Abstract
Phytochemical investigation of the ethanol extract of underground parts of Iris tenuifolia Pall. afforded five new compounds; an unusual macrolide termed moniristenulide (1), 5-methoxy-6,7-methylenedioxy-4-O-2′-cycloflavan (2), 5,7,2′,3′-tetrahydroxyflavanone (3), 5-hydroxy-6,7-dimethoxyisoflavone-2′-O-β-d-glucopyranoside (9), 5,2′,3′-dihydroxy-6,7-dimethoxyisoflavone (10), along with seven known compounds (4–8, 11–12). The structures of all purified compounds were established by analysis of 1D and 2D NMR spectroscopy and HR-ESI-MS. The antimicrobial activity of the compounds 1–3, 5, 9, and 10 was investigated using the agar diffusion method against fungi, Gram-positive and Gram-negative bacteria. In consequence, new compound 3 was found to possess the highest antibacterial activity against Enterococcus faecalis VRE and Mycobacterium vaccae. Cell proliferation and cytotoxicity tests were also applied on all isolated compounds and plant crude extract in vitro with the result of potent inhibitory effect against leukemia cells. In particular, the newly discovered isoflavone 10 was active against both of the leukemia cells K-562 and THP-1 while 4–6 of the flavanone type compounds were active against only THP-1.
1. Introduction
Iris is a genus of the family Iridaceae that consists of about 360 species of perennial herbs and sub shrubs. Iris species are widely distributed in the temperate northern hemisphere, including Euro-Asia, North Africa and North America [1]. Many members of the genus Iris have been used as traditional folk medicines for the treatment of various diseases, such as cancer, inflammation, bacterial and viral infections [2,3]. Previous phytochemical investigations on Iris plants have resulted in the isolation of a variety of secondary metabolites, which mainly include flavonoids, xanthones, terpenoids, steroids, quinones, stilbenes and simple phenolics. Moreover, the plant extracts and isolated compounds from these species were reported to have anti-inflammatory, antioxidant, antimicrobial, antitumor, antimutagenic, cytotoxic, immunomodulating, hepatoprotective, antiproliferative, estrogenic, cholinesterase inhibitory, cell proliferation stimulatory, antidiabetic and molluscicidal activities [1,4,5,6,7]. About 24 species are found in Mongolia. Specifically, Iris tenuifolia Pall., a characteristic plant of desert grasslands of Mongolia, is distributed in Mongol-Altai, Eastern Mongolia, Eastern Gobi and Gobi-Altai province [8,9]. The roots and rhizomes of I. tenuifolia are all commonly used as a traditional Mongolian medicine for treatment of kidney disorder. A root decoction of the plant has been used as a folk remedy for hypertension caused by adrenal gland diseases, ureter stones to relief renal colic and chronic nephritic [10]. This study deals with the isolation and structural elucidation of the new type of macrolide, named moniristenulide (1) and four previously unprecedented compounds: 5-methoxy-6,7-methylenedioxy-4-O-2′-cycloflavan (2), 5,7,2′,3′-tetrahydroxyflavanone (3), 5-hydroxy-6,7-dimethoxyisoflavone-2′-O-β-d-glucopyranoside (9), 5,2′,3′-trihydroxy-6,7-dimethoxyisoflavone (10), together with seven known compounds (Figure 1): four flavanone (4, 5, 6, 7), one isoflavone (8), one flavonol (11), and one sterol (12). In addition, we aimed to evaluate the effectiveness and potency of these natural compounds using antimicrobial, cell proliferation and cytotoxicity assays.
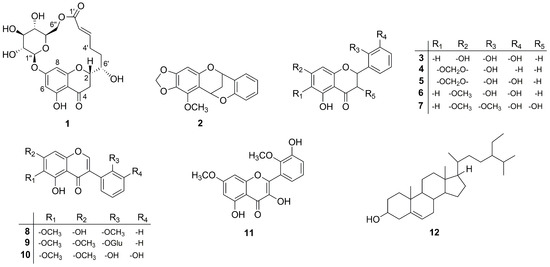
Figure 1.
The structures of compounds 1–12.
2. Results and Discussion
2.1. Structure Elucidation
The ethanol extracts from the underground parts of I. tenuifolia were subjected to repeated column chromatography followed by crystallizations leading to the isolation of five unprecedented chromane derivatives.
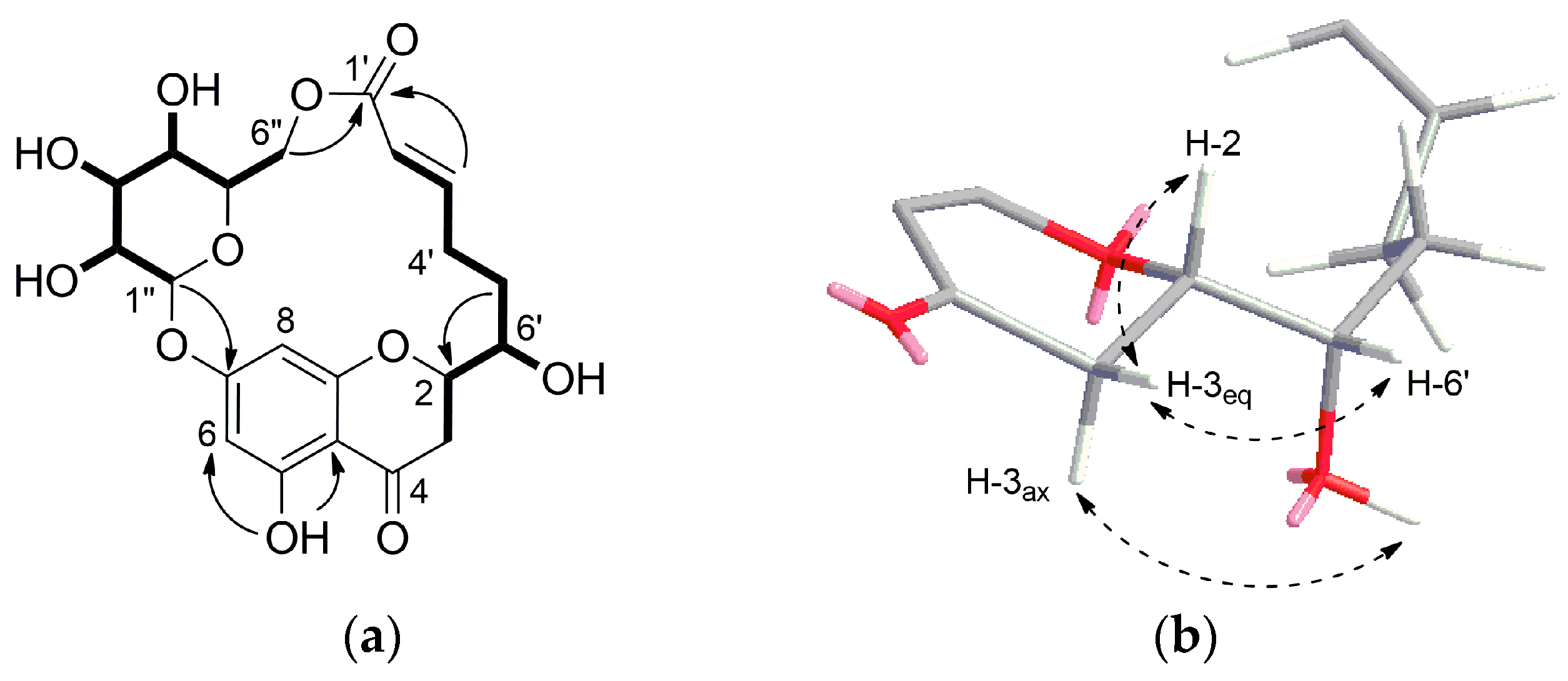
Compound 1 was isolated as white crystal. HR-ESI-MS showed an ion peak at m/z 453.1409 [M + H]+ corresponding to a molecular formula of C21H24O11. Its 1H NMR spectrum acquired in DMSO-d6 (Table 1) showed resonances for meta-coupled aromatic protons at δH 6.14 (1H, J = 2.0 Hz) and δH 5.95 (1H, J = 2.0 Hz), two olefinic protons at δH 6.94 and δH 5.75, methylene signals between δH 3.21–1.81 and number of oxygenated protons between δH 5.50–3.08 corresponding to hydroxy groups as well as oxymethines. The presence of a 2,5,7-trisubstituted chromane-4-one was identified by analysis of HMBC correlations observed for the meta-coupled aromatic doublets H-6 and H-8 as well as methylene signals H-2 and H-3 (Figure 2a). Moreover, the 1H NMR spectrum exhibited a signal of one chelated hydroxyl group (δH 12.04), which is characteristic downfield shift of a hydroxyl group at C-5 and a carbonyl group at C-4. In addition, the presence of a hydroxyl group at C-5 was supported by HMBC correlations from 5-OH (δH 12.04) to C-5 (δC 163.1), C-6 (δC 97.3) and C-10 (δC 103.4). HSQC, HMBC, and COSY data clearly revealed the existence of a glucose residue. Further analysis of the spin-spin couplings (J = 7.7 Hz) allowed the identification of β-d-glucose. Connectivity of sugar moiety with chromane ring was deduced through HMBC correlation from the anomeric proton H-1″ (δH 5.09) to C-7 (δC 164.1) of aglycon. A remaining spin system observed on the COSY spectrum comprised a series of protons H-2′–H-6′. HMBC correlations from olefinic protons H-2′ and H-3′ to a carbonyl carbon at δC 164.8 (C-1′) allowed for establishing 6-hydroxy-2-hexenoic acid residue. Trans configuration of the double bond has been identified by a large coupling constant of 15.6 Hz. This substructure has been linked to the aglycon on position C-2 by HMBC correlations from the H-5′ and H-6′ to a carbon at δC 76.9 (C-2) and in turn H-3 to a carbon at δC 70.9 (C-6′). Interestingly, a macrolide structure was established via ester linkage between sugar residue and carbonyl carbon of hexenoic acid by strong heteronuclear long-range correlation from H-6″ to C-1′. ROESY correlations between diastereotopic H-3eq (β) and H-2 and H-6′ indicated that these protons are on the same side of the chromane ring. In turn, H-3ax showed ROESY correlation to 6′-OH. From the above findings, relative configuration at C-2 and C-6′ has been established as β-configured (Figure 2b). The absolute configuration at C-2 was found to be S as it showed positive and negative Cotton effects at 305 and 287 nm, respectively in the circular dichroism (CD) spectrum (Figure S9, Supplementary Materials) [11,12]. Thus, the structure of compound 1 was established as a new type of macrolide, named moniristenulide, as shown in Figure 1.

Table 1.
1D and 2D NMR (500 MHz) data of moniristenulide (1) in DMSO-d6 (δ in ppm, J in Hz).
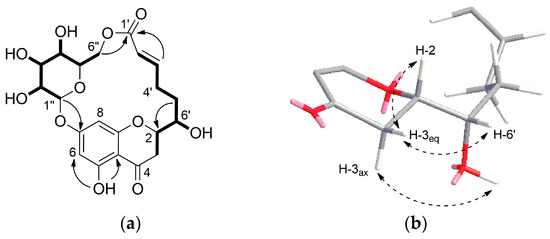
Figure 2.
(a) 1H-1H COSY (―) and key HMBC (→) correlations of moniristenulide (1); (b) ROESY correlation (dashed arrow) establishing relative configuration of 1.
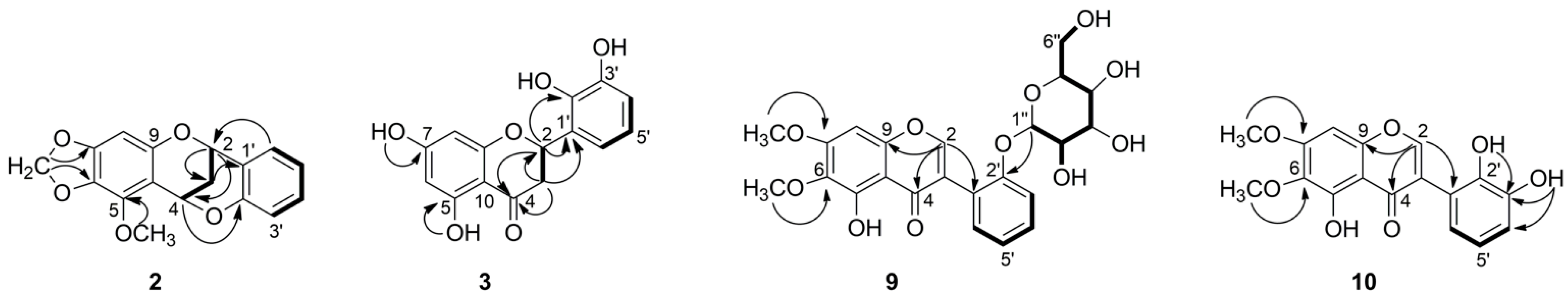
Compound 2 was isolated as colorless crystal. Its HR-ESI-MS at m/z 299.0919 [M + H]+ suggested a molecular formula of C17H14O5. Its 1H NMR spectrum showed resonances for five aromatic protons at δH 7.33 (1H, dd, J = 1.7, 7.5 Hz), 7.20 (1H, ddd, J = 1.7, 7.4, 7.4 Hz), 6.89 (1H, overlap.), 6.87 (1H, overlap.), 6.08 (1H, s), two signals of oxygenated methylene at δH 5.81 and δH 5.76 (each 1H, d, J = 1.5 Hz), two oxygenated methine protons at δH 5.64 (1H, dd, J = 2.8, 4.4 Hz) and 5.25 (1H, dd, J = 2.6, 4.4 Hz), two signals of diastereotopic methylene protons at δH 2.27 (1H, dt, J = 2.8, 13.8 Hz) and 2.15 (1H, dt, J = 2.8, 13.8 Hz) and one methoxy singlet at δH 4.09 (3H, s). Analysis of 13C NMR spectrum combined with HSQC allowed for identifying the existence of 17 carbon atoms, including 12 sp2 carbons, two methylene carbons at δC 26.5 and 100.8, two methine carbons at δC 67.5 and 62.4, and one methoxy group at δC 60.1 (Table 2). The COSY correlations of four aromatic protons at δH 7.33, 7.20, 6.89 and 6.87 (H-3′–H-6′) along with the respective HMBC correlations confirmed a presence of disubstituted aromatic B-ring, while the remaining sharp singlet at δH 6.08 (H-8) described a typical penta-substituted aromatic A-ring. Furthermore, the COSY experiment showed another spin system from H-2 to H-4 and indicated a CH-CH2-CH sequence. Additionally, the key HMBC correlation from H-6′ (δH 7.33) to C-2 (δC 67.5) revealed the flavan (2-phenylchromane) skeleton (Figure 3). The HMBC correlation from methine H-4 (δH 5.64) to C-2′ (δC 153.6) and their chemical shift indicated their linkage through an oxygen bridge. All these data deduced that compound 2 has a 4-O-2′-cycloflavan as a partial structure. The remaining two doublet signals at δH 5.81 and 5.76 had one bond correlation to a carbon atom at δC 100.8, which were assigned as a methylenedioxy group. These methylenedioxy doublet was connected to a flavan core by key HMBC correlations to C-6 and C-7. The location of methoxy group (δH 4.09) at C-5 was suggested by HMBC correlations. On the basis of the above evidence, the planar structure of compound 2 was elucidated as 5-methoxy-6,7-methylenedioxy-4-O-2′-cycloflavan and considered as an unprecedented natural product. This spectroscopic data of compound 2 is somewhat similar to the literature values for the known 4-O-2′-cycloflavan core structures, possessing a different substitution pattern in aromatic A- and B-rings [7,13,14].
Table 2.
1H NMR (500 MHz) and 13C NMR (125 MHz) data of compounds 2, 3, 9, and 10 (δ in ppm, J in Hz).
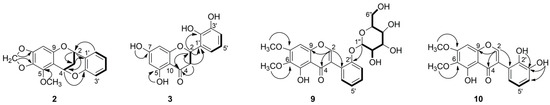
Figure 3.
1H-1H COSY (―) and key HMBC (→) correlations of 2, 3, 9, and 10.
Compound 3 was isolated as yellow solid. It showed a molecular ion peak at m/z 289.0714 [M + H]+ ascribable to a molecular formula of C15H12O6. The 1H NMR spectrum showed resonances for five aromatic proton signals at δH 5.88 (1H, d, J = 2.0 Hz), 5.90 (1H, d, J = 2.0 Hz), 6.69 (1H, t, J = 7.8 Hz), 6.79 (1H, dd, J = 1.4, 7.9 Hz), 6.88 (1H, dd, J = 1.4, 7.8 Hz), three aliphatic protons at δH 5.70 (1H, dd, J = 2.9, 12.9 Hz), 3.17 (1H, dd, J = 12.9, 17.0 Hz), and 2.69 (1H, dd, J = 3.0, 17.0 Hz), and four oxygenated protons at δH 12.12, 10.79, 9.51, and 8.71, one of which was appeared as a chelated hydroxyl group. The 13C NMR spectrum contained signals from 15 carbon atoms, which were with the full agreement of HR-MS (Table 2). Partial structure 2,5,7-trisubstituted chromane-4-one was deduced from the analysis of protons H-2 and H-3, which were existing in an AMX spin system (Figure 3). In addition, hydroxyl groups at C-5 and C-7 were supported by HMBC correlations from 5-OH (δH 12.12) to C-5 (δC 163.5), C-6 (δC 95.8) and C-10 (δC 101.7) and from 7-OH to C-6 (δC 95.8) and C-7 (δC 166.6). In addition, a COSY correlation of three aromatic protons at δH 6.69, 6.79, 6.88 along with HMBC correlations from proton H-4′ (δH 6.79) to C-2′ (δC 142.6), C-3′ (δC 145.2) and from proton H-6′ (δH 6.88) to C-2 (δC 74.0), C-2′ (δC 142.6), and C-4′ (δC 115.2) revealed a presence of a 2,3-dihydroxyphenyl moiety (ring-B) and altogether confirmed the flavanone structure. This was further supported by key long-range heteronuclear correlations from the methine proton H-2 (δH 5.70) to C-2′ (δC 142.6). The position of the remaining two hydroxyl protons at δH 9.51 and 8.71 were assigned to C-3′ and C-2′ respectively, due to observed HMBC correlations. Spectral data of 3 possess close similarity to those for the known compounds 5 and 6 [15]. The only difference was observed for the substitution on C-7, where methoxy group in 6 and methylenedioxy group in 5, while it was replaced by hydroxy group in 3. This was supported by HMBC correlations from 7-OH to C-6, C-7, and C-8 in 3 (Figure 3).
The absolute configuration at C-2 of compound 3 was determined as 2S based on its CD spectrum (Figure S25, Supplementary Materials), which displayed a positive Cotton effect at 325 nm and a negative one at 283 nm [11,12]. Consequently, compound 3 was elucidated as (2S)-5,7,2′,3′-tetrahydroxyflavanone, an undescribed member of a flavanone group of natural products.
Compound 9 was isolated as yellow crystal. The molecular formula C23H24O11 was established on the basis of the positive ion at m/z 477.1399 [M + H]+ by HR-ESI-MS. The 1H NMR spectrum revealed the presence of five aromatic protons (Table 2). The COSY spectrum showed a spin system comprising four aromatic protons at δH 7.37 (1H, overlap.), 7.27 (1H, d, J = 8.2 Hz), 7.35 (1H, overlap.), and 7.09 (1H, t, J = 7.5 Hz), characteristic for an ortho-substituted B-ring of aglycone (Figure 3). The remaining aromatic signal appeared as a sharp singlet at 6.64 (1H, s) together with chelated hydroxyl group at δH 12.68 suggested a typical penta-substituted aromatic A-ring. The singlet was assigned on C-8 according to HMBC correlation from H-8 to C-9 (δC 149.2) and C-7 (δC 158.3). The chelated hydroxyl group was positioned at C-5 by means of HMBC. In addition, a characteristic isoflavonoid signal for H-2 was appeared at δH 8.44. The isoflavone nature was supported by long-range correlations from H-2 (δH 8.44) to C-4 (δC 180.5), C-9 (δC 149.2), and C-1′ (δC 120.0) in the HMBC spectrum. Furthermore, two methoxyl singlet signals were apparent at δH 3.79 and 3.94, and they were located at C-6 and C-7 due to HMBC correlations between 6-OCH3 and C-6 (δC 128.3), as well as 7-OCH3 and C-7 (δC 158.3). Moreover, a series of COSY cross signals comprising six protons in the range of δH 3.0–4.0, four hydroxy protons δH 4.57–5.05, as well as a doublet at δH 4.89, revealed the presence of a glucose moiety (H-1″ to H-6″) [16]. The HMBC correlation from H-1″ (δH 4.89) to C-2′ (δC 155.1) revealed the sugar moiety was located at C-2′ of aglycone. The coupling constant of anomeric proton J = 7.80 Hz indicated that the sugar was β-oriented. The 1H and 13C NMR spectroscopic data of aglycone of 9 were comparable with the literature values for the irilin A [17,18]. The only difference was occurred on C-2′, where OH group of irilin A was replaced by glucopyranosyl in 9. Finally, the structure of compound 9 was elucidated as 5-hydroxy-6,7-dimethoxyisoflavone-2′-O-β-d-glucopyranoside, an unprecedented natural product.
Compound 10 was isolated as yellow crystal. HR-ESI-MS showed the [M + H]+ peak at m/z 331.0825, corresponding to the molecular formula C17H14O7. 1H NMR, 13C NMR, and HSQC data of compound 10 closely resembled to that of 9, differing only on the absence of signals corresponding to a sugar moiety and different pattern on aromatic signals of B-ring (Table 2). The COSY correlation of a triplet at δH 6.92 (1H, t, J = 7.9 Hz) with two doublets of doublets at δH 7.03 (1H, dd, J = 1.3, 7.9 Hz) and 6.71 (1H, dd, J = 1.5, 7.8 Hz), and their HMBC correlations clearly indicated ortho-dihydroxyl substitution on B-ring. The position of the hydroxyl groups, which were appeared as two singlets in the 1H NMR spectrum at δH 8.48 and 6.09, were assigned at C-2′ and C-3′, due to the HMBC correlations (Figure 3). Therefore, the structure of 10 was solved as 5,2′,3′-trihydroxy-6,7-dimethoxyisoflavone, which is an undescribed natural product so far.
Furthermore, the known compounds were readily identified by means of HR-ESI-MS, 1D, and 2D NMR data as well as in comparison with those previously reported in the literature: compound 4 as 5,2′-dihydroxy-6,7-methylenedioxyflavanone [15], 5 as 5,2′,3′-trihydroxy-6,7-methylenedioxyflavanone [15], 6 as 5,2′,3′-trihydroxy -7-methoxyflavanone [15], 7 as 3,5,3′-trihydroxy-7,2′-dimethoxyflavanone [15], 8 as 5,7-dihydroxy-6,2′-dimethoxyisoflavone [19], 11 as 3,5,3′-trihydroxy-7,2′-dimethoxy -flavonol [20], and 12 as β-sitosterol [2,21] (Figure 1). To the best of our knowledge, the known compound 11 was isolated for the first time from this plant.
2.2. Antimicrobial Activity
The newly discovered compounds 1, 2, 3, 9, and 10 were tested together with the known compound 5, which is the major component of the roots of this plant [15] for their antifungal and antibacterial activities against three fungi and eight bacterial strains (Table 3) using the agar diffusion method. All tested compounds possessed weak to moderate activity against vancomycin resistant (VRE) Enterococcus faecalis. Compounds except 1 and 9 exhibited weak and moderate activities against Bacillus subtilis and Mycobacterium vaccae, respectively, compared to ciprofloxacin. Compounds 3, 5, and 10, which contain ortho-dihydroxyl groups in B-ring at positions 2′ and 3′, were most active against bacterial strains. This supports the evidence that the ortho -dihydroxyl structural fragment in B-ring is important for antimicrobial activity [22]. Compound 2, a new cycloflavan, demonstrated activity against B. subtilis, E. faecalis, and M. vaccae. Interestingly, the new compounds 1 and 9, which have a glucose unit in their structure, showed selective activity only against E. faecalis VRE.
Table 3.
Antimicrobial activity using agar diffusion method of some isolated compounds from I. tenuifolia.
As for the fungi, the compound 5 showed moderate activity against Penicillium notatum, and compounds 5 and 10 showed weak activity against Candida albicans. It is noteworthy that compound 5, which contains a methylenedioxy group, in addition to the ortho-dihydroxyl groups in B-ring, showed broad activity against eight microorganisms out of eleven. However, no inhibition was observed with the tested compounds against bacterial strains of Escherichia coli and Pseudomonas aeruginosa (SG137 B7) or against the fungus Sporobolomyces Salmonicolor. This is the first report on the antimicrobial activity of the tested compounds.
2.3. Antiproliferative and Cytotoxic Activities
Using HUVEC, K-562, THP-1, A549, and HeLa cell lines, the antiproliferative activities and the cytotoxicity of the isolated compounds (1–12) along with plant raw extract were evaluated in vitro (Table 4). The plant extract possessed antiproliferative effects against leukemia cell lines and a cytotoxic effect on Hela cells. With the exception of 12, the compounds tested in this study were chromane derivatives. Compounds 4–6 and 10 showed inhibition effects against all of the applied cancer cell lines and exhibited potential antiproliferative activities against the THP-1 cell line with GI50 values at 16.0, 16.5, 16.9, and 9.1 µM, respectively. Moreover, compound 10 demonstrated the most potent activity (GI50 = 7.6 µM) against the K-562, followed by compounds 2 and 3 with GI50 value at 31.5 and 32.3 µM. Compound 3, the tetrahydroxy flavanone, showed selective antiproliferative inhibition effect against the K-562 cells only. These results are in a good agreement with previous studies and supports the evidence that the ortho-dihydroxyl structural fragment in B-ring is very important for anticancer activity [7,23,24,25]. Furthermore, compound 2, the cycloflavan, exhibited significant inhibitory activities against cell lines HUVEC and THP-1 with a GI50 values at 35.2 and 29.2 µM and showed the cytotoxic effect on HeLa cells with CC50 value at 42.6 µM among the tested compounds. Besides that, compound 10 showed a similar moderate inhibition effect (GI50 = 35.8 µM) on HUVEC cells. All the flavonoids examined showed their low toxicity with CC50 values of more than 100 μM on HeLa cells. In cases of compounds 7 and 8 which do not have ortho-dihydroxy substitutions on the A and B rings, reduced or no inhibitory effects were found compared to other compounds mentioned above. Compound 11 showed slightly increased activity against K-562 and moreover against THP-1 and A549 cell lines compared to compound 7. Thus, it supports the relevance of the C-2 and C-3 double bond in a flavonoid structure [23]. In contrast to 10, compound 9, which differs in its structure in the presence of a sugar moiety and a dehydroxylation on the B-ring, became completely not effective. Interestingly, compound 1, which also has a sugar residue in its structure, did not show any activity up to 100 µM against all human cancer cell lines either. Hence, the results are consistent with previous research that flavonoids glycosides were generally not effective against multiple cancer cell lines [23]. Compound 12 showed neither an antiproliferative effect nor a cytotoxicity within the tested range against all applied cell lines.
Table 4.
Antiproliferative and cytotoxic activity of the plant extract and isolated compounds from I. tenuifolia.
3. Materials and Methods
3.1. General Experimental Procedures
Solvents and reagents were purchased from Sigma-Aldrich, Deisenhofen, Germany and Qingdao Marine Chemical, China. The optical data were measured using a digital JASCO P-2000 polarimeter (Jasco, Pfungstadt, Germany). The CD spectrum was recorded on a JASCO J810 spectropolarimeter (Jasco, Pfungstadt, Germany). Ultraviolet-visible (UV-Vis) data were extracted from diode array detector (DAD) data obtained during high performance liquid chromatography-electrospray ionization-high resolution mass spectrometry (HPLC-ESI-HRMS) experiments.
NMR spectra were recorded at 298 K on 500 MHz Bruker Avance III HD spectrometer (Bruker Biospin, Rheinstetten, Germany), equipped with cryoplatforms and TCI cryoprobes (5 mm). Spectrometer control and data processing were accomplished using Bruker Topspin ver.3.2 (Bruker Biospin, Rheinstetten, Germany), and standard pulse programs were used. NMR signals were referenced to the respective solvent signals at δH 2.50 and δC 39.53 for hexadeuterodimethyl sulfoxide DMSO-d6 and δH 7.26 and δC 77.06 for deuterochloroform CDCl3. HPLC-ESI-HRMS spectra were recorded on an Agilent Infinity 1260, consisting of a combined degasser and quaternary pump, column oven, autosampler, and DAD. The DAD was coupled to a Bruker Compact quadrupole time-of-flight (QTOF) mass spectrometer (Bruker Daltonics, Bremen, Germany). Both devices were controlled by Bruker Compass ver.1.9 (Bruker Daltonics, Bremen, Germany). For HPLC separation, an Agilent Zorbax C-18 SB column (3.5 µm, 4.6 × 150 mm i.d.) was used. The mass spectrometer was operated, depending on the analyte, either in positive or negative ionization mode, employing an electrospray ionization (ESI) source. The standard settings for small molecule analysis, as provided with Bruker Compass, were used. Column chromatographic separations were performed on silica gel (200–300 mesh, Merck, Darmstadt, Germany and Qingdao Marine Chemical, China) and Sephadex LH-20 (Pharmacia Fine Chemical, Uppsala, Sweden). Thin-layer chromatography (TLC) was performed on pre-coated TLC plates with silica gel 60 F254 (Merck, Darmstadt, Germany). Spots were detected under UV absorption (λmax 254 and 364 nm) by spraying with 1% methanolic diphenylboric acid-β-ethylaminoester, 5% ethanolic polyethylene glycol or under visible light by spraying with 5% ethanolic sulfuric acid and 1% acidified methanolic vanillin.
3.2. Plant Material
Rhizomes and roots of I. tenuifolia were collected in September 2016 from Khurmen Sum of South Gobi province of Mongolia. It was identified by Urgamal Magsar, a botanist of the Institute of General and Experimental Biology, Mongolian Academy of Sciences, where voucher specimens (It 0916) of the plant have been deposited.
3.3. Extraction and Isolation
The air dried and powdered plant material (5.0 kg) was extracted three times with 95% ethanol (crude extract I) and three more times with 50% ethanol (crude extract II). The extracts were evaporated under vacuum to yield a brown residue. Crude extract I (700 g) was fractionated by column chromatography on a silica gel column, eluted with dichloromethane (CH2Cl2) and mixtures of CH2Cl2 and methanol (CH2Cl2-MeOH) (50:1, 30:1, 10:1, 5:1, 1:1, v/v) with increasing polarity. Eluates were pooled into seven fractions (A–G) on the basis of TLC analysis. Fractions A and B were subjected to column chromatography on silica gel eluted with a chloroform (CHCl3) and the mixture of CHCl3-MeOH (70:1, 50:1, 30:1, 10:1, 5:1, 1:1, v/v) with increasing polarity to give subfractions A.1–A.7 and B.1–B.6, respectively. Subfraction A.2 was further purified on silica gel column eluting with petroleum ether and ethyl acetate (pet.ether-EtOAc) (50:1, 30:1, 10:1, 5:1, 1:1, v/v) followed by Sephadex LH-20 eluting with CHCl3-MeOH (1:1, v/v) to yield compounds 4 (20.4 mg), 7 (39.9 mg), 8 (328.8 mg), 10 (85.9 mg), 11 (29.3 mg), and 12 (151.8 mg). Further subfraction B.5 was subjected to silica gel column chromatography eluting with CHCl3-MeOH (70:1, 50:1, 30:1, 10:1, 5:1, 3:1, 1:1, v/v) followed by Sephadex LH-20 with CHCl3-MeOH (1:1, v/v) to yield compounds 3 (105.6 mg), 5 (88.2 mg), and 6 (19.4 mg). These compounds were recrystallized from CHCl3-MeOH (1:1, v/v). Fraction D was separated on silica gel column chromatography using CHCl3-MeOH (50:1, 30:1, 10:1, 5:1, 1:1, v/v) as an eluent to give eight subfractions (D.1–D.8). Compound 9 (11.5 mg) was obtained in pure form by recrystallisation from CHCl3-MeOH (1:1 v/v) from subfraction D.4. Subfraction D.5 was further subjected on silica gel eluting with CHCl3-MeOH (10:1, v/v) and then purified on silica gel with pet.ether-EtOAc (5:1, 3:1, 1:1, 1:3, 1:5, v/v) to afford compound 1 (6.9 mg). Crude extract II (52 g) was fractionated by column chromatography on silica gel eluted with CHCl3 and CHCl3-MeOH (CHCl3, 50:1, 30:1, 10:1, 5:1, 3:1, 1:1, MeOH, v/v) and pooled into eight fractions (A–H). Fraction A was subjected to silica gel column eluted with a gradient of pet.ether-EtOAc (50:1, 30:1, 10:1, 5:1, 1:1, v/v) to yield compound 2 (11.0 mg) and which was further recrystallized from CHCl3-MeOH (1:1, v/v).
Moniristenulide (1): white powder, [α]D25 + 44 (c = 0.18, DMSO), CD (c = 0.22, MeOH) 305 nm (Δε + 1.25) and 287 nm (Δε − 3.96), UV (CH3CN/H2O): λmax 206, 228, 282, 326 nm; 1H NMR (500 MHz, DMSO-d6) and 13C NMR (125 MHz, DMSO-d6) data, see Table 1; HR-ESI-MS: m/z 453.1409 [M + H]+ (calcd. for C21H25O11, 453.1397).
5-Methoxy-6,7-methylenedioxy-4-O-2′-cycloflavan (2): colorless crystal, [α]D25 + 317 (c = 0.67, DMSO), UV (CH3CN/H2O): λmax 196, 206, 286 nm; 1H NMR (500 MHz, CDCl3) and 13C NMR (125 MHz, CDCl3) data, see Table 2; HR-ESI-MS: m/z 299.0919 [M + H]+ (calcd. for C17H15O5, 299.0920).
(2S)-5,7,2′,3′-Tetrahydroxyflavanone (3): yellow powder; [α]D25 + 10 (c = 0.67, DMSO), CD (c = 0.69, MeOH) 325 nm (Δε + 2.40) and 283 nm (Δε − 11.70), UV (CH3CN/H2O): λmax 202, 288 nm; 1H NMR (500 MHz, DMSO-d6) and 13C NMR (125 MHz, DMSO-d6) data, see Table 2; HR-ESI-MS: m/z 289.0714 [M + H]+ (calcd. for C15H13O6, 289.0712).
5,2′-Dihydroxy-6,7-methylenedioxyflavanone (4): yellow powder, UV (CH3CN/H2O): λmax 206, 228, 280 nm; 1H NMR (500 MHz, DMSO-d6) δH 11.86 (1H, s, 5-OH), 9.89 (1H, s, 2′-OH), 7.43 (1H, d, J = 7.5 Hz, H-6′), 7.19 (1H, t, J = 7.5 Hz, H-4′), 6.88 (1H, overlap, H-5′), 6.86 (1H, overlap, H-3′), 6.31 (1H, s, H-8), 6.06 (2H, d, J = 6.9 Hz, -CH2-), 5.72 (1H, dd, J = 2.8, 13.1 Hz, H-2), 3.26 (1H, dd, J = 13.4, 17.1 Hz, H-3a), 2.74 (1H, dd, J = 2.9, 17.1 Hz, H-3b). 13C NMR (125 MHz, DMSO-d6) δC 74.6 (C-2), 41.2 (C-3), 197.9 (C-4), 143.1 (C-5), 127.4 (C-6), 155.8 (C-7), 90.4 (C-8), 159.4 (C-9), 103.6 (C-10), 124.5 (C-1′), 154.4 (C-2′), 115.6 (C-3′), 129.6 (C-4′), 119.2 (C-5′), 127.1 (C-6′), 102.5 (-CH2-). HR-ESI-MS: m/z 301.0722 [M + H]+ (calcd. for C16H13O6, 301.0712).
5,2′,3′-Trihydroxy-6,7-methylenedioxyflavanone (5): yellow crystal, [α]D25 + 9 (c = 0.8, DMSO), UV (CH3CN/H2O): λmax 206, 244, 284 nm; 1H NMR (500 MHz, DMSO-d6) δH 11.88 (1H, s, 5-OH), 9.52 (1H, s, 3′-OH), 8.74 (1H, s, 2′-OH), 6.90 (1H, d, J = 7.8 Hz, H-6′), 6.80 (1H, dd, J = 1.2, 7.8 Hz, H-4′), 6.69 (1H, t, J = 7.8 Hz, H-5′), 6.30 (1H, s, H-8), 6.07 (2H, d, J = 6.6 Hz, -CH2-), 5.73 (1H, dd, J = 2.8, 13.2 Hz, H-2), 3.24 (1H, dd, J = 13.3, 17.2 Hz, H-3a), 2.73 (1H, dd, J = 2.9, 17.2 Hz, H-3b). 13C NMR (125 MHz, DMSO-d6) δC 74.6 (C-2), 41.2 (C-3), 197.9 (C-4), 143.1 (C-5), 127.3 (C-6), 155.7 (C-7), 90.4 (C-8), 159.4 (C-9), 103.6 (C-10), 125.2 (C-1′), 142.6 (C-2′), 145.2 (C-3′), 115.3 (C-4′), 119.0 (C-5′), 117.1 (C-6′), 102.4 (-CH2-). HR-ESI-MS: m/z 317.0666 [M + H]+ (calcd. for C16H13O7, 317.0661).
5,2′,3′-Trihydroxy-7-methoxyflavanone (6): yellow crystal; UV (CH3CN/H2O): λmax 206, 226, 286 nm; 1H NMR (500 MHz, DMSO-d6) δH 12.09 (1H, s, 5-OH), 9.56 (1H, s, 3′-OH), 8.74 (1H, s, 2′-OH), 6.89 (1H, d, J = 7.7 Hz, H-6′), 6.79 (1H, d, J = 7.7 Hz, H-4′), 6.69 (1H, t, J = 7.8 Hz, H-5′), 6.11 (1H, d, J = 2.0 Hz, H-8), 6.08 (1H, d, J = 2.0 Hz, H-6), 5.73 (1H, dd, J = 2.8, 12.9 Hz, H-2), 3.79 (3H, s, 7-OCH3), 3.23 (1H, dd, J = 12.9, 17.2 Hz, H-3a), 2.73 (1H, dd, J = 3.0, 17.2 Hz, H-3b). 13C NMR (125 MHz, DMSO- d6) δC 74.2 (C-2), 41.2 (C-3), 196.9 (C-4), 163.2 (C-5), 94.7 (C-6), 167.4 (C-7), 93.8 (C-8), 159.4 (C-9), 102.6 (C-10), 125.4 (C-1′), 143.1 (C-2′), 145.2 (C-3′), 115.2 (C-4′), 119.0 (C-5′), 117.1 (C-6′), 55.9 (7-OCH3). HR-ESI-MS: m/z 303.0877 [M + H]+ (calcd. for C16H15O6, 303.0868).
3,5,3′-Trihydroxy-7,2′-dimethoxyflavanone (7): white powder; UV (CH3CN/H2O): λmax 206, 226, 282 nm; 1H NMR (500 MHz, DMSO- d6) δH 11.88 (1H, s, 5-OH), 9.49 (1H, s, 3′-OH), 6.99 (1H, overlap, H-6′), 6.97 (1H, overlap, H-5′), 6.90 (1H, dd, J = 3.1, 6.3 Hz, H-4′), 6.13 (1H, d, J = 2.2 Hz, H-6), 6.08 (1H, d, J = 2.2 Hz, H-8), 5.94 (1H, d, J = 6.1 Hz, 3-OH), 5.46 (1H, d, J = 11.6 Hz, H-2), 4.77 (1H, dd, J = 6.1, 11.5 Hz, H-3), 3.78 (3H, s, 7-OCH3), 3.74 (3H, s, 2′-OCH3). 13C NMR (125 MHz, DMSO- d6) δC 77.7 (C-2), 70.7 (C-3), 198.4 (C-4), 163.1 (C-5), 94.9 (C-6), 167.6 (C-7), 93.8 (C-8), 162.5 (C-9), 101.4 (C-10), 130.4 (C-1′), 146.7 (C-2′), 150.2 (C-3′), 117.2 (C-4′), 123.9 (C-5′), 118.6 (C-6′), 60.5 (7-OCH3), 55.9 (2′-OCH3). HR-ESI-MS: m/z 333.0982 [M + H]+ (calcd. for C17H17O7, 333.0974).
5,7-Dihydroxy-6,2′-dimethoxyisoflavone (8): yellow crystal, UV (CH3CN/H2O): λmax 196, 226, 262 nm; 1H NMR (500 MHz, DMSO-d6) δH 12.94 (1H, s, 5-OH), 10.78 (1H, s, 7-OH), 8.24 (1H, s, H-2), 7.40 (1H, dd, J = 1.4, 7.5 Hz, H-4′), 7.24 (1H, dd, J = 1.4, 7.5 Hz, H-6′), 7.09 (1H, d, J = 8.3 Hz, H-3′), 7.00 (1H, t, J = 7.5 Hz, H-5′), 6.52 (1H, s, H-8), 3.75 (3H, s, 6-OCH3), 3.73 (3H, s, 2′-OCH3). 13C NMR (125 MHz, DMSO-d6) δC 155.3 (C-2), 119.8 (C-3), 180.1 (C-4), 152.8 (C-5), 131.5 (C-6), 157.5 (C-7), 94.0 (C-8), 153.1 (C-9), 104.7 (C-10), 120.2 (C-1′), 157.5 (C-2′), 111.3 (C-3′), 129.9 (C-4′), 120.0 (C-5′), 131.6 (C-6′), 59.9 (6-OCH3), 55.6 (7-OCH3). HR-ESI-MS: m/z 315.0888 [M + H]+ (calcd. for C17H15O6, 315.0868).
5-Hydroxy-6,7-dimethoxyisoflavone-2′-O-β-d-glucopyranoside (9): yellow crystal, UV (CH3CN/H2O): λmax 206, 216, 262, 336 nm; 1H NMR (500 MHz, DMSO-d6) and 13C NMR (125 MHz, DMSO-d6) data, see Table 2; HR-ESI-MS: m/z 477.1399 [M + H]+ (calcd. for C23H25O11, 477.1397)
5,2′,3′-Trihydroxy-6,7-dimethoxyisoflavone (10): yellow powder, UV (CH3CN/H2O): λmax 206, 222, 258, 338 nm; 1H NMR (500 MHz, CDCl3) and 13C NMR (125 MHz, CDCl3) data, see Table 2; HR-ESI-MS: m/z 331.0825 [M + H]+ (calcd. for C17H15O7, 331.0818).
3,5,3′-Trihydroxy-7,2′-dimethoxyflavonol (Irisflavone D) (11): yellow amorphous powder, UV (CH3CN/H2O): λmax 198, 256, 302, 344 nm; 1H NMR (500 MHz, DMSO-d6) δH 12.48 (1H, s, 5-OH), 9.70 (1H, s, 3′-OH), 9.14 (1H, s, 3-OH), 7.03 (1H, overlap, H-5′), 7.02 (1H, overlap, H-4′), 6.93 (1H, t, J = 4.8 Hz, H-6′), 6.61 (1H, d, J = 2.1 Hz, H-8), 6.38 (1H, d, J = 2.1 Hz, H-6), 3.84 (3H, s, 7-OCH3), 3.77 (3H, s, 2′-OCH3). 13C NMR (125 MHz, DMSO- d6) δC: 148.3 (C-2), 137.3 (C-3), 176.5 (C-4), 160.7 (C-5), 97.5 (C-6), 165.0 (C-7), 92.0 (C-8), 156.7 (C-9), 104.6 (C-10), 124.9 (C-1′), 145.9 (C-2′), 150.5 (C-3′), 123.7 (C-4′), 118.8 (C-5′), 120.9 (C-6′), 60.3 (2′-OCH3), 56.1 (7-OCH3). HR-ESI-MS: m/z 331.0835 [M + H]+ (calcd. for C17H15O7, 331.0817).
β-Sitosterol (12): white powder, 1H NMR (500 MHz, CDCl3) δH 5.35 (1H, d, J = 5.3 Hz, H-5), 3.52 (1H, m, H-3), 1.01 (3H, s, H-29), 0.92 (3H, d, J = 6.62 Hz, H-19), 0.84 (3H, t, J = 7.5 Hz, H-24), 0.82 (3H, d, J = 1.8 Hz, H-26), 0.80 (3H, d, J = 7.0 Hz, H-27), 0.68 (3H, s, H-28). 13C NMR (CDCl3, 125 MHz) δC 37.4 (C-1), 31.8 (C-2), 71.9 (C-3), 42.5 (C-4), 140.9 (C-5), 121.9 (C-8), 29.8 (C-7), 32.0 (C-8), 50.3 (C-9), 36.6 (C-10), 21.2 (C-11), 39.9 (C-12), 42.4 (C-13), 56.9 (C-14), 25.5 (C-15), 28.4 (C-16), 56.2 (C-17), 36.3 (C-18), 19.2 (C-19), 34.1 (C-20), 26.2 (C-21), 45.9 (C-22), 23.2 (C-23), 12.1 (C-24), 29.3 (C-25), 19.9 (C-26), 19.5 (C-27), 18.9 (C-28), 12.0 (C-29).
3.4. Antimicrobial Activity Assay
Compounds 1, 2, 3, 5, 9, and 10 were tested for their antimicrobial activities against S. aureus (JMRC:STI 10760), B. subtilis (JMRC:STI 10880), S. aureus MRSA (JMRC: ST 33793) E. faecalis VRE (JMRC: ST 33700), E. coli (JMRC:ST 33699), P. aeruginosa (JMRC:ST 33771), P. aeruginosa (JMRC:ST 33772), M. vaccae (JMRC:STI 10670), S. salmonicolor (JMRC:ST 35974), C. albicans (JMRC:STI 25000), and P. notatum (JMRC:STI 50164)) using agar diffusion assay as previously published [26]. Strains were obtained from the Jena Microbial Resource Collection (JMRC). The bacteria were cultivated on standard I nutrient agar in Petri dishes at 37 °C. Antifungal bioassays were conducted at 30 °C using the basidiomycetous yeast S. salmonicolor and the filamentous ascomycete P. notatum, which were cultivated on malt agar, and the ascomycetous yeast C. albicans, which was cultivated on yeast morphology agar. After inoculation of the test organisms, a disc (9 mm in diameter) was removed from the center of the Petri dish and 50 μL of the test solution (1 mg/mL in DMSO) was added to the cavity. After 18 h of incubation, the inhibiting areola were measured and documented as diameters in mm. Ciprofloxacin (5 µg/mL in deionized water) and amphotericin B (10 µg/mL in DMSO/MeOH 1:1) were used as reference substances against bacterial and fungal strains, respectively.
3.5. Antiproliferation and Cytotoxicity Assays
Compounds (1–12) were assayed against human umbilical vein endothelial cells (HUVEC), human chronic myeloid leukemia cells (K-562), human acute monocytic leukemia cells (THP-1), and human lung carcinoma cells (A549) for their antiproliferative effects and against human cervix carcinoma cells (HeLa) for their cytotoxic effect. The antiproliperative and cytotoxic effects were tested via CellTiter-Blue and methylene blue assay as previously described [27]. In this assay, K-562 (DSM ACC 10), THP-1 (DSM ACC 16), and HeLa (DSM ACC 57) were maintained in Roswell Park Memorial Institute (RPMI) 1640 medium (Cambrex 12-167F) while HUVEC (ATCC CRL-1730) and A549 (DSM ACC 107) were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) (Cambrex 12-614F). Cells that were grown in the appropriate cell culture medium were supplemented with 10 mL/L ultraglutamine 1 (Cambrex 17-605E/U1), 550 µL/L (50 mg/mL) gentamicin sulfate (Cambrex 17-518Z), and 10% heat inactivated fetal bovine serum (GIBCO Life Technologies 10270-106) at 37 °C. The tested compounds were dissolved in DMSO, and the cells were seeded in 96-well plates at a density of 1 × 104 cells/well. As for the antiproliferative effect of the compounds, the cells were incubated for 72 h, and GI50 values were evaluated to be defined as the concentration causing 50% inhibition of proliferation compared to the untreated control. With regard to the cytotoxic assay, HeLa cells were pre-incubated for 48 h without the test compounds. Then, the cells were exposed with different concentrations of compounds and incubated for 72 h. After that, the adherent HeLa cells were fixed by glutaraldehyde and stained with a 0.05% solutions of methylene blue (SERVA 29198) for 15 min. CC50 was evaluated to be defined as the concentration required for the death of 50% of the cell monolayer as compared to control groups. Under our experimental conditions, the optical density measured from the CellTiter-Blue reagent and methylene blue assay is proportional to the number of viable cells. In this experiment, absorbances were measured at 570 nm against the reference wavelength of 600 nm (CellTiter-Blue assay) and at 660 nm (methylene blue assay). Doxorubicin (Adriamycin®) and imatinib (Gleevec®) were used as positive controls for HUVEC, K-562, and HeLa cells. A repeat determination has been conducted in all experiments, and four replicates were assayed. The calculations of the different values of GI50 and CC50 were performed with software Magellan version 3.00 (Tecan Trading AG, Maennedorf, Switzerland).
4. Conclusions
The chemical investigation of the underground parts of I. tenuifolia afforded in the isolation of five unprecedented chromane derivatives (1–3, 9, 10) includes an unusual macrolide termed moniristenulide (1), together with seven known compounds (4–8, 11–12). Notably, eight out of nine isolated flavonoids have a rare 2′,3′-disubstituted configuration on the B-ring, out of which the compounds bearing ortho-dihydroxyl groups in B-ring, namely 3, 5, and 10, showed the broadest antimicrobial activity. On top of that, the molecules with methoxy or methylenedioxy substitution on the A-ring together with ortho-hydroxyl groups on the B-ring showed promising antiproliferative activities against leukemia cell lines in combination with low cytotoxicity, as shown for compounds 4–6 and 10.
Supplementary Materials
The following are available online, Figures S1–S10: 1D-, 2D-NMR, HR-ESI-MS, UV, CD, and HR-MS spectra of compound 1, Figures S11–S17: 1D-, 2D-NMR, HR-ESI-MS, and UV spectra of compound 2, Figures S18–S25: 1D-, 2D-NMR, HR-ESI-MS, UV, and CD spectra of compound 3, Figures S26–S32: 1D-, 2D-NMR, HR-ESI-MS, and UV spectra of compound 9, Figures S33–S39: 1D-, 2D-NMR, HR-ESI-MS, and UV spectra of compound 10.
Author Contributions
W.B. and E.D. designed the project and critically revised the manuscript. E.D., O.O. and S.N. performed the isolation, and structure elucidation. E.D., W.B., O.O. and S.N. drafted the manuscript. K.V. and H.-M.D., carried out the biological activity assays and statistical analysis. E.D., S.N., S.B., O.O. and C.P. recorded and did interpretation of the spectra; E.D., O.O., S.N., K.V., H.-M.D., S.B., C.P. and W.B. reviewed and edited the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Acknowledgments
O.O. is grateful for a Chinese Government Scholarship (CSC No. 2013DFH620) and to Jianqin Jiang, China Pharmaceutical University, Nanjing for supporting the isolation of some compounds. Furthermore, the authors are grateful to Khurelbaatar Luvsan, Mongolian University of Pharmaceutical Sciences, for providing the plant material. We express our gratitude to Christiane Weigel for technical assistance in the measurements of antimicrobial activities.
Conflicts of Interest
The authors declared no conflict of interest.
Sample Availability
Samples of the compounds 3–12 are available from the authors.
References
- Iwashina, T.; Mizuno, T. Flavonoids and xanthones from the genus Iris: Phytochemistry, relationships with flower colors and taxonomy, and activities and function. Nat. Prod. Commun. 2020, 15, 1–35. [Google Scholar] [CrossRef]
- Choudhary, M.I.; Hareem, S.; Siddiqui, H.; Anjum, S.; Ali, S.; Atta-ur-Rahman; Zaidi, M.I. A benzil and isoflavone from Iris tenuifolia. Phytochemistry 2008, 69, 1880–1885. [Google Scholar] [CrossRef]
- Ibrahim, S.R.M.; Mohamed, G.A.; Al-Musayeib, N.M. New constituents from the rhizomes of Egyptian Iris germanica L. Molecules 2012, 17, 2587–2598. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Cui, Y.; Zhao, C. Flavonoids of the genus Iris (Iridaceae). Mini Rev. Med. Chem. 2010, 10, 643–661. [Google Scholar] [CrossRef]
- Singab, A.N.B.; Ayoub, I.M.; El-Shazly, M.; Korinek, M.; Wu, T.Y.; Cheng, Y.-B.; Chang, F.R.; Wu, Y.C. Shedding the light on Iridaceae: Ethnobotany, phytochemistry and biological activity. Ind. Crop. Prod. 2016, 92, 308–335. [Google Scholar] [CrossRef]
- Alam, A. Pharmacology and phytochemistry of isoflavonoids from Iris species. J. Pharmacol. Clin. Res. 2017, 3, 1–6. [Google Scholar] [CrossRef]
- Cui, Y.M.; Wang, H.; Liu, Q.R.; Han, M.; Lu, Y.; Zhao, C.Q. Flavans from Iris tenuifolia and their effects on β-amyloid aggregation and neural stem cells proliferation in vitro. Bioorg. Med. Chem. Lett. 2011, 21, 4400–4403. [Google Scholar] [CrossRef]
- Urgamal, M.; Oyuntsetseg, B.; Nyambayar, D.; Dulamsuren, C. Vascular plants in Biodiversity of Mongolia: A Checklist of Plants, Fungus and Microorganisms; Mongolica Publishing: Ulaanbaatar, Mongolia, 2019. [Google Scholar]
- Urgamal, M.; Gundegmaa, V.; Baasanmunkh, S.; Oyuntsetseg, B.; Darikhand, D.; Munkh-Erdene, T. Additions to the vascular flora of Mongolia—IV. Proc. Mong. Acad. Sci. 2019, 59, 41–53. [Google Scholar]
- Ligaa, U.; Davaasuren, B.; Ninjil, N. Medicinal Plants of Mongolia Used in Western and Eastern Medicine; JKC Printing: Ulaanbaatar, Mongolia, 2005. [Google Scholar]
- Slade, D.; Ferreira, D.; Marais, J.P.J. Circular dichroism, a powerful tool for the assessment of absolute configuration of flavonoids. Phytochemistry 2005, 66, 2177–2215. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Hao, Z.; Li, M. Isolation and structure identification of flavonoids. Flavonoids Biosynth. Hum. Health 2017, 2, 18–43. [Google Scholar]
- Peng, W.; Yang, C.; Zhan, R.; Chen, Y. Two new flavans from the trunk and leaves of Horsfieldia glabra. Nat. Prod. Res. 2016, 30, 2350–2355. [Google Scholar] [CrossRef]
- Yang, X.; Zhang, W.; Ying, X.; Stien, D. New flavonoids from Portulaca oleracea L. and their activities. Fitoterapia 2018, 127, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Kojima, K.; Gombosurengyin, P.; Ondognyi, P.; Begzsurengyin, D.; Zevgeegyin, O.; Hatano, K.; Ogihara, Y. Flavanones from Iris tenuifolia. Phytochemistry 1997, 44, 711–714. [Google Scholar] [CrossRef]
- Al-Saleem, M.S.M.; Al-Wahaib, L.H.; Abdel-Mageed, W.M.; Gouda, Y.G.; Sayed, H.M. Antioxidant flavonoids from Alhagi maurorum with hepatoprotective effect. Pharmacogn. Mag. 2019, 15, 592–599. [Google Scholar]
- Hanawa, F.; Tahara, S.; Mizutani, J. Isoflavonoids produced by Iris Pseudacorus leaves treated with cupric chloride. Phytochemistry 1991, 30, 157–163. [Google Scholar] [CrossRef]
- Bergeron, C.; Marston, A.; Hakizamungu, E.; Hostettmann, K. Antifungal constituents of Chenopodium procerum. Pharm. Biol. 1995, 33, 115–119. [Google Scholar] [CrossRef]
- Shawl, A.S.; Vishwapaul; Zaman, A.; Kalla, A.K. Isoflavones of Iris spuria. Phytochemistry 1984, 23, 2405–2406. [Google Scholar] [CrossRef]
- Choudhary, M.I.; Nur-e-Alam, M.; Baig, I.; Akhtar, F.; Khan, A.M.; Ndögnii, P.Ö.; Badarchiin, T.; Purevsuren, G.; Nahar, N.; Atta-ur-Rahman. Four new flavones and a new isoflavone from Iris bungei. J. Nat. Prod. 2001, 64, 857–860. [Google Scholar] [CrossRef]
- Vitus Nyigo, A.; Peter, X.; Abiki, F.M.; Alebo, H.M.M.; Degela, R.H.M.; Gerda, F. Isolation and identification of euphol and β-sitosterol from the dichloromethane extracts of Synadenium glaucescens. J. Phytopharm. 2016, 5, 100–104. [Google Scholar] [CrossRef]
- Farhadi, F.; Khameneh, B.; Iranshahi, M.; Iranshahy, M. Antibacterial activity of flavonoids and their structure–activity relationship: An update review. Phyther. Res. 2019, 33, 13–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawaii, S.; Tomono, Y.; Katase, E.; Ogawa, K.; Yano, M. Antiproliferative activity of flavonoids on several cancer cell lines. Biosci. Biotechnol. Biochem. 1999, 63, 896–899. [Google Scholar] [CrossRef]
- Kawaii, S.; Ishikawa, Y.; Yoshizawa, Y. Relationship between the structure of methoxylated and hydroxylated flavones and their antiproliferative activity in HL60 cells. Anticancer Res. 2018, 38, 5679–5684. [Google Scholar] [CrossRef] [PubMed]
- Grigalius, I.; Petrikaite, V. Relationship between antioxidant and anticancer activity of trihydroxyflavones. Molecules 2017, 22, 2169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krieg, R.; Jortzik, E.; Goetz, A.A.; Blandin, S.; Wittlin, S.; Elhabiri, M.; Rahbari, M.; Nuryyeva, S.; Voigt, K.; Dahse, H.M.; et al. Arylmethylamino steroids as antiparasitic agents. Nat. Commun. 2017, 8, 14478. [Google Scholar] [CrossRef] [PubMed]
- Krauth, F.; Dahse, H.M.; Rüttinger, H.H.; Frohberg, P. Synthesis and characterization of novel 1,2,4-triazine derivatives with antiproliferative activity. Bioorganic Med. Chem. 2010, 18, 1816–1821. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).