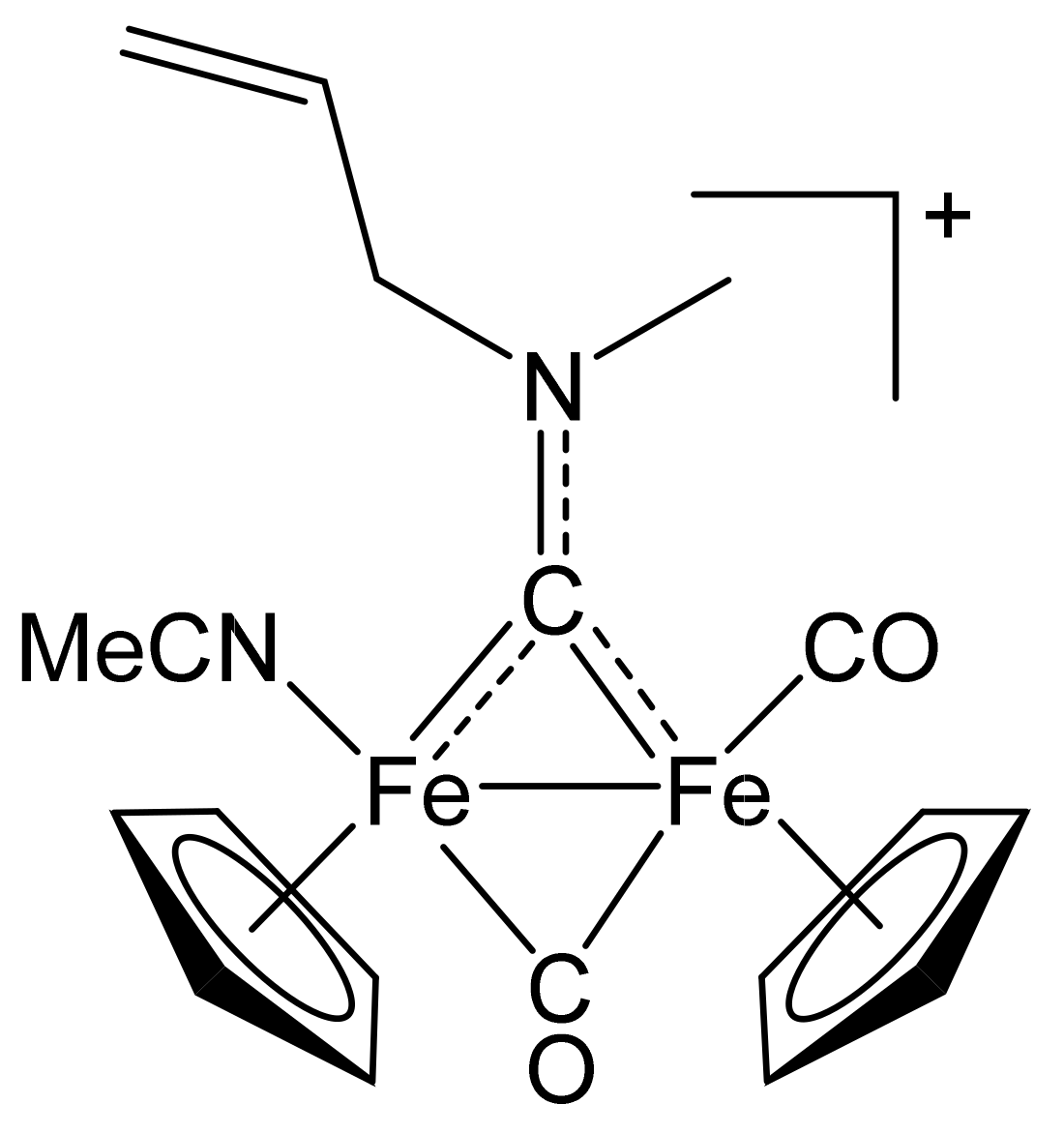
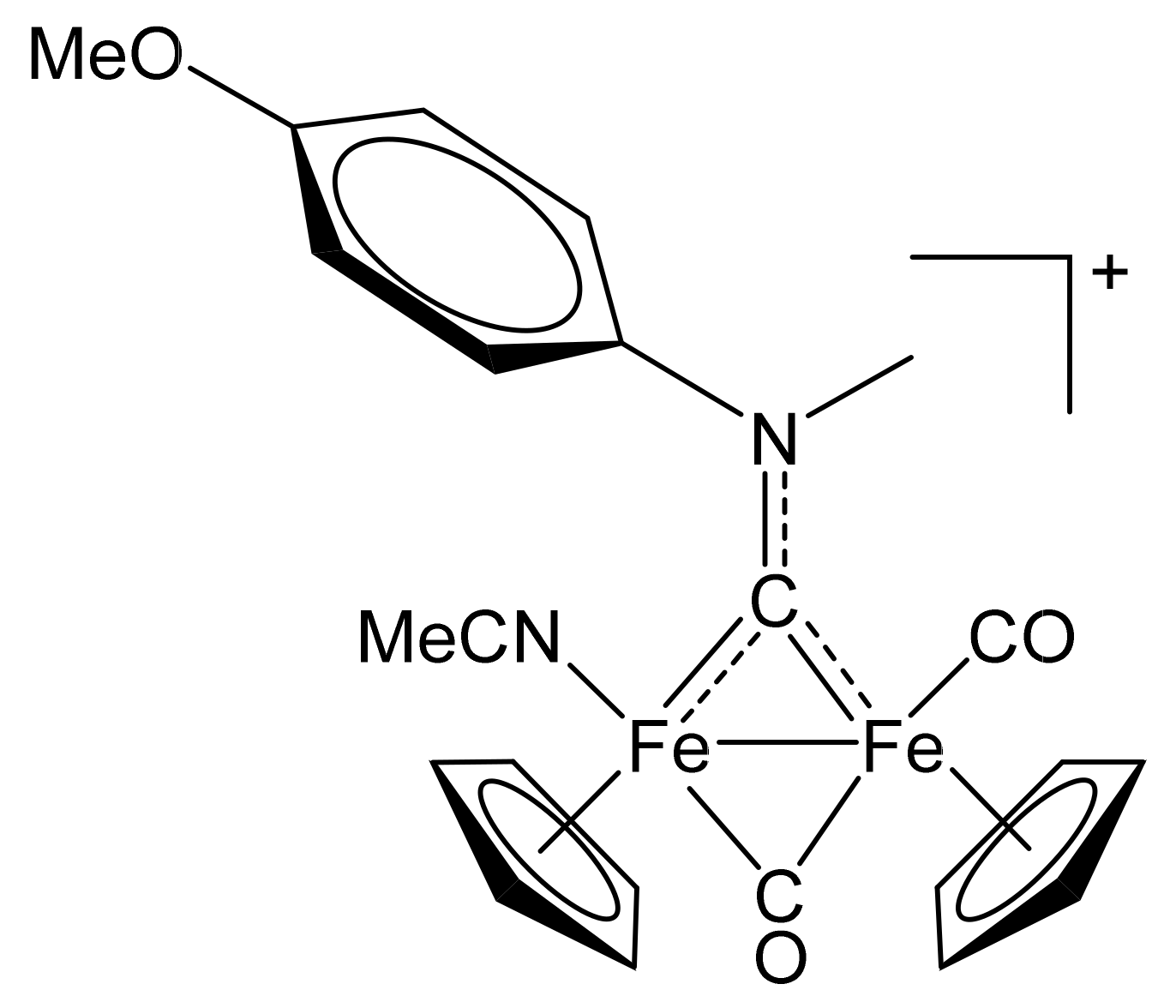
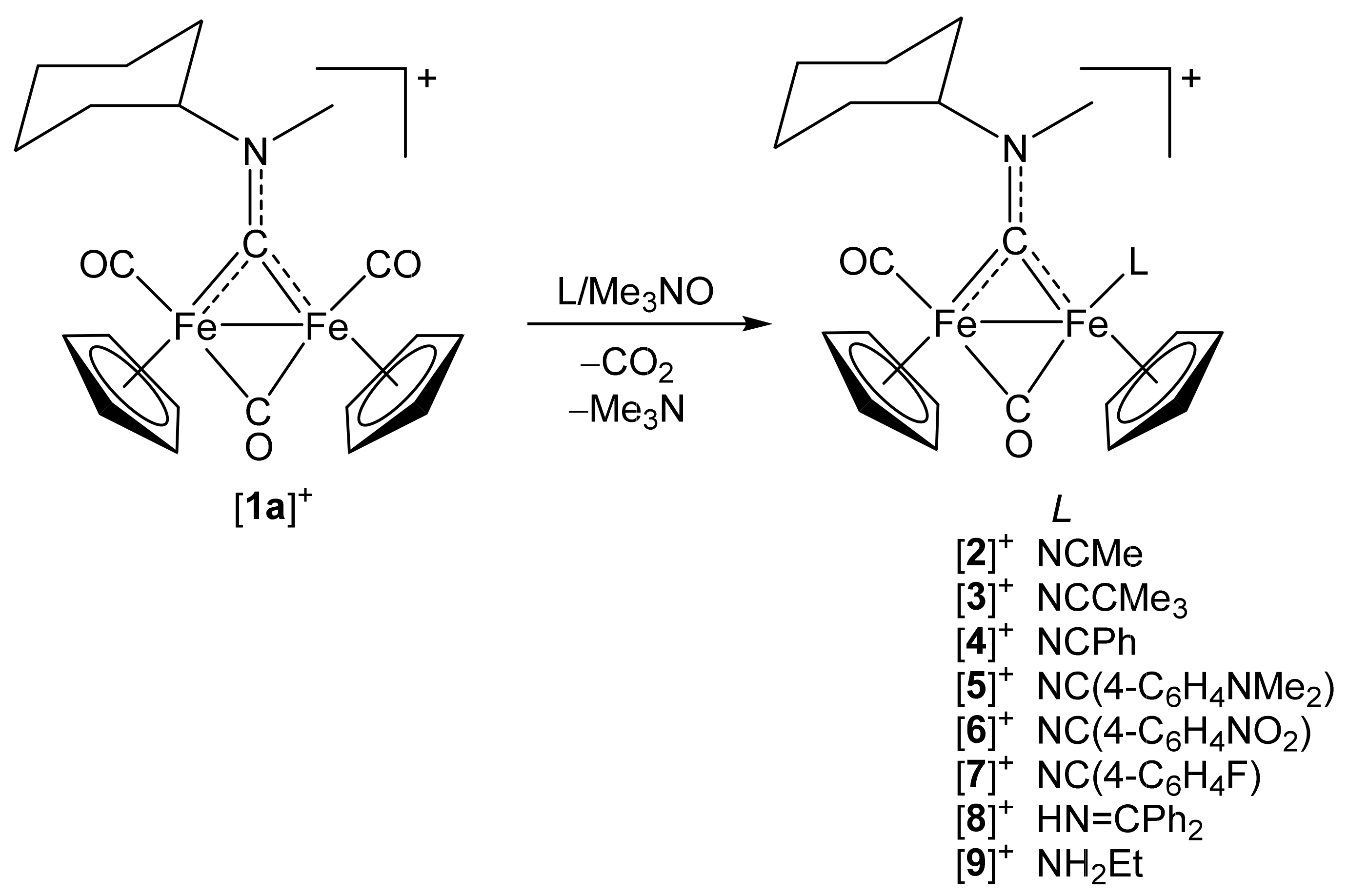
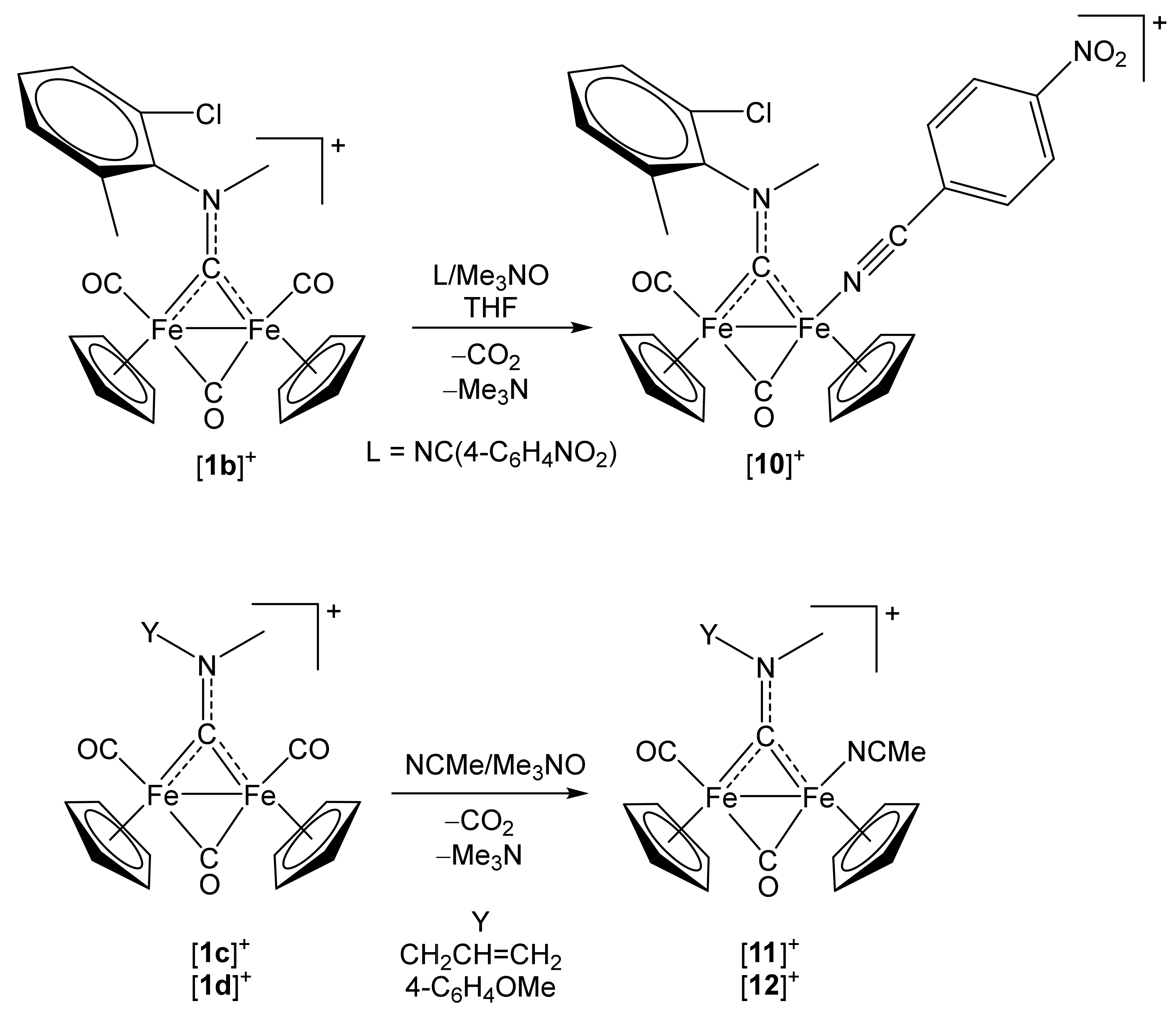
2.1. Synthesis of Diiron μ-Aminocarbyne Complexes with Nitrile- and Other Nitrogen-Ligands
2.2. Analysis of IR Spectra
2.3. Analysis of NMR Spectra
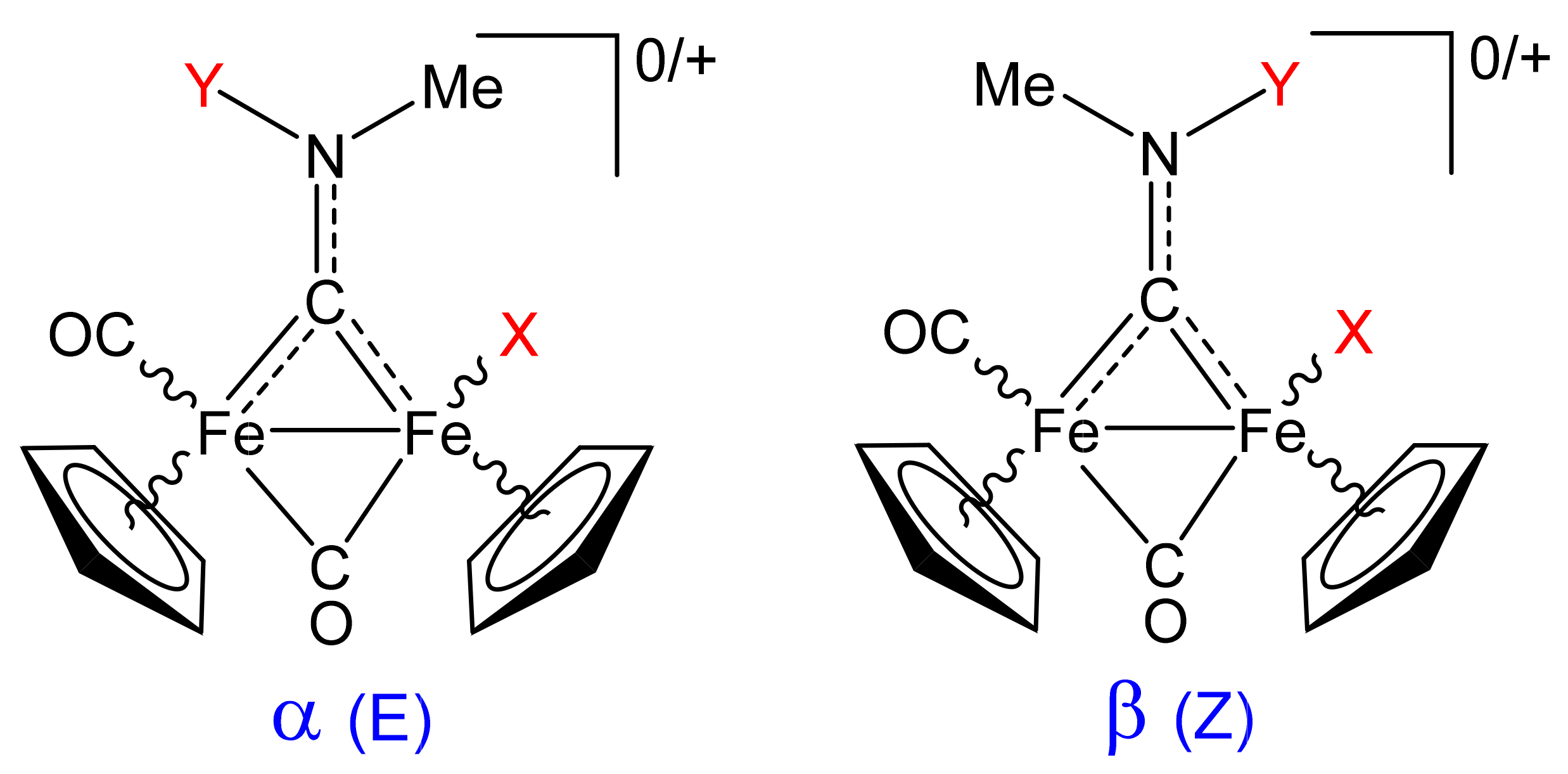
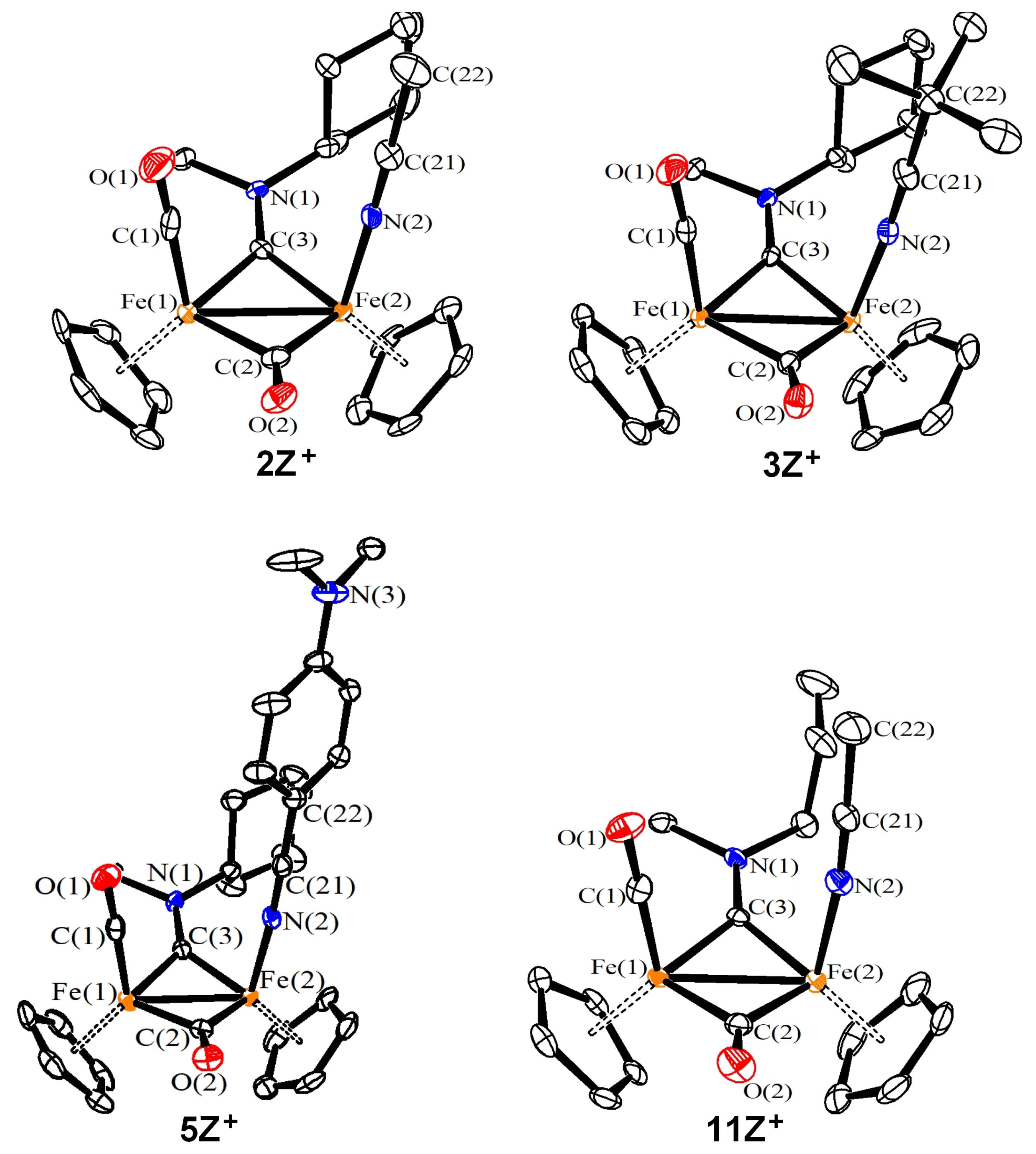
2.4. X-ray Diffraction Studies
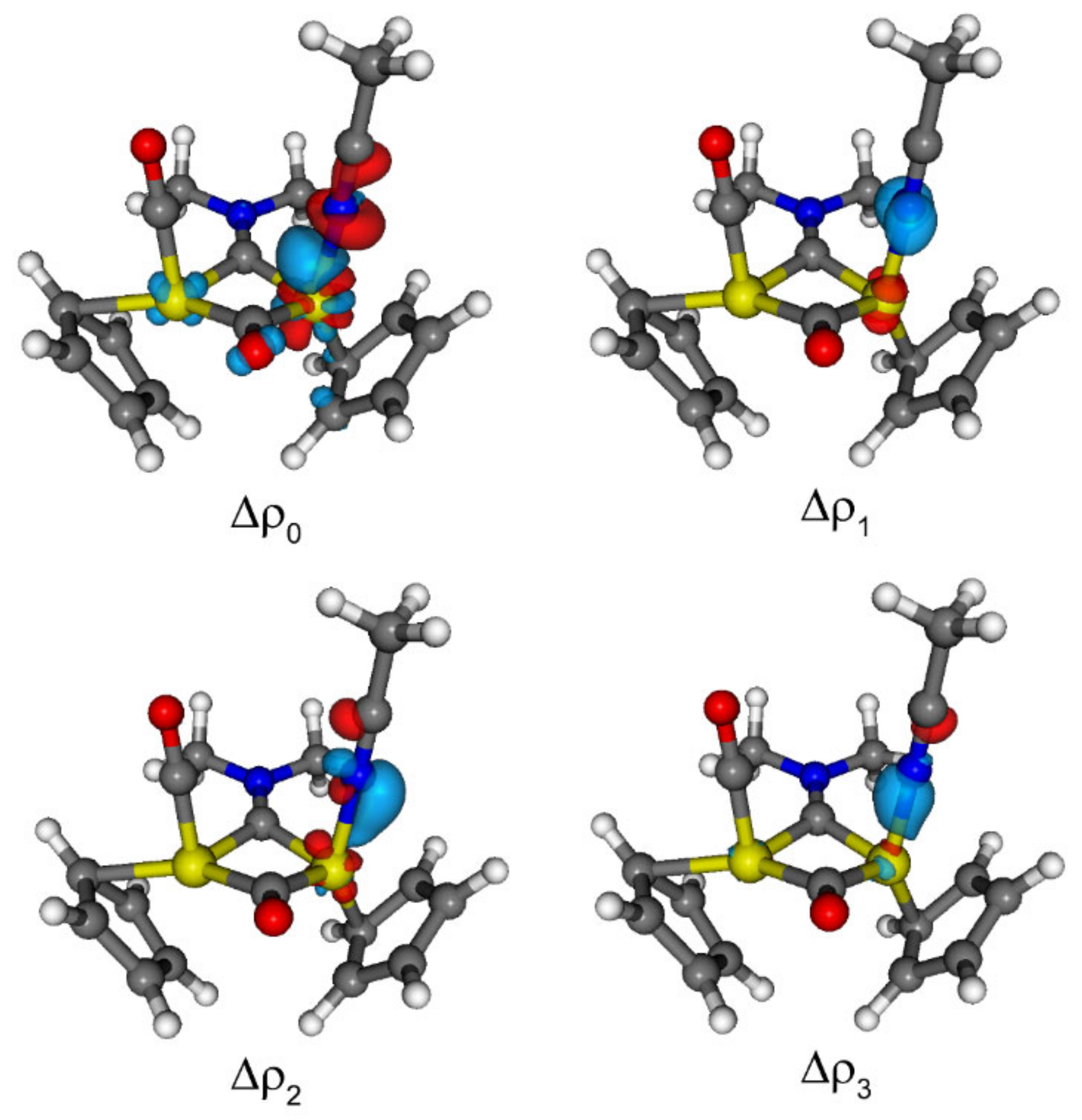
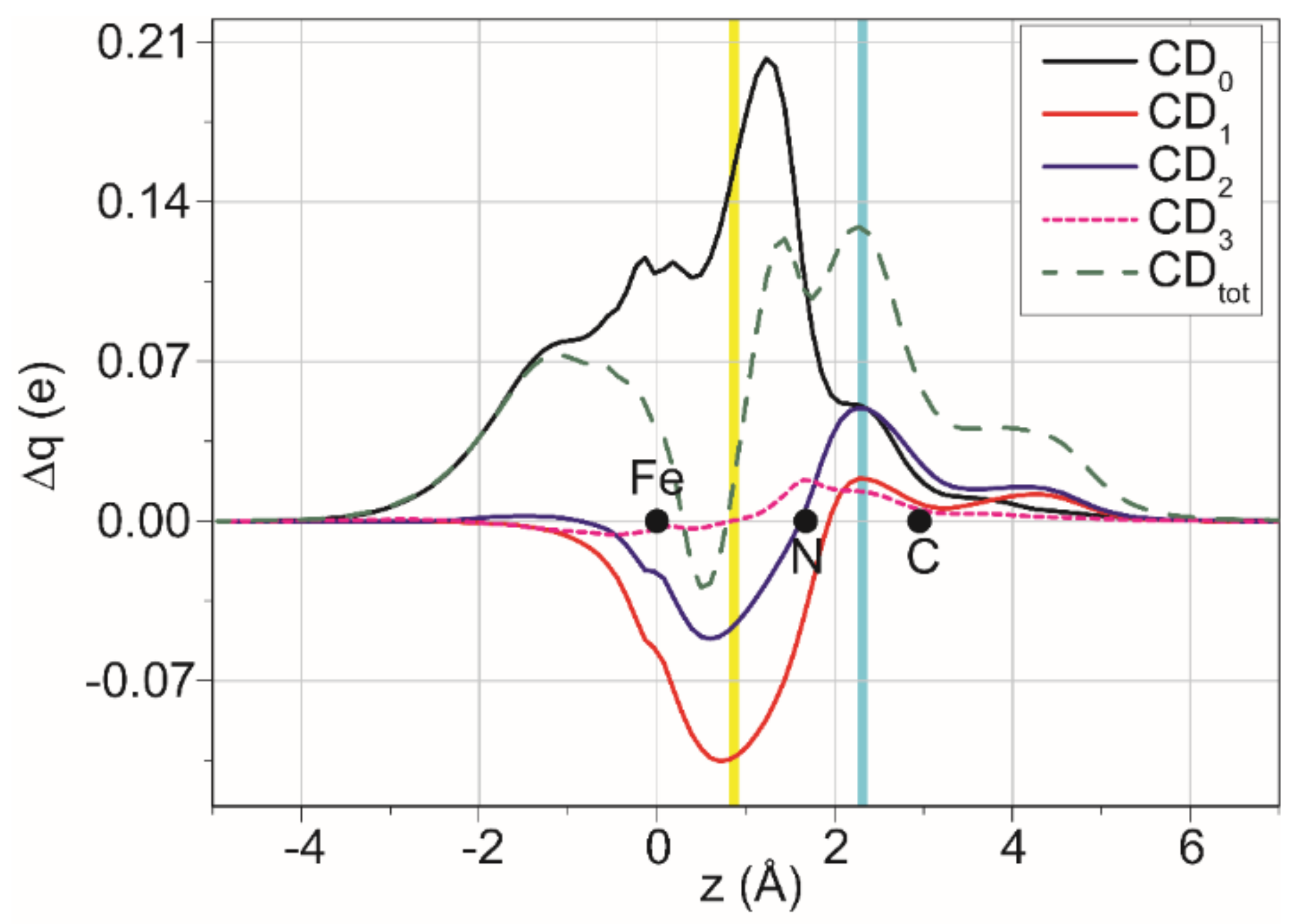
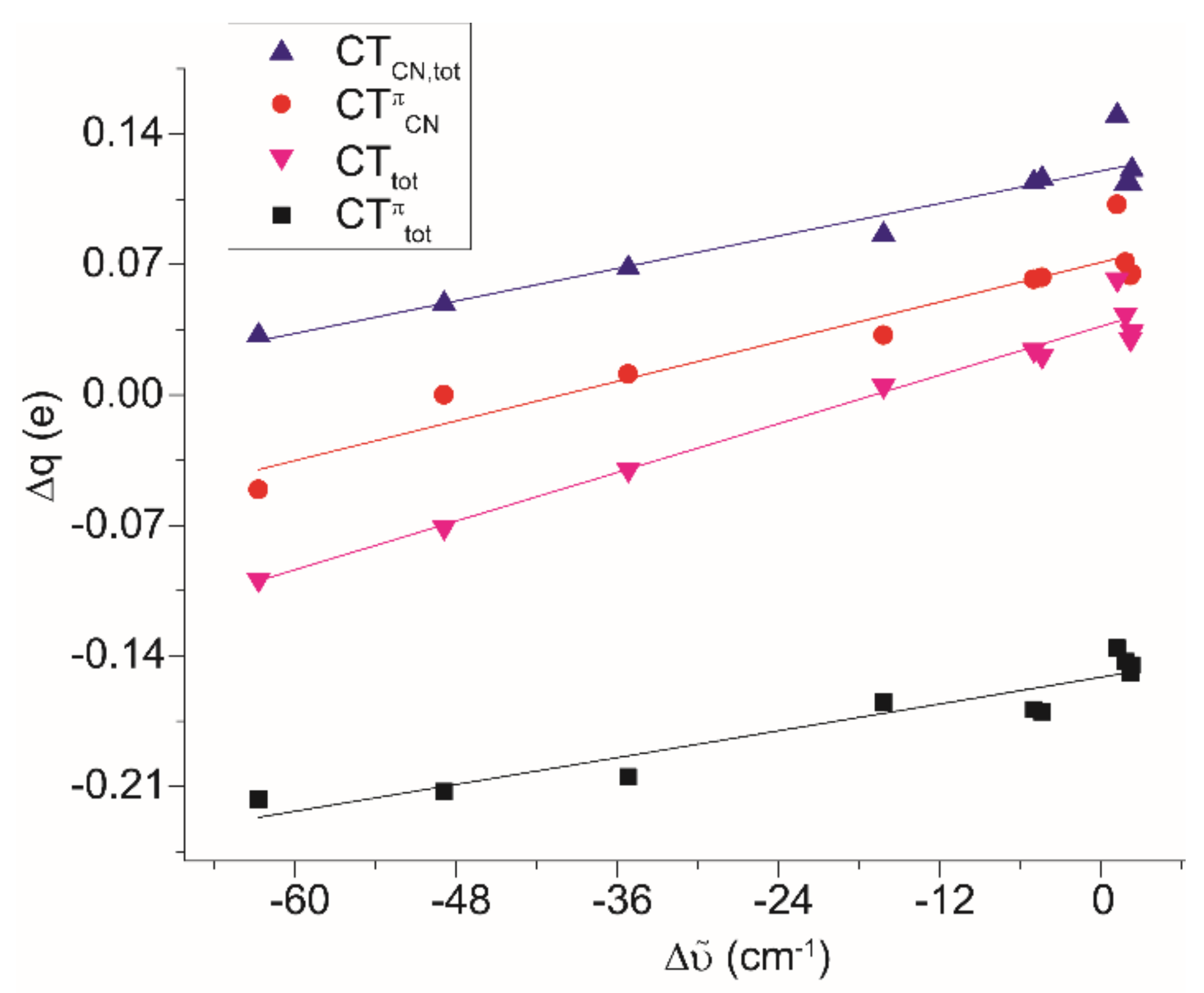
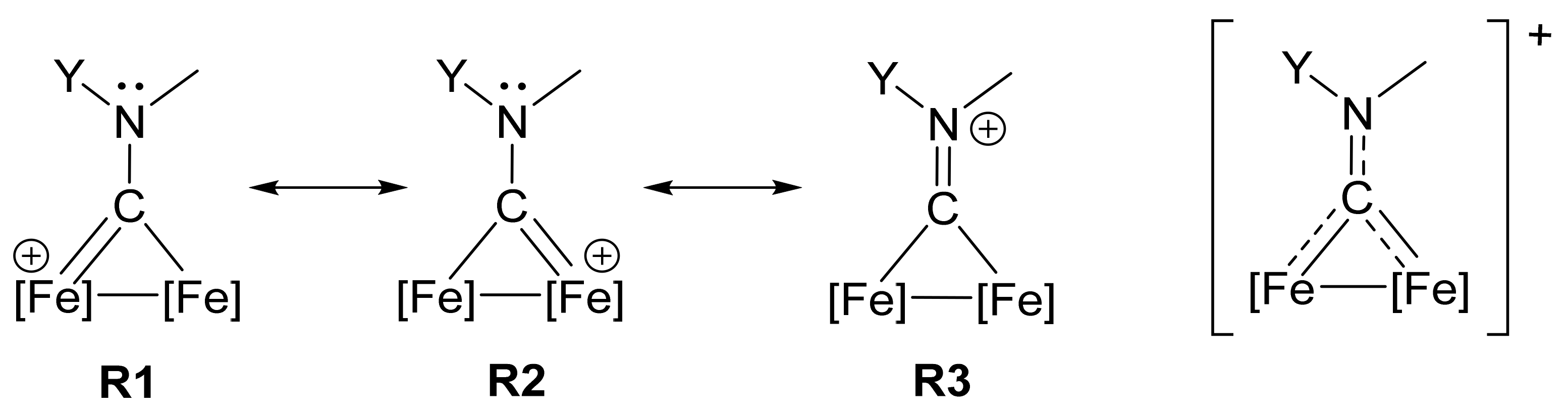
2.5. DFT Studies
3. Conclusions
4. Experimental
4.1. Materials and Methods
4.2. Synthesis and Characterization of Compounds
4.2.1. Synthesis and Characterization of [Fe2Cp2(CO)(NCMe)(µ-CO){µ-CN(Me)(Cy)}]CF3SO3, (2)CF3SO3

4.2.2. General Procedure for the Synthesis of (3–10)CF3SO3
[Fe2Cp2(CO)(NCCMe3)(µ-CO){µ-CN(Me)(Cy)}]CF3SO3, (3)CF3SO3

[Fe2Cp2(CO)(NCPh)(µ-CO){µ-CN(Me)(Cy)}]CF3SO3, (4)CF3SO3

[Fe2Cp2(CO){NC(4-C6H4NMe2)}(µ-CO){µ-CN(Me)(Cy)}]CF3SO3, (5)CF3SO3

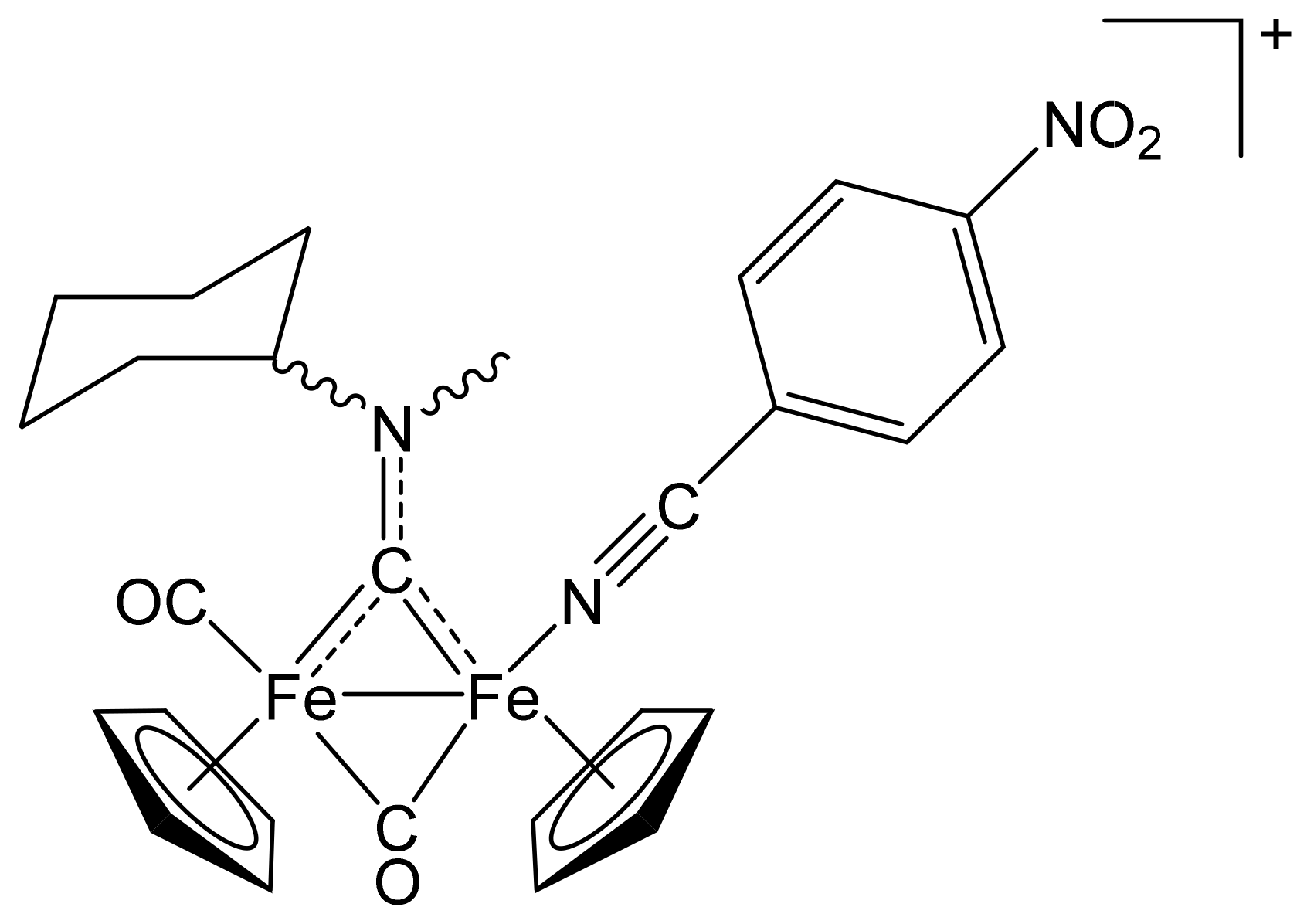
[Fe2Cp2(CO){NC(4-C6H4NO2)}(µ-CO){µ-CN(Me)(Cy)}]CF3SO3, (6)CF3SO3

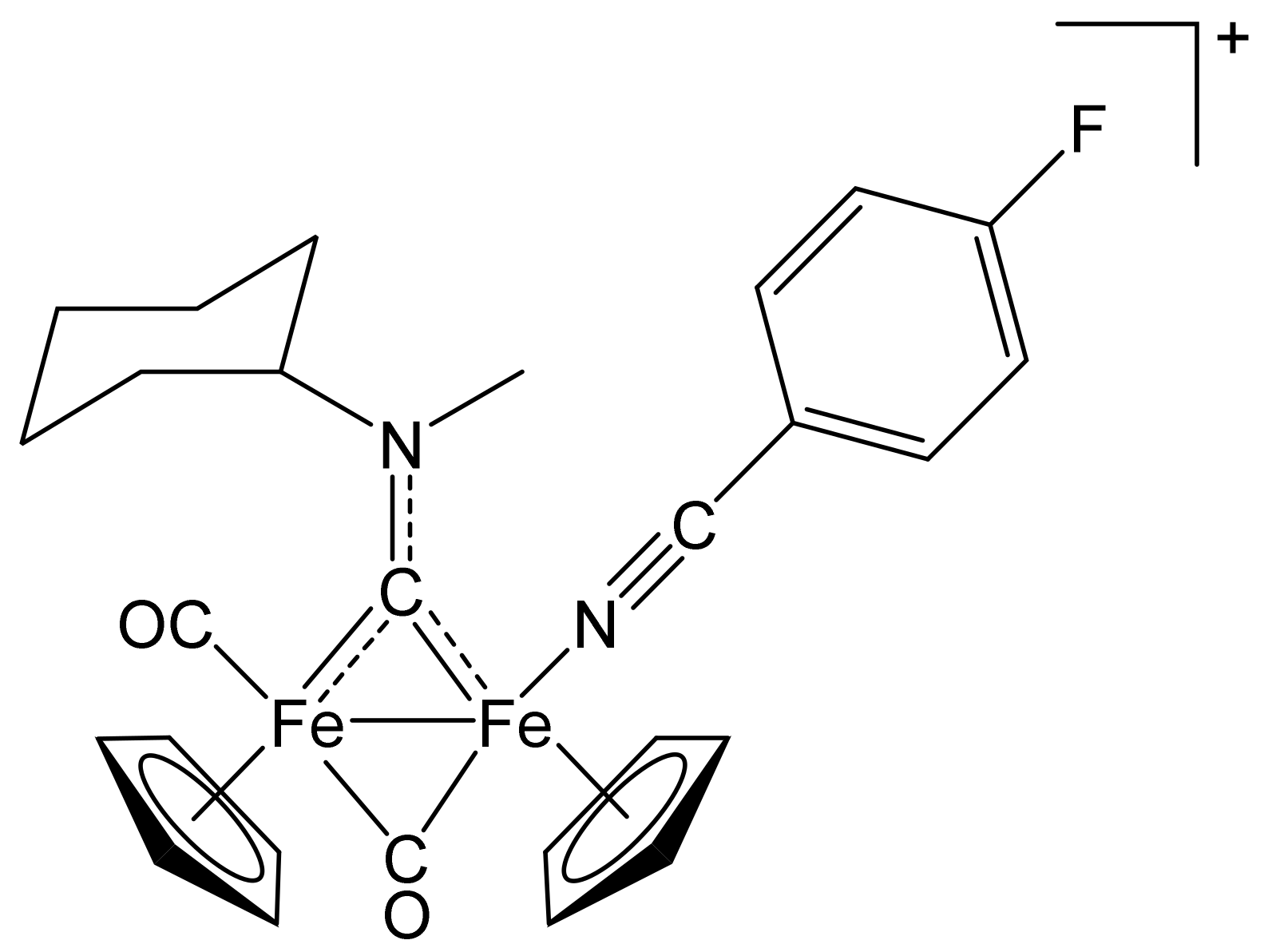
[Fe2Cp2(CO){NC(4-C6H4F)}(µ-CO){µ-CN(Me)(Cy)}]CF3SO3, (7)CF3SO3

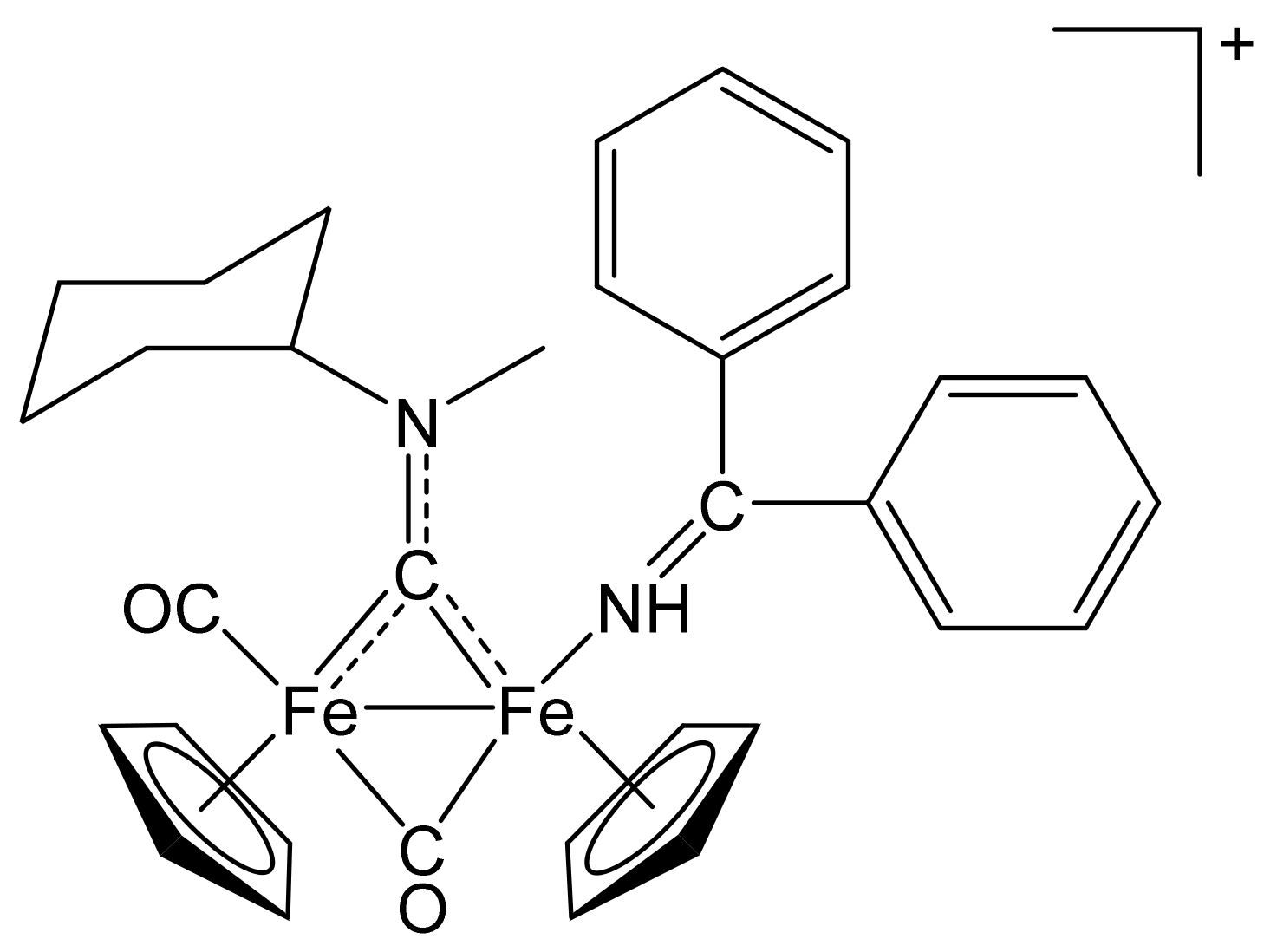
[Fe2Cp2(CO)(NH=CPh2)(µ-CO){µ-CN(Me)(Cy)}]CF3SO3, (8)CF3SO3

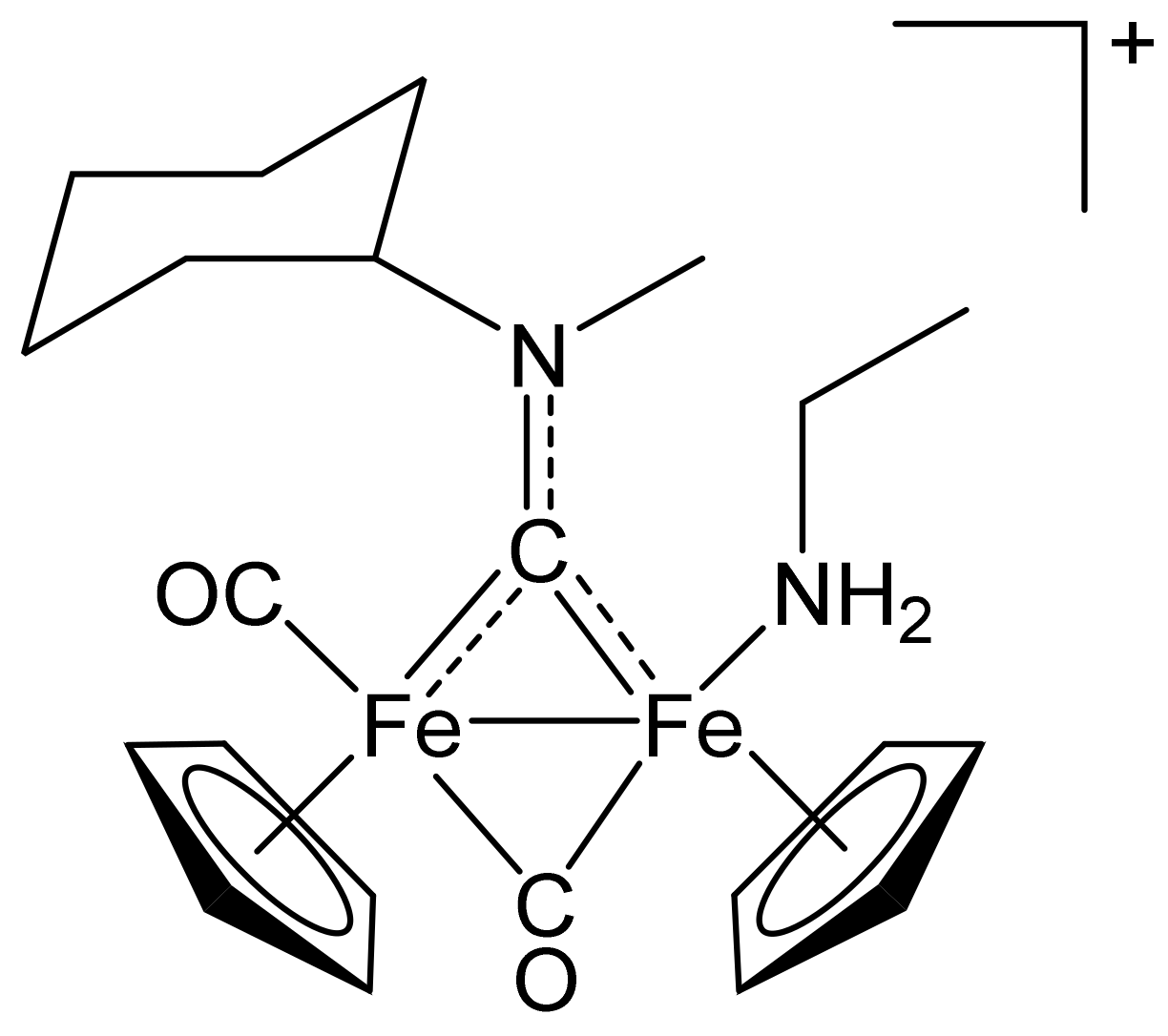
[Fe2Cp2(CO)(NH2Et)(µ-CO){µ-CN(Me)(Cy)}]CF3SO3, (9)CF3SO3

[Fe2Cp2(CO){NC(4-C6H4NO2)}(µ-CO){µ-CN(Me)(2,6-C6H3MeCl)}]CF3SO3, (10)CF3SO3

4.2.3. Synthesis and Characterization of [Fe2Cp2(CO)(NCMe)(µ-CO){µ-CN(Me)(CH2CH=CH2)}]CF3SO3, (11)CF3SO3

4.2.4. Synthesis and Characterization of [Fe2Cp2(CO)(NCMe)(µ-CO){µ-CN(Me)(4-C6H4OMe)}]CF3SO3, (12)CF3SO3

4.3. X-ray Crystallography
4.4. DFT Calculations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Storhoff, B.N.; Lewis, H.C., Jr. Organonitrile complexes of transition metals. Coord. Chem. Rev. 1977, 23, 1–29. [Google Scholar] [CrossRef]
- Pombeiro, A.J.L.; Kukushkin, V.Y. Comprehensive Coordination Chemistry II; McCleverty, J.A., Meyer, T.J., Eds.; Elsevier: Oxford, UK, 2004; Volume 1, pp. 639–660. [Google Scholar]
- Luzyanin, K.Y.; Kuznetsov, M.L. Advances in Organometallic Chemistry and Catalysis; Pombeiro, A.J.L., Ed.; Wiley: Hoboken, NJ, USA, 2014; pp. 171–183. [Google Scholar]
- Rach, S.F.; Kühn, F.E. Nitrile Ligated Transition Metal Complexes with Weakly Coordinating Counteranions and Their Catalytic Applications. Chem. Rev. 2009, 109, 2061–2080. [Google Scholar] [CrossRef]
- Pullen, E.E.; Rabinovich, D.; Incarvito, C.D.; Concolino, T.E.; Rheingold, A.L. Syntheses and Structures of Methyltris(pyrazolyl)silane Complexes of the Group 6 Metals. Inorg. Chem. 2000, 39, 1561–1567. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Zheng, Z. Dendrimers Supported by the [Re6Se8]2+ Metal Cluster Core. J. Am. Chem. Soc. 1999, 121, 3549–3550. [Google Scholar] [CrossRef]
- Fogg, D.E.; James, B.R. Chiral and Achiral Diphosphine Complexes of Ruthenium(II) Incorporating Labile Nitrile Ligands: Synthesis and Solution Chemistry of Mono- and Dinuclear Derivatives of Ru2Cl4(PP)2 (PP = Chelating Diphosphine). Inorg. Chem. 1997, 36, 1961–1966. [Google Scholar] [CrossRef]
- Huang, X.; Meggers, E. Asymmetric photocatalysis with bis-cyclometalated rhodium complexes. Acc. Chem. Res. 2019, 52, 833–847. [Google Scholar] [CrossRef]
- You, D.; Yang, H.; Sen, S.; Gabbai, F.P. Modulating the σ-Accepting Properties of an Antimony Z-type Ligand via Anion Abstraction: Remote-Controlled Reactivity of the Coordinated Platinum Atom. J. Am. Chem. Soc. 2018, 140, 9644–9651. [Google Scholar] [CrossRef]
- Benedikter, M.J.; Musso, J.V.; Frey, W.; Schowner, R.; Buchmeiser, M.R. Cationic Group VI Metal Imido Alkylidene N-Heterocyclic Carbene Nitrile Complexes: Bench-Stable, Functional-Group-Tolerant Olefin Metathesis Catalysts. Angew. Chem. Int. Ed. 2021, 60, 1374–1382. [Google Scholar] [CrossRef]
- Woodman, T.J.; Thornton-Pett, M.; Hughes, D.L.; Bochmann, M. B(C6F5)3 as C6F5 Transfer Agent in Zirconium Chemistry: Borole Sandwich and Borole-Bridged Triple-Decker Complexes. Organometallics 2001, 20, 4080–4091. [Google Scholar] [CrossRef]
- Michelin, R.A.; Mozzon, M.; Bertani, R. Reactions of transition metal-coordinated nitriles. Coord. Chem. Rev. 1996, 147, 299–338. [Google Scholar] [CrossRef]
- Viguri, M.E.; Huertos, M.A.; Perez, J.; Riera, L. Imidazole-nitrile or imidazole-isonitrile C-C coupling on rhenium tricarbonyl complexes. Chem. Eur. J. 2013, 19, 12974–12977. [Google Scholar] [CrossRef]
- Chernyshev, A.N.; Bokach, N.A.; Gushchin, P.V.; Haukka, M.; Kukushkin, V.Y. Reactions of platinum (IV)-bound nitriles with isomeric nitroanilines: Addition vs. substitution. Dalton Trans. 2012, 41, 12857–12864. [Google Scholar] [CrossRef]
- Gonzalez-Fernandez, R.; Crochet, P.; Cadierno, V. Arene-ruthenium(II) and osmium(II) complexes as catalysts for nitrilehydration and aldoxime rearrangement reactions. Inorg. Chim. Acta 2020, 517, 120180. [Google Scholar]
- Eniko Czégéni, C.; De, S.; Udvardy, A.; Derzsi, N.J.; Papp, G.; Papp, G.; Joó, F. Selective Hydration of Nitriles to Corresponding Amides in Air with Rh(I)-N-Heterocyclic Complex Catalysts. Catalysts 2020, 10, 125. [Google Scholar] [CrossRef]
- Xing, X.; Xu, C.; Chen, B.; Li, C.; Virgil, S.C.; Grubbs, R.H. Highly Active Platinum Catalysts for Nitrile and Cyanohydrin Hydration: Catalyst Design and Ligand Screening via High-Throughput Techniques. J. Am. Chem. Soc. 2018, 140, 17782–17789. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yang, K.; Ge, H. Palladium-catalyzed ligand-promoted site-selective cyanomethylation of unactivated C(sp3)–H bonds with acetonitrile. Chem. Sci. 2016, 7, 2804–2808. [Google Scholar] [CrossRef]
- Lee, K.-F.; Yang, T.; Tsang, L.-Y.; Sung, H.H.Y.; Williams, I.D.; Lin, Z.; Jia, G. Azavinylidene Complexes from Coupling Reactions of Organonitriles with Phosphines. Organometallics 2021, 40, 358–369. [Google Scholar] [CrossRef]
- Rahman, M.M.; Smith, M.D.; Peryshkov, D.V. Imido Group Interchange in Reactions of Zwitterionic Tantalum(V) Vinylimido Complexes and Nitriles. Organometallics 2018, 37, 2945–2949. [Google Scholar] [CrossRef]
- Becker, L.; Arndt, P.; Jiao, H.; Spannenberg, A.; Rosenthal, U. Nitrile-Nitrile C-C Coupling at Group 4 Metallocenes to Form 1-Metalla-2,5-diaza-cyclopenta-2,4-dienes: Synthesis and Reactivity. Angew. Chem. Int. Ed. 2013, 52, 11396–11400. [Google Scholar] [CrossRef] [PubMed]
- Alias, F.M.; Daff, P.J.; Paneque, M.; Poveda, M.L.; Carmona, E.; Perez, P.J.; Salazar, V.; Alvarado, Y.; Atencio, R.; Sanchez-Delgado, R. C-C bond-forming reactions of IrIII-alkenyls and nitriles or aldehydes: Generation of reactive hydride- and alkyl-alkylidene compounds and observation of a reversible 1,2-H shift in stable hydride-IrIII alkylidene complexes. Chem. Eur. J. 2002, 8, 5132–5146. [Google Scholar] [CrossRef]
- Braunstein, P.; Matt, D.; Dusausoy, Y.; Fischer, J. Complexes of functional phosphines. 6. Five-membered palladium or platinum metallacycles resulting from nucleophilic attack of α-phosphino carbanions on organonitriles. Synthesis and crystal structure of [cyclo] trans-Pt[Ph2PC(COOC2H5)C(Ph)NH]2 and [cyclo] Pd(C-N)Cl[.mu.-Ph2PCH2C(NH)C(CN)PPh2]Pd(C-N) [(C-N) = (o-C6H4CH2N(CH3)2)]. Organometallics 1983, 2, 1410–1417. [Google Scholar]
- Drago, R.S.J. An analysis of cobaloxime chemistry with the electrostatic-covalent model. Organomet. Chem. 1996, 512, 61–68. [Google Scholar] [CrossRef]
- Beech, G.; Marr, G.; Ashcroft, S.J. Thermochemistry of some acetonitrile and benzonitrile complexes of transition-metal halides. J. Chem. Soc. A 1970, 17, 2903–2906. [Google Scholar] [CrossRef]
- Pombeiro, A.J.L.; Kukushkin, V.Y. Reactivity of Coordinated Nitriles. In Comprehensive Organometallic Chemistry II; Elsevier: Amsterdam, The Netherlands, 1987; Volume 1. [Google Scholar]
- Kuznetsov, M.L.; Pombeiro, A.J.L. Theoretical study of redox induced isomerizations, structure and bonding of nitrile, isocyanide and carbonyl complexes of rhenium. Dalton Trans. 2003, 738–747. [Google Scholar] [CrossRef]
- Pombeiro, A.J.L. Electron-donor/acceptor properties of carbynes, carbenes, vinylidenes, allenylidenes and alkynyls as measured by electrochemical ligand parameters. J. Organomet. Chem. 2005, 690, 6021–6040. [Google Scholar] [CrossRef]
- Cordiner, R.L.; Albesa-Jove’, D.; Roberts, R.L.; Farmer, J.D.; Puschmann, H.; Corcoran, D.; Goeta, A.E.; Howard, J.A.K.; Low, P.J. Syntheses and molecular structures of group 8 benzonitrile complexes. J. Organomet. Chem. 2005, 690, 4908–4919. [Google Scholar] [CrossRef]
- Clarke, R.E.; Ford, P.C. Benzonitrile and Acetonitrile Complexes of Ruthenium Ammines. Inorg. Chem. 1970, 9, 227–235. [Google Scholar] [CrossRef]
- Eagle, C.T.; Farrar, D.G.; Holder, G.N.; Pennington, W.T.; Bailey, R.D. Structural and electronic properties of (2,2-trans)-dirhodium(II) tetrakis(N-phenylacetamidate). J. Organomet. Chem. 2000, 596, 90–94. [Google Scholar] [CrossRef]
- Garcia, M.H.; Rodrigues, J.C.; Dias, A.R.; Piedade, M.F.M.; Duarte, M.T.; Robalo, M.P.; Lopes, N. Second harmonic generation of η5-monocyclopentadienyl ruthenium p-benzonitrile derivatives by Kurtz powder technique. Crystal and molecular structure determinations of [Ru(η5-C5H5)((+)-DIOP)(p-NCC6H4NO2)][X], X=PF6−, CF3SO3− and [Ru(η5-C5H5)((+)-DIOP)(NCCH3)][PF6]. J. Organomet. Chem. 2001, 632, 133–144. [Google Scholar]
- Fernandes Alves, J.J.; Franco, D.W. Acrylonitrile and cyanoacetic acid complexes of Ru(III) and Ru(II) amines: An analysis of the σ- and π-component relevance in the Ru(II)-NCR bond. Polyhedron 1996, 15, 3299–3307. [Google Scholar] [CrossRef]
- George, A.V.; Field, L.D.; Malouf, E.Y.; McQueen, E.D.; Pike, S.R.; Purches, G.R.; Hambley, T.W.; Buys, I.E.; White, A.H.; Hockless, D.C.R.; et al. Synthesis and characterisation of nitrile complexes of iron. J. Organomet. Chem. 1997, 538, 101–110. [Google Scholar] [CrossRef]
- Winter, R.F.; Scheiring, T. Synthesis, Structures, Ligand Substitution Reactions, and Electrochemistry of the Nitrile Complexes cis-[Ru(dppm)2CI(NCR)]+ PF6− (dppm = Bis(diphenylphosphino )methane, R = CH3, C2H5, tBu, Ph). Zeitschrift für Anorganische und Allgemeine Chemie 2000, 626, 1196–1204. [Google Scholar] [CrossRef][Green Version]
- Pilon, A.; Brás, A.R.; Côrte-Real, L.; Avecilla, F.; Costa, P.J.; Preto, A.; Garcia, M.H.; Valente, A. A New Family of Iron(II)-Cyclopentadienyl Compounds Shows Strong Activity against Colorectal and Triple Negative Breast Cancer Cells. Molecules 2020, 25, 1592. [Google Scholar] [CrossRef] [PubMed]
- Brunner, H.; Tsuno, T.; Kurosawa, T.; Kitamura, H.; Ike, H. Kinetic and Thermodynamic Control of Nitrile Dissociation in the Complexes (RFe, RC)/(SFe, RC)-[CpFe(Prophos)NCR]X (X = I, PF6) by the Inductive Effect. Organometallics 2018, 37, 1892–1899. [Google Scholar] [CrossRef]
- Casella, G.; Fonseca Guerra, C.; Carlotto, S.; Sgarbossa, P.; Bertani, R.; Casarin, M. New Light on an Old Debate: Does the RCN-PtCl2 Bond Include any Back-Donation? RCN-PtCl2 Backbonding vs the IR ν C≡N Blue-shift Dichotomy in Organonitriles-platinum(II) Complexes. A thorough Density Functional Theory—Energy Decomposition Analysis study. Dalton Trans. 2019, 48, 12974–12985. [Google Scholar] [CrossRef]
- Wadepohl, H.; Arnold, U.; Pritzkow, H.; Calhorda, M.J.; Veiros, L.F. Interplay of ketenyl and nitrile ligands on d 6-transition metal centres. Acetonitrile as an end-on (two-electron) and a side-on (four-electron) ligand. J. Organomet. Chem. 1999, 587, 233–243. [Google Scholar] [CrossRef]
- Li, G.; Liu, L.; Wang, J.; Li, Q.-s.; Xie, Y.; King, R.B. Juxtaposition of the strong back-bonding carbonyl ligand and weak back-bonding acetonitrile ligand in binuclear iron complexes. Trans. Met. Chem. 2013, 38, 617–625. [Google Scholar] [CrossRef]
- Mrozek, M.F.; Wasileski, S.A.; Weaver, M.J. Periodic Trends in Electrode–Chemisorbate Bonding: Benzonitrile on Platinum-Group and Other Noble Metals as Probed by Surface-Enhanced Raman Spectroscopy Combined with Density Functional Theory. J. Am. Chem. Soc. 2001, 123, 12817–12825. [Google Scholar] [CrossRef]
- Marchetti, F. Constructing Organometallic Architectures from Aminoalkylidyne Diiron Complexes. Eur. J. Inorg. Chem. 2018, 3987–4003. [Google Scholar] [CrossRef]
- Biancalana, L.; De Franco, M.; Ciancaleoni, G.; Zacchini, S.; Pampaloni, G.; Gandin, V.; Marchetti, F. Easily Available and Amphiphilic Diiron Cyclopentadienyl Complexes Exhibit In Vitro Anticancer Activity in 2D and 3D Human Cancer Cells via Redox Modulation Triggered by CO Release. Chem. Eur. J. 2021, 27, 10169–10185. [Google Scholar] [CrossRef]
- Marchetti, F.; Zacchini, S.; Zanotti, V. Carbon monoxide–isocyanide coupling promoted by acetylide addition to a diiron complex. Chem. Commun. 2015, 51, 8101–8104. [Google Scholar] [CrossRef]
- Albano, V.G.; Busetto, L.; Marchetti, F.; Monari, M.; Zanotti, V. Acetonitrile activation in di-iron-carbyne complexes: Synthesis and structure of the cyanomethyl complex [Fe2(μ-CNMe2)(μ-CO)(CO)(CH2CN)(Cp)2]. J. Organomet. Chem. 2002, 649, 64–69. [Google Scholar] [CrossRef]
- Albano, V.G.; Bordoni, S.; Busetto, L.; Marchetti, F.; Monari, M.; Zanotti, V. C-N coupling between μ-aminocarbyne and nitrile ligands promoted by tolylacetylide addition to [Fe2{μ-CN(Me)(Xyl)}(μ-CO)(CO)(NCCMe3)(Cp)2][SO3CF3]: Formation of a novel bridging allene-diaminocarbene ligand. J. Organomet. Chem. 2003, 684, 37–43. [Google Scholar] [CrossRef]
- Busetto, L.; Marchetti, F.; Zacchini, S.; Zanotti, V.; Zoli, E. Nitrile ligands activation in dinuclear aminocarbyne complexes. J. Organomet. Chem. 2005, 690, 1959–1970. [Google Scholar] [CrossRef]
- Busetto, L.; Marchetti, F.; Zacchini, S.; Zanotti, V. Reactions of Diiron µ-Aminocarbyne Complexes Containing Nitrile Ligands. J. Braz. Chem. Soc. 2003, 14, 902–907. [Google Scholar] [CrossRef]
- IR Data for Free Nitriles, ῦ(N≡C)/cm−1. Solid Samples: N≡CPh, 2229; N≡CC6H4NMe2, 2210; N≡CC6H4NO2, 2233; N≡CC6H4F, 2234. Liquid samples: N≡CMe, 2253; N≡CCMe3, 2235. Dichloromethane solutions: N≡CPh, 2230; N≡CC6H4NMe2, 2215; N≡CC6H4NO2, 2237; N≡CC6H4F, 2233; N≡CMe, 2254; N≡CCMe3, 2233.
- 13C NMR Data for Free Nitriles in Acetone-d6 Solution: δ(N≡C)/ppm = 117.4 (N≡CMe); 126.7 (N≡CCMe3); 119.3 (N≡CPh); 121.0 (N≡CC6H4NMe2); 117.8 (N≡CC6H4NO2); 118.6 (N≡CC6H4F).
- Cox, G.; Dowling, C.; Manning, A.R.; McArdle, P.; Cunningham, D.J. A reinvestigation of the reaction of [Fe2(η-C5H5)2(CO)4-n(CNR)n] (n = 1 or 2) with strong alkylating agents. Organomet. Chem. 1992, 438, 143–158. [Google Scholar] [CrossRef]
- Albano, V.G.; Busetto, L.; Castellari, C.; Monari, M.; Palazzi, A.; Zanotti, V. Electrophilically promoted cyanide abstraction in diiron cyano(amino) alkylidene complexes: Molecular structure of [Fe2(CO)2(η-C5H5)2(µ-CO){µ-CN(CH2)4CH2}][(OC)5WCNW(CO)5]. J. Chem. Soc. Dalton Trans. 1993, 3661–3666. [Google Scholar] [CrossRef]
- Busetto, L.; Marchetti, F.; Zacchini, S.; Zanotti, V. Diiron and diruthenium aminocarbyne complexes containing pseudohalides: Stereochemistry and reactivity. Inorg. Chim. Acta 2005, 358, 1204–1216. [Google Scholar] [CrossRef]
- Albano, V.G.; Busetto, L.; Marchetti, F.; Monari, M.; Zacchini, S.; Zanotti, V. Synthesis and Characterization of New Diiron and Diruthenium µ-Aminocarbyne Complexes Containing Terminal S-, P- and C-Ligands. Zeitschrift für Naturforschung B 2007, 62b, 427–438. [Google Scholar] [CrossRef]
- Agonigi, G.; Bortoluzzi, M.; Marchetti, F.; Pampaloni, G.; Zacchini, S.; Zanotti, V. Regioselective Nucleophilic Additions to Diiron Carbonyl Complexes Containing a Bridging Aminocarbyne Ligand: A Synthetic, Crystallographic and DFT Study. Eur. J. Inorg. Chem. 2018, 960–971. [Google Scholar] [CrossRef]
- Biancalana, L.; Marchetti, F. Aminocarbyne ligands in Organometallic Chemistry. Coord. Chem. Rev. 2021, 449, 214203. [Google Scholar] [CrossRef]
- King, R.B.; Murray, R.M.; Davis, R.E.; Ross, P.K. Organonitrogen derivatives of metal carbonyls: XIII. Reactions of dialkylaminotrimethylsilylacetylenes with metal carbonyls. J. Organomet. Chem. 1987, 330, 115–132. [Google Scholar] [CrossRef]
- Boss, K.; Dowling, C.; Manning, A.R.; Cunningham, D.; McArdle, P. The reaction of [Fe2(η-C5H5)2(CO)(CNMe)(μ-CO)(μ-CNMe2)]+ and related salts with trifluoromethanesulphonic acid, HOSO2CF3: Structure of cis-[Fe2(η-C5H5)2(CO)2(μ-CNMe)(μ-CNMe2)][BPh4]. J. Organomet. Chem. 1999, 579, 252–268. [Google Scholar] [CrossRef]
- Madec, P.; Muir, K.W.; Petillon, F.Y.; Rumin, R.; Scaon, Y.; Schollhammer, P.; Talarmin, J. Generation of substrate-binding sites by electrochemical reduction of cis-[Fe2(cp)2(µ-SMe)2(MeCN)(L)]2+ (L = CO or MeCN). Reactivity of the sites toward CO and tBuNC. Crystal structure of [Fe2(cp)2(µ-SMe)2(CO)(MeCN)][BF4]2·CH2Cl2. J. Chem. Soc. Dalton Trans. 1999, 2371–2384. [Google Scholar] [CrossRef]
- Kuckmann, T.; Lerner, H.-W.; Bolte, M. Redetermination of (acetonitrile-[kappa]N)dicarbonyl([eta]5-cyclopentadienyl)iron(II) tetrafluoridoborate. Acta Crystallogr. 2012, 68E, m966. [Google Scholar]
- M’thiruaine, C.M.; Friedrich, H.B.; Changamu, E.O.; Bala, M.D. Acetonitriledicarbonyl([eta]5-pentamethylcyclopentadienyl)iron(II) tetrafluoridoborate. Acta Crystallogr. 2011, 67E, m924. [Google Scholar]
- Kalman, S.E.; Petit, A.; Brent Gunnoe, T.; Ess, D.H.; Cundari, T.R.; Sabat, M. Facile and Regioselective C–H Bond Activation of Aromatic Substrates by an Fe(II) Complex Involving a Spin-Forbidden Pathway. Organometallics 2013, 32, 1797–1806. [Google Scholar] [CrossRef]
- Chen, J.; Szalda, D.J.; Fujita, E.; Creutz, C. Iron(II) and Ruthenium(II) Complexes Containing P, N, and H Ligands: Structure, Spectroscopy, Electrochemistry, and Reactivity. Inorg. Chem. 2010, 49, 9380–9391. [Google Scholar] [CrossRef] [PubMed]
- Clegg, J.K.; Cremers, J.; Hogben, A.J.; Breiner, B.; Smulders, M.M.J.; Thoburnad, J.D.; Nitschke, J.R. A stimuli responsive system of self-assembled anion-binding Fe4L68+ cages. Chem. Sci. 2013, 4, 68–76. [Google Scholar] [CrossRef]
- Hoffbauer, M.R.; Iluc, V.M. [2+2] Cycloadditions with an Iron Carbene: A Critical Step in Enyne Metathesis. J. Am. Chem. Soc. 2021, 143, 5592–5597. [Google Scholar] [CrossRef]
- Heine, A.; Herbst-Irmer, R.; Stalke, D.; Kuhnle, W.; Zachariasse, K.A. Structure and Crystal Packing of 4-Aminobenzonitriles and 4-Amino-3,5-dimethylbenzonitriles at Various Temperatures. Acta Cryst. 1994, B50, 363–373. [Google Scholar] [CrossRef]
- Arrigoni, F.; Bertini, L.; De Gioia, L.; Cingolani, A.; Mazzoni, R.; Zanotti, V.; Zampella, G. Mechanistic Insight into Electrocatalytic H2 Production by [Fe2(CN){μ-CN(Me)2}(μ-CO)(CO)(Cp)2]: Effects of Dithiolate Replacement in [FeFe] Hydrogenase Models. Inorg. Chem. 2017, 56, 13852–13864. [Google Scholar] [CrossRef] [PubMed]
- Ortel, A.M.; Ritleng, V.; Chetcuti, M.J.; Veiros, L.F. C-H Activation of Acetonitrile at Nickel: Ligand Flip and Conversion of N-Bound Acetonitrile into a C-Bound Cyanomethyl Ligand. J. Am. Chem. Soc. 2010, 132, 13588–13589. [Google Scholar] [CrossRef] [PubMed]
- Tanabe, T.; Evans, M.E.; Brennessel, W.W.; Jones, W.D. C-H and C-CN Bond Activation of Acetonitrile and Succinonitrile by [Tp'Rh(PR3)]. Organometallics 2011, 30, 834–843. [Google Scholar] [CrossRef]
- Garcia, M.H.; Robalo, M.P.; Teixeira, A.P.S.; Dias, A.R.; Piedade, M.F.M.; Duarte, M.T. Synthesis of new donor/acceptor η5-cyclopentadienyl and η5-indenyliron(II) complexes with p-benzonitrile derivatives. Crystal structures of [Fe(η5-C5H5)(CO)(P(OC6H5)3)(p-NCC6H4NO2)][BF4]·CH2Cl2 and [Fe(η5-C9H7)(CO)(P(OC6H5)3)(p-NCC6H4NO2)][BF4]. J. Organomet. Chem. 2001, 632, 145–156. [Google Scholar] [CrossRef][Green Version]
- Bengu, B.; Boerio, F.J. Interaction of epoxy/dicyandiamide adhesives with metal substrates. J. Adhes. 2006, 82, 1133–1155. [Google Scholar] [CrossRef]
- Bistoni, G.; Rampino, S.; Scafuri, N.; Ciancaleoni, G.; Zuccaccia, D.; Belpassi, L.; Tarantelli, F. How π back-donation quantitatively controls the CO stretching response in classical and non-classical metal carbonyl complexes. Chem. Sci. 2016, 7, 1174–1184. [Google Scholar] [CrossRef]
- Lupinetti, A.J.; Fau, S.; Frenking, G.; Strauss, S.H. Nonclassical Metal Carbonyls: [Ag(CO)]+ and [Ag(CO)2]+. J. Phys. Chem. A 1997, 101, 9551–9559. [Google Scholar] [CrossRef]
- Frenking, G.; Loschen, C.; Krapp, A.; Fau, S.; Strauss, S.H. Electronic structure of CO—An exercise in modern chemical bonding theory. J. Comput. Chem. 2007, 28, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Menges, F. Spectragryph—Optical Spectroscopy Software. Version 1.2.5, @2016–2017. Available online: http://www.effemm2.de/spectragryph (accessed on 1 November 2021).
- Fulmer, G.R.; Miller, A.J.M.; Sherden, N.H.; Gottlieb, H.E.; Nudelman, A.; Stoltz, B.M.; Bercaw, J.E.; Goldberg, K.I. NMR Chemical Shifts of Trace Impurities: Common Laboratory Solvents, Organics, and Gases in Deuterated Solvents Relevant to the Organometallic Chemist. Organometallics 2010, 29, 2176–2179. [Google Scholar] [CrossRef]
- Willker, W.; Leibfritz, D.; Kerssebaum, R.; Bermel, W. Gradient selection in inverse heteronuclear correlation spectroscopy. Magn. Reson. Chem. 1993, 31, 287–292. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Spek, A.L. PLATON, A Multipurpose Crystallographic Tool; Utrecht University: Utrecht, The Netherlands, 2005. [Google Scholar]
- Neese, F. Software Update: The ORCA Program System, Version 4.0. WIREs Comput. Mol. Sci. 2017, 8, e1327. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Van Lenthe, E.; Baerends, E.J.; Snijders, J. Relativistic regular two-component Hamiltonian. J. Chem. Phys. 1993, 99, 4597–4610. [Google Scholar] [CrossRef]
- Van Lenthe, E.; Ehlers, A.; Baerends, E.-J. Geometry optimizations in the zero order regular approximation for relativistic effects. J. Chem. Phys. 1999, 110, 8943–8953. [Google Scholar] [CrossRef]
- Hopffgarten, M.; von Frenking, G. Energy decomposition analysis. WIREs Comput. Mol. Sci. 2012, 2, 43–62. [Google Scholar] [CrossRef]
- Mitoraj, M.P.; Michalak, A.; Ziegler, T. A Combined Charge and Energy Decomposition Scheme for Bond Analysis. J. Chem. Theory Comput. 2009, 5, 962–975. [Google Scholar] [CrossRef]
- Radoń, M. On the properties of natural orbitals for chemical valence. Theor. Chem. Acc. 2008, 120, 337–339. [Google Scholar] [CrossRef]
- Nalewajski, R.F.; Mrozek, J.; Michalak, A. Two-electron valence indices from the Kohn-Sham orbitals. Int. J. Quantum Chem. 1997, 61, 589–601. [Google Scholar] [CrossRef]
- Nalewajski, R.F.; Ozek, J. Modified valence indices from the two-particle density matrix. Int. J. Quantum Chem. 1994, 51, 187–200. [Google Scholar] [CrossRef]
- Nalewajski, R.F.; Köster, A.M.; Jug, K. Chemical valence from the two-particle density matrix. Theor. Chim. Acta 1993, 85, 463–484. [Google Scholar] [CrossRef]
- Ciancaleoni, G.; Nunzi, F.; Belpassi, L. Charge Displacement Analysis—A Tool to Theoretically Characterize the Charge Transfer Contribution of Halogen Bonds. Molecules 2020, 25, 300. [Google Scholar] [CrossRef] [PubMed]
- Bistoni, G.; Rampino, S.; Tarantelli, F.; Belpassi, L. Charge-displacement analysis via natural orbitals for chemical valence: Charge transfer effects in coordination chemistry. J. Chem. Phys. 2015, 142, 084112. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IR (CH2Cl2), ῦ/cm−1 | IR (Solid), ῦ/cm−1 | 13C NMR (Acetone-d6), δ/ppm | E/Z. Ratio | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Comp. | ῦ (t-CO) | ῦ (μ-CO) | ῦ (μ-CN) | ῦ (N≡C) | Δῦ (N≡C) | ῦ (N≡C) | Δῦ (N≡C) | δ (μ-CN) a | δ (μ-CN) b | δ (N≡C) a | δ (N≡C) b | Δδ(N≡C) a | Δδ(N≡C) b | |
| (2)+ | 1985 | 1818 | 1561 | 2279 | +25 | 2279 | +26 | 330.5 | 132.0 | 131.8 | +14.6 | +14.4 | 1.9 | |
| (3)+ | 1984 | 1820 | 1559 | 2264 | +31 | 2262 | +27 | 330.7 | 330.2 | 139.8 | 140.0 | +13.1 | +13.3 | 2.3 |
| (4)+ | 1984 | 1820 | 1560 | 2238 | +8 | 2238 | +9 | 329.7 | 330.0 | 131.2 | 131.6 | +11.9 | +12.3 | 1.2 |
| (5)+ | 1985 | 1819 | 1561 | 2227 | +12 | 2227 | +17 | 330.9 | 330.4 | 133.4 | 133.9 | +12.4 | +12.9 | 1.1 |
| (6)+ | 1982 | 1821 | 1561 | 2230 | −7 | 2230 | −3 | 329.2 | 329.0 | 129.2 | 129.6 | +11.4 | +11.8 | 1.3 |
| (7)+ | 1983 | 1819 | 1559 | 2240 | +7 | 2240 | +6 | 329.9 | 329.7 | 130.4 | 130.7 | +11.8 | +12.1 | 1.0 |
| (8)+ | 1974 | 1810 | 1558 | 334.5 | 1.7 | |||||||||
| (9)+ | 1965 | 1800 | 1532 | 331.7 | 332.0 | 1.3 | ||||||||
| (10)+ | 1984 | 1820 | 1560 | 2235 | −2 | 2235 | +2 | 342.0 c | 341.1 c | 129.1 c | 130.0 c | +11.3 c | +12.2 c | 2.3 |
| (11)+ | 1987 | 1818 | 1568 | 2277 | +23 | 2277 | +24 | 333.4 | 132.1 | +14.7 | 0.15 | |||
| (12)+ | 1984 | 1817 | 1525 | 2281 | +27 | 2281 | +28 | 338.2 | 338.1 | 132.1 | 132.3 | +14.7 | +14.9 | 0.82 |
| (2Z)+ | (3Z)+ | (5Z)+ | (11Z)+ | |
|---|---|---|---|---|
| Fe(1)-Fe(2) | 2.5100(10) | 2.5091(9) | 2.5023(7) | 2.5107(7) |
| Fe(1)-C(1) | 1.792(7) | 1.761(3) | 1.759(4) | 1.754(4) |
| Fe(2)-N(2) | 1.924(5) | 1.928(3) | 1.912(3) | 1.918(3) |
| Fe(1)-C(2) | 1.969(6) | 1.964(3) | 1.960(4) | 1.947(4) |
| Fe(2)-C(2) | 1.887(6) | 1.910(3) | 1.896(4) | 1.914(4) |
| Fe(1)-C(3) | 1.884(5) | 1.880(3) | 1.875(4) | 1.880(3) |
| Fe(2)-C(3) | 1.861(5) | 1.863(3) | 1.863(4) | 1.871(4) |
| Fe(1)-Cpav | 2.111(13) | 2.112(7) | 2.110(9) | 2.110(7) |
| Fe(2)-Cpav | 2.095(12) | 2.112(7) | 2.107(9) | 2.102(7) |
| C(1)-O(1) | 1.092(7) | 1.141(4) | 1.146(5) | 1.143(5) |
| C(2)-O(2) | 1.157(7) | 1.171(4) | 1.166(5) | 1.166(5) |
| N(1)-C(3) | 1.288(7) | 1.297(4) | 1.295(5) | 1.290(5) |
| N(2)-C(21) | 1.126(7) | 1.140(4) | 1.148(5) | 1.133(5) |
| Fe(1)-C(1)-O(1) | 177.4(6) | 176.6(3) | 176.9(3) | 179.7(4) |
| Fe(2)-N(2)-C(21) | 177.3(5) | 175.6(2) | 172.8(3) | 173.6(3) |
| Fe(1)-C(2)-Fe(2) | 81.2(2) | 80.73(11) | 80.91(14) | 81.13(14) |
| Fe(1)-C(3)-Fe(2) | 84.2(2) | 84.18(12) | 84.03(15) | 84.03(14) |
| N(2)-C(21)-C(22) | 178.3(6) | 174.9(3) | 178.7(4) | 177.7(4) |
| Sum at N(1) | 359.6(8) | 360.0(3) | 360.0(5) | 360.0(5) |
| Sum at C(3) | 359.5(6) | 359.8(3) | 359.3(4) | 359.6(4) |
| Complex | Fe–N (Å) | N≡C (Å) | Ref. |
|---|---|---|---|
 | 1.924(3) | 1.141(4) | [59] |
 | 1.935(4) | 1.147(6) | [60] |
 | 1.924(3) | 1.143(5) | [61] |
 | 1.9025(11) | 1.1468(17) | [62] |
 | 1.9099(19) | 1.134(3) | [63] |
 | 2.1510(12) | 1.1369(17) | [64] |
 | 1.945(2) | 1.147(3) | [65] |
 | 1.906(5) | 1.146(8) | [16] |
 | 1.145(3) | [66] |
| Comp. | Δῦ | BDE | ΔE0 (CT0) | ΔE1 (CT1) | ΔE2 (CT2) | CTtot (CTπtot) | CTCN (CTπCN) | % b.d. |
|---|---|---|---|---|---|---|---|---|
| (M1)+ | +2.3 | −46.1 | −33.2 (0.167) | −11.1 (−0.102) | −9.6 (−0.043) | 0.034 (−0.145) | 0.121 (0.065) | 34.7 |
| (2Z)+exp a | − | − | −30.2 (0.158) | −9.1 (−0.085) | −7.6 (−0.034) | 0.048 (−0.119) | 0.108 (0.063) | 31.9 |
| (2Z)+ | +2.5 | −46.3 | −33.7 (0.167) | −11.5 (−0.107) | −9.6 (−0.038) | 0.029 (−0.145) | 0.111 (0.054) | 34.8 |
| (2E)+ | +2.2 | −46.6 | −33.6 (0.168) | −11.4 (−0.106) | −9.6 (−0.043) | 0.030 (−0.149) | 0.113 (0.064) | 35.0 |
| (3E)+ | +1.8 | −50.0 | −34.1 (0.173) | −11.2 (−0.100) | −9.5 (−0.043) | 0.043 (−0.143) | 0.113 (0.071) | 33.7 |
| (4E)+ | −4.4 | −50.9 | −34.5 (0.176) | −13.5 (−0.126) | −10.1 (−0.044) | 0.021 (−0.170) | 0.116 (0.063) | 36.6 |
| (5E)+ | 1.2 | −56.1 | −35.4 (0.182) | −11.9 (−0.096) | −9.4 (−0.040) | 0.062 (−0.136) | 0.150 (0.102) | 33.2 |
| (6E)+ | −16.2 | −47.7 | −33.9 (0.169) | −15.7 (−0.119) | −10.9 (−0.046) | 0.005 (−0.165) | 0.086 (0.032) | 39.8 |
| (7E)+ | −5.0 | −50.0 | −34.5 (0.178) | −13.4 (−0.124) | −10.2 (−0.045) | 0.024 (−0.169) | 0.114 (0.062) | 36.5 |
| (M2)+ | −62.7 | −42.5 | −32.5 (0.108) | −18.1 (−0.178) | −15.0 (−0.039) | −0.099 (−0.217) | 0.032 (−0.051) | 46.6 |
| (M3)+ | −48.9 | −44.1 | −33.2 (0.132) | −16.7 (−0.169) | −13.7 (−0.044) | −0.071 (−0.213) | 0.049 (0.00) | 44.0 |
| (M4)+ | −35.2 | −46.0 | −33.5 (0.154) | −16.1 (−0.159) | −11.0 (−0.046) | −0.040 (−0.205) | 0.068 (0.011) | 40.8 |
| (2)CF3SO3 | (3)CF3SO3 | (5)CF3SO3 | (11)CF3SO3 | |
|---|---|---|---|---|
| Formula | C23H27F3Fe2N2O5S | C26H33F3Fe2N2O5S | C30H34F3Fe2N3O5S | C20H21F3Fe2N2O5S |
| FW | 612.22 | 654.30 | 717.36 | 570.15 |
| T, K | 100(2) | 100(2) | 100(2) | 100(2) |
| λ, Å | 0.71073 | 0.71073 | 0.71073 | 0.71073 |
| Crystal system | Monoclinic | Monoclinic | Monoclinic | Orthorhombic |
| Space group | P21/n | P21/n | C2/c | Pbca |
| a, Å | 10.9065(9) | 12.081(4) | 28.694(3) | 15.1677(7) |
| b, Å | 14.8064(11) | 17.491(6) | 10.6013(9) | 13.5839(6) |
| c, Å | 15.9959(12) | 13.857(4) | 23.210(2) | 22.1751(10) |
| β | 101.382(3) | 107.13(2) | 116.562(3) | 90 |
| Cell Volume, Å3 | 2532.3(3) | 2798.2(16) | 6315.0(10) | 4568.9(4) |
| Z | 4 | 4 | 8 | 8 |
| Dc, g∙cm−3 | 1.606 | 1.553 | 1.509 | 1.658 |
| µ, mm−1 | 1.287 | 1.170 | 1.046 | 1.420 |
| F(000) | 1256 | 1352 | 2960 | 2320 |
| Crystal size, mm | 0.16 × 0.14 × 0.11 | 0.22 × 0.20 × 0.10 | 0.18 × 0.16 × 0.12 | 0.18 × 0.16 × 0.11 |
| θ limits | 1.892–25.027 | 1.929–27.000 | 1.962–26.000 | 1.837–25.999 |
| Reflections collected | 44,496 | 37,875 | 39,670 | 59,100 |
| Independent reflections | 4453 [Rint = 0.1492] | 6110 [Rint = 0.0453] | 6200 [Rint = 0.0435] | 4467 [Rint = 0.0805] |
| Data/restraints/parameters | 4453/0/328 | 6110/24/356 | 6200/175/454 | 4467/0/300 |
| Goodness on fit on F2 | 1.054 | 1.127 | 1.084 | 1.252 |
| R1 (I > 2σ(I)) | 0.0597 | 0.0485 | 0.0581 | 0.0532 |
| wR2 (all data) | 0.1606 | 0.1056 | 0.16030 | 0.1085 |
| Largest diff. peak and hole, e Å−3 | 0.966/–0.694 | 0.830/–0.579 | 2.211/–0.881 | 0.803/–0.469 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bresciani, G.; Biancalana, L.; Pampaloni, G.; Zacchini, S.; Ciancaleoni, G.; Marchetti, F. A Comprehensive Analysis of the Metal–Nitrile Bonding in an Organo-Diiron System. Molecules 2021, 26, 7088. https://doi.org/10.3390/molecules26237088
Bresciani G, Biancalana L, Pampaloni G, Zacchini S, Ciancaleoni G, Marchetti F. A Comprehensive Analysis of the Metal–Nitrile Bonding in an Organo-Diiron System. Molecules. 2021; 26(23):7088. https://doi.org/10.3390/molecules26237088
Chicago/Turabian StyleBresciani, Giulio, Lorenzo Biancalana, Guido Pampaloni, Stefano Zacchini, Gianluca Ciancaleoni, and Fabio Marchetti. 2021. "A Comprehensive Analysis of the Metal–Nitrile Bonding in an Organo-Diiron System" Molecules 26, no. 23: 7088. https://doi.org/10.3390/molecules26237088
APA StyleBresciani, G., Biancalana, L., Pampaloni, G., Zacchini, S., Ciancaleoni, G., & Marchetti, F. (2021). A Comprehensive Analysis of the Metal–Nitrile Bonding in an Organo-Diiron System. Molecules, 26(23), 7088. https://doi.org/10.3390/molecules26237088