
Electrochemical Organic Synthesis of Electron-Rich Biaryl Scaffolds: An Update

 ,
,  ,
,
Abstract
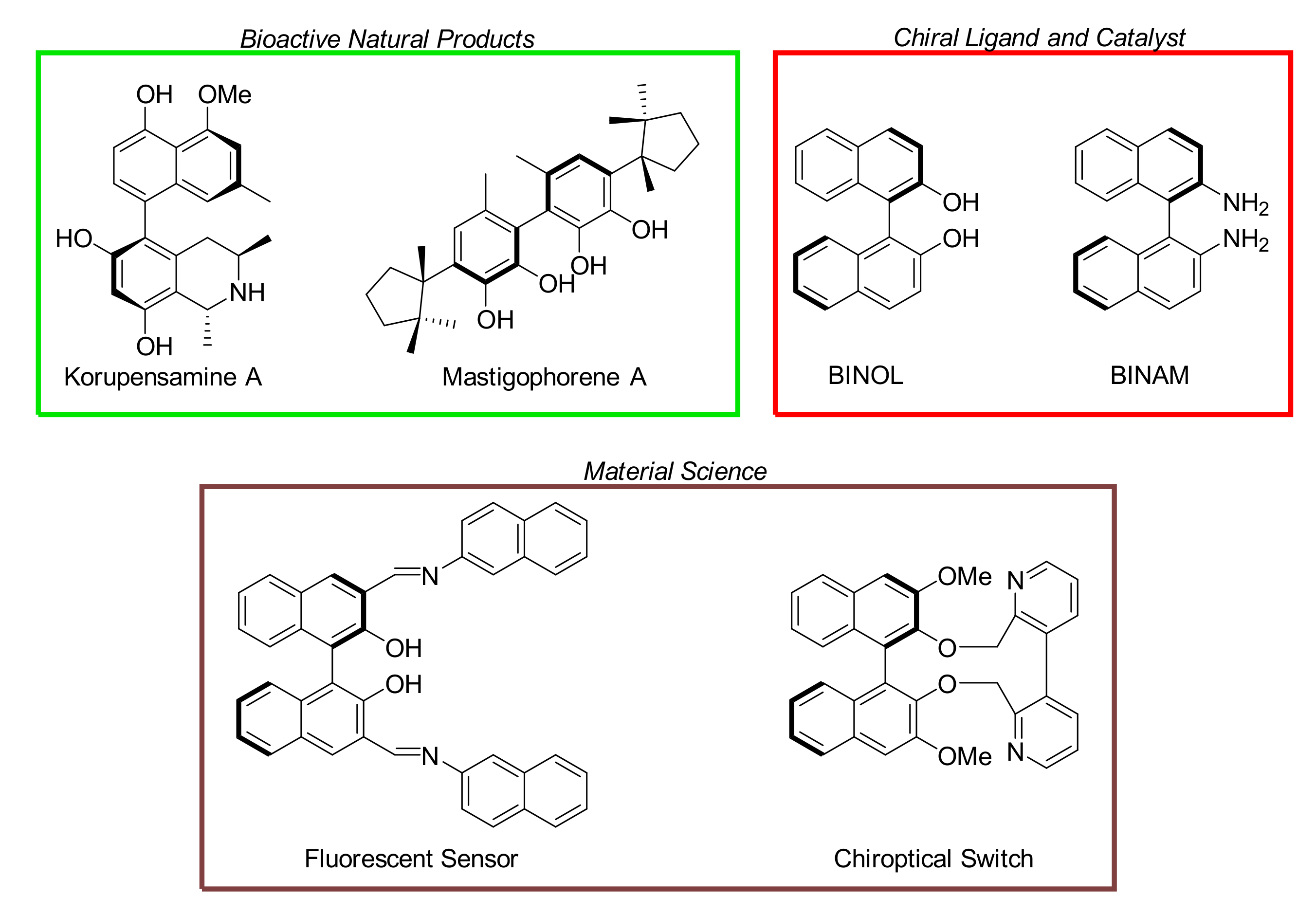
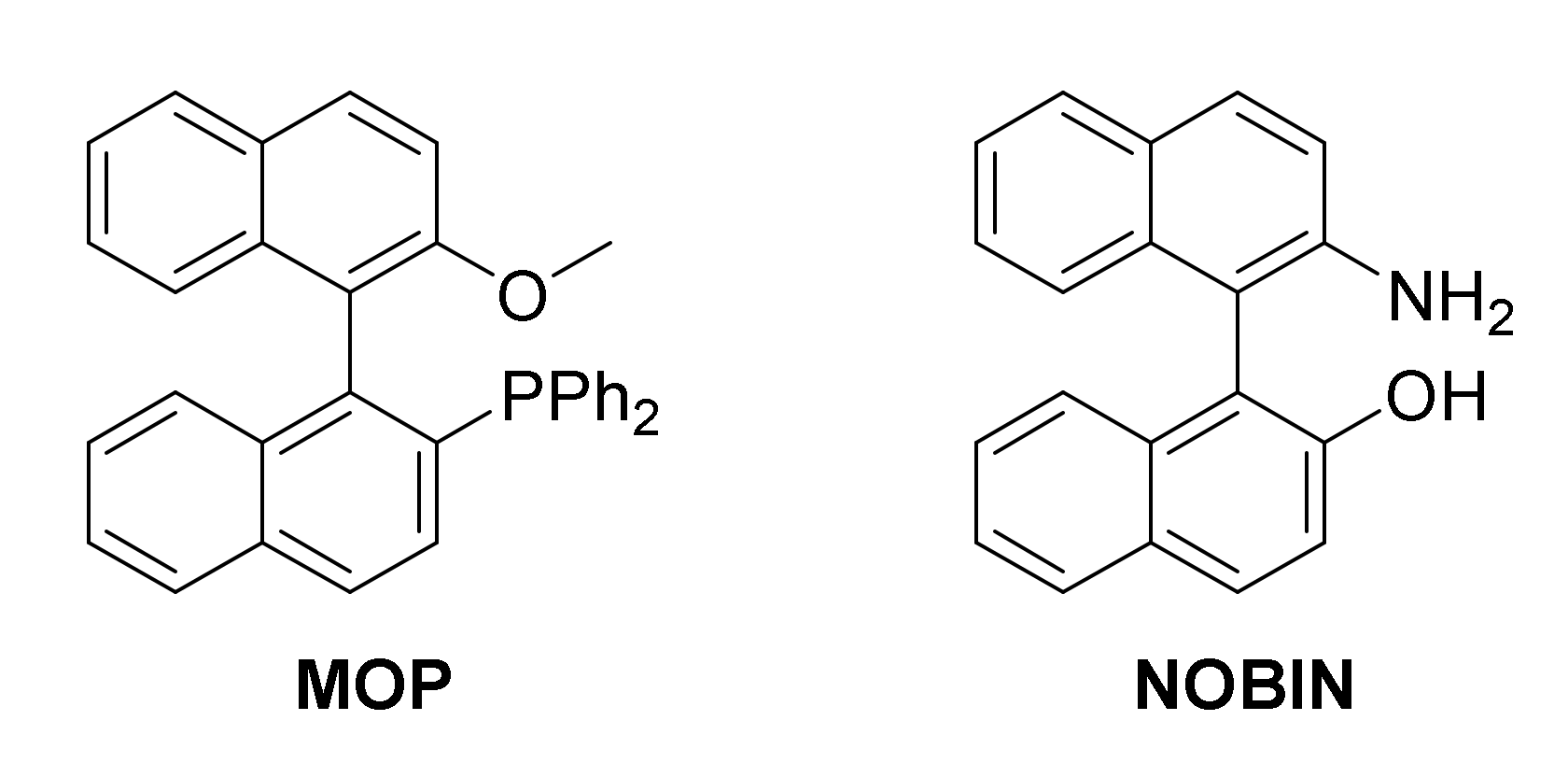
1. Introduction
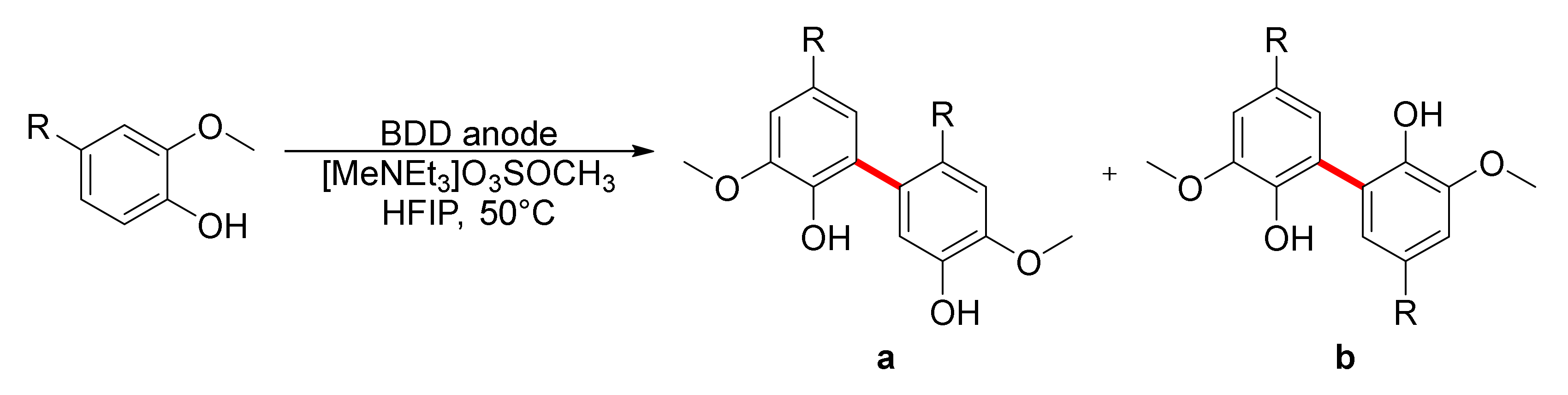
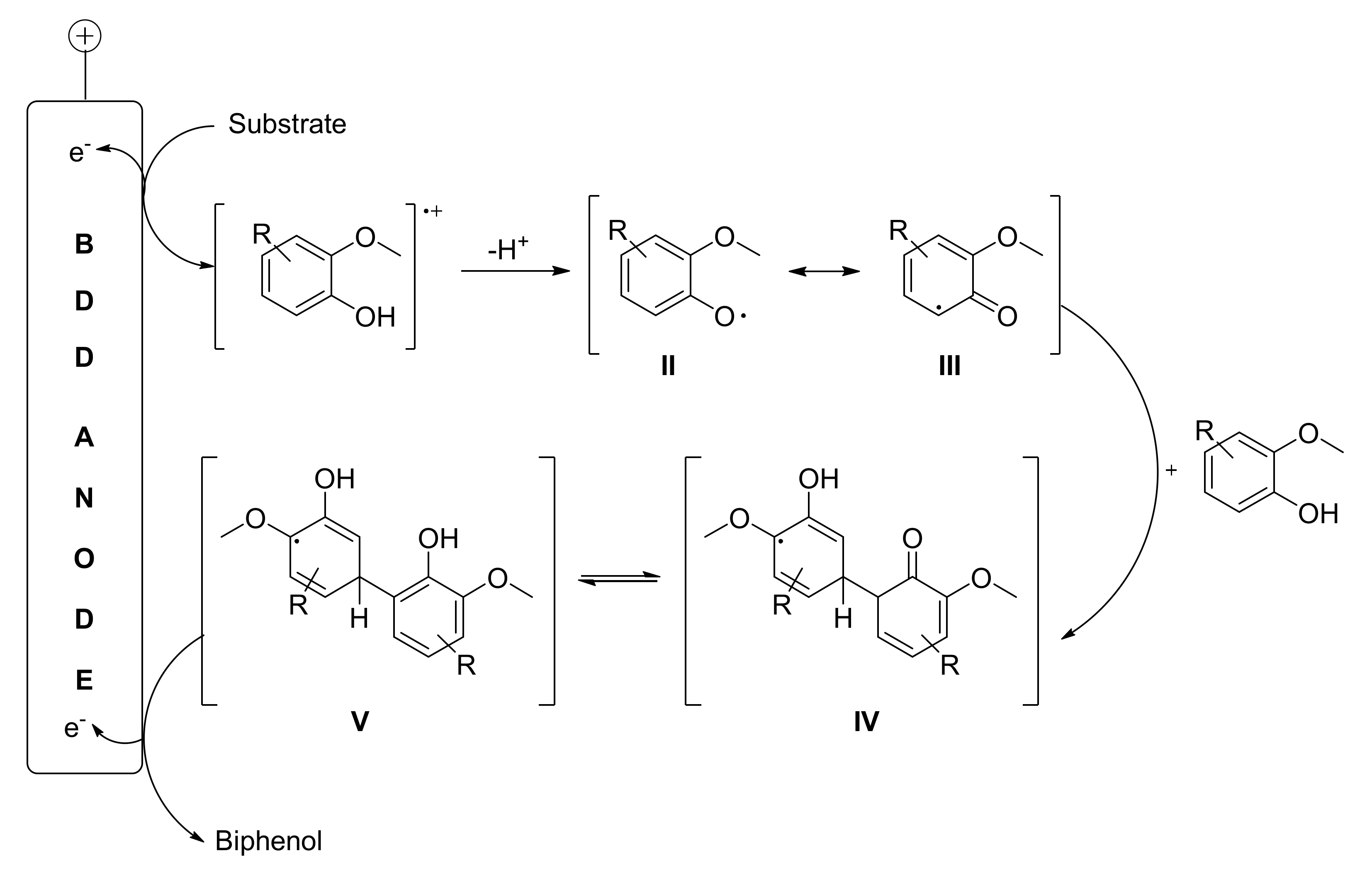
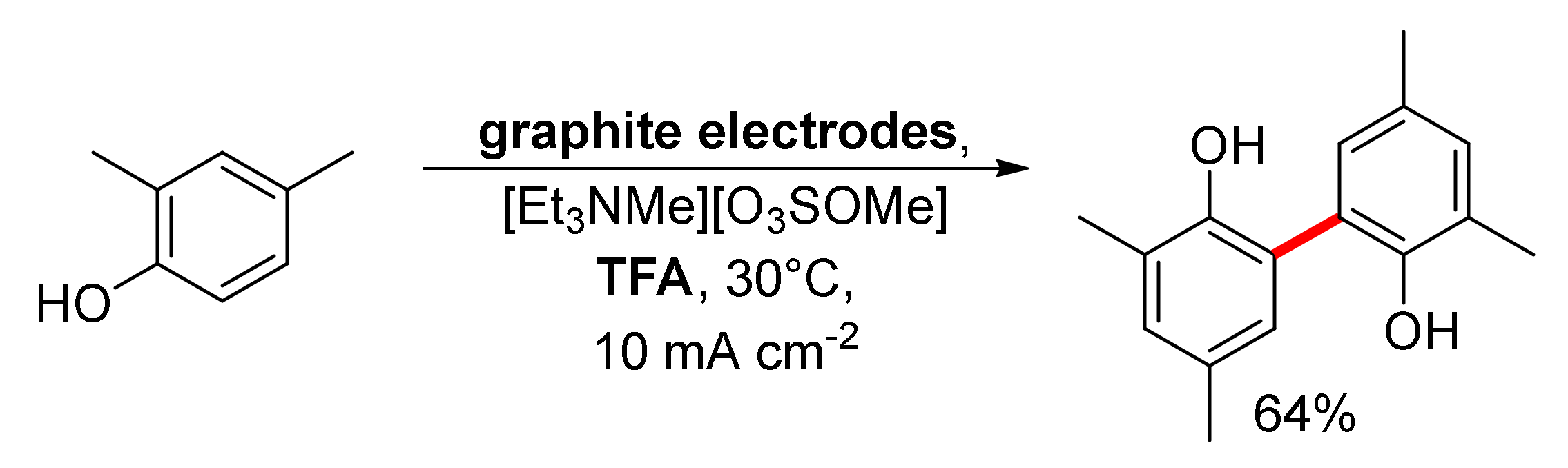
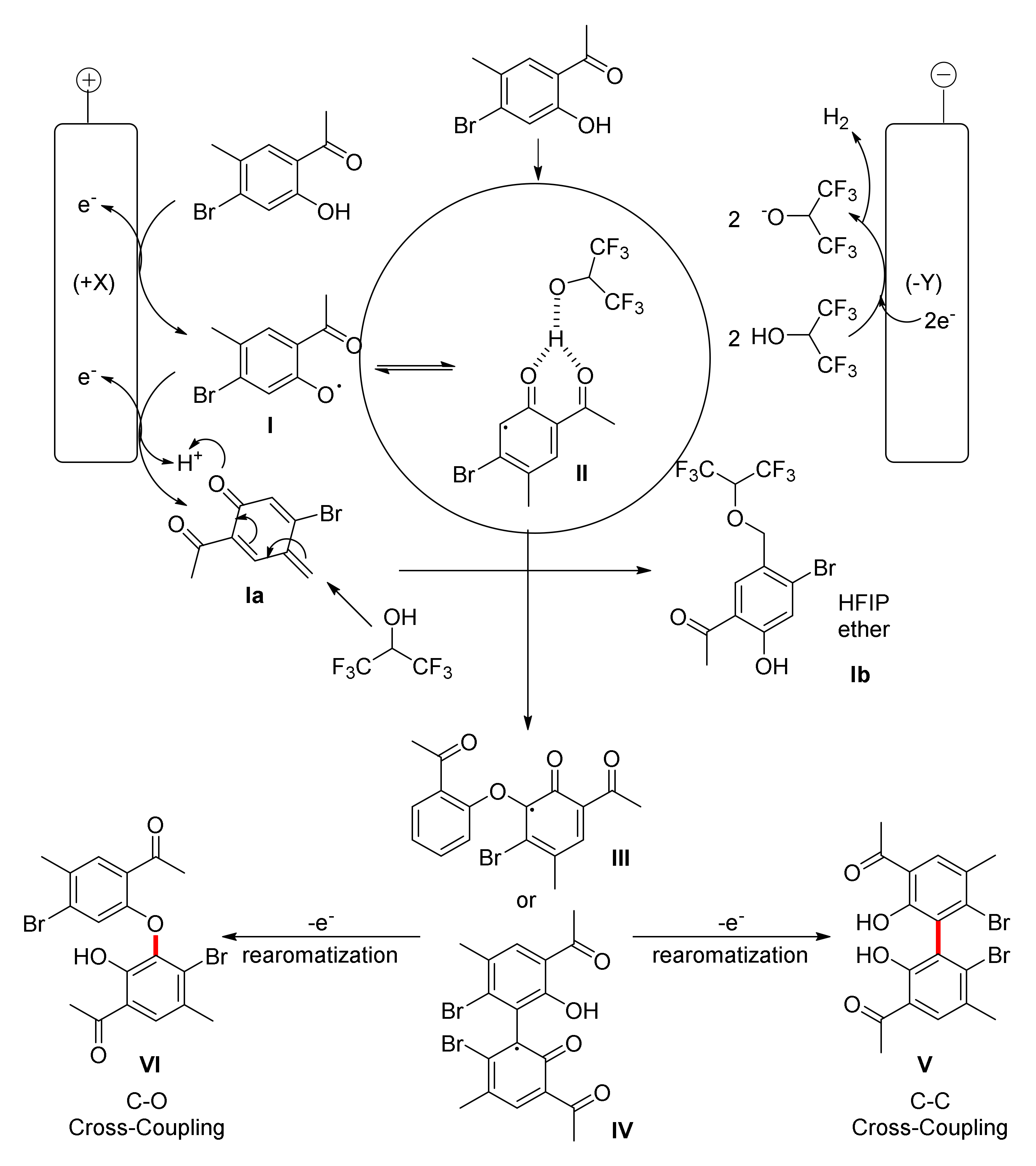
2. Biphenols Electrosynthesis
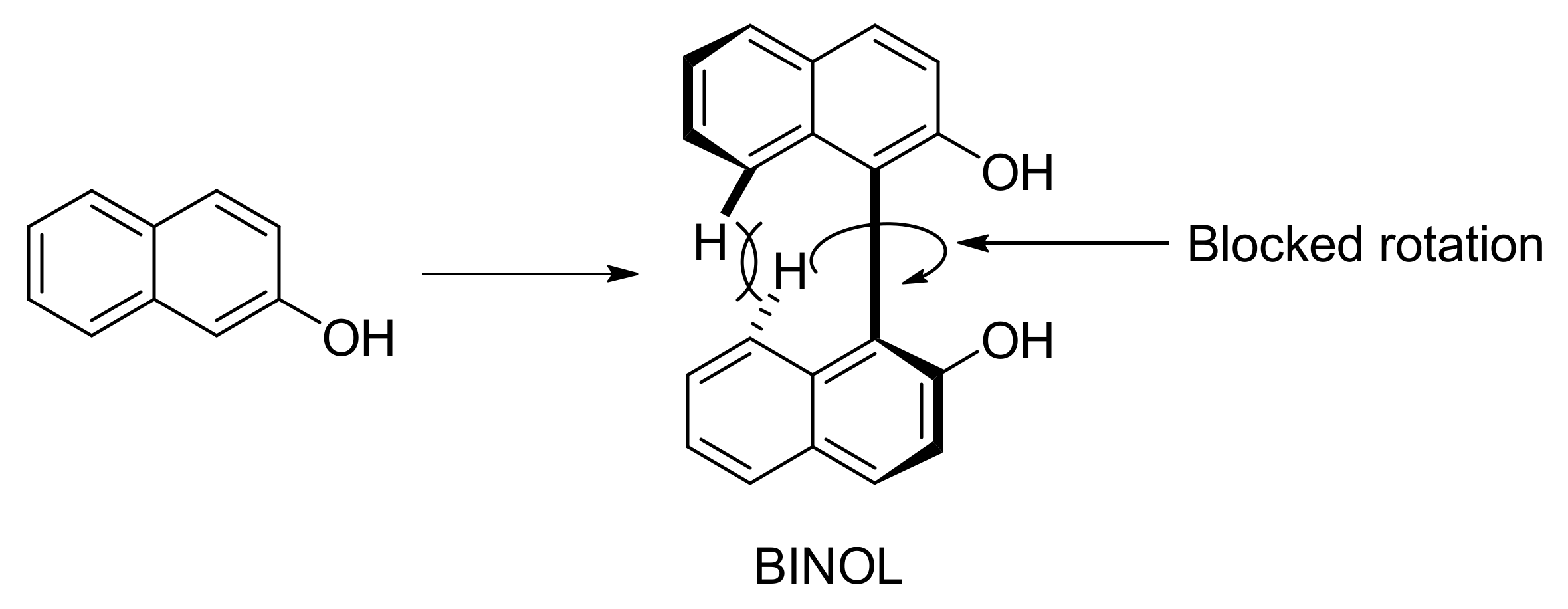
3. Naphthols Electrocoupling
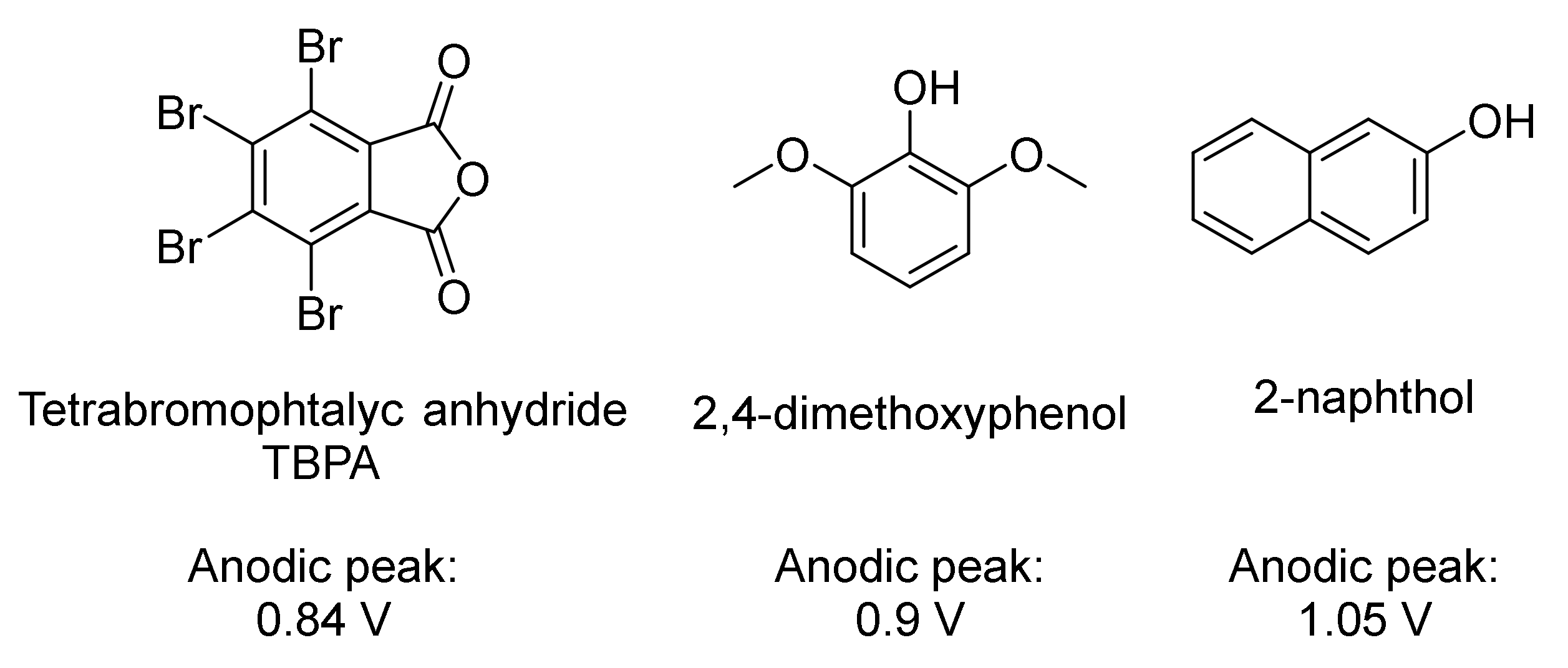
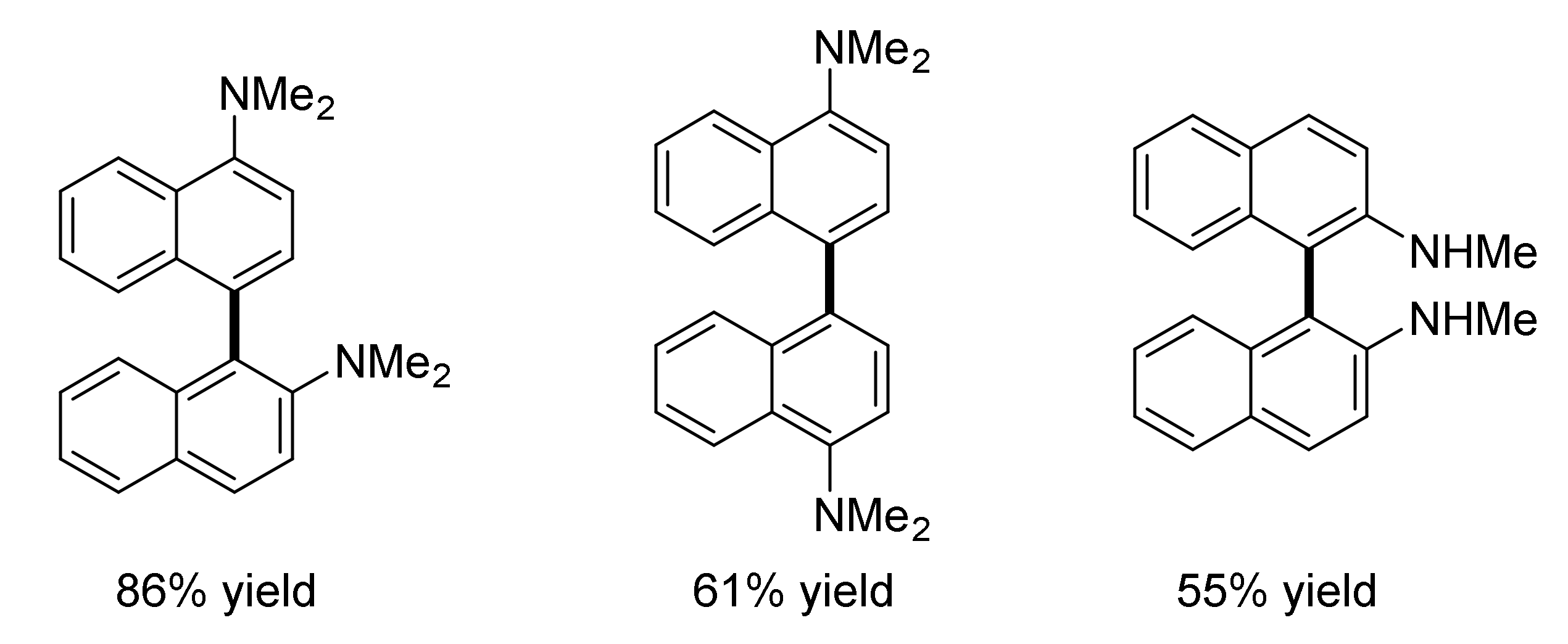
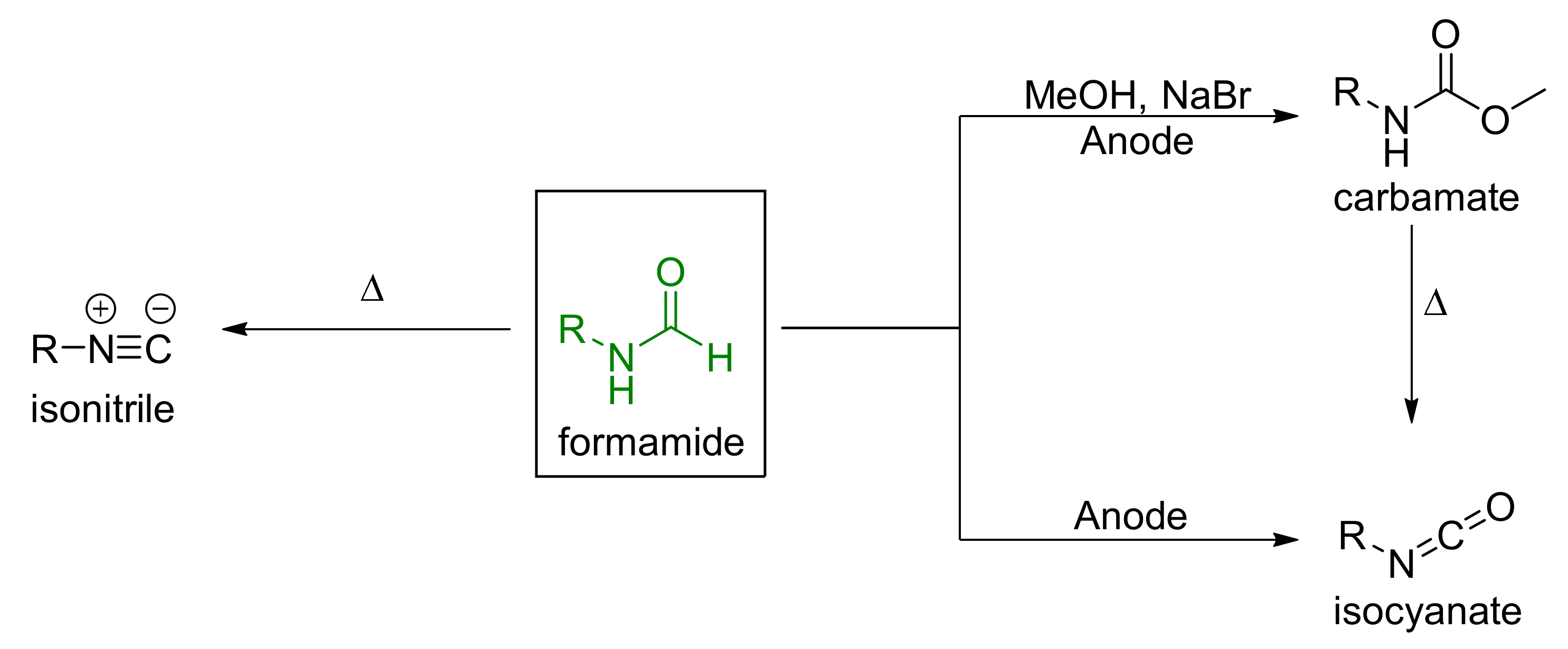
4. Aniline Electrocoupling
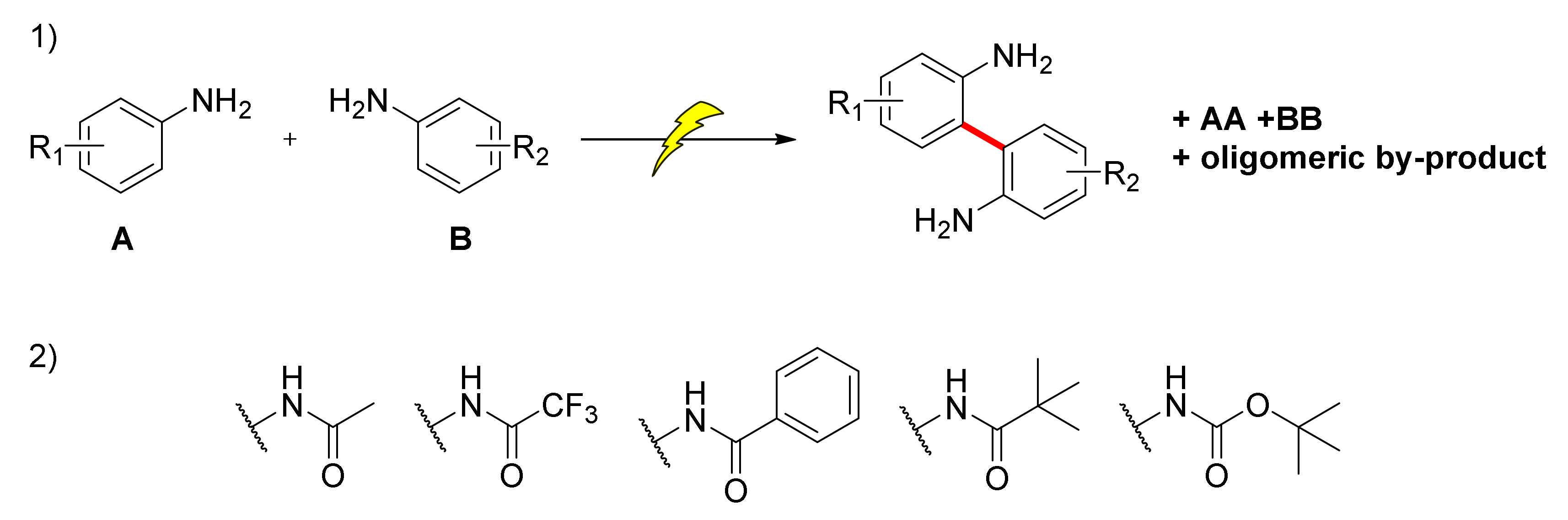
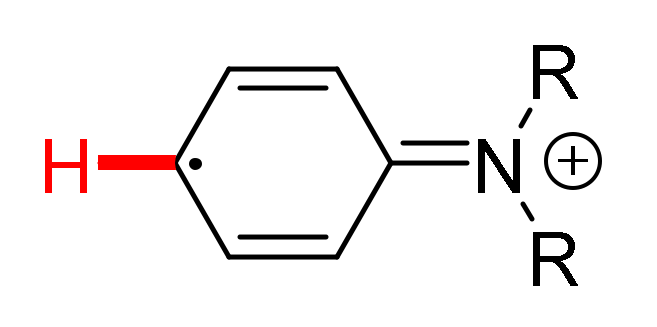
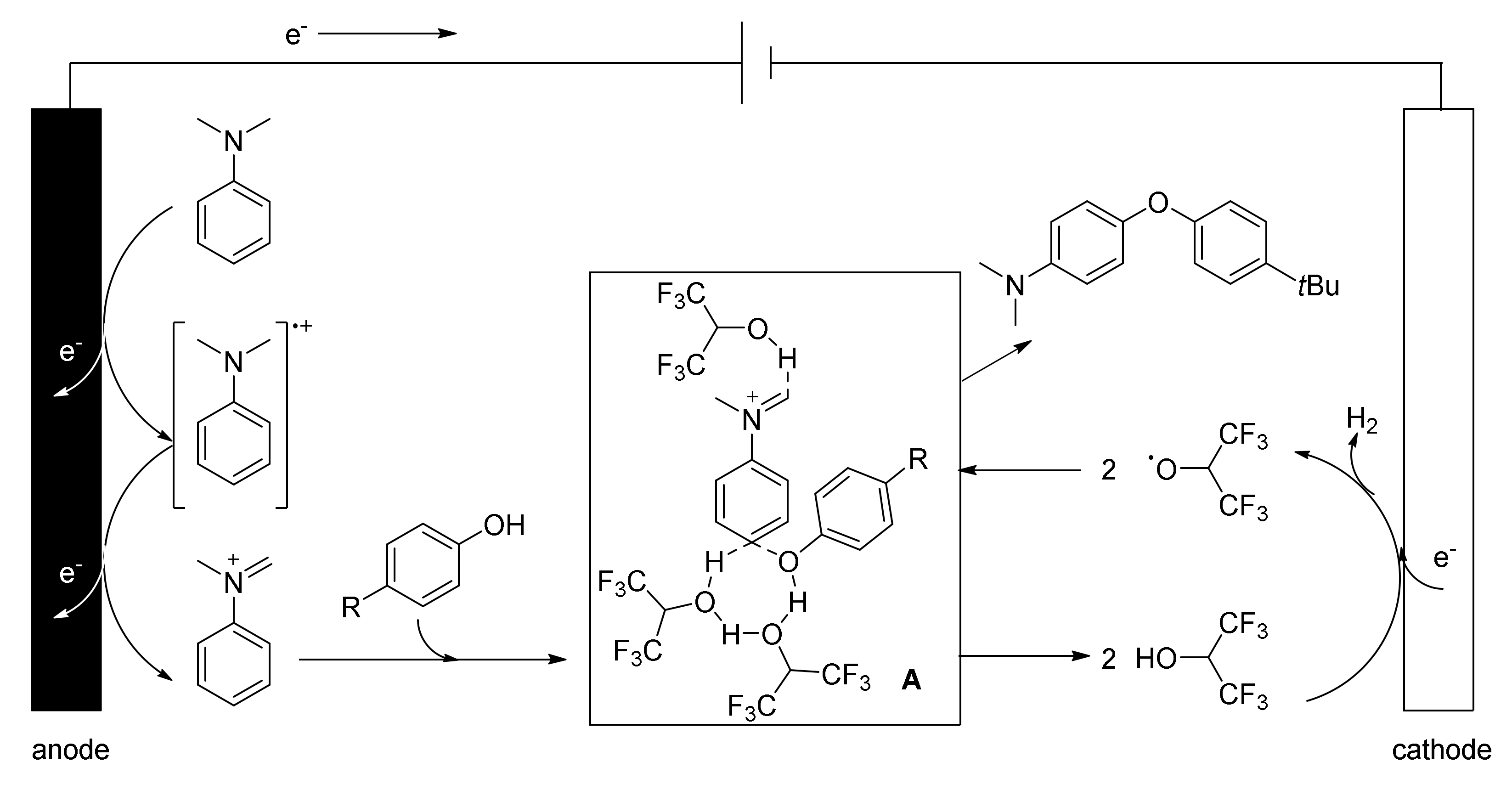
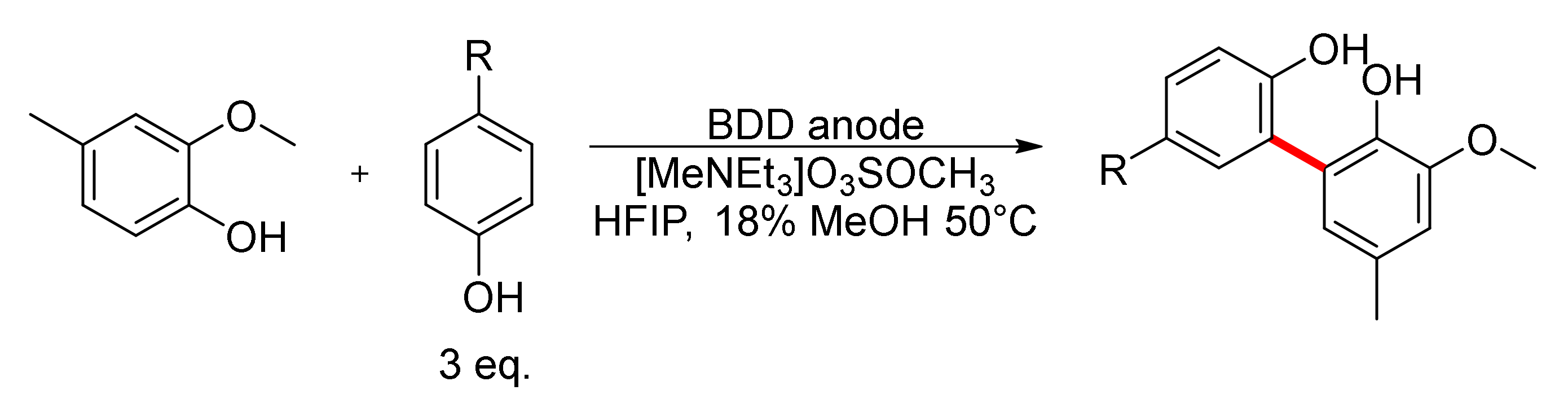
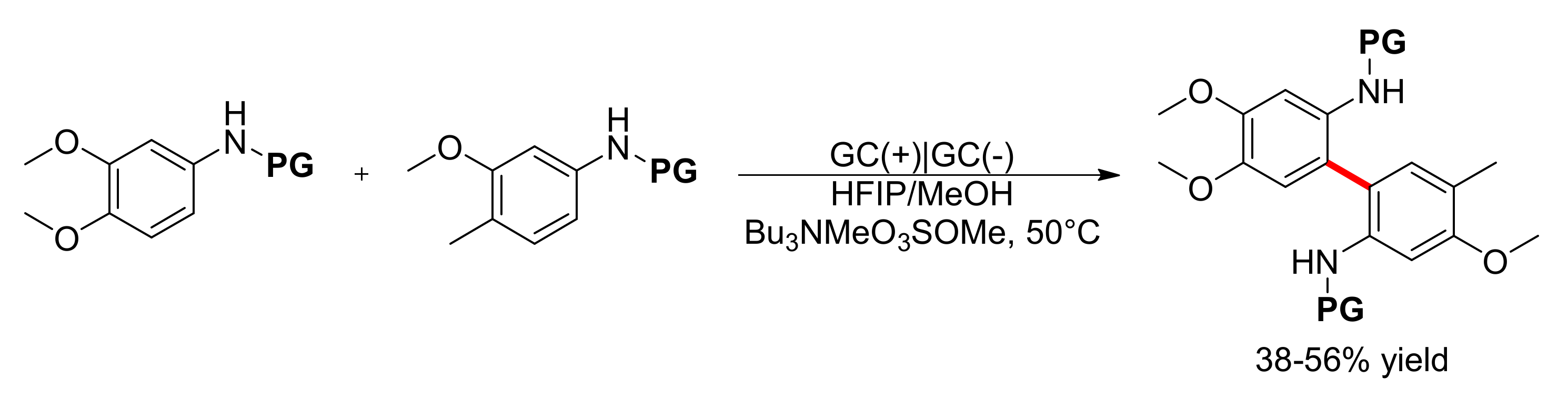
5. Unsymmetrical Biaryl Electrosynthesis
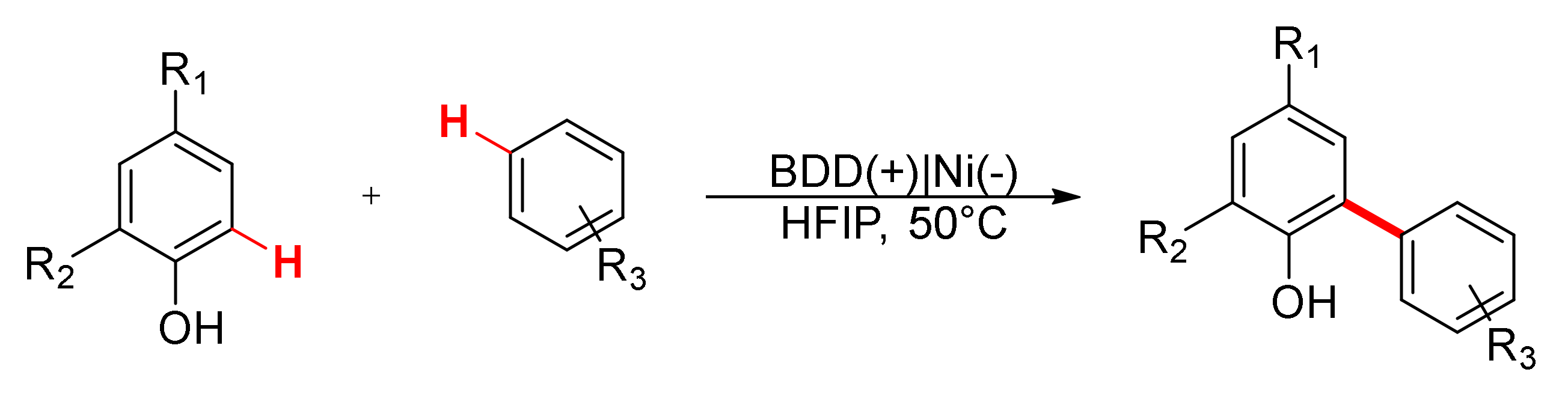
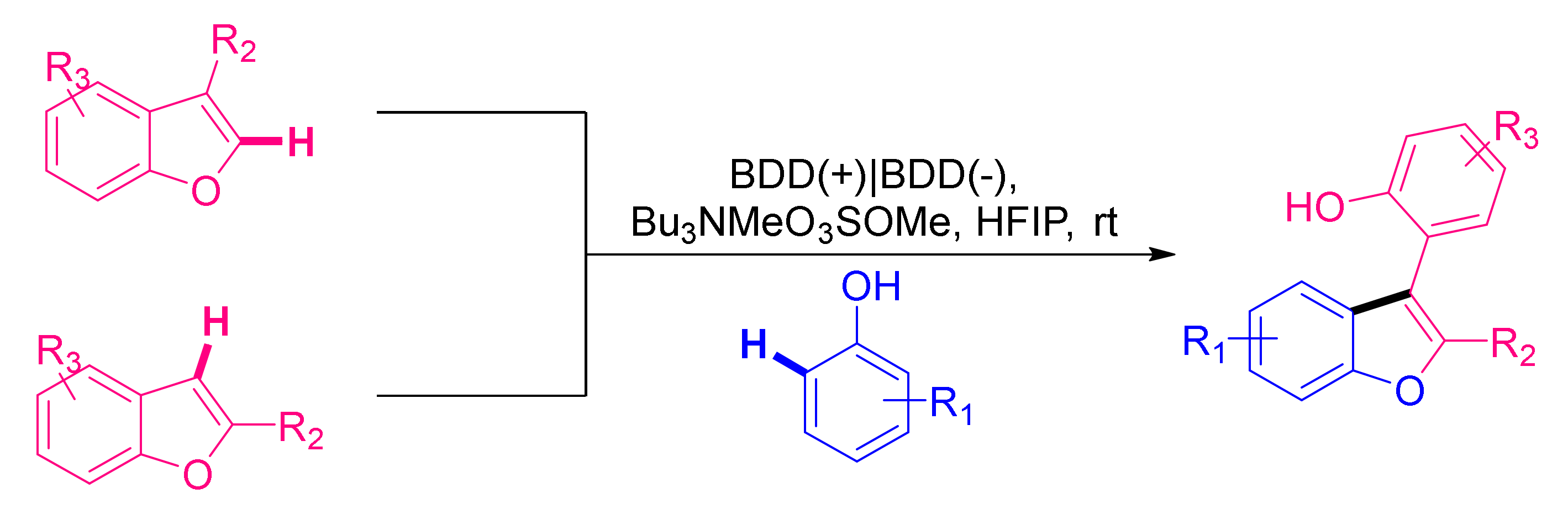
6. Miscellaneous
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yuan, S.; Chang, J.; Yu, B. Construction of Biologically Important Biaryl Scaffolds through Direct C–H Bond Activation: Advances and Prospects. Top. Curr. Chem. Z 2020, 378, 23. [Google Scholar] [CrossRef]
- Ali, S.; Asad, M.H.H.B.; Khan, F.; Murtaza, G.; Rizvanov, A.A.; Iqbal, J.; Babak, B.; Hussain, I. Biological Evaluation of Newly Synthesized Biaryl Guanidine Derivatives to Arrest β-Secretase Enzymatic Activity Involved in Alzheimer’s Disease. BioMed Res. Int. 2020, 2020, e8934289. [Google Scholar] [CrossRef] [PubMed]
- Biosca, M.; Pàmies, O.; Diéguez, M. Ir–Biaryl Phosphite–Oxazoline Catalyst Libraries: A Breakthrough in the Asymmetric Hydrogenation of Challenging Olefins. Catal. Sci. Technol. 2020, 10, 613–624. [Google Scholar] [CrossRef]
- Fuentes-Rivera, J.J.; Zick, M.E.; Düfert, M.A.; Milner, P.J. Overcoming Halide Inhibition of Suzuki–Miyaura Couplings with Biaryl Monophosphine-Based Catalysts. Org. Process Res. Dev. 2019, 23, 1631–1637. [Google Scholar] [CrossRef]
- Yoganathan, S.; Alagaratnam, A.; Acharekar, N.; Kong, J. Ellagic Acid and Schisandrins: Natural Biaryl Polyphenols with Therapeutic Potential to Overcome Multidrug Resistance in Cancer. Cells 2021, 10, 458. [Google Scholar] [CrossRef] [PubMed]
- Flick, A.C.; Ding, H.X.; Leverett, C.A.; Kyne, R.E.; Liu, K.K.-C.; Fink, S.J.; O’Donnell, C.J. Synthetic Approaches to the New Drugs Approved During 2015. J. Med. Chem. 2017, 60, 6480–6515. [Google Scholar] [CrossRef]
- Zhu, S.S.; Swager, T.M. Design of Conducting Redox Polymers: A Polythiophene-Ru(Bipy)3;N⊕Hybrid Material. Adv. Mater. 1996, 8, 497–500. [Google Scholar] [CrossRef]
- Baudoin, O.; Cesario, M.; Guénard, D.; Guéritte, F. Application of the Palladium-Catalysed Borylation/Suzuki Coupling (BSC) Reaction to the Synthesis of Biologically Active Biaryl Lactams. J. Org. Chem. 2002, 67, 1199–1207. [Google Scholar] [CrossRef] [PubMed]
- Gong, X.; Wu, J.; Meng, Y.; Zhang, Y.; Ye, L.-W.; Zhu, C. Ligand-Free Palladium Catalyzed Ullmann Biaryl Synthesis: ’Household’ Reagents and Mild Reaction Conditions. Green Chem. 2019, 21, 995–999. [Google Scholar] [CrossRef]
- Ullmann, F.; Bielecki, J. Ueber Synthesen in der Biphenylreihe. Ber. Dtsch. Chem. Ges. 1901, 34, 2174–2185. [Google Scholar] [CrossRef]
- Amaya, T.; Jin, Y.; Tobisu, M. Recent Advances in Gomberg-Backmann Biaryl Synthesis. Tetrahedron Lett. 2019, 60, 151062. [Google Scholar] [CrossRef]
- Qiu, H.; Shuai, B.; Wang, Y.-Z.; Liu, D.; Chen, Y.-G.; Gao, P.-S.; Ma, H.-X.; Chen, S.; Mei, T.-S. Enantioselective Ni-Catalysed Electrochemical Synthesis of Biaryl Atropisomers. J. Am. Chem. Soc. 2020, 142, 9872–9878. [Google Scholar] [CrossRef] [PubMed]
- Liao, G.; Zhou, T.; Yao, Q.-J.; Shi, B.-F. Recent Advances in the Synthesis of Axially Chiral Biaryls via Transition Metal-Catalysed Asymmetric C–H Functionalization. Chem. Commun. 2019, 55, 8514–8523. [Google Scholar] [CrossRef] [PubMed]
- Kloss, F.; Neuwirth, T.; Haensch, V.G.; Hertweck, C. Metal-Free Synthesis of Pharmaceutically Important Biaryls by Photosplicing. Angew. Chem. Int. Ed. 2018, 57, 14476–14481. [Google Scholar] [CrossRef] [PubMed]
- Pollok, D.; Waldvogel, S.R. Electro-Organic Synthesis—A 21st Century Technique. Chem. Sci. 2020, 11, 12386–12400. [Google Scholar] [CrossRef]
- Pletcher, D. Organic Electrosynthesis—A Road to Greater Application. A Mini Review. Electrochem. Commun. 2018, 88, 1–4. [Google Scholar] [CrossRef]
- Utley, J. Trends in Organic Electrosynthesis. Chem. Soc. Rev. 1997, 26, 157–167. [Google Scholar] [CrossRef]
- Yuan, Y.; Lei, A. Is Electrosynthesis Always Green and Advantageous Compared to Traditional Methods? Nat. Commun. 2020, 11, 802. [Google Scholar] [CrossRef]
- Bringmann, G.; Gulder, T.; Gulder, T.A.M.; Breuning, M. Atroposelective Total Synthesis of Axially Chiral Biaryl Natural Products. Chem. Rev. 2011, 111, 563–639. [Google Scholar] [CrossRef]
- Hua, Z.; Vassar, V.C.; Choi, H.; Ojima, I. New Biphenol-Based, Fine-Tunable Monodentate Phosphoramidite Ligands for Catalytic Asymmetric Transformations. Proc. Natl. Acad. Sci. USA 2004, 101, 5411–5416. [Google Scholar] [CrossRef]
- Van Leeuwen, P.W.N.M.; Kamer, P.C.J.; Claver, C.; Pàmies, O.; Diéguez, M. Phosphite-Containing Ligands for Asymmetric Catalysis. Chem. Rev. 2011, 111, 2077–2118. [Google Scholar] [CrossRef]
- Cobb, S.J.; Ayres, Z.J.; Macpherson, J.V. Boron Doped Diamond: A Designer Electrode Material for the Twenty-First Century. Annu. Rev. Anal. Chem. 2018, 11, 463–484. [Google Scholar] [CrossRef] [PubMed]
- Macpherson, J.V. A Practical Guide to Using Boron Doped Diamond in Electrochemical Research. Phys. Chem. Chem. Phys. 2015, 17, 2935–2949. [Google Scholar] [CrossRef] [PubMed]
- Colomer, I.; Chamberlain, A.E.R.; Haughey, M.B.; Donohoe, T.J. Hexafluoroisopropanol as a Highly Versatile Solvent. Nat. Rev. Chem. 2017, 1, 1–12. [Google Scholar] [CrossRef]
- Kirste, A.; Schnakenburg, G.; Waldvogel, S.R. Anodic Coupling of Guaiacol Derivatives on Boron-Doped Diamond Electrodes. Org. Lett. 2011, 13, 3126–3129. [Google Scholar] [CrossRef]
- Hussain, I.; Singh, T. Synthesis of Biaryls through Aromatic C- Bond Activation: A Review of Recent Developments. Adv. Synth. Catal. 2014, 356, 1661–1696. [Google Scholar] [CrossRef]
- Kirste, A.; Hayashi, S.; Schnakenburg, G.; Malkowsky, I.M.; Stecker, F.; Fischer, A.; Fuchigami, T.; Waldvogel, S.R. Highly Selective Electrosynthesis of Biphenols on Graphite Electrodes in Fluorinated Media. Chem. Eur. J. 2011, 17, 14164–14169. [Google Scholar] [CrossRef] [PubMed]
- Degner, D. Organic electrosyntheses in industry. In Proceedings of the Electrochemistry III; Stekchan, E., Ed.; Springer: Berlin/Heidelberg, Germany, 1988; pp. 1–95. [Google Scholar]
- Baumgärtel, H.F. Beck: Elektroorganische Chemie, Verlag Chemie, Weinheim 1974, 391 Seiten, Preis: DM 88,—. Ber. Bunsenges. Phys. Chem. 1975, 79, 716. [Google Scholar] [CrossRef]
- Selt, M.; Mentizi, S.; Schollmeyer, D.; Franke, R.; Waldvogel, S.R. Selective and Scalable Dehydrogenative Electrochemical Synthesis of 3,3′,5,5′-Tetramethyl-2,2′-Biphenol. Synlett 2019, 30, 2062–2067. [Google Scholar] [CrossRef]
- Elsler, B.; Schollmeyer, D.; Dyballa, K.M.; Franke, R.; Waldvogel, S.R. Metal- and Reagent-Free Highly Selective Anodic Cross-Coupling Reaction of Phenols. Angew. Chem. Int. Ed. 2014, 53, 5210–5213. [Google Scholar] [CrossRef] [PubMed]
- Atobe, M.; Tateno, H.; Matsumura, Y. Applications of Flow Microreactors in Electrosynthetic Processes. Chem. Rev. 2018, 118, 4541–4572. [Google Scholar] [CrossRef]
- Wiebe, A.; Schollmeyer, D.; Dyballa, K.M.; Franke, R.; Waldvogel, S.R. Selective Synthesis of Partially Protected Nonsymmetric Biphenols by Reagent- and Metal-Free Anodic Cross-Coupling Reaction. Angew. Chem. Int. Ed. 2016, 55, 11801–11805. [Google Scholar] [CrossRef] [PubMed]
- Ruecker, C. The Triisopropylsilyl Group in Organic Chemistry: Just a Protective Group, or More? Chem. Rev. 1995, 95, 1009–1064. [Google Scholar] [CrossRef]
- Li, X.; Hewgley, J.B.; Mulrooney, C.A.; Yang, J.; Kozlowski, M.C. Enantioselective Oxidative Biaryl Coupling Reactions Catalysed by 1,5-Diazadecalin Metal Complexes: Efficient Formation of Chiral Functionalized BINOL Derivatives. J. Org. Chem. 2003, 68, 5500–5511. [Google Scholar] [CrossRef] [PubMed]
- Röckl, J.L.; Schollmeyer, D.; Franke, R.; Waldvogel, S.R. Dehydrogenative Anodic C−C Coupling of Phenols Bearing Electron-Withdrawing Groups. Angew. Chem. Int. Ed. 2020, 59, 315–319. [Google Scholar] [CrossRef]
- Röckl, J.L.; Imada, Y.; Chiba, K.; Franke, R.; Waldvogel, S.R. Dehydrogenative Anodic Cyanation Reaction of Phenols in Benzylic Positions. ChemElectroChem 2019, 6, 4184–4187. [Google Scholar] [CrossRef]
- Imada, Y.; Röckl, J.L.; Wiebe, A.; Gieshoff, T.; Schollmeyer, D.; Chiba, K.; Franke, R.; Waldvogel, S.R. Metal- and Reagent-Free Dehydrogenative Formal Benzyl–Aryl Cross-Coupling by Anodic Activation in 1,1,1,3,3,3-Hexafluoropropan-2-Ol. Angew. Chem. Int. Ed. 2018, 57, 12136–12140. [Google Scholar] [CrossRef]
- Terada, M.; Gupta, Y.; Kikuchi, J. Bis-Phosphoric Acid Derived from BINOL Dimer as a Chiral Brønsted Acid Catalyst for Enantioselective Transformations. Chem. Lett. 2019, 48, 260–263. [Google Scholar] [CrossRef]
- Sasai, H.; Suzuki, T.; Arai, S.; Arai, T.; Shibasaki, M. Basic Character of Rare Earth Metal Alkoxides. Utilisation in Catalytic Carbon-Carbon Bond-Forming Reactions and Catalytic Asymmetric Nitroaldol Reactions. J. Am. Chem. Soc. 1992, 114, 4418–4420. [Google Scholar] [CrossRef]
- Wu, Y.; Yang, L.; Wu, B.; Li, J.; Liu, B.; Ke, G.; Dong, F.; Zhou, Y.; He, H. Accurate Understanding the Catalytic Role of MnO2 in the Oxidative-Coupling of 2-Naphthols into 1,1′-Bi-2-Naphthols. Catal. Lett. 2021, 151, 901–908. [Google Scholar] [CrossRef]
- Hayashi, H.; Ueno, T.; Kim, C.; Uchida, T. Ruthenium-Catalyzed Cross-Selective Asymmetric Oxidative Coupling of Arenols. Org. Lett. 2020, 22, 1469–1474. [Google Scholar] [CrossRef] [PubMed]
- Shirakawa, S.; Maruoka, K. A New Strategy for Organocatalyzed Asymmetric Synthesis of BINOL Derivatives. Chem 2017, 2, 329–331. [Google Scholar] [CrossRef][Green Version]
- Takizawa, S.; Katayama, T.; Kameyama, C.; Onitsuka, K.; Suzuki, T.; Yanagida, T.; Kawai, T.; Sasai, H. Chiral Dinuclear Vanadium(V) Catalysts for Oxidative Coupling of 2-Naphthols. Chem. Commun. 2008, 15, 1810–1812. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.; Liu, Q.; Gong, L.; Cui, X.; Mi, A.; Jiang, Y. Novel Achiral Biphenol-Derived Diastereomeric Oxovanadium(IV) Complexes for Highly Enantioselective Oxidative Coupling of 2-Naphthols. Angew. Chem. Int. Ed. 2002, 41, 4532–4535. [Google Scholar] [CrossRef]
- Anjalin, M.; Kanagathara, N.; Baby Suganthi, A.R. A Brief Review on Aniline and Its Derivatives. Mater. Today Proc. 2020, 33, 4751–4755. [Google Scholar] [CrossRef]
- Amini, B.; Lowenkron, S. Aniline and its derivatives. In Kirk-Othmer Encyclopedia of Chemical Technology; American Cancer Society: New York, NY, USA, 2003; ISBN 978-0-471-23896-6. [Google Scholar]
- Nakatsuka, H.; Yamamura, T.; Shuto, Y.; Tanaka, S.; Yoshimura, M.; Kitamura, M. Mechanism of Asymmetric Hydrogenation of Aromatic Ketones Catalysed by a Combined System of Ru(π-CH2C(CH3)CH2)2(Cod) and the Chiral Sp2N/Sp3NH Hybrid Linear N4 Ligand Ph-BINAN-H-Py. J. Am. Chem. Soc. 2015, 137, 8138–8149. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.-H.; Kim, H.-K.; Lee, G.H.; Kang, D.-S.; Park, J.-A.; Kim, K.M.; Chang, Y.; Kim, T.-J. Gd Complexes of Macrocyclic Diethylenetriaminepentaacetic Acid (DTPA) Biphenyl-2,2′-Bisamides as Strong Blood-Pool Magnetic Resonance Imaging Contrast Agents. J. Med. Chem. 2011, 54, 5385–5394. [Google Scholar] [CrossRef] [PubMed]
- Letheby, H. XXIX.—On the Production of a Blue Substance by the Electrolysis of Sulphate of Aniline. J. Chem. Soc. 1862, 15, 161–163. [Google Scholar] [CrossRef]
- Long, C.-Y.; Ni, S.-F.; Su, M.-H.; Wang, X.-Q.; Tan, W. Highly Chemoselective Access to 2,2′-Diaminobiaryls via Ni-Catalyzed Protecting-Group-Free Coupling of 2-Haloanilines. ACS Catal. 2020, 10, 13641–13649. [Google Scholar] [CrossRef]
- Hiroto, S. Synthesis of π-Functional Molecules through Oxidation of Aromatic Amines. Chem. Asian J. 2019, 14, 2514–2523. [Google Scholar] [CrossRef]
- Schulz, L.; Enders, M.; Elsler, B.; Schollmeyer, D.; Dyballa, K.M.; Franke, R.; Waldvogel, S.R. Reagent- and Metal-Free Anodic C−C Cross-Coupling of Aniline Derivatives. Angew. Chem. Int. Ed. 2017, 56, 4877–4881. [Google Scholar] [CrossRef] [PubMed]
- Souto, J.A.; Martínez, C.; Velilla, I.; Muñiz, K. Defined Hypervalent Iodine(III) Reagents Incorporating Transferable Nitrogen Groups: Nucleophilic Amination through Electrophilic Activation. Angew. Chem. Int. Ed. 2013, 52, 1324–1328. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Kubo, H.; Itani, I.; Morimoto, K.; Dohi, T.; Kita, Y. Organocatalytic C–H/C–H′ Cross-Biaryl Coupling: C-Selective Arylation of Sulfonanilides with Aromatic Hydrocarbons. J. Am. Chem. Soc. 2013, 135, 14078–14081. [Google Scholar] [CrossRef]
- Schulz, L.; Franke, R.; Waldvogel, S.R. Direct Anodic Dehydrogenative Cross- and Homo-Coupling of Formanilides. ChemElectroChem 2018, 5, 2069–2072. [Google Scholar] [CrossRef]
- Kohlmeyer, C.; Schäfer, A.; Huy, P.H.; Hilt, G. Formamide-Catalyzed Nucleophilic Substitutions: Mechanistic Insight and Rationalisation of Catalytic Activity. ACS Catal. 2020, 10, 11567–11577. [Google Scholar] [CrossRef]
- Jin, Z.; Li, Y.-J.; Ma, Y.-Q.; Qiu, L.-L.; Fang, J.-X. Biphenyl-Based Diaminophosphine Oxides as Air-Stable Preligands for the Nickel-Catalyzed Kumada–Tamao–Corriu Coupling of Deactivated Aryl Chlorides, Fluorides, and Tosylates. Chem. Eur. J. 2012, 18, 446–450. [Google Scholar] [CrossRef]
- Martinez, J.; Laur, J. Active Esters of Formic Acid as Useful Formylating Agents: Improvements in the Synthesis of Formyl-Amino Acid Esters, N-α-Formyl-Met-Leu-Phe-OH, and Formyl-Met-Lys-Pro-Arg, a Phagocytosis Stimulating Peptide. Synthesis 1982, 1982, 979–981. [Google Scholar] [CrossRef]
- Liu, X.; Cai, T.-C.; Guo, D.; Wang, B.-B.; Ying, S.; Wang, H.; Tang, S.; Shen, Q.; Gui, Q.-W. Electrochemical Synthesis of Symmetrical Benzidines through Dehydrogenative Cross-Coupling Reaction. Tetrahedron Lett. 2021, 70, 153021. [Google Scholar] [CrossRef]
- Ibrahim, D.H.; Dunet, J.; Robert, F.; Landais, Y. Oxidation of 1-Arylcyclohexa-2,5-Dienes and Subsequent Double Michael Addition. A Rapid Access to the Büchi Ketone and the Pentacyclic Core of Aspidosperma Alkaloids. Heterocycles 2018, 97, 459–477. [Google Scholar] [CrossRef]
- Koguchi, Y.; Kohno, J.; Nishio, M.; Takahashi, K.; Okuda, T.; Ohnuki, T.; Komatsubara, S. TMC-95A, B, C, and D, Novel Proteasome Inhibitors Produced by Apiospora Montagnei Sacc. TC 1093 Taxonomy, Production, Isolation, and Biological Activities. J. Antibiot. 2000, 53, 105–109. [Google Scholar] [CrossRef]
- Wencel-Delord, J.; Panossian, A.; Leroux, F.R.; Colobert, F. Recent Advances and New Concepts for the Synthesis of Axially Stereoenriched Biaryls. Chem. Soc. Rev. 2015, 44, 3418–3430. [Google Scholar] [CrossRef]
- Uozumi, Y.; Hayashi, T. Catalytic Asymmetric Synthesis of Optically Active 2-Alkanols via Hydrosilylation of 1-Alkenes with a Chiral Monophosphine-Palladium Catalyst. J. Am. Chem. Soc. 1991, 113, 9887–9888. [Google Scholar] [CrossRef]
- Smrčina, M.; Lorenc, M.; Hanuš, V.; Kočovský, P. A Facile Synthesis of 2-Amino-2′-Hydroxy-1,1′-Binaphthyl and 2,2′-Diamino-1,1′-Binaphthyl by Oxidative Coupling Using Copper(II) Chloride. Synlett 1991, 1991, 231–232. [Google Scholar] [CrossRef]
- Gavrilov, K.N.; Shiryaev, A.A.; Zheglov, S.V.; Bochelyuk, M.S.; Chuchelkin, I.V.; Tafeenko, V.A.; Chernyshev, V.V.; Zamilatskov, I.A.; Mikhel, I.S. NOBIN-Based Chiral Phosphite-Type Ligands and Their Application in Asymmetric Catalysis. Tetrahedron Lett. 2015, 56, 4756–4761. [Google Scholar] [CrossRef]
- Zhang, D.; Wang, Q. Palladium Catalysed Asymmetric Suzuki–Miyaura Coupling Reactions to Axially Chiral Biaryl Compounds: Chiral Ligands and Recent Advances. Coord. Chem. Rev. 2015, 286, 1–16. [Google Scholar] [CrossRef]
- Wang, G.; Shi, Q.; Hu, W.; Chen, T.; Guo, Y.; Hu, Z.; Gong, M.; Guo, J.; Wei, D.; Fu, Z.; et al. Organocatalytic Asymmetric N-Sulfonyl Amide C-N Bond Activation to Access Axially Chiral Biaryl Amino Acids. Nat. Commun. 2020, 11, 946. [Google Scholar] [CrossRef] [PubMed]
- Benmahdjoub, S.; Ibrahim, N.; Benmerad, B.; Alami, M.; Messaoudi, S. One-Pot Assembly of Unsymmetrical Biaryl Thioglycosides through Chemoselective Palladium-Catalyzed Three-Component Tandem Reaction. Org. Lett. 2018, 20, 4067–4071. [Google Scholar] [CrossRef]
- Yamamura, S.; Nishiyama, S. Anodic Oxidation of Phenols Towards the Synthesis of Bioactive Natural Products. Synlett 2002, 2002, 533–543. [Google Scholar] [CrossRef]
- Kirste, A.; Schnakenburg, G.; Stecker, F.; Fischer, A.; Waldvogel, S.R. Anodic Phenol–Arene Cross-Coupling Reaction on Boron-Doped Diamond Electrodes. Angew. Chem. Int. Ed. 2010, 49, 971–975. [Google Scholar] [CrossRef] [PubMed]
- Kirste, A.; Elsler, B.; Schnakenburg, G.; Waldvogel, S.R. Efficient Anodic and Direct Phenol-Arene C,C Cross-Coupling: The Benign Role of Water or Methanol. J. Am. Chem. Soc. 2012, 134, 3571–3576. [Google Scholar] [CrossRef]
- Kashiwagi, T.; Elsler, B.; Waldvogel, S.R.; Fuchigami, T.; Atobe, M. Reaction Condition Screening by Using Electrochemical Microreactor: Application to Anodic Phenol-Arene C,C Cross-Coupling Reaction in High Acceptor Number Media. J. Electrochem. Soc. 2013, 160, G3058. [Google Scholar] [CrossRef]
- Simcik, M.F. Aquatic Processes and Systems in Perspective. Global Transport and Fate of Perfluorochemicals. J. Environ. Monit. 2005, 7, 759–763. [Google Scholar] [CrossRef]
- Mayer, U.; Gutmann, V.; Gerger, W. The Acceptor Number—A Quantitative Empirical Parameter for the Electrophilic Properties of Solvents. Mon. Chem. 1975, 106, 1235–1257. [Google Scholar] [CrossRef]
- Cozens, F.L.; Kanagasabapathy, V.M.; McClelland, R.A.; Steenken, S. Lifetimes and UV-Visible Absorption Spectra of Benzyl, Phenethyl, and Cumyl Carbocations and Corresponding Vinyl Cations. A Laser Flash Photolysis Study. Can. J. Chem. 1999, 77, 2069–2082. [Google Scholar] [CrossRef]
- Dahms, B.; Franke, R.; Waldvogel, S.R. Metal- and Reagent-Free Anodic Dehydrogenative Cross-Coupling of Naphthylamines with Phenols. ChemElectroChem 2018, 5, 1249–1252. [Google Scholar] [CrossRef]
- Yasukouchi, K.; Taniguchi, I.; Yamaguchi, H.; Yokoyama, M.; Murasaki, M. Anodic Cyclization of Di-2-Naphthylamine. Chem. Lett. 1979, 8, 1167–1170. [Google Scholar] [CrossRef]
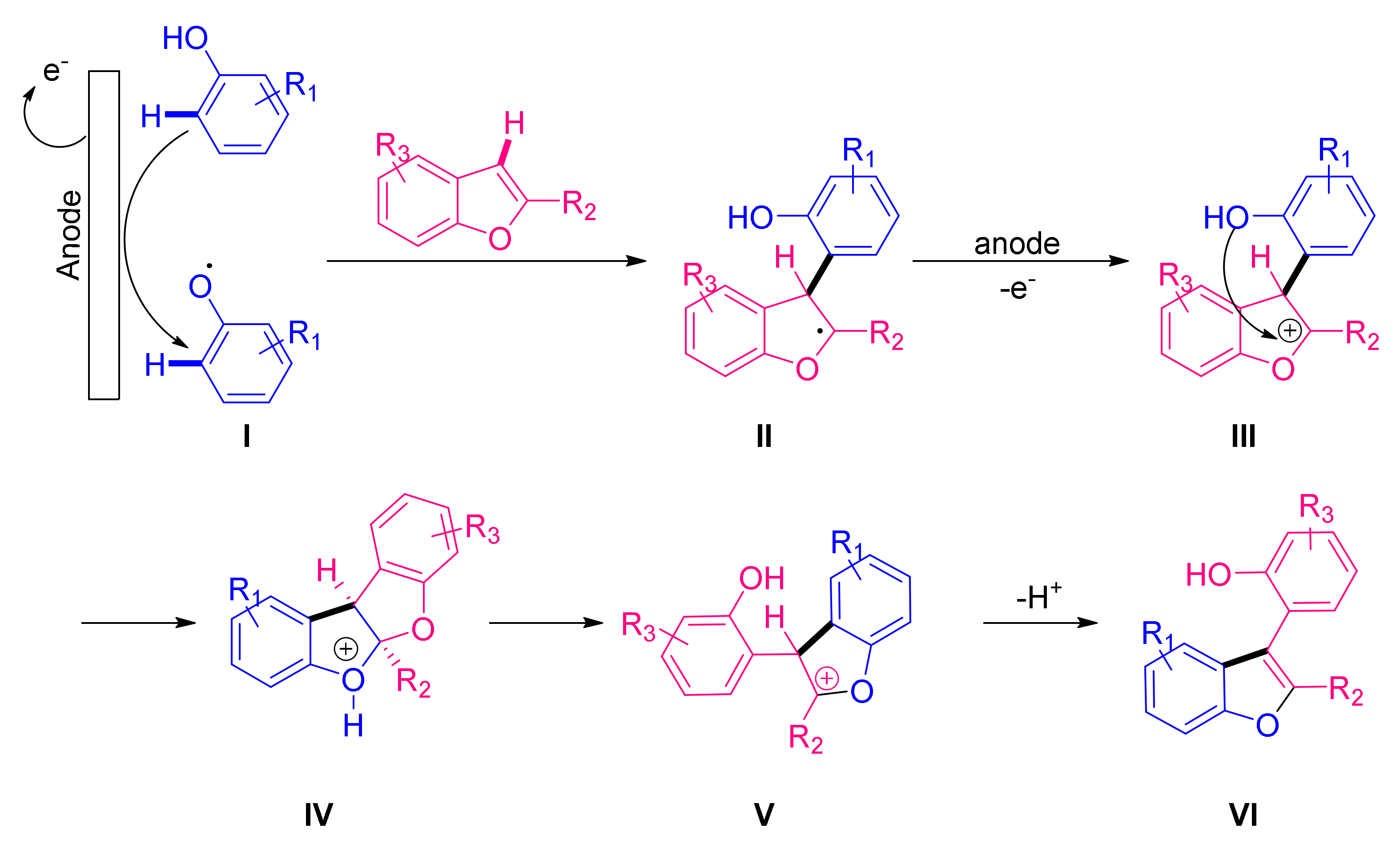
- Lips, S.; Frontana-Uribe, B.A.; Dörr, M.; Schollmeyer, D.; Franke, R.; Waldvogel, S.R. Metal- and Reagent-Free Anodic C−C Cross-Coupling of Phenols with Benzofurans Leading to a Furan Metathesis. Chem. Eur. J. 2018, 24, 6057–6061. [Google Scholar] [CrossRef]
- Miao, Y.; Hu, Y.; Yang, J.; Liu, T.; Sun, J.; Wang, X. Natural Source, Bioactivity and Synthesis of Benzofuran Derivatives. RSC Adv. 2019, 9, 27510–27540. [Google Scholar] [CrossRef]
- Khanam, H. Shamsuzzaman Bioactive Benzofuran Derivatives: A Review. Eur. J. Med. Chem. 2015, 97, 483–504. [Google Scholar] [CrossRef]
- Khodarahmi, G.; Asadi, P.; Hassanzadeh, F.; Khodarahmi, E. Benzofuran as a Promising Scaffold for the Synthesis of Antimicrobial and Antibreast Cancer Agents: A Review. J. Res. Med. Sci. 2015, 20, 1094–1104. [Google Scholar] [CrossRef]
- Wang, Q.-Q.; Jiang, Y.-Y.; Zeng, C.-C.; Sun, B.-G. Electrocatalytic Synthesis of Non-Symmetric Biphenols Mediated by Tri(p-Bromophenyl)Amine: Selective Oxidative Cross-Coupling of Different Phenols and Naphthols. Chin. J. Chem. 2019, 37, 352–358. [Google Scholar] [CrossRef]
- Luo, M.-J.; Li, Y.; Ouyang, X.-H.; Li, J.-H.; He, D.-L. Electrochemical Dehydrogenative Cross-Coupling of Two Anilines: Facile Synthesis of Unsymmetrical Biaryls. Chem. Commun. 2020, 56, 2707–2710. [Google Scholar] [CrossRef]
- Vettorazzi, N.; Silber, J.J.; Sereno, L. Anodic Oxidation of 1-Naphthylamine in Acetonitrile. J. Electroanal. Chem. Interfacial Electrochem. 1981, 125, 459–475. [Google Scholar] [CrossRef]
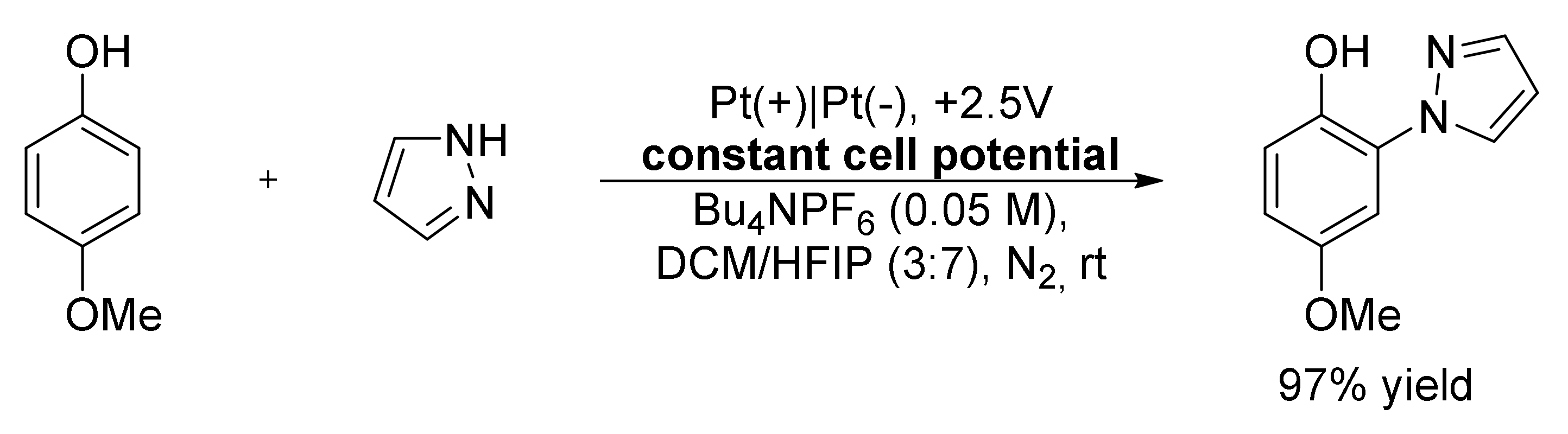
- Feng, P.; Ma, G.; Chen, X.; Wu, X.; Lin, L.; Liu, P.; Chen, T. Electrooxidative and Regioselective C−H Azolation of Phenol and Aniline Derivatives. Angew. Chem. Int. Ed. 2019, 58, 8400–8404. [Google Scholar] [CrossRef] [PubMed]
- Jabir, N.R.; Firoz, C.K.; Bhushan, A.; Tabrez, S.; Kamal, M.A. The Use of Azoles Containing Natural Products in Cancer Prevention and Treatment: An Overview. Anti Cancer Agents Med. Chem. 2018, 18, 6–14. [Google Scholar] [CrossRef]
- Fontana, G. Current Bioactive Azole-Containing Natural Products. Curr. Bioact. Compd. 2010, 6, 284–308. [Google Scholar] [CrossRef]
- Gao, H.; Shreeve, J.M. Azole-Based Energetic Salts. Chem. Rev. 2011, 111, 7377–7436. [Google Scholar] [CrossRef]
- Lips, S.; Schollmeyer, D.; Franke, R.; Waldvogel, S.R. Regioselective Metal- and Reagent-Free Arylation of Benzothiophenes by Dehydrogenative Electrosynthesis. Angew. Chem. Int. Ed. 2018, 57, 13325–13329. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Zhou, Y.; Zhou, Y.; Chiang, C.-W.; Lei, A. Electro-Oxidative C(Sp3)–H Amination of Azoles via Intermolecular Oxidative C(Sp3)–H/N–H Cross-Coupling. ACS Catal. 2017, 7, 8320–8323. [Google Scholar] [CrossRef]
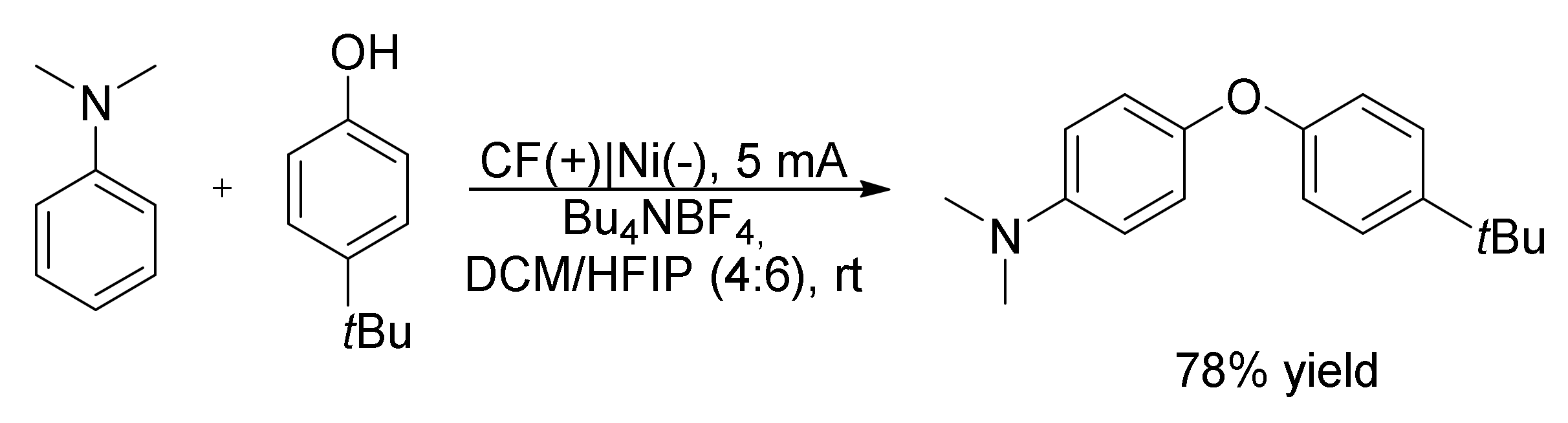
- Tang, H.; Smolders, S.; Li, Y.; Vos, D.D.; Vercammen, J. Electro-Oxidative C(Sp2)–H/O–H Cross-Dehydrogenative Coupling of Phenols and Tertiary Anilines for Diaryl Ether Formation. Catal. Sci. Technol. 2021, 11, 3925–3930. [Google Scholar] [CrossRef]
- Bedos-Belval, F.; Rouch, A.; Vanucci-Bacqué, C.; Baltas, M. Diaryl Ether Derivatives as Anticancer Agents—A Review. Med. Chem. Commun. 2012, 3, 1356–1372. [Google Scholar] [CrossRef]
- Lee, C.-C.; Leung, M.; Lee, P.-Y.; Chiu, T.-L.; Lee, J.-H.; Liu, C.; Chou, P.-T. Synthesis and Properties of Oxygen-Linked N-Phenylcarbazole Dendrimers. Macromolecules 2012, 45, 751–765. [Google Scholar] [CrossRef]
- Roughley, S.D.; Jordan, A.M. The Medicinal Chemist’s Toolbox: An Analysis of Reactions Used in the Pursuit of Drug Candidates. J. Med. Chem. 2011, 54, 3451–3479. [Google Scholar] [CrossRef]
- Osa, T.; Kashiwagi, Y.; Yanagisawa, Y.; Bobbitt, J.M. Enantioselective, electrocatalytic oxidative coupling of naphthol, naphthyl ether and phenanthrol on a TEMPO-modified graphite felt electrode in the presence of (–)-sparteine. J. Chem. Soc. Chem. Commun. 1994, 21, 2535–2537. [Google Scholar] [CrossRef]
- Pletcher, D. Electrolysis Cells for Laboratory Organic Synthesis. Curr. Opin. Electrochem. 2020, 24, 1–5. [Google Scholar] [CrossRef]
- Noël, T.; Cao, Y.; Laudadio, G. The Fundamentals Behind the Use of Flow Reactors in Electrochemistry. Acc. Chem. Res. 2019, 52, 2858–2869. [Google Scholar] [CrossRef] [PubMed]
- Elsherbini, M.; Wirth, T. Electroorganic Synthesis under Flow Conditions. Acc. Chem. Res. 2019, 52, 3287–3296. [Google Scholar] [CrossRef] [PubMed]
- Marino, D.D.; Stöckmann, D.; Kriescher, S.; Stiefel, S.; Wessling, M. Electrochemical Depolymerisation of Lignin in a Deep Eutectic Solvent. Green Chem. 2016, 18, 6021–6028. [Google Scholar] [CrossRef]
- Nkuku, C.A.; LeSuer, R.J. Electrochemistry in Deep Eutectic Solvents. J. Phys. Chem. B 2007, 111, 13271–13277. [Google Scholar] [CrossRef]






























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
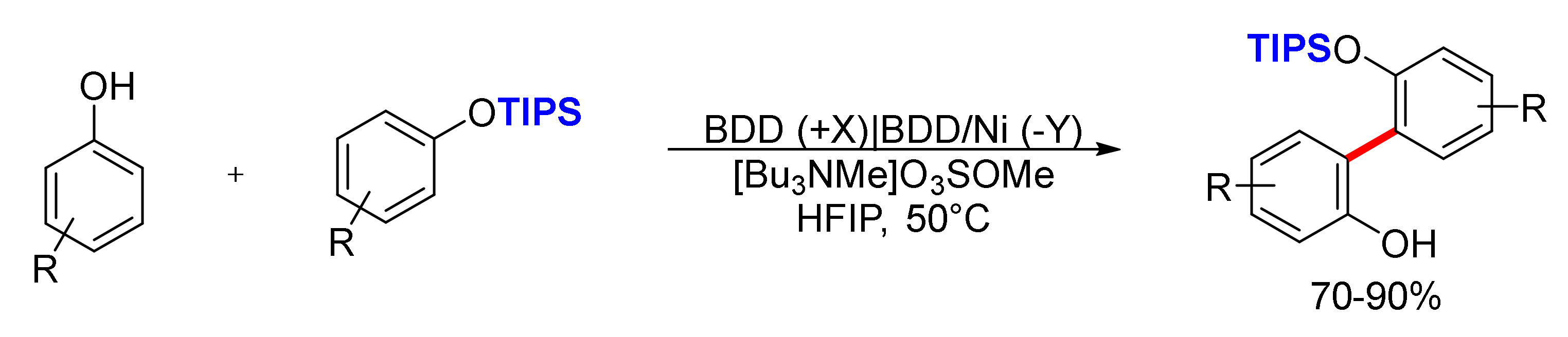{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
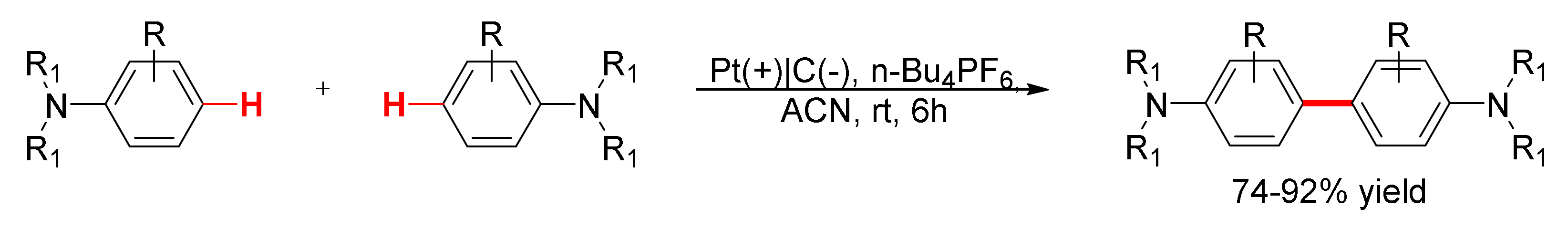{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Solvent | Acceptor Number |
|---|---|
| HFIP | 88 |
| AcOH | 52.9 |
| HCOOH | 83.6 |
| TFA | 105 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Medici, F.; Resta, S.; Puglisi, A.; Rossi, S.; Raimondi, L.; Benaglia, M. Electrochemical Organic Synthesis of Electron-Rich Biaryl Scaffolds: An Update. Molecules 2021, 26, 6968. https://doi.org/10.3390/molecules26226968
Medici F, Resta S, Puglisi A, Rossi S, Raimondi L, Benaglia M. Electrochemical Organic Synthesis of Electron-Rich Biaryl Scaffolds: An Update. Molecules. 2021; 26(22):6968. https://doi.org/10.3390/molecules26226968
Chicago/Turabian StyleMedici, Fabrizio, Simonetta Resta, Alessandra Puglisi, Sergio Rossi, Laura Raimondi, and Maurizio Benaglia. 2021. "Electrochemical Organic Synthesis of Electron-Rich Biaryl Scaffolds: An Update" Molecules 26, no. 22: 6968. https://doi.org/10.3390/molecules26226968
APA StyleMedici, F., Resta, S., Puglisi, A., Rossi, S., Raimondi, L., & Benaglia, M. (2021). Electrochemical Organic Synthesis of Electron-Rich Biaryl Scaffolds: An Update. Molecules, 26(22), 6968. https://doi.org/10.3390/molecules26226968