
Artificial Intelligence for Autonomous Molecular Design: A Perspective


Abstract
:
1. Introduction
2. Results and Highlights
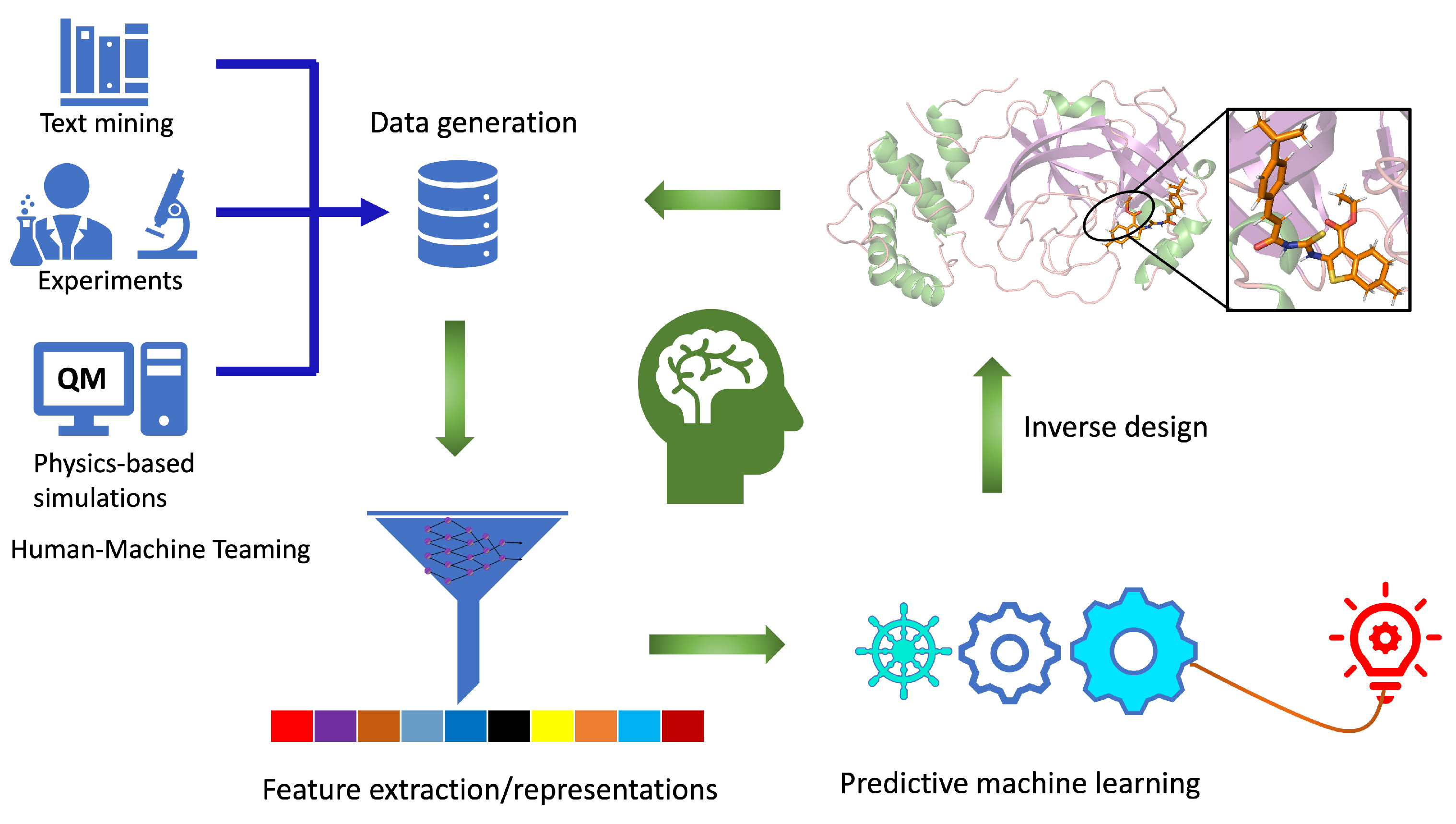
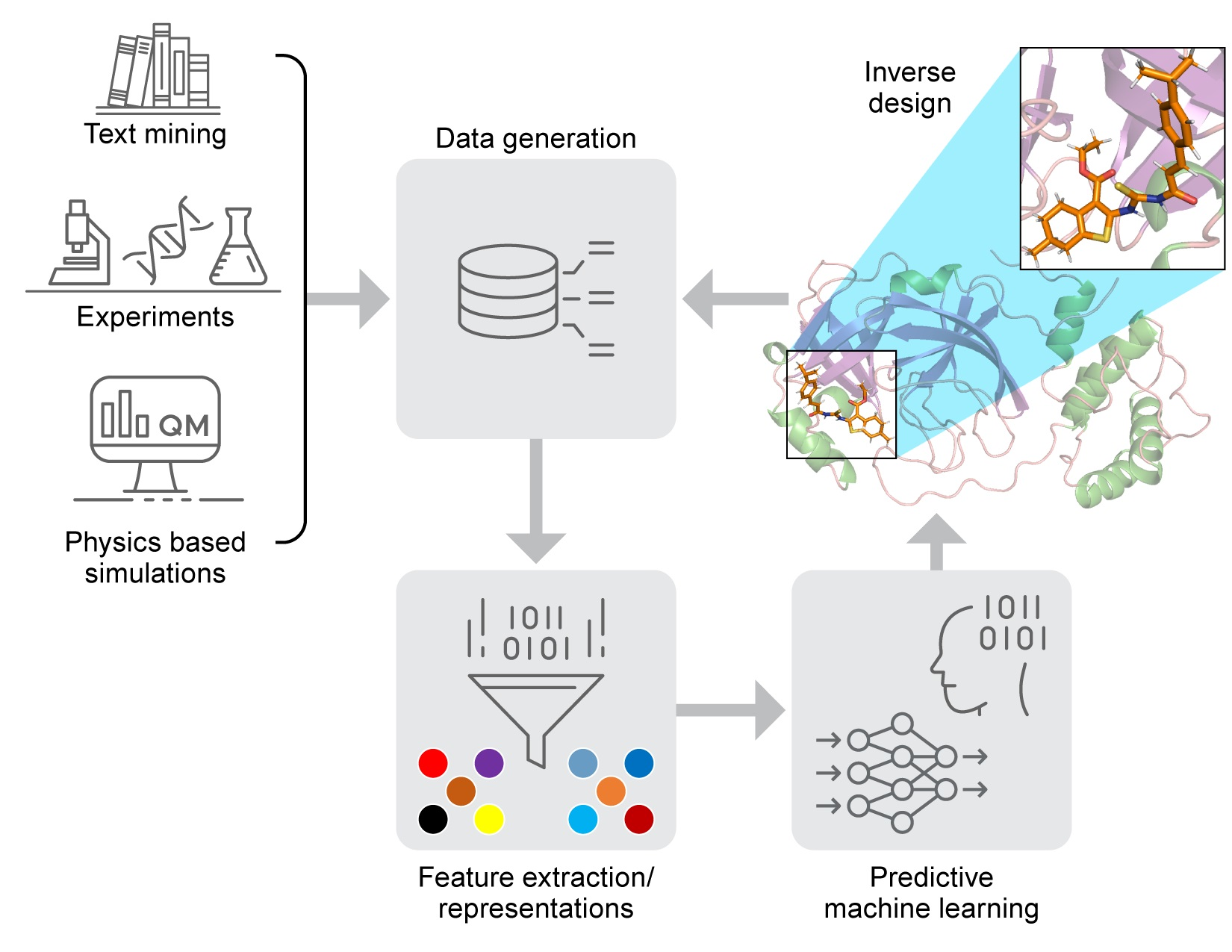
2.1. Components of Computational Autonomous Molecular Design Workflow
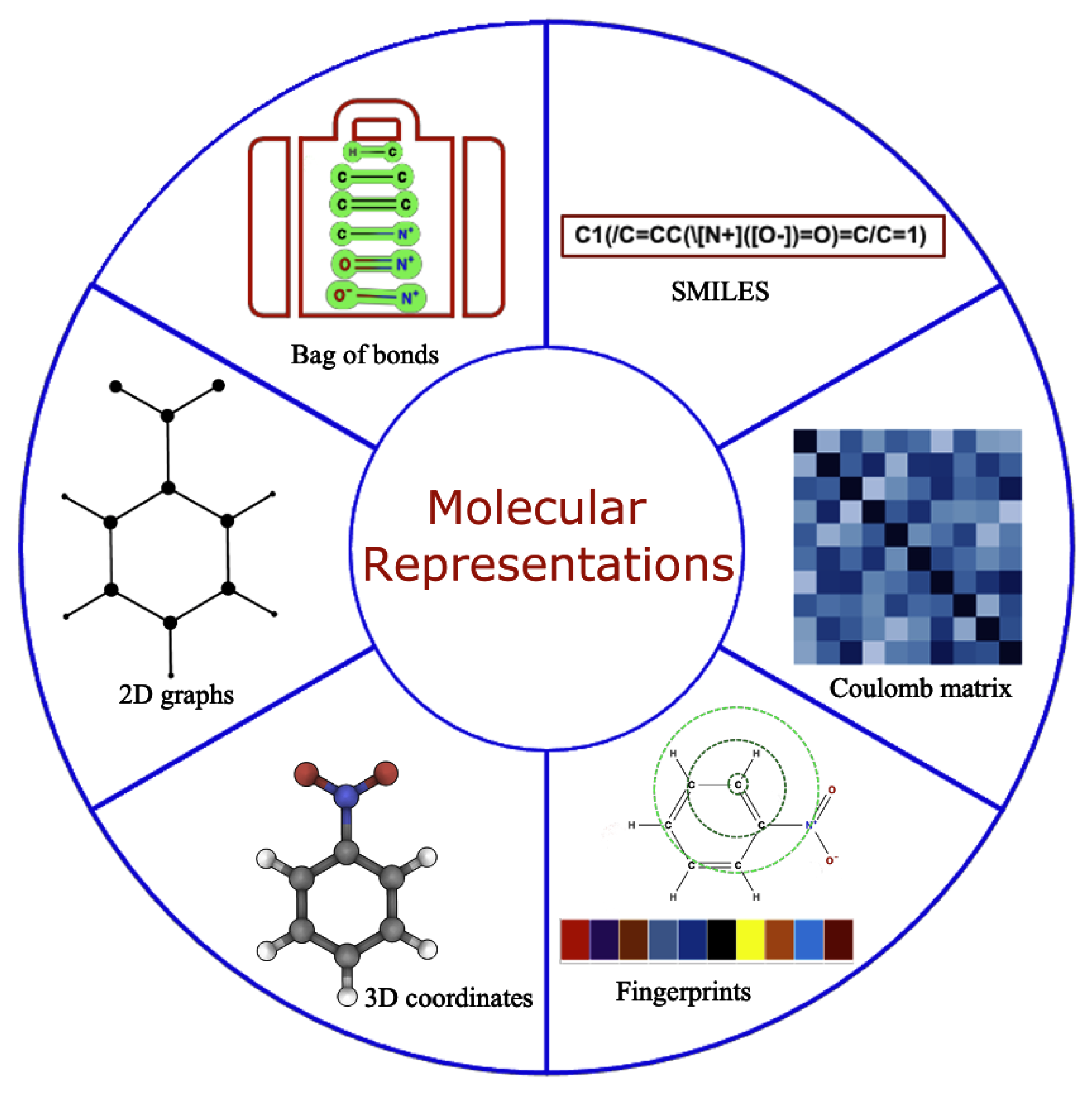
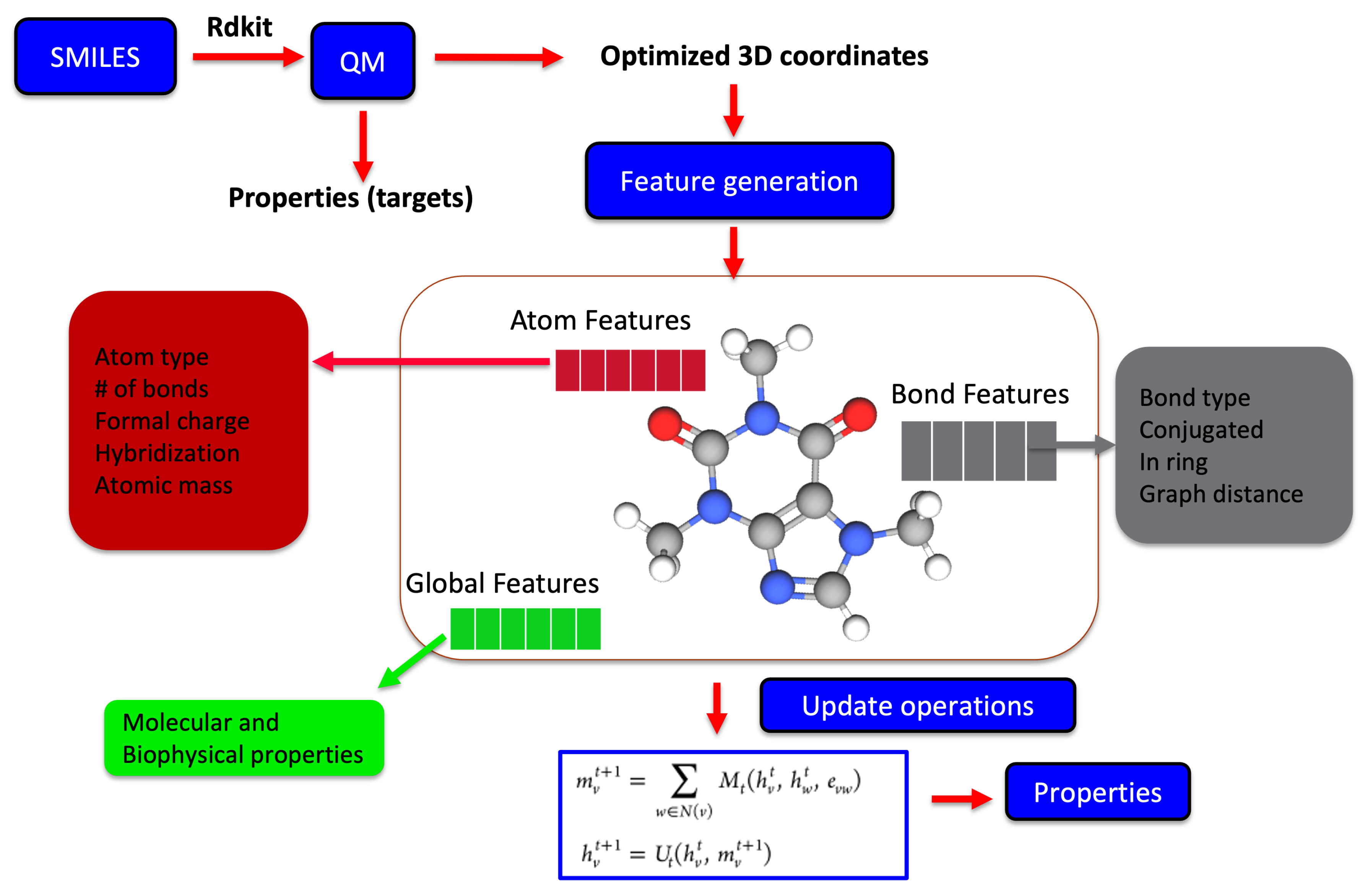
2.2. Data Generation and Molecular Representation
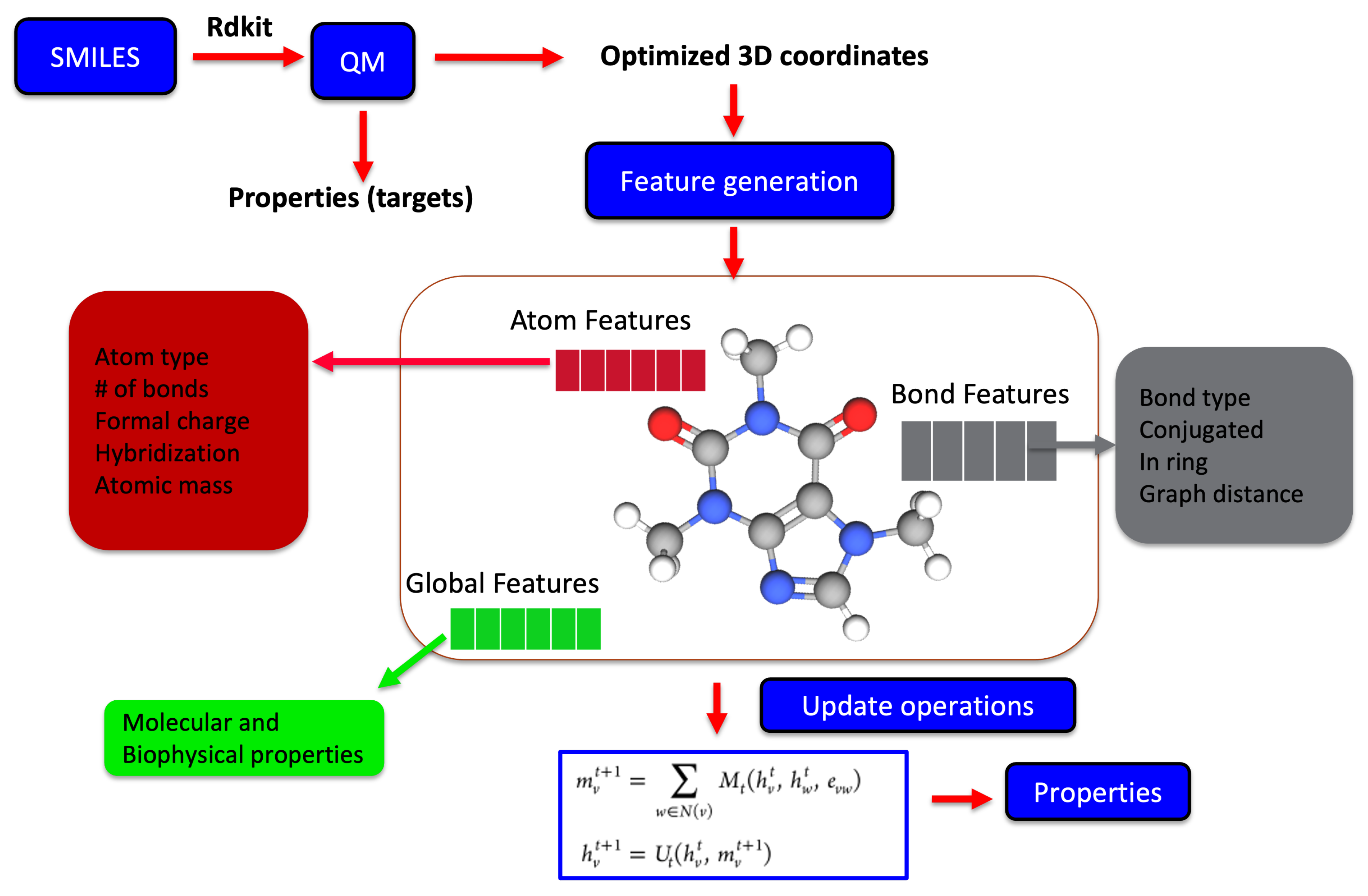
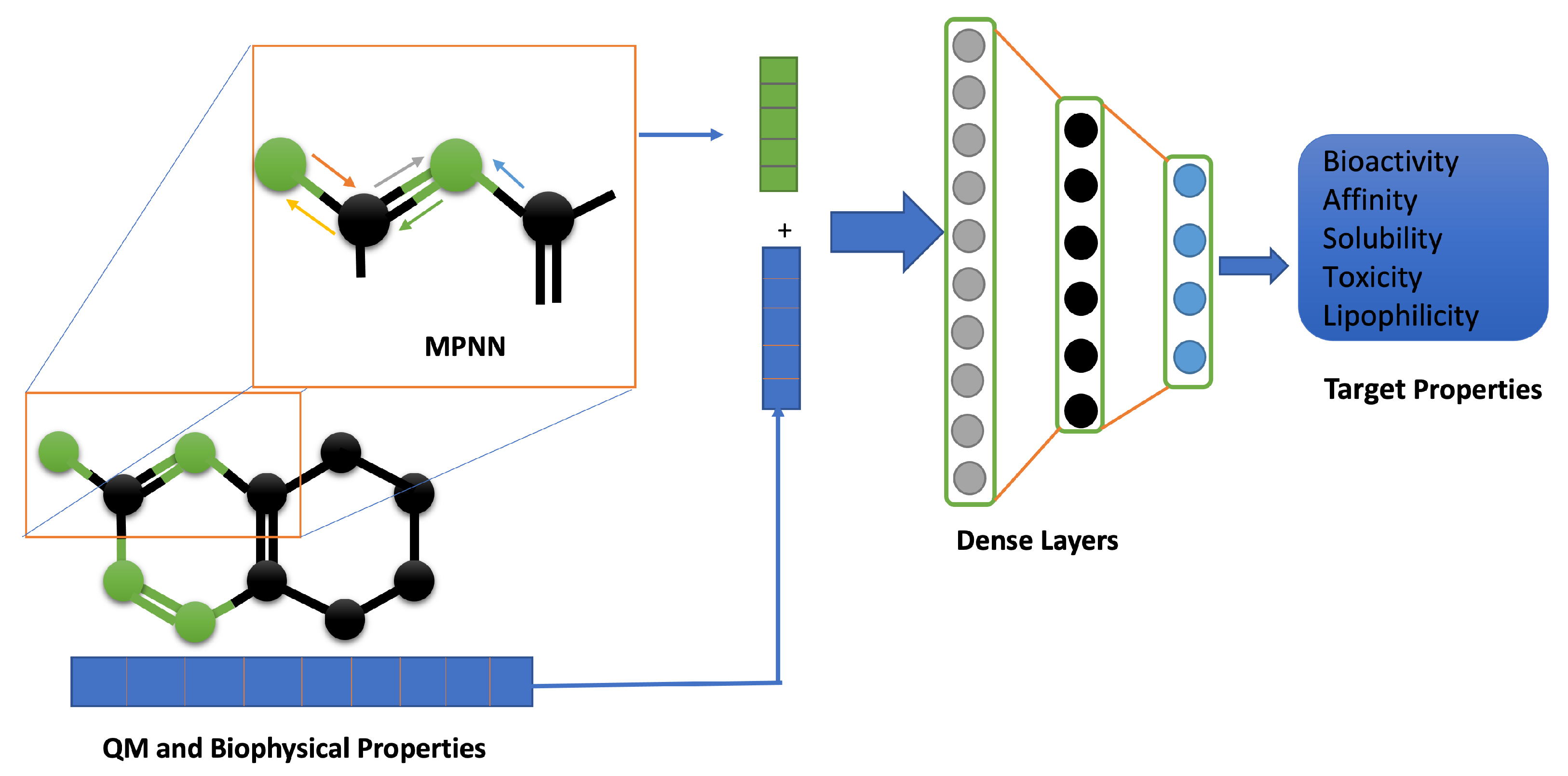
2.3. Molecular Representation in Automated Pipelines
2.4. Physics-Informed Machine Learning
2.5. Inverse Molecular Design
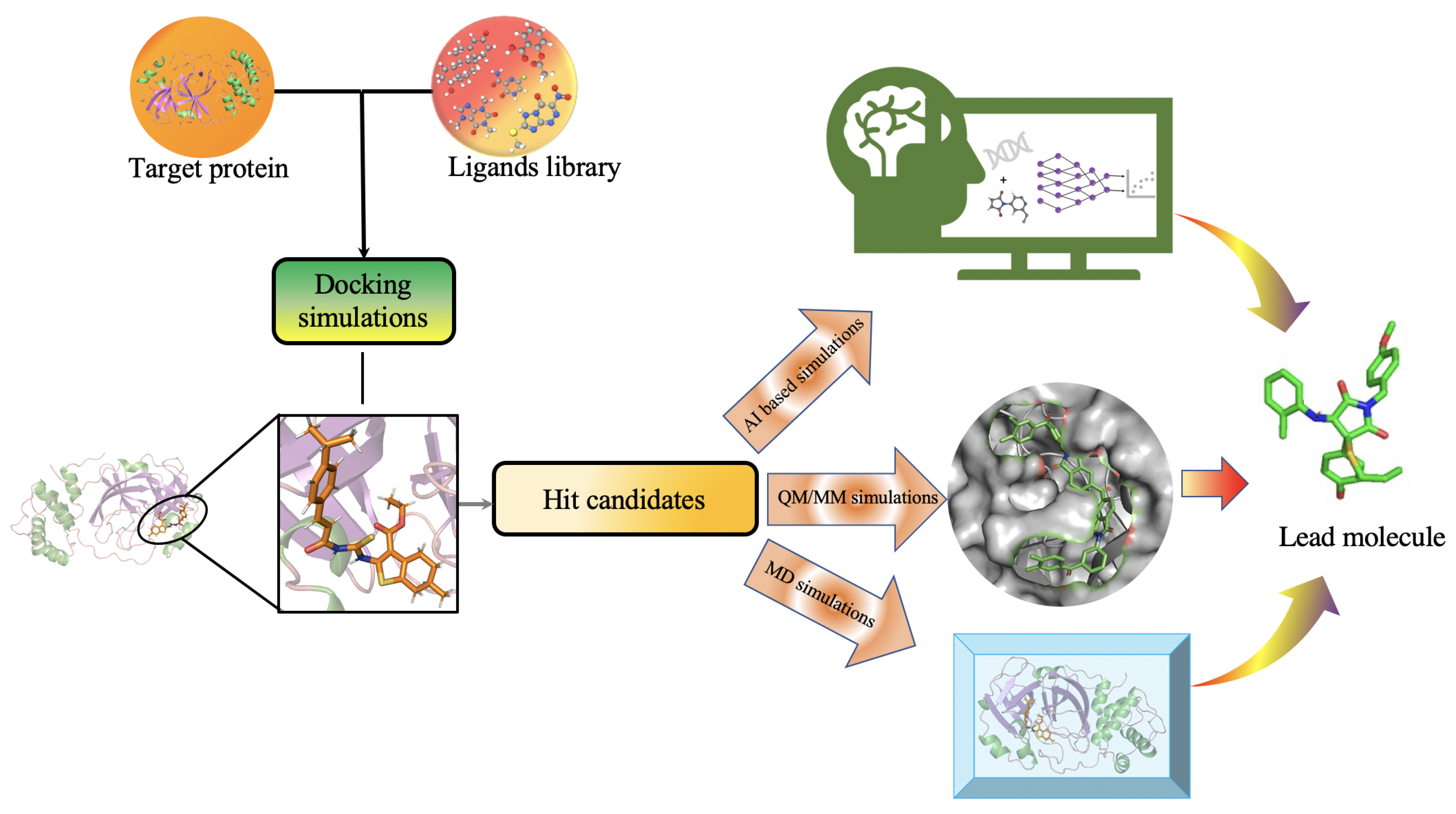
2.6. Protein Target Specific Molecular Design
3. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Zhavoronkov, A.; Ivanenkov, Y.A.; Aliper, A.; Veselov, M.S.; Aladinskiy, V.A.; Aladinskaya, A.V.; Terentiev, V.A.; Polykovskiy, D.A.; Kuznetsov, M.D.; Asadulaev, A.; et al. Deep learning enables rapid identification of potent DDR1 kinase inhibitors. Nat. Biotechnol. 2019, 37, 1038–1040. [Google Scholar] [CrossRef] [PubMed]
- Kackar, R.N. Off-Line Quality Control, Parameter Design, and the Taguchi Method. J. Qual. Technol. 1985, 17, 176–188. [Google Scholar] [CrossRef]
- Kim, E.; Huang, K.; Tomala, A.; Matthews, S.; Strubell, E.; Saunders, A.; McCallum, A.; Olivetti, E. Machine-learned and codified synthesis parameters of oxide materials. Sci. Data 2017, 4, 1–9. [Google Scholar] [CrossRef]
- Leelananda, S.P.; Lindert, S. Computational methods in drug discovery. Beilstein J. Org. Chem. 2016, 12, 2694–2718. [Google Scholar] [CrossRef] [Green Version]
- DiMasi, J.A.; Grabowski, H.G.; Hansen, R.W. Innovation in the pharmaceutical industry: New estimates of R&D costs. J. Health Econ. 2016, 47, 20–33. [Google Scholar]
- Murcko, M.A. Envisioning the future: Medicine in the year 2050. Disruptive Sci. Technol. 2012, 1, 89–99. [Google Scholar] [CrossRef] [Green Version]
- Paul, S.M.; Mytelka, D.S.; Dunwiddie, C.T.; Persinger, C.C.; Munos, B.H.; Lindborg, S.R.; Schacht, A.L. How to improve R&D productivity: The pharmaceutical industry’s grand challenge. Nat. Rev. Drug Discov. 2010, 9, 203–214. [Google Scholar] [PubMed]
- Nicolaou, C.A.; Humblet, C.; Hu, H.; Martin, E.M.; Dorsey, F.C.; Castle, T.M.; Burton, K.I.; Hu, H.; Hendle, J.; Hickey, M.J.; et al. Idea2Data: Toward a new paradigm for drug discovery. ACS Med. Chem. Lett. 2019, 10, 278–286. [Google Scholar] [CrossRef]
- Vidler, L.R.; Baumgartner, M.P. Creating a virtual assistant for medicinal chemistry. ACS Med. Chem. Lett. 2019, 10, 1051–1055. [Google Scholar] [CrossRef]
- Struble, T.J.; Alvarez, J.C.; Brown, S.P.; Chytil, M.; Cisar, J.; DesJarlais, R.L.; Engkvist, O.; Frank, S.A.; Greve, D.R.; Griffin, D.J.; et al. Current and Future Roles of Artificial Intelligence in Medicinal Chemistry Synthesis. J. Med. Chem. 2020, 63, 8667–8682. [Google Scholar] [CrossRef] [Green Version]
- Godfrey, A.G.; Masquelin, T.; Hemmerle, H. A remote-controlled adaptive medchem lab: An innovative approach to enable drug discovery in the 21st Century. Drug Discov. Today 2013, 18, 795–802. [Google Scholar] [CrossRef]
- Farrant, E. Automation of Synthesis in Medicinal Chemistry: Progress and Challenges. ACS Med. Chem. Lett. 2020, 11, 1506–1513. [Google Scholar] [CrossRef]
- Winicov, H.; Schainbaum, J.; Buckley, J.; Longino, G.; Hill, J.; Berkoff, C. Chemical process optimization by computer—A self-directed chemical synthesis system. Anal. Chim. Acta 1978, 103, 469–476. [Google Scholar] [CrossRef]
- Marklund, E.; Degiacomi, M.; Robinson, C.; Baldwin, A.; Benesch, J. Collision Cross Sections for Structural Proteomics. Structure 2015, 23, 791–799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Kuhn, M.; Gavin, A.C.; Bork, P. Identification of metabolites from tandem mass spectra with a machine learning approach utilizing structural features. Bioinformatics 2019, 36, 1213–1218. [Google Scholar] [CrossRef] [Green Version]
- Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef] [Green Version]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef] [Green Version]
- Jain, A.; Hautier, G.; Moore, C.J.; Ong, S.P.; Fischer, C.C.; Mueller, T.; Persson, K.A.; Ceder, G. A high-throughput infrastructure for density functional theory calculations. Comput. Mater. Sci. 2011, 50, 2295–2310. [Google Scholar] [CrossRef]
- Qu, X.; Jain, A.; Rajput, N.N.; Cheng, L.; Zhang, Y.; Ong, S.P.; Brafman, M.; Maginn, E.; Curtiss, L.A.; Persson, K.A. The Electrolyte Genome project: A big data approach in battery materials discovery. Comput. Mater. Sci. 2015, 103, 56–67. [Google Scholar] [CrossRef] [Green Version]
- Qiao, Z.; Welborn, M.; Anandkumar, A.; Manby, F.R.; Miller, T.F. OrbNet: Deep learning for quantum chemistry using symmetry-adapted atomic-orbital features. J. Chem. Phys. 2020, 153, 124111. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.R.; Husch, T.; Ding, F.; Miller, T.F. Analytical Gradients for Molecular-Orbital-Based Machine Learning. arXiv 2020, arXiv:2012.08899. [Google Scholar]
- Dral, P.O. Quantum Chemistry in the Age of Machine Learning. J. Phys. Chem. Lett. 2020, 11, 2336–2347. [Google Scholar] [CrossRef] [PubMed]
- Bogojeski, M.; Vogt-Maranto, L.; Tuckerman, M.E.; Müller, K.R.; Burke, K. Quantum chemical accuracy from density functional approximations via machine learning. Nat. Commun. 2020, 11, 5223. [Google Scholar] [CrossRef]
- Joshi, R.P.; McNaughton, A.; Thomas, D.G.; Henry, C.S.; Canon, S.R.; McCue, L.A.; Kumar, N. Quantum Mechanical Methods Predict Accurate Thermodynamics of Biochemical Reactions. ACS Omega 2021, 6, 9948–9959. [Google Scholar] [CrossRef]
- Ramakrishnan, R.; Dral, P.O.; Rupp, M.; von Lilienfeld, O.A. Quantum chemistry structures and properties of 134 kilo molecules. Sci. Data 2014, 1, 140022. [Google Scholar] [CrossRef]
- Ruddigkeit, L.; van Deursen, R.; Blum, L.C.; Reymond, J.L. Enumeration of 166 Billion Organic Small Molecules in the Chemical Universe Database GDB-17. J. Chem. Inf. Model. 2012, 52, 2864–2875. [Google Scholar] [CrossRef]
- Shen, V.; Siderius, D.; Krekelberg, W.; Mountain, R.D.; Hatch, H.W. NIST Standard Reference Simulation Website, NIST Standard Reference Database Number 173; National Institute of Standards and Technology: Gaithersburg, MD, USA, 2017.
- Seaver, S.M.; Liu, F.; Zhang, Q.; Jeffryes, J.; Faria, J.P.; Edirisinghe, J.N.; Mundy, M.; Chia, N.; Noor, E.; Beber, M.E.; et al. The ModelSEED Biochemistry Database for the Integration of Metabolic Annotations and the Reconstruction, Comparison and Analysis of Metabolic Models for Plants, Fungi and Microbes. Nucleic Acids Res. 2021, 49, D575–D588. [Google Scholar] [CrossRef] [PubMed]
- Kononova, O.; Huo, H.; He, T.; Rong, Z.; Botari, T.; Sun, W.; Tshitoyan, V.; Ceder, G. Text-mined dataset of inorganic materials synthesis recipes. Sci. Data 2019, 6, 1–11. [Google Scholar] [CrossRef]
- Zheng, S.; Dharssi, S.; Wu, M.; Li, J.; Lu, Z. Text Mining for Drug Discovery. In Bioinformatics and Drug Discovery; Larson, R.S., Oprea, T.I., Eds.; Springer: New York, NY, USA, 2019; pp. 231–252. [Google Scholar]
- Singhal, A.; Simmons, M.; Lu, Z. Text mining for precision medicine: Automating disease-mutation relationship extraction from biomedical literature. J. Am. Med. Inform. Assoc. 2016, 23, 766–772. [Google Scholar] [CrossRef] [Green Version]
- Krallinger, M.; Rabal, O.; Lourenço, A.; Oyarzabal, J.; Valencia, A. Information Retrieval and Text Mining Technologies for Chemistry. Chem. Rev. 2017, 117, 7673–7761. [Google Scholar] [CrossRef]
- Huang, B.; von Lilienfeld, O.A. Communication: Understanding molecular representations in machine learning: The role of uniqueness and target similarity. J. Chem. Phys. 2016, 145, 161102. [Google Scholar] [CrossRef]
- Chen, C.; Ye, W.; Zuo, Y.; Zheng, C.; Ong, S.P. Graph Networks as a Universal Machine Learning Framework for Molecules and Crystals. Chem. Mater. 2019, 31, 3564–3572. [Google Scholar] [CrossRef] [Green Version]
- Elton, D.C.; Boukouvalas, Z.; Fuge, M.D.; Chung, P.W. Deep learning for molecular design—A review of the state of the art. Mol. Syst. Des. Eng. 2019, 4, 828–849. [Google Scholar] [CrossRef] [Green Version]
- Bjerrum, E.J. SMILES Enumeration as Data Augmentation for Neural Network Modeling of Molecules. arXiv 2017, arXiv:1703.07076. [Google Scholar]
- Gilmer, J.; Schoenholz, S.S.; Riley, P.F.; Vinyals, O.; Dahl, G.E. Neural Message Passing for Quantum Chemistry. arXiv 2017, arXiv:1704.01212. [Google Scholar]
- Hamilton, W.L.; Ying, R.; Leskovec, J. Representation Learning on Graphs: Methods and Applications. arXiv 2017, arXiv:1709.05584. [Google Scholar]
- Kearnes, S.; McCloskey, K.; Berndl, M.; Pande, V.; Riley, P. Molecular graph convolutions: Moving beyond fingerprints. J. Comput. Aided Mol. Des. 2016, 30, 595–608. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Ramsundar, B.; Feinberg, E.; Gomes, J.; Geniesse, C.; Pappu, A.S.; Leswing, K.; Pande, V. MoleculeNet: A benchmark for molecular machine learning. Chem. Sci. 2018, 9, 513–530. [Google Scholar] [CrossRef] [Green Version]
- Rupp, M.; Tkatchenko, A.; Muller, K.R.; Von Lilienfeld, O.A. Fast and accurate modeling of molecular atomization energies with machine learning. Phys. Rev. Lett. 2012, 108, 058301. [Google Scholar] [CrossRef] [PubMed]
- Hansen, K.; Biegler, F.; Ramakrishnan, R.; Pronobis, W.; Von Lilienfeld, O.A.; Muller, K.R.; Tkatchenko, A. Machine learning predictions of molecular properties: Accurate many-body potentials and nonlocality in chemical space. J. Phys. Chem. Lett. 2015, 6, 2326–2331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weininger, D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Comput. Sci. 1988, 28, 31–36. [Google Scholar] [CrossRef]
- Heller, S.; McNaught, A.; Stein, S.; Tchekhovskoi, D.; Pletnev, I. InChI—The worldwide chemical structure identifier standard. J. cheminform. J. Cheminform. 2013, 5, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grethe, G.; Goodman, J.; Allen, C. International chemical identifier for chemical reactions. J. Cheminform. 2013, 5, O16. [Google Scholar] [CrossRef]
- Elton, D.C.; Boukouvalas, Z.; Butrico, M.S.; Fuge, M.D.; Chung, P.W. Applying machine learning techniques to predict the properties of energetic materials. Sci. Rep. 2018, 8, 9059. [Google Scholar] [CrossRef]
- Landrum, G. RDKit: Open-Source Cheminformatics Software. 2016. Available online: http://rdkit.org/ (accessed on 20 December 2020).
- Cxcalc, ChemAxon. Available online: https://www.chemaxon.com (accessed on 20 December 2020).
- Krenn, M.; Häse, F.; Nigam, A.; Friederich, P.; Aspuru-Guzik, A. Self-referencing embedded strings (SELFIES): A 100% robust molecular string representation. Mach. Learn. Sci. Technol. 2020, 1, 045024. [Google Scholar] [CrossRef]
- Available online: https://aspuru.substack.com/p/molecular-graph-representations-and (accessed on 20 December 2020).
- Koichi, S.; Iwata, S.; Uno, T.; Koshino, H.; Satoh, H. Algorithm for advanced canonical coding of planar chemical structures that considers stereochemical and symmetric information. J. Chem. Inf. Model. 2007, 47, 1734–1746. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.M. Towards a Universal SMILES representation—A standard method to generate canonical SMILES based on the InChI. J. Cheminform. 2012, 4, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Daylight Chemical Information Systems Inc. Available online: http://www.daylight.com/dayhtml/doc/theory/theory.smarts.html (accessed on 20 December 2020).
- O’Boyle, N.; Dalke, A. DeepSMILES: An Adaptation of SMILES for Use in Machine-Learning of Chemical Structures. Chemrxiv 2018, 1–9. [Google Scholar] [CrossRef]
- Maragakis, P.; Nisonoff, H.; Cole, B.; Shaw, D.E. A Deep-Learning View of Chemical Space Designed to Facilitate Drug Discovery. J. Chem. Inf. Model. 2020, 60, 4487–4496. [Google Scholar] [CrossRef]
- Nigam, A.; Friederich, P.; Krenn, M.; Aspuru-Guzik, A. Augmenting Genetic Algorithms with Deep Neural Networks for Exploring the Chemical Space. arXiv 2020, arXiv:1909.11655. [Google Scholar]
- Gebauer, N.; Gastegger, M.; Schütt, K. Symmetry-adapted generation of 3d point sets for the targeted discovery of molecules. In Advances in Neural Information Processing Systems; Wallach, H., Larochelle, H., Beygelzimer, A., d’Alché-Buc, F., Fox, E., Garnett, R., Eds.; Curran Associates, Inc.: Vancouver, BC, Canada, 2019; Volume 32, pp. 7566–7578. [Google Scholar]
- Schütt, K.T.; Kessel, P.; Gastegger, M.; Nicoli, K.A.; Tkatchenko, A.; Müller, K.R. SchNetPack: A Deep Learning Toolbox For Atomistic Systems. J. Chem. Theory Comput. 2019, 15, 448–455. [Google Scholar] [CrossRef] [Green Version]
- Minnich, A.J.; McLoughlin, K.; Tse, M.; Deng, J.; Weber, A.; Murad, N.; Madej, B.D.; Ramsundar, B.; Rush, T.; Calad-Thomson, S.; et al. AMPL: A Data-Driven Modeling Pipeline for Drug Discovery. J. Chem. Inf. Model. 2020, 60, 1955–1968. [Google Scholar] [CrossRef]
- St. John, P.C.; Phillips, C.; Kemper, T.W.; Wilson, A.N.; Guan, Y.; Crowley, M.F.; Nimlos, M.R.; Larsen, R.E. Message-passing neural networks for high-throughput polymer screening. J. Chem. Phys. 2019, 150, 234111. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Swanson, K.; Jin, W.; Coley, C.; Eiden, P.; Gao, H.; Guzman-Perez, A.; Hopper, T.; Kelley, B.; Mathea, M.; et al. Analyzing Learned Molecular Representations for Property Prediction. J. Chem. Inf. Model. 2019, 59, 3370–3388. [Google Scholar] [CrossRef] [Green Version]
- Göller, A.H.; Kuhnke, L.; Montanari, F.; Bonin, A.; Schneckener, S.; ter Laak, A.; Wichard, J.; Lobell, M.; Hillisch, A. Bayer’s in silico ADMET platform: A journey of machine learning over the past two decades. Drug Discov. Today 2020, 25, 1702–1709. [Google Scholar] [CrossRef] [PubMed]
- Schütt, K.T.; Arbabzadah, F.; Chmiela, S.; Müller, K.R.; Tkatchenko, A. Quantum-chemical insights from deep tensor neural networks. Nat. Commun. 2017, 8, 13890. [Google Scholar] [CrossRef] [Green Version]
- Schütt, K.T.; Sauceda, H.E.; Kindermans, P.J.; Tkatchenko, A.; Müller, K.R. SchNet—A deep learning architecture for molecules and materials. J. Chem. Phys. 2018, 148, 241722. [Google Scholar] [CrossRef] [PubMed]
- Schütt, K.; Kindermans, P.J.; Sauceda Felix, H.E.; Chmiela, S.; Tkatchenko, A.; Müller, K.R. SchNet: A continuous-filter convolutional neural network for modeling quantum interactions. In Advances in Neural Information Processing Systems 30; Guyon, I., Luxburg, U.V., Bengio, S., Wallach, H., Fergus, R., Vishwanathan, S., Garnett, R., Eds.; Curran Associates, Inc.: Vancouver, BC, Canada, 2017; pp. 991–1001. [Google Scholar]
- Axelrod, S.; Gomez-Bombarelli, R. GEOM: Energy-annotated molecular conformations for property prediction and molecular generation. arXiv 2020, arXiv:2006.05531. [Google Scholar]
- Yue, S.; Muniz, M.C.; Calegari Andrade, M.F.; Zhang, L.; Car, R.; Panagiotopoulos, A.Z. When do short-range atomistic machine-learning models fall short? J. Chem. Phys. 2021, 154, 034111. [Google Scholar] [CrossRef]
- Matlock, M.K.; Dang, N.L.; Swamidass, S.J. Learning a Local-Variable Model of Aromatic and Conjugated Systems. ACS Cent. Sci. 2018, 4, 52–62. [Google Scholar] [CrossRef] [Green Version]
- Joshi, R.P.; Gebauer, N.W.A.; Bontha, M.; Khazaieli, M.; James, R.M.; Brown, J.B.; Kumar, N. 3D-Scaffold: A Deep Learning Framework to Generate 3D Coordinates of Drug-like Molecules with Desired Scaffolds. J. Phys. Chem. B 2021. [Google Scholar] [CrossRef] [PubMed]
- Gertrudes, J.; Maltarollo, V.; Silva, R.; Oliveira, P.; Honorio, K.; da Silva, A. Machine Learning Techniques and Drug Design. Curr. Med. Chem. 2012, 19, 4289–4297. [Google Scholar] [CrossRef] [PubMed]
- Talevi, A.; Morales, J.F.; Hather, G.; Podichetty, J.T.; Kim, S.; Bloomingdale, P.C.; Kim, S.; Burton, J.; Brown, J.D.; Winterstein, A.G.; et al. Machine Learning in Drug Discovery and Development Part 1: A Primer. CPT Pharmacomet. Syst. Pharmacol. 2020, 9, 129–142. [Google Scholar] [CrossRef]
- Lo, Y.C.; Rensi, S.E.; Torng, W.; Altman, R.B. Machine learning in chemoinformatics and drug discovery. Drug Discov. Today 2018, 23, 1538–1546. [Google Scholar] [CrossRef]
- Agarwal, S.; Dugar, D.; Sengupta, S. Ranking Chemical Structures for Drug Discovery: A New Machine Learning Approach. J. Chem. Inf. Model. 2010, 50, 716–731. [Google Scholar] [CrossRef]
- Rodrigues, T.; Bernardes, G.J. Machine learning for target discovery in drug development. Curr. Opin. Chem. Biol. 2020, 56, 16–22. [Google Scholar] [CrossRef]
- Gao, D.; Chen, Q.; Zeng, Y.; Jiang, M.; Zhang, Y. Applications of Machine Learning in Drug Target Discovery. Curr. Drug Metab. 2020, 21, 790–803. [Google Scholar] [CrossRef]
- Dahal, K.; Gautam, Y. Argumentative Comparative Analysis of Machine Learning on Coronary Artery Disease. Open J. Stat. 2020, 10, 694–705. [Google Scholar] [CrossRef]
- Duvenaud, D.; Maclaurin, D.; Aguilera-Iparraguirre, J.; Gómez-Bombarelli, R.; Hirzel, T.; Aspuru-Guzik, A.; Adams, R.P. Convolutional Networks on Graphs for Learning Molecular Fingerprints. arXiv 2015, arXiv:1509.09292. [Google Scholar]
- Faber, F.A.; Hutchison, L.; Huang, B.; Gilmer, J.; Schoenholz, S.S.; Dahl, G.E.; Vinyals, O.; Kearnes, S.; Riley, P.F.; von Lilienfeld, O.A. Prediction Errors of Molecular Machine Learning Models Lower than Hybrid DFT Error. J. Chem. Theory Comput. 2017, 13, 5255–5264. [Google Scholar] [CrossRef]
- Fung, V.; Zhang, J.; Juarez, E.; Sumpter, B.G. Benchmarking graph neural networks for materials chemistry. NPJ Comput. Mater. 2021, 7, 84. [Google Scholar] [CrossRef]
- Jørgensen, P.; Jacobsen, K.; Schmidt, M. Neural Message Passing with Edge Updates for Predicting Properties of Molecules and Materials. In Proceedings of the 32nd Conference on Neural Information Processing Systems, NIPS 2018, Montréal, QC, Canada, 3–8 December 2018. [Google Scholar]
- Polishchuk, P.G.; Madzhidov, T.I.; Varnek, A. Estimation of the size of drug-like chemical space based on GDB-17 data. J. Comput. Aided Mol. Des. 2013, 27, 675–679. [Google Scholar] [CrossRef]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A.; et al. PubChem Substance and Compound databases. Nucleic Acids Res. 2015, 44, D1202–D1213. [Google Scholar] [CrossRef]
- Coley, C.W. Defining and Exploring Chemical Spaces. Trends Chem. 2021, 3, 133–145. [Google Scholar] [CrossRef]
- Zunger, A. Inverse design in search of materials with target functionalities. Nat. Rev. Chem. 2018, 2, 1–16. [Google Scholar] [CrossRef]
- Kuhn, C.; Beratan, D.N. Inverse strategies for molecular design. J. Phys. Chem. 1996, 100, 10595–10599. [Google Scholar] [CrossRef]
- Gómez-Bombarelli, R.; Wei, J.N.; Duvenaud, D.; Hernández-Lobato, J.M.; Sánchez-Lengeling, B.; Sheberla, D.; Aguilera-Iparraguirre, J.; Hirzel, T.D.; Adams, R.P.; Aspuru-Guzik, A. Automatic Chemical Design Using a Data-Driven Continuous Representation of Molecules. ACS Cent. Sci. 2018, 4, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Zaremba, W.; Sutskever, I.; Vinyals, O. Recurrent Neural Network Regularization. arXiv 2015, arXiv:1409.2329. [Google Scholar]
- Schmidhuber, J. Deep learning in neural networks: An overview. Neural Netw. 2015, 61, 85–117. [Google Scholar] [CrossRef] [Green Version]
- Kusner, M.J.; Paige, B.; Hernández-Lobato, J.M. Grammar Variational Autoencoder. In Proceedings of the 34th International Conference on Machine Learning—Volume 70, ICML’17, Sydney, Australia, 6–11 August 2017; pp. 1945–1954. [Google Scholar]
- Liu, Q.; Allamanis, M.; Brockschmidt, M.; Gaunt, A.L. Constrained Graph Variational Autoencoders for Molecule Design. arXiv 2018, arXiv:1805.09076. [Google Scholar]
- Jin, W.; Yang, K.; Barzilay, R.; Jaakkola, T. Learning Multimodal Graph-to-Graph Translation for Molecular Optimization. arXiv 2018, arXiv:1812.01070. [Google Scholar]
- Jin, W.; Barzilay, R.; Jaakkola, T.S. Multi-Resolution Autoregressive Graph-to-Graph Translation for Molecules. Chemrxiv 2019, 1–13. [Google Scholar] [CrossRef]
- Bian, Y.; Wang, J.; Jun, J.J.; Xie, X.Q. Deep Convolutional Generative Adversarial Network (dcGAN) Models for Screening and Design of Small Molecules Targeting Cannabinoid Receptors. Mol. Pharm. 2019, 16, 4451–4460. [Google Scholar] [CrossRef]
- Cao, N.D.; Kipf, T. MolGAN: An implicit generative model for small molecular graphs. arXiv 2018, arXiv:1805.11973. [Google Scholar]
- Kadurin, A.; Nikolenko, S.; Khrabrov, K.; Aliper, A.; Zhavoronkov, A. druGAN: An Advanced Generative Adversarial Autoencoder Model for de Novo Generation of New Molecules with Desired Molecular Properties in Silico. Mol. Pharm. 2017, 14, 3098–3104. [Google Scholar] [CrossRef] [PubMed]
- Guimaraes, G.L.; Sanchez-Lengeling, B.; Outeiral, C.; Farias, P.L.C.; Aspuru-Guzik, A. Objective-Reinforced Generative Adversarial Networks (ORGAN) for Sequence Generation Models. arXiv 2017, arXiv:1705.10843. [Google Scholar]
- Sanchez-Lengeling, B.; Outeiral, C.; Guimaraes, G.L.; Aspuru-Guzik, A. Optimizing distributions over molecular space. An Objective-Reinforced Generative Adversarial Network for Inverse-design Chemistry (ORGANIC). Chemrxiv 2017, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Putin, E.; Asadulaev, A.; Ivanenkov, Y.; Aladinskiy, V.; Sanchez-Lengeling, B.; Aspuru-Guzik, A.; Zhavoronkov, A. Reinforced Adversarial Neural Computer for de Novo Molecular Design. J. Chem. Inf. Model. 2018, 58, 1194–1204. [Google Scholar] [CrossRef]
- Méndez-Lucio, O.; Baillif, B.; Clevert, D.A.; Rouquié, D.; Wichard, J. De Novo Generation of Hit-like Molecules from Gene Expression Signatures Using Artificial Intelligence. Nat. Comm. 2020, 11, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Popova, M.; Isayev, O.; Tropsha, A. Deep reinforcement learning for de novo drug design. Sci. Adv. 2018, 4, 7885. [Google Scholar] [CrossRef] [Green Version]
- Olivecrona, M.; Blaschke, T.; Engkvist, O.; Chen, H. Molecular De Novo Design through Deep Reinforcement Learning. J. Cheminform. 2017, 9, 1758–2946. [Google Scholar] [CrossRef] [Green Version]
- Ståhl, N.; Falkman, G.; Karlsson, A.; Mathiason, G.; Boström, J. Deep Reinforcement Learning for Multiparameter Optimization in de novo Drug Design. J. Chem. Inf. Model. 2019, 59, 3166–3176. [Google Scholar] [CrossRef] [Green Version]
- O’Boyle, N.M.; Campbell, C.M.; Hutchison, G.R. Computational Design and Selection of Optimal Organic Photovoltaic Materials. J. Phys. Chem. C 2011, 115, 16200–16210. [Google Scholar] [CrossRef] [Green Version]
- Virshup, A.M.; Contreras-García, J.; Wipf, P.; Yang, W.; Beratan, D.N. Stochastic Voyages into Uncharted Chemical Space Produce a Representative Library of All Possible Drug-Like Compounds. J. Am. Chem. Soc. 2013, 135, 7296–7303. [Google Scholar] [CrossRef] [Green Version]
- Rupakheti, C.; Virshup, A.; Yang, W.; Beratan, D.N. Strategy To Discover Diverse Optimal Molecules in the Small Molecule Universe. J. Chem. Inf. Model. 2015, 55, 529–537. [Google Scholar] [CrossRef]
- Jensen, J.H. A graph-based genetic algorithm and generative model/Monte Carlo tree search for the exploration of chemical space. Chem. Sci. 2019, 10, 3567–3572. [Google Scholar] [CrossRef] [Green Version]
- Paszkowicz, W. Properties of a genetic algorithm equipped with a dynamic penalty function. Comput. Mater. Sci. 2009, 45, 77–83. [Google Scholar] [CrossRef]
- Simm, G.N.C.; Pinsler, R.; Csányi, G.; Hernández-Lobato, J.M. Symmetry-Aware Actor-Critic for 3D Molecular Design. arXiv 2020, arXiv:2011.12747. [Google Scholar]
- Simm, G.N.C.; Pinsler, R.; Hernández-Lobato, J.M. Reinforcement Learning for Molecular Design Guided by Quantum Mechanics. arXiv 2020, arXiv:2002.07717. [Google Scholar]
- Li, Y.; Hu, J.; Wang, Y.; Zhou, J.; Zhang, L.; Liu, Z. DeepScaffold: A Comprehensive Tool for Scaffold-Based De Novo Drug Discovery Using Deep Learning. J. Chem. Inf. Model. 2020, 60, 77–91. [Google Scholar] [CrossRef]
- Lim, J.; Hwang, S.Y.; Moon, S.; Kim, S.; Kim, W.Y. Scaffold-based molecular design with a graph generative model. Chem. Sci. 2020, 11, 1153–1164. [Google Scholar] [CrossRef] [Green Version]
- Arús-Pous, J.; Patronov, A.; Bjerrum, E.J.; Tyrchan, C.; Reymond, J.L.; Chen, H.; Engkvist, O. SMILES-based deep generative scaffold decorator for de-novo drug design. J. Cheminform. 2020, 12, 38. [Google Scholar] [CrossRef]
- Zhang, K.Y.J.; Milburn, M.V.; Artis, D.R. Scaffold-Based Drug Discovery. In Structure-Based Drug Discovery; Springer: Dordrecht, The Netherlands, 2007; pp. 129–153. [Google Scholar]
- Scott, O.B.; Edith Chan, A.W. ScaffoldGraph: An open-source library for the generation and analysis of molecular scaffold networks and scaffold trees. Bioinformatics 2020, 36, 3930–3931. [Google Scholar] [CrossRef]
- Romero, J.; Olson, J.P.; Aspuru-Guzik, A. Quantum autoencoders for efficient compression of quantum data. Quantum Sci. Technol. 2017, 2, 045001. [Google Scholar] [CrossRef] [Green Version]
- Allcock, J.; Zhang, S. Quantum machine learning. Natl. Sci. Rev. 2018, 6, 26–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gentile, F.; Agrawal, V.; Hsing, M.; Ton, A.T.; Ban, F.; Norinder, U.; Gleave, M.E.; Cherkasov, A. Deep Docking: A Deep Learning Platform for Augmentation of Structure Based Drug Discovery. ACS Cent. Sci. 2020, 6, 939–949. [Google Scholar] [CrossRef]
- Stein, R.M.; Kang, H.J.; McCorvy, J.D.; Glatfelter, G.C.; Jones, A.J.; Che, T.; Slocum, S.; Huang, X.P.; Savych, O.; Moroz, Y.S.; et al. Virtual discovery of melatonin receptor ligands to modulate circadian rhythms. Nature 2020, 579, 609–614. [Google Scholar] [CrossRef]
- Lyu, J.; Wang, S.; Balius, T.E.; Singh, I.; Levit, A.; Moroz, Y.S.; O’Meara, M.J.; Che, T.; Algaa, E.; Tolmachova, K.; et al. Ultra-large library docking for discovering new chemotypes. Nature 2019, 566, 224–229. [Google Scholar] [CrossRef] [PubMed]
- Guterres, H.; Im, W. Improving Protein-Ligand Docking Results with High-Throughput Molecular Dynamics Simulations. J. Chem. Inform. Model. 2020, 60, 2189–2198. [Google Scholar] [CrossRef]
- Wallach, I.; Dzamba, M.; Heifets, A. AtomNet: A deep convolutional neural network for bioactivity prediction in structure-based drug discovery. arXiv 2015, arXiv:1510.02855. [Google Scholar]
- Ragoza, M.; Hochuli, J.; Idrobo, E.; Sunseri, J.; Koes, D.R. Protein–ligand scoring with convolutional neural networks. J. Chem. Inf. Model. 2017, 57, 942–957. [Google Scholar] [CrossRef] [Green Version]
- Jiménez, J.; Skalic, M.; Martinez-Rosell, G.; De Fabritiis, G. K deep: Protein–ligand absolute binding affinity prediction via 3d-convolutional neural networks. J. Chem. Inf. Model. 2018, 58, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Stepniewska-Dziubinska, M.M.; Zielenkiewicz, P.; Siedlecki, P. Development and evaluation of a deep learning model for protein–ligand binding affinity prediction. Bioinformatics 2018, 34, 3666–3674. [Google Scholar] [CrossRef] [Green Version]
- Karimi, M.; Wu, D.; Wang, Z.; Shen, Y. DeepAffinity: Interpretable deep learning of compound-protein affinity through unified recurrent and convolutional neural networks. Bioinformatics 2019, 35, 3329–3338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Wan, F.; Shu, H.; Jiang, T.; Zhao, D.; Zeng, J. MONN: A Multi-objective Neural Network for Predicting Compound-Protein Interactions and Affinities. Cell Syst. 2020, 10, 308–322.e11. [Google Scholar] [CrossRef]
- Gao, K.Y.; Fokoue, A.; Luo, H.; Iyengar, A.; Dey, S.; Zhang, P. Interpretable Drug Target Prediction Using Deep Neural Representation. In Proceedings of the 2018 International Joint Conference on Artificial Intelligence, IJCAI, Stockholm, Schweden, 13–18 July 2018; Volume 2018, pp. 3371–3377. [Google Scholar]
- Lim, J.; Ryu, S.; Park, K.; Choe, Y.J.; Ham, J.; Kim, W.Y. Predicting Drug–Target Interaction Using a Novel Graph Neural Network with 3D Structure-Embedded Graph Representation. J. Chem. Inf. Model. 2019, 59, 3981–3988. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Rangarajan, S. Designing compact training sets for data-driven molecular property prediction through optimal exploitation and exploration. Mol. Syst. Des. Eng. 2019, 4, 1048–1057. [Google Scholar] [CrossRef]
- Warmuth, M.K.; Rätsch, G.; Mathieson, M.; Liao, J.; Lemmen, C. Active Learning in the Drug Discovery Process. In Advances in Neural Information Processing Systems 14; Dietterich, T.G., Becker, S., Ghahramani, Z., Eds.; MIT Press: Cambridge, MA, USA, 2002; pp. 1449–1456. [Google Scholar]
- Fusani, L.; Cabrera, A.C. Active learning strategies with COMBINE analysis: New tricks for an old dog. J. Comput. Aided Mol. Des. 2019, 33, 287–294. [Google Scholar] [CrossRef]
- Green, D.V.S.; Pickett, S.; Luscombe, C.; Senger, S.; Marcus, D.; Meslamani, J.; Brett, D.; Powell, A.; Masson, J. BRADSHAW: A system for automated molecular design. J. Comput. Aided Mol. Des. 2019, 34, 747–765. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Li, R.; Zeng, T.; Sun, Q.; Kumar, S.; Ye, J.; Ji, S. Deep Model Based Transfer and Multi-Task Learning for Biological Image Analysis. IEEE Trans. Big Data 2016, 6, 322–333. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Methods | Key Feature | Advantage | Drawbacks |
|---|---|---|---|
| MPNN [60] |
|
|
|
| d-MPNN [61] |
|
|
|
| SchNet [58] |
|
|
|
| MEGNet [34] |
|
|
|
| SchNet-edge [80] |
|
|
|
| Property | Units | MPNN | SchNet-Edge | SchNet | MegNet | Target |
|---|---|---|---|---|---|---|
| HOMO | eV | 0.043 | 0.037 | 0.041 | 0.038 ± 0.001 | 0.043 |
| LUMO | eV | 0.037 | 0.031 | 0.034 | 0.031 ± 0.000 | 0.043 |
| band gap | eV | 0.069 | 0.058 | 0.063 | 0.061 ± 0.001 | 0.043 |
| ZPVE | meV | 1.500 | 1.490 | 1.700 | 1.400 ± 0.060 | 1.200 |
| dipole moment | Debye | 0.030 | 0.029 | 0.033 | 0.040 ± 0.001 | 0.100 |
| polarizability | Bohr | 0.092 | 0.077 | 0.235 | 0.083 ± 0.001 | 0.100 |
| R | Bohr | 0.180 | 0.072 | 0.073 | 0.265 ± 0.001 | 1.200 |
| U | eV | 0.019 | 0.011 * | 0.014 | 0.009 ± 0.000 * | 0.043 |
| U | eV | 0.019 | 0.016 * | 0.019 | 0.010 ± 0.000 * | 0.043 |
| H | eV | 0.017 | 0.011 * | 0.014 | 0.010 ± 0.000 * | 0.043 |
| G | eV | 0.019 | 0.012 * | 0.014 | 0.010 ± 0.000 * | 0.043 |
| C | cal (mol K) | 0.040 | 0.032 | 0.033 | 0.030 ± 0.000 | 0.050 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Joshi, R.P.; Kumar, N. Artificial Intelligence for Autonomous Molecular Design: A Perspective. Molecules 2021, 26, 6761. https://doi.org/10.3390/molecules26226761
Joshi RP, Kumar N. Artificial Intelligence for Autonomous Molecular Design: A Perspective. Molecules. 2021; 26(22):6761. https://doi.org/10.3390/molecules26226761
Chicago/Turabian StyleJoshi, Rajendra P., and Neeraj Kumar. 2021. "Artificial Intelligence for Autonomous Molecular Design: A Perspective" Molecules 26, no. 22: 6761. https://doi.org/10.3390/molecules26226761
APA StyleJoshi, R. P., & Kumar, N. (2021). Artificial Intelligence for Autonomous Molecular Design: A Perspective. Molecules, 26(22), 6761. https://doi.org/10.3390/molecules26226761